

Abstract
The MTA1 protein contributes to the process of cancer progression and metastasis through multiple genes and protein targets and interacting proteins with roles in transformation, anchorage-independent growth, invasion, survival, DNA-repair, angiogenesis, hormone-independence, metastasis and therapeutic resistance. MTA proteins control a spectrum of cancer promoting processes by modulating the expression of target genes and/or the activity of MTA-interacting proteins. In the case of MTA1, these functions are manifested through post-translational modifications of MTA1 in response to upstream signals, MTA1 interaction with binding proteins and the expression of target gene products. The MTA1 coregulator interacts with nucleosomes through modified histones and is an integrator of extracellular signaling and gene activator. Studies delineating the molecular basis of dual functionality of MTA1 reveal that the functions of MTA1-chromatin modifying complexes in the context of target gene regulation are dynamic in nature. The nature and targets of MTA1-chromatin modifying complexes are also governed by the dynamic plasticity of the nucleosome landscape as well as kinetics of activation and inactivation of enzymes responsible for post-translational modifications on the MTA1 protein. These broadly applicable functions also explain why MTA1 may be a ‘hub’ gene, whose current understanding is limited to selective influences on gene with roles in cancer but further research may reveal a more global influence. Because the deregulation of enzymes and their substrates with roles in MTA1-biology is not necessarily limited to cancer, we speculate that the lessons from MTA1 as a prototype dual master coregulator will be relevant for other human diseases. In this context, the concept of the dynamic nature of corepressor versus coactivator complexes and the MTA1 proteome as a function of time to signal is likely to be generally applicable to other multi-proteins regulatory complexes in living systems.
Keywords: MTA1, Chromatin remodeling, Gene expression, Cancer and metastasis, Signaling, Epigenetics, Therapeutic resistance
1 Introduction
Since the identification of mta1 as a metastasis relevant gene in 1994, MTA1 has emerged as one of the highly deregulated oncogenes in human cancer [1], and its elevated levels correlate well with tumor aggressiveness and unfavorable outcomes for cancer patients in general [2–7]. Because of the functional significance of MTA1 in cancer cells, there has been a general and growing scientific interest in the MTA family of proteins.
Although the MTA family of proteins shares many similar characteristics, the family exhibits many significant differences as well. MTA1, 2 and 3 proteins are encoded by genes with distinct chromosomal localization in humans, and two MTA1 isoforms (i.e., MTA1s, ZG29p) and one MTA3 isoform (i.e., MTA3L) (Fig. 1). In general, MTA1 and MTA2 exhibits a similar expression pattern while MTA3’s expression is somewhat opposing to MTA1. A comparative analysis of MTA proteins suggests a high homology in the N-terminal regions and divergent features in the C-terminal regions of MTA proteins. All MTA proteins with the exception of MTA1-ZG29p variant contains one of each of BAH-, ELM- and SANT- domains [2–7]. The structural functional relationships of these domains are elegantly reviewed by Millard et al in this issue. Although the MTA proteins also contain a GATA-like zinc-finger motif, the MTA proteins have so far not been shown to directly bind to DNA. Among the MTA proteins, only MTA1 contains a unique proline-rich region in its C-terminus, which might explain MTA1’s interactions with a range of signaling molecules. The MTA proteins have been shown to localize in the nucleus as well as other sub-cellular compartments [8–10].
Figure 1.
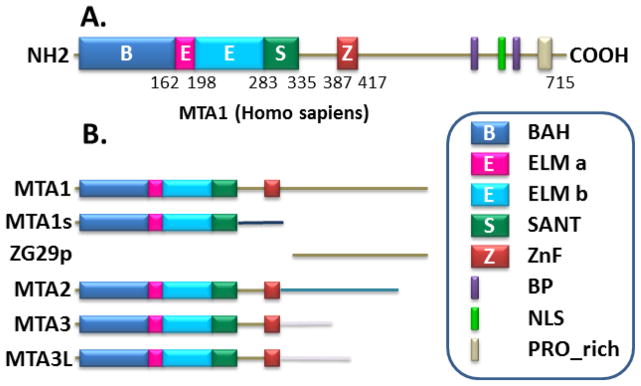
Schematic representation of structural domains of MTA proteins.
The MTA proteins modulate a spectrum of cancer promoting processes, including transformation, anchorage-independence, invasion, EMT, survival, DNA-damage response, angiogenesis, inflammation, metastasis and chemo-resistance. Mechanistically, these varied roles of MTA proteins in cancer cells are largely the functional outcome of the modulation of target genes’ expression and/or the activity of MTA-interacting proteins. For example, MTA1 upregulation favors the process of oncogenesis through stimulating the Ras, the Wnt1 and the STAT3 signaling pathways [11–14] when it acts as a downstream effector of cMyc-mediated transformation [15] and by antagonizing tumor suppressors, such as p53 [16–18]. Because the roles and clinical significance of MTA proteins in human cancer are discussed by other contributors in this issue, this review will focus on our current understanding of MTA1’s mechanisms and underlying principles of action behind biological effects of MTA1.
2 MTA1-chromatin remodeling complexes in breast cancer biology
Among the mechanisms of cancer progression and metastasis, the regulation of expression of underlying target genes by coordinated interplays of chromatin remodeling complexes in response to upstream signaling events occupy a central place [3, 7]. In general, chromatin remodeling complexes interact with proteins with the ATPase activity to remodel the chromatin in an energy-dependent manner. The functional output of chromatin remodeling complexes depends upon the nature of extracellular signals as well as enzymes, coactivators and corepressors in a given complex. One such group of chromatin remodeling complexes is the MTA-containing NuRD family of complexes.
Although the Mta1 was initially cloned as a metastasis-associated gene in 1994 [1], the nature of its biochemical activity remained unknown until MTA1 and MTA2 peptides were detected in the NuRD complexes [19, 20]. These observations provided the first clue about an undisclosed role of the MTA family of proteins in chromatin remodeling. Further studies suggested the various MTA proteins do not co-exist in the same NuRD complex and this separation might contribute to functional specialization of the NuRD complexes [21]. Among the MTA proteins, only MTA1 exhibits a dual coregulatory activity, as it acts both as a coactivator or corepressor, due to its engagement with distinct chromatin remodeling components in the context of target gene nucleosome landscape and upstream signaling.
The next phase of discovering MTA biology was driven by the identification of the first direct target of the MTA1-NuRD complex, the estrogen receptor-alpha (ERα) in breast cancer cells [8]. A large body of work has shown that MTA1 represses ERα-transactivation function and promotes the development of ER-negative phenotypes as well as resistance to anti-estrogens, such as tamoxifen [8, 22, 23]. Further, a number of MTA1-interacting proteins have been shown to impair the transcription of estrogen regulated genes through different mechanisms and, thereby, contribute to the development of hormone-independence of breast cancer cells (Fig. 2). For example, heat-shock factor 1 (HSF1) utilizes HSF1-MTA1/NuRD complex to inhibit the transcription of ER-responsive genes [24]; MTA1 interaction with ERα coactivators such as NRIF3 or MICoA inhibits transcription from ER-responsive genes and contributes to the development of hormone-independent phenotypes [25, 26]; and MTA1 interaction with MAT1–a component of the cyclin dependent kinase activating kinase (CAK) complex—impairs the transcription from ER- regulated genes, leading to hormone-independence [27].
Figure 2.

Regulatory cross-talk between the estrogen receptor alpha and MTA family members. A, Inhibition of ER-signaling by MTA1 and MTA1s through distinct mechanisms. B, E2-MTA3 pathway promotes EMT. C, Opposing expression patterns of MTA proteins in during cancer progression in a murine model. See text for details.
Similar to MTA1, the MTA2 also represses ERα transactivation and promotes ER-negative phenotypes [28, 29], while MTA3 exhibits a distinct set of functions as compared to MTA1 and MTA2 in breast cancer [30, 31]. Accordingly, the expression and subcellular localization of MTA1, MTA2 and MTA3 are differentially regulated in a murine model of breast cancer progression: expression of MTA1 and MTA2 gradually increases from the normal duct to MIN stage (premalignant lesions, hyperplasia and ductal carcinoma) to invasive carcinoma, while MTA3 expression decreases as a function of cancer progression [32]. While MTA1 and MTA2 repress ERα functions, the MTA3 is an estrogen-inducible gene itself and MTA3-NuRD complex inhibits Snail repression of E-cadherin [30], leading to epithelial-to-mesenchymal transition (EMT) and invasion of breast cancer cells and tumors (Fig. 2). In brief, all MTA proteins, namely MTA1, MTA2 and MTA3 as well as MTA1s (see below), modulate ERα activity and functions using different mechanisms, and, thus, are expected to influence breast cancer biology in a significant manner. Because of the paramount significance of ERα in the pathobiology and targeted therapeutic of breast cancer, the scientific community has increased its interest in the field of MTA proteins.
3 The MTA1s variant in breast cancer
The MTA1s is a naturally occurring spliced isoform of MTA1, generated by differential splicing at a cryptic splice site in exon 14 followed by a frame-shift, which adds 33 novel amino acids to the translated protein [33]. These 33 novel amino acids contain a nuclear receptor (NR) interacting motif, called LXXLL. As compared to the full-length MTA1, MTA1s predominantly resides in the cytoplasm and sequesters ERα in the cytoplasm. Thus, MTA1s redirects ERα signaling away from the nucleus to the cytoplasm [33]. The significance of MTA1s in the cytoplasm was revealed by its ability to stimulate non-genomic ERα signaling, such as stimulation of ERK-signaling [33]. MTA1s also interacts with receptor-tyrosine kinase signaling via the GRB2/SoS complex. Similar to the full-length MTA1, although MTA1s also promotes hormone-independent phenotypes, it does so through a different mechanism wherein it inhibits the nuclear translocation of activated ERα (Fig. 2). Further, MTA1s is an interacting substrate of casein kinase I-γ2, a cytoplasmic kinase downstream of estrogen signaling. CKI-γ2-mediated MTA1s phosphorylation on serine-321 potentiates the ability of MTA1s to redirect and inhibit ERα nuclear functions [34].
In addition to ERα-pathway, MTA1s also cooperate with the Wnt1 pathway by stimulating Wnt1 signaling via promoting ERK-dependent phosphorylation of GSK-3β and nuclear translocation of β-catenin and through stimulation of the Wnt1 target genes in breast cancer cells [12, 13]. Accordingly, MTA1s overexpression in a transgenic murine model stimulates the Wnt1 signaling in virgin mammary glands as well as phenotypic changes similar to that of Wnt1-transgenic mammary glands. In contrast, MTA1 deletion in knockout mice downregulates the Wnt1 signaling and leads to ductal hypobranching in mammary glands as compared to ductal branching in wild-type mice [13].
4 MTA1’s mechanism of action in cancer cells
The cancer relevant functions of MTA1 may be the result of one or more of the following mechanisms: 1) post-translational modifications of MTA1 in response to upstream signaling pathways; 2) MTA1 interactions with its binding proteins, including oncogenes; and 3) modulation of expression of a large number of target genes due to its dual coregulatory activity. These mechanisms are further influenced by dynamic changes in response to upstream signals—including growth factors, oncogenes and genotoxic signals—as well as by adaptive needs of cancer cells during oncogenesis and metastasis. Because Li et al have reviewed the role of MTA1 in the DNA damage response elsewhere in this issue, we have not summarized here the mechanisms of MTA1 protein stability by genotoxic stress.
4.1 Post-translational modifications of MTA1 protein
The functions of the MTA1 protein are largely regulated by its post-translational modifications (PTM), which in turn influence its ability to interact with its interacting proteins and contribute to the formation of functional chromatin remodeling complexes. For example, MTA1’s acetylation plays an important role in conferring its transforming activity; MTA1’s stability is governed by its ubiquitination; MTA1’s SUMOylation potentiates its corepressor activity; and MTA1’s methylation is required for its corepressor activity (Fig. 3).
Figure 3.
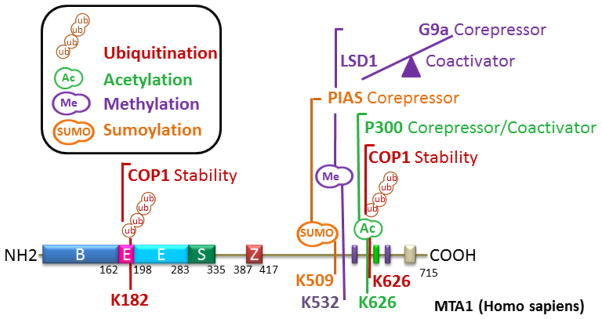
Current understanding of post-translational modifications of MTA1.
4.1.1 Acetylation
The MTA1 protein is acetylated by histone acetyl-transferase (HAT) p300 on lysine 626, a PTM signature important for its ability to transform the Rat1 fibroblasts, as acetylation-inactive MTA1 mutant lacks oncogenic activity [11]. One of the mechanisms of MTA1-mediated transformation include stimulation of the Ras-Raf pathway via reciprocal transcriptional repression of Gαi2 (GTP binding protein alpha subunit), a negative regulator of Ras. The acetylated MTA1 allows its interaction with HDAC – presumably the acetylated substrate serves as a docking platform for HDAC – to form the MTA1-HDAC/NuRD complex to repress Gαi2-transcription. Interestingly, there are also situations wherein MTA1 acetylation on lysine 626 is also required for its coactivator activity to stimulate the transcription of Breast Cancer Amplified Sequence 3 (Bcas3) in ERα positive breast cancer cells [35]. However, the underlying basis of the requirement of MTA1-lysine 626 acetylation for both corepressor and coactivator activity in the context of distinct target genes remains unknown. These findings suggest that acetylated MTA1 may have a broader significance in ER-positive breast cancer. Similar to MTA1, the MTA2 also undergoes acetylation on lysine 152 by p300 HAT [36]. The acetylation of MTA2 impacts its tumorigenic potential, as an acetylation inactive MTA2 mutant inhibits the growth of colorectal cancer cells. These observations suggest that the acetylation of MTA proteins and not merely its overexpression may be important for the action of MTA1 in cancer cells and that it might be possible to target MTA-acetylation for clinical development.
4.1.2 Ubiquitination
The aberrant MTA1 expression in human cancer also raises questions regarding the potential stability of MTA1 by the endogenous proteosomal system in cancer. Indeed, MTA1 undergoes ubiquitination and degradation via ubiquitin-proteasome pathway. MTA1 is ubiquitinated by the RING finger E3 ubiquitin ligase COP1 (constitutive photomorphogenesis protein1) on lysine 626 and lysine 182. COP1 degradation by ionizing radiation or experimentally by siRNA consistently results in enhanced MTA1 stability [37]. Interestingly, MTA1 also exerts a negative feedback on the COP1 by promoting its autoubiquitination and degradation. It has been proposed that ionizing radiation leads to DNA damage by acting as a circuit breaker for this feedback loop by releasing MTA1 from the MTA1-COP1 complex through stimulating ATM-phosphorylation dependent COP1 degradation [37]. Because MTA1 is essential for the ATM-mediated DNA damage response, targeting MTA1 is likely to increase the cellular sensitivity to ionizing-induced cell death.
4.1.3 SUMOylation
SUMOylation is another post-translational mark important for MTA1 activity. MTA1 is a substrate of the small ubiquitin-related modifier 2/3 (SUMO2/3) and its SUMOylation is promoted by PIAS (Protein inhibitor of activated STAT) protein [38]. MTA1 contains a specific SUMO interacting motif (SIM) for the PIAS-mediated conjugation of SUMO moiety on lysine 509 located within the SUMO consensus in the SIM region. As expected, MTA1’s SUMOylation increases its corepressor activity by supporting the recruitment of HDAC2 to the target promoters. Additionally, MTA1 acts as a coactivator of SUMO2 transcription. The significance of MTA1 in cancer biology is also evident by its ability to interact with SUMOlyated HIC1 (hypermethylated in cancer 1, a tumor suppressor gene) to promote the tumor-suppressor activity of HIC [39]. These observations suggest that SUMOylation of MTA1 may serve as an endogenous regulator of MTA1 corepressive activity.
4.1.4 Methylation
The MTA1 is methylated by G9a methyltransferase at lysine 532, and methylation helps in the orchestration of a functional MTA1-NuRD repressor complex. In contrast, MTA1’s lysine 532 demethylation by LSD1 demethylase drives its interaction with bivalent histone H3K4-AcK9 marks and facilitates in the formation of a MTA1-containing NURF-coactivator complex [40]. Therefore, MTA1’s lysine 532 methylation and demethylation modifications are believed to together be a molecular switch of MTA1’s dual activity to act as a corepressor or coactivator. These observations suggest that MTA1’s alliance with the components of the NuRD complex is dynamic and not, as previously thought, static. The unique methylation status of MTA1 modulated by cognitive actions of methyltransferase and demethylase enzymes is expected to play a key role in chromosomal remodeling and the fate of transcription.
4.2 MTA1-interacting proteins
The C-terminal region of MTA1 protein contains a number of structural features, including a proline-rich Src-homology 3 domain-interacting region, allowing it to interact with a variety of molecules with established roles in cancer relevant processes. Accordingly, MTA1 interacts with a large number of regulatory proteins, as illustrated by an early MTA1-proteome analysis by the NURSA organization [41]. Owing to the ability of MTA1 upon a spectrum of cellular processes important for oncogenesis, metastasis or both, MTA1 is expected to regulate a given cancer-modulating process through interacting with multiple interacting proteins in the context of cell-types, signals and the dynamic nature of chromatin (fig. 4). Due to a large number of MTA1-interacting proteins in cancer cells, we chose to briefly review only a few proteins as examples of MTA1-interacting proteins with roles in estrogen signaling, DNA repair, angiogenesis and metastasis.
Figure 4.

MTA1 contributes to the process of cancer progression and metastasis through multiple genes and protein targets and interacting proteins. Representative examples of cancer relevant functions modulated by MTA1 through multiple gene or protein targets.
MTA1 modulates ERα-transactivation activity and hormone-independence of breast cancer cells by interacting with HSF1, NRIF3, MICoA, MAT1, LMO4, or HDACs following distinct mechanisms (Fig. 4). Similarly, MTA1 contributes to DNA damage response signaling and DNA-repair through multiple mechanisms involving interactions with p53, COP1, ATR, or PARP etc. (see Li et al in this issue).
One of the key cancer promoting activities of MTA1 is its strong association with angiogenesis in human cancer [42–46]. The underlying basis of MTA1’s role in angiogenesis includes its upregulation under hypoxic conditions in hypoxia-inducible factor-1α (HIF1α) - a key modifier of angiogenesis. MTA1 also stabilizes HIF1α by promoting its deacetylation on lysine 532 via the MTA1/HDAC1 complex. Interestingly, MTA1 also stimulates the transcription of HIF1α and in turn increases the expression of downstream targets of HIF1α to promote and/or support angiogenesis [42]. In this context, targeting MTA has been recently proposed as a therapeutic approach to inhibit angiogenesis in prostate cancer [47].
MTA1 interacts with the E-cadherin chromatin to repress its transcription, leading to increased invasion [48]. The mechanism of MTA1 regulation of E-cadherin is dependent on MTA1 interaction with Snail and Slug proteins, the two upstream regulators of E-cadherin gene expression. The MTA1-Slug-Snail complex binds to E-boxes at the E-cadherin chromatin and utilizes HDAC’s deacetylation activity to downregulate E-cadherin expression and, thereby, provides a novel mechanism for cancer invasiveness.
Genetic ablation of Mta1 in a mouse model of breast-lung cancer metastasis shows that MTA1’s status acts as a modifier of breast-to-lung secondary metastases without affecting the primary mammary gland tumors [14]. Further, MTA1 interacts with STAT3 and forms a coactivator on the Stat3 chromatin to promote its own transcription. These observations suggest that MTA1 may influence the function of another major oncogene in human cancer, the STAT3. These observations raise the possibility that while overexpression of MTA1 is oncogenic in nature, the physiologic level of MTA1 is likely to act as a modifier of metastatic potential of other oncogenes in addition to their transforming activities.
4.3 MTA1 and therapeutic resistance
Due to chromatin remodeling activity of MTA-containing complexes and because of MTA1’s dual coregulatory activity, MTA’s cancer phenotypes are the result of modulation of target genes’ expression, here we will illustrate the role of MTA1 in a few representative clinically relevant examples.
Resistance to therapy continues to be a major bottle-neck in cancer therapeutics and is likely the result of modulation of candidate regulatory gene products. For example, MTA1 overexpression leads to transcriptional repression of ERα in ER-positive breast cancer and development of ER-negative phenotypes as well as resistance to tamoxifen in model systems. Therefore, clinical outcome of tamoxifen therapy in breast cancer patients is likely to be influenced by the status of upstream modifiers of ERα signaling, such as MTA1 and/or functions of its downstream target gene products such as ESR1, in addition to other pathways. Interestingly, the status of ESR1 is differentially affected in ERα-positive and -negative breast cancer cell lines, and this has been linked with differential action of the transcription factor TFAP2C and IFN-γ inducible IFI16 – both part of distinct MTA1 complexes [49]. The level of ESR1 is also affected by the status of MTA1, as its depletion also decreases ESR1 expression. These results suggest that the balance between the recruitment of MTA1-TFAP2C and the MTA1-IFI16 complexes to the ESR1 promoter alongside the ERα levels are the critical modulators of ER-signaling and anti-estrogenic response [49].
The loss of ERα signaling in breast cancer gives rise to elevated metastasis in patients. MTA3, a component of the NuRD complex, has been suggested as another possible mechanism of the loss of ERα signaling. ERα regulates the expression of MTA3, which in turn represses the expression of Snail, a transcription factor linked to EMT process during metastasis [50]. Interestingly, a loss of ERα signaling may be also accompanied by a compensatory increase in TGFβ signaling, which may also feed into aggressively of the EMT process [50].
Because MTA1 can increase cancer aggressiveness by promoting metastasis, MTA1’s connection to TGF-β signaling—another pathway responsible for aggressive tumor formation—is also important. MTA1 has been shown to regulate SMAD7, an important component of TGF-β signaling in breast cancer cells [51]. SMAD7 acts as a negative regulator of the TGF-β signaling pathway by modulating signal transduction to the nucleus. Additionally SMAD7 can inhibit SMAD2 and SMAD3 activity, thus blocking TGF-β signaling and its effect on cancer promotion. Interestingly, MTA1 depletion or HDAC inhibitor treatment increases the level of SMAD7, suggesting that MTA1 can recruit HDACs to the SMAD7 promoter and consequently repress SMAD7 transcription [51]. Results suggested that MTA1 is a positive modulator of TGF-β signaling and reveals another mechanism for MTA1 mediated cancer promotion.
One of the mechanisms of action of tamoxifen includes tamoxifen-triggered recruitment of the MTA1- and MTA2-containing NuRD complexes to the estrogen responsive genes through the tamoxifen-ERα complex [52]. These results suggest that MTA1 and/or its target gene products are expected to correlate with the tamoxifen sensitivity in breast cancer patients. In this context, expression of MTA1 target gene BCAS3 in premenopausal women treated with adjuvant tamoxifen revealed that breast tumors with high BCAS3 expression did not show any difference between tamoxifen treated or untreated groups in their recurrence-free survival, indicating no beneficial effect of antiestrogenic therapy [53]. This was in contrast to tamoxifen treated breast cancer patients with low or moderate levels of BCAS3 expression. In brief, the status of MTA1 and/or targets may contribute to the chemo-sensitivity of breast cancer tumors to anti-estrogenic therapy.
Similar to breast cancer, MTA1 expression correlates well with prostate cancer progression from the benign to metastatic prostate cancer [54]. Clinical relevance of MTA1 in prostate cancer pathobiology was revealed by the observation that targeting MTA1 by pterostilbene sensitizes prostate cancer cells to growth inhibition, apoptosis, compromises angiogenesis and metastasis in xenograft-based studies [55]. In another potentially clinically relevant study, high level of MTA1 correlates well with chemo-resistance of prostate cancer cells to Docetaxel (DTX) [56]. Mechanistically, chemo-resistance of prostate cancer cells to DTX has been shown to be associated with the transcription repression of a nuclear receptor NR4A1 by the MTA1-NuRD complex [56]. Additionally, the experimental repression of NR4A1 attenuates apoptosis in response to genotoxic stress, resulting in potential therapeutic resistance. In another experimental strategy, targeting MTA1 has been shown to be effective in inhibiting the growth and invasion of prostate cancer cells in xenograft studies and this has been attributed to suppression of angiogenesis [47].
More recently, MTA1 overexpression has been linked with resistance to cisplatin – one of the most commonly used chemotherapeutic agents – in nasopharyngeal carcinoma cell lines. Because of cancer supporting functions of MTA1, the observed chemoresistance was associated with the ability of MTA1 to promote cancer stem cell-like phenotypes [57]. To address the possibility of targeting MTA1 to restore chemosensitivity to cisplatin, Feng et al stably downregulated MTA1 in nasopharyngeal carcinoma cells and found that indeed, MTA1 downregulation inhibits tumorigenesis and sensitizes xenograft tumors to cisplatin [57].
5 Global MTA1-nucleosome interactions
The MTA1 is a global modifier of gene expression and believed to act as a “hub” gene because it is at the inter-junction of multiple cellular pathways [58, 59]. Because MTA1 has not been shown to directly bind to DNA despite the presence a zinc finger motif, we hypothesized that MTA1 may modulate gene expression via interacting with chromatin remodeling machinery. The connection of MTA1 with the nucleosome was also somewhat expected due to the nature of the bromo-, the SANT- and the Elm- domains of MTA1, as well as the presence of MTA1 and MTA2 in the NuRD complexes. Consistent with this notion, we found that MTA1 binds to chromatin with a high affinity, as most of the MTA1 remained bound to chromatin in the presence of a high salt extraction buffer. However, treatment of cells with a pharmacological inhibitor of protein methylation rapidly dissociated MTA1 from the chromatin by a substantially lower salt concentration [40], thus revealing a potential role of MTA1 methylation in its chromatin affinity. In fact, MTA1 is methylated on lysine 532 by G9a methyltransferase.
The MTA1’s methylation is believed to be important for its high-affinity interaction with chromatin and highlights the significance of post-translation modifications in MTA1-nucleosome interactions. Indeed, methylated MTA1 binds to the histone H3 amino acid 12–21 peptide but not amino acid 1–11 and 22–44 [40]. MTA1 also interacts with the histone H3-K9 and H3-K4 mono-methyl peptides as well as with the histone H3-K9 acetyl peptide [40]. The extension of these peptide-bindings studies to MTA1 domains revealed that MTA1 uses two distinct MTA1 domains to interact with modified histones: the ELM2-SANT domain interacts with histone-H3K9 and -H3K4-methylated marks, while the C-terminal MTA1 domain interacts with hisone-H3-K9-acetyl mark [40].
MTA1 binds to the histone H3 much more strongly than with histone H4 or H2B peptides [40]. The MTA1 also interact with the histone H3 tail, but this interaction is dispensable for the association of the NuRD complex with chromatin [60]. Interestingly, an esophageal carcinomas based biomarker study demonstrated an inverse relationship between the levels of MTA1 upregulation and acetylation status of histone H4 [61]. Considering that the acetylated proteins are the target of the MTA1-NuRD complex, the authors found that MTA1 overexpressing cancer correlates well with metastasis to the lymphatic system and poorer prognosis, while histone H4 acetylation status inversely correlates with cancer invasiveness and patients with increased acetylated histone H4 associate an improved clinical prognosis as compared to patients with low acetylated H4 [61]. In brief, MTA1 may contribute to chromatin remodeling activity owning to its ability to read specific histone marks – either due to its own structural features or interacting proteins or both.
In line of a high affinity interaction with chromatin [40], MTA1 has recently been found to act as an organizer of higher-order chromatin with a suggested role in chromatin decondensation [62]. Liu et al have found that MTA1 interactions with nucleosomes are dynamic in nature and fluctuate during the cell cycle progression. MTA1 affects chromatin’s interaction with the histone H1 – which is known to confer stability to the DNA-Chromatin complexes. Interestingly, this study found that the decondensation of chromatin during interphase by MTA1 is independent of the Mi-2/NuRD activity [63]. This implies that the prevalence of MTA1 functions that might be independent of the NuRD complex, as is widely demonstrated by the activity of MTA1 as a coactivator. Together, these observations reinforce a broader mechanistic significance of the MTA1 master coregulator in shaping the landscape of the target chromatin in cancer cells. The authors expect that these functions of MTA1 (and perhaps of other MTA proteins) are likely to be deregulated in cancer cells due to hyperstimulation of signaling cascades as well as hypersensitivity of signaling components to extracellular signals, which in turn is expected to modify the nature of post-translational modifications in MTA1 and/or its interacting nucleosomes (Fig. 5).
Figure 5.

Dual functionality of MTA1 master coregulator – a working model.
6 Molecular basis of corepressor versus coactivator activity of MTA1
Although MTA1 was initially identified as a transcription repressor of ERα–– the first recognized direct target of the MTA1-NuRD complex—a large number of studies have established that MTA1 is also a potent activator of target genes with roles in cancer [7, 58]. In addition, genetic depletion of MTA1 in the murine embryonic fibroblasts (MEF) from the MTA1-kockout mice leads to repression as well as stimulation of a large number of genes as compared to gene expression in wild-type MEF [58], revealing that MTA1 status affects the expression of genes in a dual manner. In general, corepressor or coactivator activities of MTA1 are NuRD-complex dependent or -independent, respectively. Further the functionality of MTA1-complexes is expected to be further modulated in the context of target gene chromatin, the nature of upstream stimulating signals, and the activation or deactivation of enzymes responsible for various post-translational modifications on MTA1 protein and nucleosome (Fig. 5). These observations suggest that MTA1’s dual functionality may have a global impact on gene expression.
One of most critical post-translational modifications of MTA1 includes its methylation on lysine 532 by G9a enzyme [40]. The methylated MTA1 preferentially interacts with the CHD4 ATPase. The Chromatin remodeling complexes containing the wild-type MTA1 but not lysine 532-methylation deficient MTA1 mutant or non-methylated MTA1 participate in the hydrolysis of ATP [40]. This may be important for generating the remodeling of the target chromatin by the core complex. MTA1-CHD4 interaction facilitates the formation of a functional NuRD compressor complex. It appears that the ability of MTA1 to remodel chromatin might be modulated by its ELM2-SANT and C-terminal amino acid 442–715 regions, which interact with the modified methylated-H3K9 plus H3K4 and with modified acetyl-H3K9, respectively [40].
The relevance of MTA1-lysine 532 methylation is further evident by its demethylation by Lysine specific demethylase-1 (LSD1) to destabilize the NuRD repressive complex as well as formation of MTA1-chromatin remodeling complexes distinct from the MTA1-NuRD complex [40]. Similar to MTA1, LSD1 has previously been shown to act either as a coactivator or corepressor [63], probably due to its differential interactions with its modified substrates. MTA1’s demethylation by LSD1 plays an important role in antagonizing its corepressive activity. Thus, MTA1 demethylation might be required for MTA1 to interact with a different set of interacting proteins and histone marks, which subsequently lead to the formation of distinct functional MTA1-coactivator complexes [40]. Consistent with this notion, MTA1 demethylation leads to its preferred interaction with acetylated histone H3K9 and BPTF chromatin remodeling protein on the chromatin of stimulated target genes. This process of transition of coactivator activity of MTA1 may be further facilitated by the ability of MTA1 to interact with the modified H3K4-AcK9 marks on the target gene chromatin.
These observations suggest that MTA1 methylation might act as a switch for modifying the activity of MTA1 from a corepressor to coactivator, due to modulating the nature of MTA1-interacting proteins leading to the formation of distinct chromatin remodeling complexes (Fig. 5). Because of the co-existence of enzymes responsible for modifying various post-translational modifications on the MTA1 as well as nucleosomes in vivo, we speculated that the nature of MTA1 modifications and resulting MTA1-containing chromatin remodeling complexes are likely to be differentially regulated as a function of time through a series of intermediate complexes. In fact, methylated-MTA1 and non-methylated MTA1 were found to be an integral component of the MTA1-NuRD corepressor and MTA1-NuRF coactivator complexes in a cyclic manner, respectively. Interestingly, these complexes oscillate as a function of time in a signal-dependent manner [40].
7 Conclusions and perspectives
In summary, signal-dependent post-translational modifications of MTA1, as illustrated by using MTA1’s lysine 532 methylation as an example, plays an important role in the dual functionality of the MTA1 master coregulator. Cancer cells are continuously subjected to a variety of autocrine or paracrine polypeptides that, in turn, stimulate enzymes responsible for various MTA1’s post-translational modifications. Further, many of these regulatory enzymes and signaling molecules are constitutively activated in cancer cells due to inherent mutations and/or deregulation of interacting-protein pathways. In a nut shell, MTA1 functions are likely to be the outcome of cumulative effects of the kinetic changes in the upstream activating or repressive signaling cascades. Therefore, we believe that the ultimate composition of MTA1-containing chromatin remodeling complexes is likely to be regulated by the kinetics of upstream modifying and de-modifying enzymes as well as the nature of intermediate and/or transient protein complexes.
The functions of MTA1-containing regulatory complexes are also modulated by the dynamic plasticity of the target chromatin landscape and its cross-talk with upstream signaling events. These MTA1 protein-centered cellular changes will be ultimately manifested in gene expression and functions of resulting gene products. Because some of the signaling cascades may be dominant in cancer-types specific manner while other signaling cascades may be shared in cancer at-large, MTA1 may exhibit functions that are specific to a given cancer as well as shared across multiple cancer types. The mechanistic intricacies of upstream modifiers of MTA1’s biochemistry are likely to continue to provide us with clues about the cell-type specific and shared functions of MTA1.
Because deregulation of co-existing enzymes with roles in epigenetic modifications of histones and non-histone proteins is not necessarily limited to cancer but prevalent in multiple human diseases, the lessons learned from the MTA1 coregulator biology in cancer cells will be also relevant for other human diseases. Another concept that emerges from the MTA1 coregulator biology is that the proteome of a given protein may be dynamic and not static in nature. For example, due to quantitative differences in the composition of the MTA1-coactivator and -corepressive complexes, we postulate that the nature of MTA1 proteome could be distinct under these two opposing functional states of the MTA1 master coregulator and could be further modified as a function of time to signal. The authors also predict that the concept of dynamic nature of corepressor versus coactivator complexes as a function of time and the nature and strength of signal(s) may be generally equally applicable for other multi-proteins regulatory complexes in living systems. Future research will help us and our collaborators and colleagues to experimentally test the validity of these and many other fruitful avenues that have originated while studying MTA1 as a prototype master coregulator in cancer medicine.
Acknowledgments
We apologize for not citing many other deserving studies from our colleagues due to space limitations. We thank fellows, research staff and colleagues in the Kumar laboratory for their contribution to the biology of MTA1 in human cancer. The MTA1 project in Kumar’s lab is supported by NIH grant CA098823.
Footnotes
Conflict of Interest Statement: The authors declare no any potential conflict of interest.
References
- 1.Toh Y, Pencil SD, Nicolson GL. A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J Biol Chem. 1994;269(37):22958–22963. [PubMed] [Google Scholar]
- 2.Kumar R, Wang RA, Bagheri-Yarmand R. Emerging roles of MTA family members in human cancers. Semin Oncol. 2003;30(5 Suppl 16):30–37. doi: 10.1053/j.seminoncol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Bowen NJ, Fujita N, Kajita M, Wade PA. Mi-2/NuRD: multiple complexes for many purposes. Biochim Biophys Acta. 2004;1677(1–3):52–57. doi: 10.1016/j.bbaexp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Manavathi B, Kumar R. Metastasis tumor antigens, an emerging family of multifaceted master coregulators. J Biol Chem. 2007;282(3):1529–1533. doi: 10.1074/jbc.R600029200. [DOI] [PubMed] [Google Scholar]
- 5.Singh RR, Kumar R. MTA family of transcriptional metaregulators in mammary gland morphogenesis and breast cancer. J Mammary Gland Biol Neoplasia. 2007;12(2–3):115–125. doi: 10.1007/s10911-007-9043-7. [DOI] [PubMed] [Google Scholar]
- 6.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26(37):5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 7.Li DQ, Pakala SB, Nair SS, Eswaran J, Kumar R. Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer Res. 72(2):387–394. doi: 10.1158/0008-5472.CAN-11-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M, et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3(1):30–37. doi: 10.1038/35050532. [DOI] [PubMed] [Google Scholar]
- 9.Li W, Ma L, Zhao J, Liu X, Li Z, Zhang Y. Expression profile of MTA1 in adult mouse tissues. Tissue Cell. 2009;41(6):390–399. doi: 10.1016/j.tice.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Xu D, Wang H, Zhang Y, Chang Y, Zhang J, et al. The subcellular distribution and function of MTA1 in cancer differentiation. Oncotarget. 5(13):5153–5164. doi: 10.18632/oncotarget.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohshiro K, Rayala SK, Wigerup C, Pakala SB, Natha RS, Gururaj AE, et al. Acetylation-dependent oncogenic activity of metastasis-associated protein 1 co-regulator. EMBO Rep. 11(9):691–697. doi: 10.1038/embor.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar R, Balasenthil S, Manavathi B, Rayala SK, Pakala SB. Metastasis-associated protein 1 and its short form variant stimulates Wnt1 transcription through promoting its derepression from Six3 corepressor. Cancer Res. 70(16):6649–6658. doi: 10.1158/0008-5472.CAN-10-0909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar R, Balasenthil S, Pakala SB, Rayala SK, Sahin AA, Ohshiro K. Metastasis-associated protein 1 short form stimulates Wnt1 pathway in mammary epithelial and cancer cells. Cancer Res. 70(16):6598–6608. doi: 10.1158/0008-5472.CAN-10-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pakala SB, Rayala SK, Wang RA, Ohshiro K, Mudvari P, Reddy SD, et al. MTA1 promotes STAT3 transcription and pulmonary metastasis in breast cancer. Cancer Res. 73(12):3761–3770. doi: 10.1158/0008-5472.CAN-12-3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang XY, DeSalle LM, Patel JH, Capobianco AJ, Yu D, Thomas-Tikhonenko A, et al. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc Natl Acad Sci U S A. 2005;102(39):13968–13973. doi: 10.1073/pnas.0502330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li DQ, Divijendra Natha Reddy S, Pakala SB, Wu X, Zhang Y, Rayala SK, et al. MTA1 coregulator regulates p53 stability and function. J Biol Chem. 2009;284(50):34545–34552. doi: 10.1074/jbc.M109.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li DQ, Pakala SB, Reddy SD, Ohshiro K, Peng SH, Lian Y, et al. Revelation of p53-independent function of MTA1 in DNA damage response via modulation of the p21 WAF1-proliferating cell nuclear antigen pathway. J Biol Chem. 285(13):10044–10052. doi: 10.1074/jbc.M109.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li DQ, Kumar R. Mi-2/NuRD complex making inroads into DNA-damage response pathway. Cell Cycle. 9(11):2071–2079. doi: 10.4161/cc.9.11.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2(6):851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13(15):1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao YL, Yang WM. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J Biol Chem. 2003;278(43):42560–42568. doi: 10.1074/jbc.M302955200. [DOI] [PubMed] [Google Scholar]
- 22.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem. 2004;279(15):15050–15058. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 23.Singh RR, Barnes CJ, Talukder AH, Fuqua SA, Kumar R. Negative regulation of estrogen receptor alpha transactivation functions by LIM domain only 4 protein. Cancer Res. 2005;65(22):10594–10601. doi: 10.1158/0008-5472.CAN-05-2268. [DOI] [PubMed] [Google Scholar]
- 24.Khaleque MA, Bharti A, Gong J, Gray PJ, Sachdev V, Ciocca DR, et al. Heat shock factor 1 represses estrogen-dependent transcription through association with MTA1. Oncogene. 2008;27(13):1886–1893. doi: 10.1038/sj.onc.1210834. [DOI] [PubMed] [Google Scholar]
- 25.Mishra SK, Mazumdar A, Vadlamudi RK, Li F, Wang RA, Yu W, et al. MICoA, a novel metastasis-associated protein 1 (MTA1) interacting protein coactivator, regulates estrogen receptor-alpha transactivation functions. J Biol Chem. 2003;278(21):19209–19219. doi: 10.1074/jbc.M301968200. [DOI] [PubMed] [Google Scholar]
- 26.Talukder AH, Gururaj A, Mishra SK, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 interacts with NRIF3, an estrogen-inducible nuclear receptor coregulator. Mol Cell Biol. 2004;24(15):6581–6591. doi: 10.1128/MCB.24.15.6581-6591.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talukder AH, Mishra SK, Mandal M, Balasenthil S, Mehta S, Sahin AA, et al. MTA1 interacts with MAT1, a cyclin-dependent kinase-activating kinase complex ring finger factor, and regulates estrogen receptor transactivation functions. J Biol Chem. 2003;278(13):11676–11685. doi: 10.1074/jbc.M209570200. [DOI] [PubMed] [Google Scholar]
- 28.Covington KR, Brusco L, Barone I, Tsimelzon A, Selever J, Corona-Rodriguez A, et al. Metastasis tumor-associated protein 2 enhances metastatic behavior and is associated with poor outcomes in estrogen receptor-negative breast cancer. Breast Cancer Res Treat. doi: 10.1007/s10549-013-2709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui Y, Niu A, Pestell R, Kumar R, Curran EM, Liu Y, et al. Metastasis-associated protein 2 is a repressor of estrogen receptor alpha whose overexpression leads to estrogen-independent growth of human breast cancer cells. Mol Endocrinol. 2006;20(9):2020–2035. doi: 10.1210/me.2005-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113(2):207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 31.Kumar R. Another tie that binds the MTA family to breast cancer. Cell. 2003;113(2):142–143. doi: 10.1016/s0092-8674(03)00274-5. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Stephens LC, Kumar R. Metastasis tumor antigen family proteins during breast cancer progression and metastasis in a reliable mouse model for human breast cancer. Clin Cancer Res. 2006;12(5):1479–1486. doi: 10.1158/1078-0432.CCR-05-1519. [DOI] [PubMed] [Google Scholar]
- 33.Kumar R, Wang RA, Mazumdar A, Talukder AH, Mandal M, Yang Z, et al. A naturally occurring MTA1 variant sequesters oestrogen receptor-alpha in the cytoplasm. Nature. 2002;418(6898):654–657. doi: 10.1038/nature00889. [DOI] [PubMed] [Google Scholar]
- 34.Mishra SK, Yang Z, Mazumdar A, Talukder AH, Larose L, Kumar R. Metastatic tumor antigen 1 short form (MTA1s) associates with casein kinase I-gamma2, an estrogen-responsive kinase. Oncogene. 2004;23(25):4422–4429. doi: 10.1038/sj.onc.1207569. [DOI] [PubMed] [Google Scholar]
- 35.Gururaj AE, Singh RR, Rayala SK, Holm C, den Hollander P, Zhang H, et al. MTA1, a transcriptional activator of breast cancer amplified sequence 3. Proc Natl Acad Sci U S A. 2006;103(17):6670–6675. doi: 10.1073/pnas.0601989103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Zhan S, Tan W, Cheng R, Gong H, Zhu Q. P300 binds to and acetylates MTA2 to promote colorectal cancer cells growth. Biochem Biophys Res Commun. 444(3):387–390. doi: 10.1016/j.bbrc.2014.01.062. [DOI] [PubMed] [Google Scholar]
- 37.Li DQ, Ohshiro K, Reddy SD, Pakala SB, Lee MH, Zhang Y, et al. E3 ubiquitin ligase COP1 regulates the stability and functions of MTA1. Proc Natl Acad Sci U S A. 2009;106(41):17493–17498. doi: 10.1073/pnas.0908027106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cong L, Pakala SB, Ohshiro K, Li DQ, Kumar R. SUMOylation and SUMO-interacting motif (SIM) of metastasis tumor antigen 1 (MTA1) synergistically regulate its transcriptional repressor function. J Biol Chem. 286(51):43793–43808. doi: 10.1074/jbc.M111.267237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Rechem C, Boulay G, Pinte S, Stankovic-Valentin N, Guerardel C, Leprince D. Differential regulation of HIC1 target genes by CtBP and NuRD, via an acetylation/SUMOylation switch, in quiescent versus proliferating cells. Mol Cell Biol. 30(16):4045–4059. doi: 10.1128/MCB.00582-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nair SS, Li DQ, Kumar R. A core chromatin remodeling factor instructs global chromatin signaling through multivalent reading of nucleosome codes. Mol Cell. 49(4):704–718. doi: 10.1016/j.molcel.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.http://www.nursa.org/
- 42.Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. 2006;25(6):1231–1241. doi: 10.1038/sj.emboj.7601025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng X, Du L, Wang C, Yang Y, Li J, Liu H, et al. Close association of metastasis-associated protein 1 overexpression with increased angiogenesis and poor survival in patients with histologically node-negative gastric cancer. World J Surg. 37(4):792–798. doi: 10.1007/s00268-012-1898-0. [DOI] [PubMed] [Google Scholar]
- 44.Jang KS, Paik SS, Chung H, Oh YH, Kong G. MTA1 overexpression correlates significantly with tumor grade and angiogenesis in human breast cancers. Cancer Sci. 2006;97(5):374–379. doi: 10.1111/j.1349-7006.2006.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li SH, Tian H, Yue WM, Li L, Gao C, et al. Metastasis-associated protein 1 nuclear expression is closely associated with tumor progression and angiogenesis in patients with esophageal squamous cell cancer. World J Surg. 2012;36(3):623–631. doi: 10.1007/s00268-011-1421-z. [DOI] [PubMed] [Google Scholar]
- 46.Li SH, Tian H, Yue WM, Li L, Li WJ, Chen ZT, et al. Overexpression of metastasis-associated protein 1 is significantly correlated with tumor angiogenesis and poor survival in patients with early-stage non-small cell lung cancer. Ann Surg Oncol. 2011;18(7):2048–2056. doi: 10.1245/s10434-010-1510-5. [DOI] [PubMed] [Google Scholar]
- 47.Kai L, Wang J, Ivanovic M, Chung YT, Laskin WB, Schulze-Hoepfner F, et al. Targeting prostate cancer angiogenesis through metastasis-associated protein 1 (MTA1) Prostate. 71(3):268–280. doi: 10.1002/pros.21240. [DOI] [PubMed] [Google Scholar]
- 48.Weng W, Yin J, Zhang Y, Qiu J, Wang X. Metastasis-associated protein 1 promotes tumor invasion by downregulation of E-cadherin. Int J Oncol. 44(3):812–818. doi: 10.3892/ijo.2014.2253. [DOI] [PubMed] [Google Scholar]
- 49.Kang HJ, Lee MH, Kang HL, Kim SH, Ahn JR, Na H, et al. Differential regulation of estrogen receptor alpha expression in breast cancer cells by metastasis-associated protein 1. Cancer Res. 74(5):1484–1494. doi: 10.1158/0008-5472.CAN-13-2020. [DOI] [PubMed] [Google Scholar]
- 50.Dhasarathy A, Kajita M, Wade PA. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol Endocrinol. 2007;21(12):2907–2918. doi: 10.1210/me.2007-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salot S, Gude R. MTA1-mediated transcriptional repression of SMAD7 in breast cancer cell lines. Eur J Cancer. 49(2):492–499. doi: 10.1016/j.ejca.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 52.Liu XF, Bagchi MK. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biological Chemistry. 2004;279(15):15050–15058. doi: 10.1074/jbc.M311932200. [DOI] [PubMed] [Google Scholar]
- 53.Gururaj AE, Holm C, Landberg G, Kumar R. Breast cancer-amplified sequence 3, a target of metastasis-associated protein 1, contributes to tamoxifen resistance in premenopausal patients with breast cancer. Cell Cycle. 2006;5(13):1407–1410. doi: 10.4161/cc.5.13.2924. [DOI] [PubMed] [Google Scholar]
- 54.Hofer MD, Kuefer R, Varambally S, Li H, Ma J, Shapiro GI, et al. The role of metastasis-associated protein 1 in prostate cancer progression. Cancer Research. 2004;64(3):825–829. doi: 10.1158/0008-5472.can-03-2755. [DOI] [PubMed] [Google Scholar]
- 55.Li K, Dias SJ, Rimando AM, Dhar S, Mizuno CS, Penman AD, et al. Pterostilbene acts through metastasis-associated protein 1 to inhibit tumor growth, progression and metastasis in prostate cancer. PLoS One. 2013;8(3):e57542. doi: 10.1371/journal.pone.0057542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu L, Su YS, Zhao J, Wang H, Li W. Repression of NR4A1 by a chromatin modifier promotes docetaxel resistance in PC-3 human prostate cancer cells. FEBS Lett. 587(16):2542–2551. doi: 10.1016/j.febslet.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 57.Feng X, Zhang Q, Xia S, Xia B, Zhang Y, Deng X, et al. MTA1 Overexpression Induces Cisplatin Resistance Innasopharyngeal Carcinoma by Promoting Cancer Stem Cells Properties. Molecules and Cells. 2014 doi: 10.14348/molcells.2014.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghanta KS, Li DQ, Eswaran J, Kumar R. Gene profiling of MTA1 identifies novel gene targets and functions. PLoS One. 6(2):e17135. doi: 10.1371/journal.pone.0017135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38(8):896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 60.Wu M, Wang L, Li Q, Li J, Qin J, Wong J. The MTA family proteins as novel histone H3 binding proteins. Cell Biosci. 2013;3(1):1. doi: 10.1186/2045-3701-3-1. 2045-3701-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toh Y, Ohga T, Endo K, Adachi E, Kusumoto H, Haraguchi M, et al. Expression of the metastasis-associated MTA1 protein and its relationship to deacetylation of the histone H4 in esophageal squamous cell carcinomas. Int J Cancer. 2004;110(3):362–367. doi: 10.1002/ijc.20154. [DOI] [PubMed] [Google Scholar]
- 62.Liu J, Wang H, Ma F, Xu D, Chang Y, Zhang J, et al. MTA1 regulates higher-order chromatin structure and histone H1-chromatin interaction in-vivo. Mol Oncol. 2014 doi: 10.1016/j.molonc.2014.08.007. S1574-7891(14)00202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]