

Abstract
Human cytomegalovirus (HCMV) infection can cause severe illnesses, including encephalopathy and mental retardation, in immunocompromised and immunologically immature patients. Current pharmacotherapies for treating systemic HCMV infections include ganciclovir, cidofovir, and foscarnet. However, long-term administration of these agents can result in serious adverse effects (myelosuppression and/or nephrotoxicity) and the development of viral strains with reduced susceptibility to drugs. The deoxyribosylindole (indole) nucleosides demonstrate a 20-fold greater activity in vitro (the drug concentration at which 50% of the number of plaques was reduced with the presence of drug compared to the number in the absence of drug [EC50] = 0.34 μM) than ganciclovir (EC50 = 7.4 μM) without any observed increase in cytotoxicity. Based on structural similarity to the benzimidazole nucleosides, we hypothesize that the indole nucleosides target the HCMV terminase, an enzyme responsible for packaging viral DNA into capsids and cleaving the DNA into genome-length units. To test this hypothesis, an indole nucleoside-resistant HCMV strain was isolated, the open reading frames of the genes that encode the viral terminase were sequenced, and a G766C mutation in exon 1 of UL89 was identified; this mutation resulted in an E256Q change in the amino acid sequence of the corresponding protein. An HCMV wild-type strain, engineered with this mutation to confirm resistance, demonstrated an 18-fold decrease in susceptibility to the indole nucleosides (EC50 = 3.1 ± 0.7 μM) compared to that of wild-type virus (EC50 = 0.17 ± 0.04 μM). Interestingly, this mutation did not confer resistance to the benzimidazole nucleosides (EC50 for wild-type HCMV = 0.25 ± 0.04 μM, EC50 for HCMV pUL89 E256Q = 0.23 ± 0.04 μM). We conclude, therefore, that the G766C mutation that results in the E256Q substitution is unique for indole nucleoside resistance and distinct from previously discovered substitutions that confer both indole and benzimidazole nucleoside resistance (D344E and A355T).
INTRODUCTION
Human cytomegalovirus (HCMV), a prototypical member of the Betaherpesvirinae- subfamily, is one of the most successful parasites in the human population. Following infection, HCMV can remain latent as a subclinical, lifelong infection in an immunocompetent human host (1, 2). However, congenital HCMV infection, affecting 1 in every 100 to 150 newborns, causes permanent neurological damages, including hearing impairment and mental retardation (3, 4). Similarly, HCMV infection is the most common cause of sight-threatening retinitis in HIV-infected patients and graft rejection in postallograft patients (5, 6). The current FDA-approved agents for the systemic treatment of HCMV infections are ganciclovir (GCV), its prodrug (valganciclovir), cidofovir, and foscarnet (7, 8). GCV, the first-line therapy option for HCMV, is a deoxyguanosine nucleoside analog selectively phosphorylated by the viral kinase pUL97 to a monophosphate and further phosphorylated by endogenous kinases to GCV triphosphate, which inhibits the viral DNA polymerase and/or competes with dGTP for incorporation into DNA, resulting in inhibition of chain elongation (9–14). Cidofovir, an acyclic nucleoside phosphonate, and foscarnet, a pyrophosphate analogue, also target the viral polymerase (8, 15). Because these compounds share a similar mechanism of action, the incidence of cross-resistance is a growing concern (16, 17). In addition, long-term administration of these agents can lead to toxic side effects ranging from hematological disorders (granulocytopenia, anemia, neutropenia) to central nervous system events (hallucination, psychosis) and nephrotoxicity (18–20). Considering the disadvantages of current therapies, a new class of compounds with a better toxicity profile and a novel mechanism of action for the treatment of systemic HCMV infections is warranted.
HCMV terminase is the viral enzyme responsible for the packaging of the high-molecular-weight DNA concatemer into capsids, followed by the cleavage of the viral DNA into functional, genome-sized pieces (21–25). The enzyme, comprised of three subunits (pUL51, pUL56, and pUL89), is not expressed in mammalian cells, thus making terminase an attractive new target for anti-HCMV pharmacotherapy (24, 26). Previous studies have demonstrated that the benzimidazole ribonucleosides inhibit the maturation of the polygenomic concatemeric HCMV DNA by interacting with and inhibiting the HCMV terminase (21, 27, 28). In addition, the benzimidazoles demonstrate a toxicity profile more favorable than that of GCV (29). However, these drugs are readily metabolized in vivo to an inactive aglycone metabolite with an estimated half-life of 0.6 h, which is too rapid for these compounds to be considered clinically viable (30).
The deoxyribosylindole nucleoside UMJD 1896 (Fig. 1), chemically related to benzimidazole nucleosides, may serve as a viable option for HCMV pharmacotherapy due to its potent and selective antiviral activity. It has previously been demonstrated that this indole nucleoside elicits 20-fold greater activity (the drug concentration at which 50% of the number of plaques was reduced with the presence of drug compared to the number in the absence of drug [EC50] = 0.34 μM) than GCV (EC50 = 7.4 μM) without any observed increase in cytotoxicity (31). In addition, it has been established that certain indole nucleosides exert an antiviral effect in the replication cycle that is later than that of GCV but similar to that of the benzimidazole nucleosides (31). Furthermore, a benzimidazole nucleoside-resistant HCMV isolate containing both the D344E and A355T substitutions in pUL89 also exhibits decreased susceptibility to the indole nucleosides (31, 32). Thus, the goal of this study was to test the hypothesis that the indole nucleosides target the HCMV terminase, which results in inhibition of the normal viral maturation process.
FIG 1.
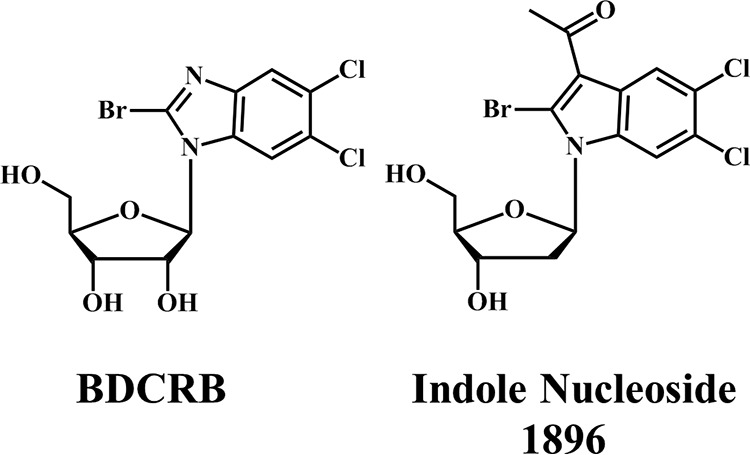
Structures of 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole (BDCRB) and 3-acetyl-2-bromo-5,6-dichloro-1-(2-deoxy-β-d-ribofuranosyl)indole (UMJD 1896, indole nucleoside 1896).
MATERIALS AND METHODS
Chemicals.
Deoxyribosylindole nucleoside UMJD 1896 [3-acetyl-2-bromo-5,6-dichloro-1-(2-deoxy-β-d-ribofuranosyl)indole] and the benzimidazole ribonucleoside 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole (BDCRB) (Fig. 1) were synthesized in the laboratory of L. B. Townsend as described previously (31, 33). GCV was kindly provided by Hoffmann-La Roche (Palo Alto, CA).
Cell culture procedures.
Human foreskin fibroblasts (HFFs) were grown in minimum essential medium (Eagle) [MEM(E)] with Earle's salts and 10% fetal bovine serum. They were grown at 37°C under a humidified atmosphere of 3 to 5% CO2 and 97 to 95% air and were regularly passaged at 1:2 dilutions using conventional procedures with 0.05% trypsin and 0.02% EDTA in HEPES-buffered saline (34).
Virus strains.
Wild-type (wt) HCMV strain Towne was kindly provided by M. F. Stinski, University of Iowa. A bacterial artificial chromosome (BAC) clone of strain AD169 (AD169rv) was generously provided by U. H. Koszinowski, Ludwig Maximilian University (Munich, Germany) (35).
HCMV plaque reduction assay.
HFFs were plated in 24-well cluster dishes at 80,000 cells in a total volume of 1 ml of medium per well and incubated overnight. Each plate was infected with 100 PFU. The virus was allowed to absorb for 2 h at 37°C in a CO2 incubator with intermittent rocking of the dishes. Immediately postinfection, the plates were treated with serial dilutions of drug (0.01 to 100 μM). After incubation for 10 days, the plates were stained with crystal violet (0.1% in 20% methanol), plaques were enumerated using an inverted microscope at 20- to 30-fold magnification, and EC50s were calculated from graphing of the data on a semilog plot.
Selection of indole nucleoside-resistant virus.
HFFs were infected with HCMV Towne at a multiplicity of infection of 0.01 and grown in a 25-cm2 tissue culture flask in the presence of 0.5 μM indole nucleoside 1896 for 2 weeks. Supernatant progeny virus was then passaged in the presence of, first, a 1.0 μM concentration and then a 2.0 μM concentration of indole nucleoside 1896; the duration for each passage was 2 weeks. The resulting virus was further passaged in 2.0 μM indole nucleoside 1896 for a 3-month period and frozen in liquid nitrogen, and the titer was determined. The resulting virus stock was purified using the Klein limiting dilution method (36) and termed 1896r.
DNA sequencing.
Primers for PCR amplification and DNA sequencing were deduced on the basis of the published sequence of the wild-type Towne strain of HCMV (GenBank accession number FJ616285.1). The terminase subunits encoded by viral genes UL51, UL56, and UL89 were selectively amplified by PCR. Following amplification, the products were run on a 1% agarose gel, extracted, and purified using a QIAquick gel extraction kit (Qiagen, Valencia, CA). The amplified products were sequenced using an Applied Biosystems 3730xl DNA analyzer (ISU DNA Facility, Office of Biotechnology). Sequences were analyzed and aligned using Clustal Omega software (Cambridgeshire, United Kingdom).
Marker transfer studies.
To transfer the 1896r mutation hypothesized to cause drug resistance to strain AD169, en passant mutagenesis (37, 38) was performed, as described elsewhere (25, 39, 40), on an infectious BAC clone of HCMV strain AD169, AD169rv (a gift of Ulrich Koszinowski, Munich, Germany) (35). A glutamate codon (GAG) corresponding to amino acid position number 256 in the UL89 open reading frame was mutated to a glutamine codon, CAG, by introducing a C-to-G transversion mutation at nucleotide position 83022 of the sequence with GenBank accession number AC146999.1 Briefly, an excisable kanamycin resistance marker (I-SceI–aphAI) was amplified from a plasmid template (pEPkan-S2) by using oligonucleotide primers UL89 E256Q Fw (5′-GCA CCA AAA GCA CGT GTC GCA GTT TGT GCT CAA AGA GGT CCA GTT CCG CTG CCG CCA CAC CTA GGG ATA ACA GGG TAA TCG ATT T-3′) and UL89 E256Q Rv (5′-ACC ACG TAG TCG CGC GCG AAG GTG TGG CGG CAG CGG AAC TGG ACC TCT TTG AGC ACA AAC GCC AGT GTT ACA ACC AAT TAA CC-3′), which had been custom synthesized, and was purified via polyacrylamide gel electrophoresis (PAGE) by Integrated DNA Technologies, Inc. (Coralville, IA). The resulting PCR product was gel purified using a NucleoSpin gel and PCR cleanup kit (Macherey-Nagel, Inc., Bethlehem, PA) and was then electroporated into Escherichia coli strain GS1783 (a gift of Greg Smith, Northwestern University, Chicago, IL) harboring the AD169rv BAC. Single Kanr colonies were picked, and Red recombinase and I-SceI homing endonuclease activities were induced to remove the I-SceI–aphAI cassette. A kanamycin-sensitive colony containing the recombinant BAC, AD169rv UL89 E256Q, was isolated. The overall genetic integrity of the recombinant BAC was confirmed by restriction enzyme digestion analysis (not shown). The presence of the intended single-base-pair mutation and the absence of spurious changes were verified by DNA sequencing (not shown) of the modified region (Genewiz, Inc., South Plainfield, NJ).
Virus reconstitution.
Infectious virus was reconstituted from BAC DNA as described previously (25, 39, 40) by cotransfection of ∼0.7 × 105 human foreskin fibroblasts in a 24-well cluster plate (Corning, Inc.) with a total of 1 μg DNA containing 750 ng BAC DNA, 200 ng of pp71 expression plasmid pSG5-pp71 (a gift of Robert Kalejta, University of Wisconsin, Madison, WI) (41), and 50 ng of the Cre recombinase expression plasmid pCAGGS-nlsCre (a gift of Michael I. Kotlikoff, Cornell University, Ithaca, NY) (42) by using 10 μl of Superfect transfection reagent (Qiagen, Inc., Valencia, CA) according to the manufacturer's instructions.
Data analysis.
Results were graphed and analyzed using Prism software (version 5.0; GraphPad Software, San Diego, CA) to determine the standard deviation and statistical significance (Student's t test).
RESULTS
Initial characterization of indole nucleoside 1896-resistant HCMV isolate (1896r).
To determine whether the HCMV terminase is the target of the deoxyribosylindole nucleosides, wild-type HCMV Towne was grown in the presence of increasing concentrations of indole nucleoside 1896. An isolate (plaque purified and termed 1896r) was subjected to increasing concentrations of drug to confirm resistance (Fig. 2; Table 1). As expected, 1896r was approximately 5-fold more resistant to indole nucleoside 1896 (EC50 = 2.3 ± 0.8 μM) than wild-type virus (HCMV Towne) (EC50 = 0.46 ± 0.17 μM). In addition, 1896r was subjected to increasing concentrations of BDCRB in order to determine if the virus is cross-resistant to this drug. Interestingly, 1896r exhibited no significant change in susceptibility to BDCRB (EC50 = 6.0 ± 2.1 μM) compared to that of wild-type virus (HCMV Towne) (EC50 = 3.0 ± 1.1 μM). As expected, 1896r did not exhibit any change in susceptibility to GCV (1896r EC50 = 8.7 ± 2.1 μM; wild-type HCMV EC50 = 10.9 ± 4.5 μM). These results demonstrate that 1896r, while resistant to indole nucleoside 1896, does not exhibit cross-resistance to BDCRB or GCV.
FIG 2.
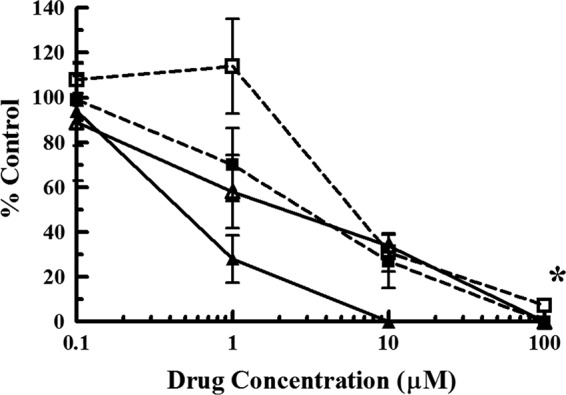
Initial characterization of an indole nucleoside 1896-resistant HCMV isolate (1896r). HFFs infected with an indole nucleoside 1896-resistant HCMV isolate (open symbols) were subjected to increasing concentrations of either BDCRB (dashed lines, ■ and □) or indole nucleoside 1896 (solid lines, ▲ and Δ) and compared to HFFs infected with wild-type HCMV (Towne; closed symbols) subjected to the same concentrations of drug. The y axis represents the percentage of plaques compared to the amount for a no-drug control. The values represent the mean ± standard deviation from at least three experiments. *, P < 0.001, in which the replication of 1896r was significantly different from that of wild-type virus (for indole nucleoside 1896 only; no statistically significant difference was found for BDCRB).
TABLE 1.
Antiviral activity of GCV, BDCRB, and indole nucleoside 1896 against 1896r and HCMV UL89 E256Q
| Antiviral | Comparison of Towne and 1896r |
Comparison of AD169 and HCMV UL89 E256Q |
||||
|---|---|---|---|---|---|---|
| EC50 (μM) |
Fold resistance of 1896r | EC50 (μM) |
Fold resistance of HCMV UL89 E256Q | |||
| wt (Towne) | 1896r | wt (AD169) | HCMV UL89 E256Q | |||
| GCV | 10.9 ± 4.5 | 8.7 ± 2.1 | 0.80 | 1.5 ± 0.7 | 0.65 ± 0.21 | 0.43 |
| BDCRB | 3.0 ± 1.1 | 6.0 ± 2.1 | 2 | 0.25 ± 0.04 | 0.23 ± 0.04 | 0.92 |
| 1896 | 0.46 ± 0.17 | 2.3 ± 0.8 | 5a | 0.17 ± 0.04 | 3.1 ± 0.7 | 18b |
P < 0.05.
P < 0.001.
DNA sequencing of 1896r.
The benzimidazole nucleoside-resistant HCMV isolates reportedly exhibit cross-resistance to indole nucleoside 1896 (31). This resistance was mapped to mutations in UL56 and UL89, two subunits of the viral terminase enzyme (27, 32). In addition to pUL56 and pUL89, pUL51 may be another subunit that comprises the viral terminase enzyme (24). Therefore, the open reading frames of UL51, UL56, and UL89 in 1896r were sequenced. The results demonstrated that both UL51 and UL56 of 1896r contain no mutations that would affect the amino acid sequence of the translated protein. However, UL89 contains a single-base-pair mutation (G to C) at position 766 (G766C), which is located within the first exon of the gene. The mutation would result in a glutamate-to-glutamine change in the amino acid residue at position 256 of the pUL89 protein (E256Q) (Table 2). We hypothesize that this point mutation is responsible for the resistance of HCMV to indole nucleoside 1896 but not to BDCRB.
TABLE 2.
Amino acid sequence of pUL89
| HCMV strain | Amino acid sequence at the following amino acid positiona: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 241 | 251 | 261 | 271 | 281 | 291 | 301 | 311 | 321 | 331 | 341 | 351 | |
| Towne | VAHQKHVSQF | VLKEVEFRCR | HTFARDYVVE | NKDNVISIDH | RGAKSTALFA | SCYNTNSIRG | QNFHLLLVDE | AHFIKKEAFN | TILGFLAQNT | TKIIFISSTN | TTSDATCFLT | RLNNAPFDML |
| AD169 | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- |
| 1038rBb | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---E------ | ----T----- |
| 1896r | ---------- | -----Q---- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- |
| E256Q | ---------- | -----Q---- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- |
The amino acid sequence of pUL89 was inferred from the UL89 open reading frame. Only the portion of the sequence which contained the substitutions that confer resistance is presented. Amino acid changes are shown in boldface. Polymorphisms in UL89 of HCMV Towne and AD169 were reported in a previous study (53). The position number is the first amino acid in the sequence of 10 residues.
A BDCRB-resistant HCMV isolate containing both D344E and A355T (32).
Recombinant HCMV pUL89 E256Q exhibits resistance to indole nucleoside 1896 with no change in benzimidazole nucleoside susceptibility.
To determine whether the G766C transversion mutation in UL89 was the mutation that confers antiviral drug resistance in 1896r and corroborate the hypothesis that this drug targets the HCMV terminase, the mutation was engineered into wild-type strain AD169, resulting in HCMV pUL89 E256Q. HFFs infected with this engineered virus were exposed to increasing concentrations of GCV, BDCRB, or indole nucleoside 1896 and were compared to HFFs infected with wild-type strain AD169 under the same conditions. As shown in Fig. 3 and Table 1, HCMV pUL89 E256Q was approximately 18-fold more resistant to indole nucleoside 1896 (EC50 = 3.1 ± 0.7 μM) than wild-type virus (EC50 = 0.17 ± 0.04 μM). Interestingly, and confirming the results obtained with 1896r, the recombinant virus did not exhibit any cross-resistance to BDCRB (EC50 for wt HCMV AD169 = 0.25 ± 0.04 μM, EC50 for HCMV UL89 E256Q = 0.23 ± 0.04 μM). Furthermore, and similar to the findings for 1896r, there was no significant change in the susceptibility of HCMV pUL89 E256Q to GCV (EC50 = 0.65 ± 0.21 μM) compared to that of wild-type AD169 virus (EC50 = 1.5 ± 0.7 μM). We therefore conclude that the G766C mutation in UL89, which results in an E256Q amino acid change in pUL89 and confers deoxyribosylindole nucleoside analog resistance, is distinct from previously identified amino acid substitutions (D344E and A355T) which confer resistance to both indole and benzimidazole nucleosides.
FIG 3.
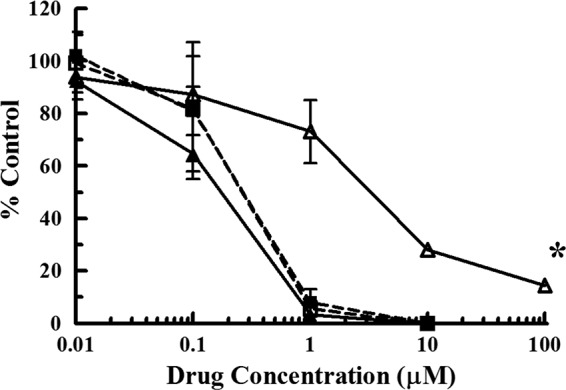
Decreased susceptibility of HCMV pUL89 E256Q to indole nucleoside 1896. HFFs infected with a wild-type HCMV strain (AD169) engineered with the UL89 G766C point mutation (open symbols) were subjected to increasing concentrations of either BDCRB (dashed lines, ■ and □) or indole nucleoside 1896 (solid lines, ▲ and Δ) and compared to HFFs infected with wild-type HCMV (AD169; closed symbols) subjected to the same concentrations of drug. The y axis represents the percentage of plaques compared to the amount for a no-drug control. The values represent the mean ± standard deviation from at least four experiments. *P < 0.0001, in which the replication of HCMV E256Q was significantly different from that of wild-type virus (for indole nucleoside 1896 only; no statistically significant difference was found for BDCRB).
DISCUSSION
The deoxyribosylindole nucleosides elicit an antiviral effect late in the HCMV replication cycle (31). Since it has previously been demonstrated that mutations in the HCMV genome that confer resistance to the chemically related benzimidazole ribonucleosides also confer resistance to the indole nucleosides (31) and that those mutations occur in the open reading frames of the viral terminase enzyme (24, 27, 43), we hypothesized that the molecular mechanism by which the indole nucleosides inhibit viral replication is targeting of the HCMV terminase. Our current results demonstrate that a previously uncharacterized point mutation in UL89, which results in a single amino acid residue change at position 256 of the encoded protein (Table 2), renders the virus resistant to the indole nucleosides but, interestingly, not to the benzimidazole nucleoside BDCRB (Table 1). These results establish that pUL89 is a target of the indole nucleosides.
Previous studies have demonstrated that certain mutations can decrease the replication capacity of virus upon infection (39, 44). However, mutations that confer resistance to the chemically related benzimidazole nucleosides did not affect the replicative fitness of those viruses (27). Although the replicative fitness of the viruses presented herein was not directly compared, wild-type virus engineered with the G766C mutation in UL89 (HCMV pUL89 E256Q) replicated to titers that were indistinguishable from those observed for wild-type AD169 HCMV (data not shown). This general observation leads us to speculate that viral fitness was not reduced due to the introduction of the G766C mutation that confers resistance to indole nucleoside 1896.
GCV and cidofovir have been found to elicit an antiviral effect through direct inhibition of the viral DNA polymerase and/or incorporation into elongating viral DNA, resulting in chain termination (7). Therefore, it is not surprising that many mutations in the viral DNA polymerase that result in resistance to one of these drugs also result in decreased susceptibility to the other (45). Since it has been previously established that mutations that result in the resistance of HCMV to the benzimidazole nucleosides confer resistance to the indole nucleosides (31), the implication is that these compounds elicit an antiviral effect in a similar manner. As such, we contend that the most likely mechanism of action for the indole nucleosides is inhibition of pUL89 nuclease activity, particularly because the benzimidazole nucleosides inhibit HCMV terminase function in that manner (21). In addition, it has also been established that the benzimidazole nucleosides inhibit the interaction of pUL56 with the viral portal protein (pUL104), thereby preventing insertion of the viral genome into the procapsid (28). Therefore, it seems likely that the indole nucleosides would also inhibit the insertion of the viral genome into the procapsid by preventing the interaction of pUL56 and pUL104. Nonetheless, while pUL104 does associate with the viral terminase for processing and packaging and previous studies have demonstrated there is a mutation in UL104 common in benzimidazole-resistant HCMV isolates (46), this mutation does not in and of itself confer resistance. Thus, we did not sequence UL104 in 1896r, especially since the mutation discovered in UL89 was sufficient for drug resistance.
Despite their many similarities, however, there are important differences between the benzimidazole and indole nucleosides. We hypothesize that key chemical differences found in the indole nucleosides but not the benzimidazole nucleosides confer enough structural dissimilarity between the two compounds to allow them to bind their targets in a similar but not precisely the same manner. Chemically, they differ by the presence of an acetyl group at the 3 position of the indole ring, the lack of nitrogen at the 3 position of the indole compared to the benzimidazole, and the lack of a hydroxyl group at the 2′ position of the sugar (31). Our results presented herein demonstrating that a mutation in pUL89 that confers resistance to the indole nucleosides but not the benzimidazole nucleosides provides evidence for this hypothesis (Table 1). In addition, we speculate that these key chemical differences that allow the indole nucleosides to bind to the viral terminase in a different manner may also allow in vivo stability greater than what has been observed with the benzimidazole nucleosides. Although previous studies have demonstrated that the benzimidazole nucleosides elicit good antiviral activity and selectivity in vitro (33), they are metabolized via glycosidic bond degradation too rapidly in vivo to be considered viable clinical candidates (30). The chemical differences between the indole and the benzimidazole nucleosides result in a stronger glycosidic bond, and we therefore hypothesize that the indole nucleosides will not be as readily metabolized in vivo (31). Further investigation into the metabolism of the indole nucleosides is warranted if these compounds are to be considered viable for clinical use.
Current pharmacotherapies (GCV, cidofovir, foscarnet) and some currently in clinical trials (brincidofovir, cyclopropavir) for the treatment of systemic HCMV infections target the viral DNA polymerase (pUL54). Previous studies have demonstrated high levels of cross-resistance between these compounds (47–49). Since long-term pharmacotherapy is generally required due to the recurrence of infection upon cessation of treatment, the development of HCMV strains with reduced susceptibility to multiple drugs is a major concern. In addition to the increased safety profile of the indole nucleosides, the main advantage to this pharmacotherapy is the distinct mechanism of action by which this compound elicits an antiviral effect. As such, no significant levels of cross-resistance should exist between the indole nucleosides and other pharmacotherapies that target the viral DNA polymerase. Consistent with this is our results presented herein demonstrating no significant change in the susceptibility of mutant (HCMV pUL89 E256Q) virus to GCV compared to that of wild-type AD169 virus subjected to the same concentrations of drug (Table 1).
Interest in the HCMV terminase, comprised of subunits pUL51, pUL56, and pUL89, as a potential target for antiviral pharmacotherapy is increasing (24, 25). In addition to the benzimidazole and the indole nucleosides, BAY 38-4766, letermovir, and the anti-HIV integrase inhibitor raltegravir are a few compounds that elicit an antiviral effect against HCMV through inhibition of the terminase function (50–52). Although it appears that all these compounds affect the terminase by different and distinct mechanisms, the result is the same: impairment of viral genome processing and packaging. While we consider it unlikely that any significant levels of cross-resistance between currently approved pharmacotherapies and the indole nucleosides would occur, it remains plausible that mutations conferring resistance to the indole nucleosides could confer cross-resistance to other compounds that target the HCMV terminase. This possibility, however, was not addressed in the present study.
With the use of monotherapy for the treatment of HCMV infections, the development of drug resistance (and possibly cross-resistance) can occur within 2 weeks of initiation of therapy (17). It is therefore important to develop new pharmacotherapies that have not only a greater safety profile but also a different mechanism of action so as to minimize the possibility of drug resistance and/or cross-resistance. Previous results have demonstrated that the indole nucleosides have greater antiviral activity than GCV without any observed increase in cytotoxicity (31). Our results presented herein demonstrate a distinct mechanism of action for the indole nucleosides compared to the mechanisms of action of currently approved pharmacotherapies. Further development and examination of the molecular mechanism of action of these compounds are warranted.
ACKNOWLEDGMENTS
This project was supported by grants from the American Heart Association (12GRNT11890012, 14GRNT19970013), the National Institute of General Medical Sciences (P30GM110703), and the Iowa Space Grant Consortium (NASA grant number NNX10AK63H) and funds from the University of Michigan and Drake University.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
REFERENCES
- 1.Landolfo S, Gariglio M, Gribaudo G, Lembo D. 2003. The human cytomegalovirus. Pharmacol Ther 98:269–297. doi: 10.1016/S0163-7258(03)00034-2. [DOI] [PubMed] [Google Scholar]
- 2.Sinclair J, Sissons P. 2006. Latency and reactivation of human cytomegalovirus. J Gen Virol 87(Pt 7):1763–1779. doi: 10.1099/vir.0.81891-0. [DOI] [PubMed] [Google Scholar]
- 3.Dollard SC, Grosse SD, Ross DS. 2007. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol 17:355–363. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 4.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitwalli AH, Nazmi A, Al Ghonaim M, Shaheen F, Kfoury H. 2013. Cytomegalovirus disease in a renal transplant recipient: the importance of pre-transplant screening of the donor and recipient. Saudi J Kidney Dis Transpl 24:80–85. doi: 10.4103/1319-2442.106250. [DOI] [PubMed] [Google Scholar]
- 6.Heiden D, Ford N, Wilson D, Rodriguez WR, Margolis T, Janssens B, Bedelu M, Tun N, Goemaere E, Saranchuk P, Sabapathy K, Smithuis F, Luyirika E Drew WL. 2007. Cytomegalovirus retinitis: the neglected disease of the AIDS pandemic. PLoS Med 4:e334. doi: 10.1371/journal.pmed.0040334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrei G, De Clercq E, Snoeck R. 2009. Drug targets in cytomegalovirus infection. Infect Disord Drug Targets 9:201–222. doi: 10.2174/187152609787847758. [DOI] [PubMed] [Google Scholar]
- 8.Biron KK. 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res 71:154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Littler E, Stuart AD, Chee MS. 1992. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 358:160–162. doi: 10.1038/358160a0. [DOI] [PubMed] [Google Scholar]
- 10.Matthews T, Boehme R. 1988. Antiviral activity and mechanism of action of ganciclovir. Rev Infect Dis 10:S490–S494. doi: 10.1093/clinids/10.Supplement_3.S490. [DOI] [PubMed] [Google Scholar]
- 11.Freitas VR, Smee DF, Chernow M, Boehme R, Matthews TR. 1985. Activity of 9-(1,3-dihydroxy-2-propoxymethyl)guanine compared with that of acyclovir against human, monkey, and rodent cytomegaloviruses. Antimicrob Agents Chemother 28:240–245. doi: 10.1128/AAC.28.2.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamzeh FM, Lietman PS. 1991. Intranuclear accumulation of subgenomic noninfectious human cytomegalovirus DNA in infected cells in the presence of ganciclovir. Antimicrob Agents Chemother 35:1818–1823. doi: 10.1128/AAC.35.9.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamzeh FM, Lietman PS, Gibson W, Hayward GS. 1990. Identification of the lytic origin of DNA replication in human cytomegalovirus by a novel approach utilizing ganciclovir-induced chain termination. J Virol 64:6184–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Z, He YS, Kim Y, Chu L, Ohmstede C, Biron KK, Coen DM. 1997. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J Virol 71:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong X, Smith JL, Chen MS. 1997. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob Agents Chemother 41:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drew WL, Paya CV, Emery V. 2001. Cytomegalovirus (CMV) resistance to antivirals. Am J Transplant 1:307–312. doi: 10.1034/j.1600-6143.2001.10403.x. [DOI] [PubMed] [Google Scholar]
- 17.Drew WL, Miner RC, Busch DF, Follansbee SE, Gullett J, Mehalko SG, Gordon SM, Owen WF Jr, Matthews TR, Buhles WC, DeArmond B. 1991. Prevalence of resistance in patients receiving ganciclovir for serious cytomegalovirus infection. J Infect Dis 163:716–719. doi: 10.1093/infdis/163.4.716. [DOI] [PubMed] [Google Scholar]
- 18.Walmsley S, Tseng A. 1999. Comparative tolerability of therapies for cytomegalovirus retinitis. Drug Saf 21:203–224. doi: 10.2165/00002018-199921030-00005. [DOI] [PubMed] [Google Scholar]
- 19.Danziger-Isakov L, Mark Baillie G. 2009. Hematologic complications of anti-CMV therapy in solid organ transplant recipients. Clin Transplant 23:295–304. doi: 10.1111/j.1399-0012.2008.00942.x. [DOI] [PubMed] [Google Scholar]
- 20.Lea A, Bryson H. 1996. Cidofovir. Drugs 52:225–230. doi: 10.2165/00003495-199652020-00006. [DOI] [PubMed] [Google Scholar]
- 21.Scheffczik H, Savva CGW, Holzenburg A, Kolesnikova L, Bogner E. 2002. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res 30:1695–1703. doi: 10.1093/nar/30.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholz B, Rechter S, Drach JC, Townsend LB, Bogner E. 2003. Identification of the ATP-binding site in the terminase subunit of pUL56 of human cytomegalovirus. Nucleic Acids Res 31:1426–1433. doi: 10.1093/nar/gkg229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bogner E, Radsak K, Stinski MF. 1998. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J Virol 72:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borst EM, Kleine-Albers J, Gabaev I, Babic M, Wagner K, Binz A, Degenhardt I, Kalesse M, Jonjic S, Bauerfeind R, Messerle M. 2013. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J Virol 87:1720–1732. doi: 10.1128/JVI.01955-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Li G, Schauflinger M, Nguyen CC, Hall ED, Yurochko AD, von Einem J, Kamil JP. 2013. The ULb′ region of the human cytomegalovirus genome confers an increased requirement for the viral protein kinase UL97. J Virol 87:6359–6376. doi: 10.1128/JVI.03477-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogner E. 2002. Human cytomegalovirus terminase as a target for antiviral chemotherapy. Rev Med Virol 12:115–127. doi: 10.1002/rmv.344. [DOI] [PubMed] [Google Scholar]
- 27.Krosky PM, Underwood MR, Turk SR, Feng KW-H, Jain RG, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J Virol 72:4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dittmer A, Drach JC, Townsend LB, Fischer A, Bogner E. 2005. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-d-ribonucleosides. J Virol 79:14660–14667. doi: 10.1128/JVI.79.23.14660-14667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nassiri MR, Emerson SG, Devivar RV, Townsend LB, Drach JC, Taichman RS. 1996. Comparison of benzimidazole nucleosides and ganciclovir on the in vitro proliferation and colony formation of human bone marrow progenitor cells. Br J Haematol 93:273–279. doi: 10.1046/j.1365-2141.1996.5231066.x. [DOI] [PubMed] [Google Scholar]
- 30.Good SS, Owens BS, Townsend LB, Drach JC. 1994. The disposition in rats and monkeys of 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)-benzimidazole (BDCRB) and its 2,5,6-trichloro congener (TCRB). Antiviral Res 23(Suppl):103. doi: 10.1016/0166-3542(94)90251-8. [DOI] [Google Scholar]
- 31.Williams JD, Ptak RG, Drach JC, Townsend LB. 2004. Synthesis, antiviral activity, and mode of action of some 3-substituted 2,5,6-trichloroindole 2′- and 5′-deoxyribonucleosides. J Med Chem 47:5773–5782. doi: 10.1021/jm0400606. [DOI] [PubMed] [Google Scholar]
- 32.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J Virol 72:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsend LB, Devivar RV, Turk SR, Nassiri MR, Drach JC. 1995. Design, synthesis, and antiviral activity of certain 2,5,6-trihalo-1-(β-d-ribofuranosyl)benzimidazoles. J Med Chem 38:4098–4105. doi: 10.1021/jm00020a025. [DOI] [PubMed] [Google Scholar]
- 34.Shipman C., Jr 1969. Evaluation of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) as a tissue culture buffer. Proc Soc Exp Biol Med 130:305–310. doi: 10.3181/00379727-130-33543. [DOI] [PubMed] [Google Scholar]
- 35.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol 74:7720–7729. doi: 10.1128/JVI.74.17.7720-7729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klein RJ. 1975. Isolation of herpes simplex virus clones and drug resistant mutants in microcultures. Arch Virol 49:73–80. doi: 10.1007/BF02175598. [DOI] [PubMed] [Google Scholar]
- 37.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless Red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 38.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 39.Gentry BG, Vollmer LE, Hall ED, Borysko KZ, Zemlicka J, Kamil JP, Drach JC. 2013. Resistance of human cytomegalovirus to cyclopropavir maps to a base pair deletion in the open reading frame of UL97. Antimicrob Agents Chemother 57:4343–4348. doi: 10.1128/AAC.00214-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li G, Rak M, Nguyen CC, Umashankar M, Goodrum FD, Kamil JP. 2014. An epistatic relationship between the viral protein kinase UL97 and the UL133-UL138 latency locus during the human cytomegalovirus lytic cycle. J Virol 88:6047–6060. doi: 10.1128/JVI.00447-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol 23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarosinski KW, Hunt HD, Osterrieder N. 2010. Down-regulation of MHC class I by the Marek's disease virus (MDV) UL49.5 gene product mildly affects virulence in a haplotype-specific fashion. Virology 405:457–463. doi: 10.1016/j.virol.2010.06.041. [DOI] [PubMed] [Google Scholar]
- 43.Evers DL, Komazin G, Ptak RG, Shin D, Emmer BT, Townsend LB, Drach JC. 2004. Inhibition of human cytomegalovirus replication by benzimidazole nucleosides involves three distinct mechanisms. Antimicrob Agents Chemother 48:3918–3927. doi: 10.1128/AAC.48.10.3918-3927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. 1999. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J Virol 73:5663–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drew WL. 2010. Cytomegalovirus resistance testing: pitfalls and problems for the clinician. Clin Infect Dis 50:733–736. doi: 10.1086/650463. [DOI] [PubMed] [Google Scholar]
- 46.Komazin G, Townsend LB, Drach JC. 2004. Role of a mutation in human cytomegalovirus gene UL104 in resistance to benzimidazole ribonucleosides. J Virol 78:710–715. doi: 10.1128/JVI.78.2.710-715.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chou S, Lurain NS, Thompson KD, Miner RC, Drew WL. 2003. Viral DNA polymerase mutations associated with drug resistance in human cytomegalovirus. J Infect Dis 188:32–39. doi: 10.1086/375743. [DOI] [PubMed] [Google Scholar]
- 48.Chou S, Marousek G, Bowlin TL. 2012. Cyclopropavir susceptibility of cytomegalovirus DNA polymerase mutants selected after antiviral drug exposure. Antimicrob Agents Chemother 56:197–201. doi: 10.1128/AAC.05559-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erice A. 1999. Resistance of human cytomegalovirus to antiviral drugs. Clin Microbiol Rev 12:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, Graeper S, Klenk HD, Ruebsamen-Waigmann H, Hallenberger S. 2001. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J Virol 75:9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verghese PS, Schleiss MR. 2013. Letermovir treatment of human cytomegalovirus infection antiinfective agent. Drugs Future 38:291–298. doi: 10.1358/dof.2013.038.05.1946425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nadal M, Mas PJ, Blanco AG, Arnan C, Sola M, Hart DJ, Coll M. 2010. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc Natl Acad Sci U S A 107:16078–16083. doi: 10.1073/pnas.1007144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krosky PM, Ptak RG, Underwood MR, Biron KK, Townsend LB, Drach JC. 2000. Differences in DNA packaging genes and sensitivity to benzimidazole ribonucleosides between human cytomegalovirus strains AD169 and Towne. Antivir Chem Chemother 11:349–352. [DOI] [PubMed] [Google Scholar]