

Abstract
Non-reducing polyketide synthases (NR-PKSs) are unique among PKSs in their domain structure, notably including a starter unit:acyl-carrier protein (ACP) transacylase (SAT) domain that selects an acyl group as the primer for biosynthesis, most commonly acetyl-CoA from central metabolism. This clan of mega-enzymes resembles fatty acid synthases (FASs) by both sharing their central chain elongation steps and their capacity for iterative catalysis. In this mode of synthesis, catalytic domains involved in chain extension exhibit substrate plasticity to accommodate growing chains as small as two carbons to 20 or more. PksA is the NR-PKS central to the biosynthesis of the mycotoxin aflatoxin B1 and whose SAT domain accepts an unusual hexanoyl starter from a dedicated yeast-like FAS. Explored in this paper is the ability of PksA to utilize a selection of potential starter units as substrates to initiate and sustain extension and cyclization to on-target, programmed polyketide synthesis. Most of these starter units were successfully accepted and properly processed by PksA to achieve biosynthesis of the predicted naphthopyrone product. Analysis of the on-target and derailment products revealed trends of tolerance by individual PksA domains to alternative starter units. In addition, natural and unnatural variants of the active site cysteine were examined and found to be capable of biosynthesis, suggesting possible direct loading of starter units onto the β-ketoacyl synthase (KS) domain. In light of the data assembled here, the predictable synthesis of un-natural products by NR-PKSs is more fully defined.
INTRODUCTION
Fatty acid synthases (FASs) and all but one clan of the related polyketide synthases (PKSs) exemplify the rare catalytic strategy of iteration; that is, the catalytic domains of these large proteins are re-used, typically a fixed number of times, and can accommodate substrates as small as two carbons to as large as 20 or more(1–3). Thus, unlike the acute substrate discrimination associated with highly evolved primary metabolic enzymes, the potential synthetic hazards of substrate promiscuity are overcome by deeply buried active sites accessible to intermediates borne on carrier proteins and intraprotein transfers that confer high effective concentrations. Such intrinsic plasticity in iterative systems raises the fundamental question of whether these active sites can more readily accommodate alternative building blocks to synthesize non-native products. Previous studies of modular PKSs, where each module canonically catalyzes a single set of elongation and tailoring steps during biosynthesis, have shown limited tolerance to a narrow range of both non-native starter(4–8) and extender units(9, 10). The fungal non-reducing PKSs (NR-PKSs) mediate the simplest chain extension chemistry to a classical poly-β-ketone intermediate with no processing at the β-carbon during elongation, which distinguishes the other partially and highly reducing PKSs and all FASs(11–14).
NR-PKSs contain two domains unique among PKSs: a starter unit: acyl-carrier protein (ACP) transferase (SAT) that brings the chain initiating component onto the enzyme(15), and a product template (PT) domain that controls the cyclization of the fully elongated, highly reactive poly-β-ketone intermediate to specific aromatic and fused aromatic products(3, 16, 17). Combinatorial assemblies of heterodomains from a selection of NR-PKSs have shown that polyketide chain length is largely controlled by the β-ketoacyl synthase (KS) domain, and the cyclization pattern is determined by the PT domain(18, 19). The enzyme responsible for the biosynthesis of norsolorinic acid anthrone in Aspergillus parasiticus, the precursor to aflatoxin B1 (1), PksA(20), accepts an unusual hexanoyl starter unit from a dedicated pair of yeast-like FASs (HexA/B, Scheme 1A)(21–23). The SAT domain of most NR-PKSs prefers acetyl-CoA from common cellular metabolism and shows strong selection against even propionyl-CoA(24). However, there are similar enzyme pairs such as Hpm8 and Hpm3, where the highly-reducing PKS Hpm8 synthesizes a relatively large partially-reduced starter unit, which is then transferred to the NR-PKS Hpm3 for further elongation(25).
Scheme 1.

Biosynthetic reactions catalyzed by PksA. (A) Early steps of aflatoxin B1 biosynthesis by HexA, HexB and PksA in Aspergillus parasiticus. (B) In vitro assays of alternative and native starter units performed in this study. SAT: starter-unit:acyl-carrier protein (ACP) transacylase, KS: β-ketoacyl synthase, MAT: malonyl:ACP transacylase, PT: product template, TE: thioesterase
In this paper we combine the SAT-KS-MAT (malonyl-CoA:ACP transacylase) tridomain and the PT-ACP didomain of PksA to examine the ability not only of the SAT to accept nonnative starter units but also the KS and the PT to perform their central tasks in product synthesis as a more complete measure of both starter unit tolerance and NR-PKS synthetic function. To simplify the analysis, the thioesterase (TE) domain, which catalyzes a further (Dieckmann) cyclization to anthrone 2, is not present and we rely on the facile self-enol lactonization of the PT-cyclized product 3 to release pyrone 4 or its starter unit-dependent structural variant from the ACP (Scheme 1B).
RESULTS AND DISCUSSION
Alternative starter unit in vitro assays
The C-terminal His6-tagged PksA SAT–KS–MAT tridomain and PT–ACP didomain were over-produced in E. coli and purified by Co2+-affinity chromatography as previously described(18). Potential starter units were presented as N-acetylcysteamine (SNAC) thioesters (Figure 1), which were prepared by coupling of N-acetylcysteamine to commercially-available or readily synthesized carboxylic acids (see Supplemental Information). Each substrate (1 mM) was assayed under uniform conditions in phosphate buffer at pH 7 containing glycerol (10%), malonyl-SNAC (1 mM), TCEP (1 mM), PksA SAT-KS-MAT and PT-ACP (10 μM), and incubated at room temperature for 4 hours. Products were extracted into ethyl acetate, separated by HPLC and the UV-vis spectrum of each was recorded (200–800 nm). Exact masses were determined by UPLC/ESI-MS. In all instances the naphthopyrone products gave a characteristic absorbance signature.
Figure 1.

Acyl-SNAC starter units and their products (A) Potential acyl-SNAC (N-acetylcysteamine) starter units assayed in this study, and their observed naphthopyrone products. (B) Starred products that do not conform to the template shown in (A) due to unusual number of ketide extensions (also see Supporting Information).
Linear acyl-SNAC starter units
Early examination of PksA starter unit preferences of the SAT domain indicated modest selectivity for hexanoyl over butanoyl or octanoyl(15). In a first set of experiments, systematic examination of the homologous series of linear C4–C8 SNAC thioesters (5–9) was undertaken in the synthesis of norpyrone 4 and its structural variants. The non-native C5 fatty acid thioester was incorporated with comparable efficiency to the native hexanoyl starter (Figures 2, S1, S3), but reactions of C4, C7 and C8 chain lengths were still evident. To facilitate comparison among the C4–C8 thioesters and, indeed, all potential starter units examined, the naphthopyrone product peak intensities were plotted relative to that of hexanoyl in Figure 2. Starter units longer than C8 were not tested owing to their limited solubility under the assay conditions. Reactions of 6, 8 and 9 were less efficient in naphthopyrone product formation and correspondingly gave increased shunt and truncation products appearing at early retention times in the HPLC chromatogram (Figures S2, S4, S5). Recent deconstruction and domain “swapping” or heterodomain combinatorial experiments have revealed tight control of overall polyketide chain length to largely ± one ketide (C2) extension, while the PT domain exerts strict programming of ring formation despite receiving chain lengths both shorter and longer than wild-type(18, 19). Reactions utilizing the C8 starter unit 9 exemplify this tight chain length control, as one fewer ketide extension was performed to give a naphthopyrone product with the native C20 chain length. Similarly, one extra ketide extension occurred in the reaction of the C4 starter unit 6 to yield a C20 product. In the case of the C7 starter unit 8, both C21 and C19 products were observed, whereas only the C19 product was produced from the C5 starter unit 7. This overall chain-length constraint in PksA likely amplifies the apparent narrow range of well-accepted linear starter units.
Figure 2.
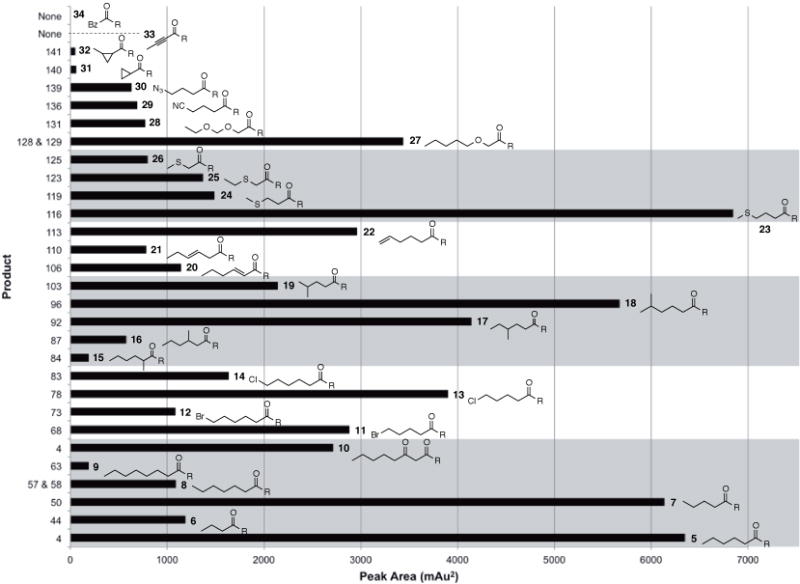
Relative production of on-target naphthopyrones. Product numbers are indicated on the y-axis and starter units are indicated on the right side of each bar. In the case of starter units 8 and 27, where two naphthopyrone products of different chain lengths were produced, the peak areas of both products have been combined for this chart.
To analyze the pattern of truncation and derailment products observed in all reactions, these off-target products have been categorized by basic core structures corresponding to known chromophores(19, 26)(Figure 3A, see Scheme S1 for full structures of all characterized products). Control reactions containing malonyl-SNAC as the sole substrate produced primarily the naphthopyrone 42 (Figure 3B) in which decarboxylated malonyl-SNAC was used as the starter unit. Monitoring the steady-state population of intermediates bound to the PksA ACP by mass spectrometry revealed acetyl in addition to the expected hexanoyl and malonyl(27). One can visualize that the appearance and inefficient utilization of an acetyl starter could arise by direct transfer from acetyl-SNAC, but more likely by transfer from malonyl-SNAC to the MAT domain and then to the ACP, followed by decarboxylation. In the case of the linear C4–C8 SNAC thioesters 5–9, the primary derailment products observed were the tri- and tetra-ketide lactones having core structure C, while only small amounts of improperly cyclized but fully extended and cyclodehydrated SEK4 and SEK4b analogs were produced (cores D and E). Therefore, extension by the KS domain may be the limiting factor in productive biosynthesis with linear starter units shorter or longer than the native hexanoyl primer, rather than cyclization in the proper register by the PT domain. As expected, the product profile for the reaction of C8 β-ketothioester 10 is similar to that of the native hexanoyl starter unit. While the observed products were nearly identical for the two reactions, efficiency was lower in the non-native case (Figure S6). As this starter unit represents the first intermediate product of elongation, the reason for decreased productivity presumably owes to reduced binding efficiency to the SAT (or KS domain, see below) domain before transfer to the ACP.
Figure 3.

Core structures of observed derailment products. (A) Derailment product cores were determined based on exact mass and known chromophores. (B) Major product observed in control reactions containing malonyl-SNAC as the sole substrate.
Halogenated acyl-SNAC starter units
Experiments conducted to evaluate the efficiency of naphthopyrone production by PksA utilizing substituted acyl-SNAC thioesters demonstrated that modifications at the distal end of the thioester were well tolerated by the enzyme, particularly halogenation and methylation. Starter units halogenated at the remote end of the acyl chain (11–14) were relatively well accepted, resulting in high product formation, albeit accompanied by significant amounts of derailment products, primarily the triacetic acid lactone analog (core C). The product profiles among the four tested halogenated starter units were similar, although relative and absolute amounts of on-target and derailment products varied depending on chain length and identity of the halogen substituent. Similar to the linear, un-substituted starter units, reactions of the longer 7-atom halogenated substrates resulted in higher derailment production and lower naphthopyrone production. Additionally, brominated products were produced in smaller amounts than chlorinated products, presumably owing to the relative steric bulk of the halogen substituent (Figures 2, S7–S10).
Methylated acyl-SNAC starter units
Biosynthetic production and catalytic efficiency decrease with chain branching close to the thioester, as exemplified with reactions utilizing methylated starter units 15, 16, 17, and 18. Starter units with substituents at the 2- and 3- position were particularly unfavored, while starter units substituted at the 4- and 5- position were significantly better tolerated. In the case of 2-branched 15, the major product was 42 (Figures 3B, S11), suggesting this starter unit is so poorly tolerated that inefficient priming and extension using decarboxylated malonyl-SNAC took place. Similarly, the reaction utilizing 16 also produced 42, albeit in a smaller proportion. The fully extended but improperly cyclized SEK4/SEK4b analogs 86a,b (cores D and E) were the major derailment products in reactions with 16, suggesting the KS domain is significantly more tolerant of substitution at the C-3 position than the PT (Figure S12). Although only a small difference in naphthopyrone production was observed between reactions utilizing 17 and 18 (Figures 2, S13, S14), there was a marked difference in the ratio of naphthopyrone to derailment products. Thioester 17 gave a significant amount of the SEK4/SEK4b analogs 91a,b (Figure S13). While it was expected that a five-carbon long starter unit branched near its terminus, 19, would give an outcome comparable to 17 based on the location of the methyl group, production of naphthopyrone product was significantly lower (Figures 2, S15). These results indicate that branched starter units, especially if proximal to the thioester, can impede the ability of the PT domain to properly cyclize the mature polyketide chain.
Unsaturated acyl-SNAC starter units
Analogous to the methyl-branched starter units, desaturation was relatively well tolerated at the alkyl terminus of the acyl chain (starter unit 22), while the α,β-unsaturated SNAC thioester 20 reacted poorly. This reduced reactivity may owe to the intrinsically greater stability of a conjugated thioester slowing each acyl transfer step, or enzyme inhibition by this electrophilic species. Three unsaturated starter units were tested for incorporation into naphthopyrone products. The reaction utilizing 22 resulted in moderate production of the on-target final product and few derailment products (Figures 2, S18). Conversely, reactions of 20 produced in the main the spontaneously cyclized SEK4/SEK4b analogs 104a,b (Figure S16). No significant peaks representing truncation products were observed in this reaction, suggesting the α,β-alkene was problematic for processing by the PT domain, but not the KS domain. Unexpectedly, production of the naphthopyrone in the reaction of 21 was approximately equal to that of 20, but a more diverse profile of derailment products was observed with 20 (Figures 2, S17). These included both improperly or partially cyclized, but fully extended products as well as truncation products. As was the case of the methyl-branched starter units, the relative rigidity afforded by the alkene interferes more with proper biosynthetic processing when located closer to the thioester.
Heteroatom-substitued acyl-SNAC starter units
PksA is capable of incorporating oxyether- and thioether-containing starter units into naphthopyrone products (e.g. 23–28), but significant decreases in production and efficiency were observed. Reactions utilizing methylthioether 23 resulted in product formation comparable to the analogous six-carbon starter unit 5, with only a slight increase in derailment products (Figures 2, S19). The major derailment product observed for this reaction was the triketide lactone 114 (core C), while no significant peak intensities correlating to the SEK4/SEK4b analogs were observed. Processing of 23 into the naphthopyrone product 116 (Figures 2, S19) is, therefore, as efficient as that of the native starter unit. Reactions of five- and four-atom thioethers (24, 25, 26) resulted in lower product formation than the comparable carbon-only starter units (Figures 2, S20–S22). This effect is particularly evident with starter unit 26, which produced significantly less product than its analogous carbon-only starter unit 6, reactions utilizing four-atom thioethers underwent one extra extension to maintain the native C20 chain length, providing further evidence for tight chain length control by the KS domain. Additionally, an eight-atom oxyether starter unit 27 produced a C20 chain length product, analogous to the C8 alkane starter unit 9. Surprisingly, production of the naphthopyrone product 129 (Figures 2, S23) containing this starter unit was significantly greater than that from starter unit 9, with correspondingly lower levels of derailment products. The seven-atom oxyether starter unit 28 produced much less naphthopyrone product than 27, and increased amounts of the spontaneously cyclized derailment product 130a,b (cores D and E) (Figure S24).
Starter units amenable to downstream coupling strategies, such as those containing terminal cyano- and azido- groups (e.g. 29 and 30), were accepted by the enzyme, albeit with relatively low targeted product formation. In both cases, significant amounts of triketide and tetraketide truncation products were produced (Figures S25, S26). In the case of starter unit 29, fully extended but improperly cyclized products 134a,b (Figure S25) were observed as well.
Sterically constrained acyl-SNAC starter units
To test the tolerance of PksA to bulkier, more rigid substrates, SNAC-thioesters containing cyclopropyl groups (31, 32), an alkyne (33) and a benzyl group (34) were assayed. In the case of 31 and 32, a small amount of naphthopyrone product containing the cyclopropyl group was detected in both reactions. Production with these starter units was extremely low, and all domains of the enzyme presumably had difficulty accommodating the cyclopropyl group within their active sites. Compound 42 was the dominant product of these reactions, suggesting decarboxylated malonyl-SNAC is a more favorable primer for these reactions than 31 and 32 (Figures S27, S28). This product was also observed in other reactions where production is poor, including starter units 6 and 15, as noted above, and 2-butynoyl-SNAC (33) (Figure S29). Control reactions where malonyl-SNAC alone was provided to the enzyme in the absence of a candidate acyl-SNAC starter unit gave similar results (Figure S31). Conversely, benzoyl-SNAC reactions did not produce naphthopyrone product (Figure S30). However, a very small amount of the triketide lactone (core C) was observed in the reaction of benzoyl-SNAC, suggesting some tolerance of this starter unit by the KS domain.
SAT active site mutants
A series of SAT active site (Cys117) mutants was tested for product formation to further evaluate the role of the SAT domain in starter unit selection. Mutants were initially selected to mimic the other two naturally-occuring SAT active site motifs (GXSXG as in Giberella zeae Pks13 and GXGXG as in Aspergillus terreus ACAS)(28, 29) as well as another non-nucleophilic mutant (C117A). Against expectation, presumably inactive C117A formed comparable levels of product to wild-type enzyme as depicted in Figure 4. Furthermore, the C117G mutant produced notably more product than wild-type. Reactions with C117S, however, resulted in extremely low product formation. This pattern was consistent among all starter units tested and presumably owes to hydrolysis of SNAC thioesters before inefficient acyl transfer to the ACP can take place.
Figure 4.

Comparison of hexyl-naphthopyrone (4) production by PksA-SAT active site mutants.
Given the largely unaltered reaction profiles with C117A and C117G variants, other SAT active site mutants were constructed to probe whether non-covalent binding was responsible for increased production in C117A and C117G mutants. C117L, C117Q and C117N mutations were attempted in an effort to partially occlude the active site. Soluble SAT-C117N-KS-MAT was obtained and evaluated for naphthopyrone product formation. Production by this mutant was comparable to that of the wild-type enzyme and the C117A mutant, suggesting any non-covalent binding to the active site likely plays a small role, if any (Figure 4). It is worth noting, however, that production from C117G was still higher than the other variants, consistent with the possibility that non-covalent recognition and transfer may yet play a role in bringing starters onto the enzyme.
Conclusions
PksA can accept and utilize a circumscribed selection of acyl-SNAc substrates as starter units to support naphthopyrone biosynthesis. The enzyme is tolerant to minor modifications in chain length and constitution of the thioester starter unit, especially at the distal end of the acyl chain, including substituents that allow subsequent modification of the resulting tricyclic product; for example “Click” chemistry(30). However, significant losses in production efficiency are observed with longer chain lengths or substitutions proximal to the thioester.
Recent experimental advances have allowed production of full-length NR-PKSs, albeit in much lower yields than smaller multi-domain constructs(19). As a control to measure the synthetic penalty, if any, exacted by dissection into a 2-part combination of SAT-KS-MAT and PT-ACP tri- and didomains, the corresponding fully intact 5-domain construct of PksA was prepared. Owing to low protein yields, in vitro reactions were performed with 1 μM PksA SAT-KS-MAT-PT-ACP, but under otherwise identical conditions to the previous experiments. Three starter units (hexanoyl-SNAC [5], pentanoyl-SNAC [7], and benzoyl-SNAC [34]) were selected to compare production from both efficient and inefficient substrates. For the former two efficient substrates, presuming reasonably that product formation tracks linearly with pentadomain enzyme concentration, production by the two-part combination lagged the intact construct by only a factor of 1.5 in the case of 5 and 1.2 in the case of 7 (Figure S32). As seen above in experiments with the two-part system, no naphthopyrone product containing the benzyl ring was observed in reactions of 34. This finding was in complete accord with similar comparisons of intact vs. domain combinations determined recently, and confirms the validity of the deconstruction method for the purposes of the experiments described herein(19).
The SAT active site mutants tested in this study gave unanticipated results. While some native SAT domains contain a glycine substitution of the active site cysteine, these have previously been presumed to be inactive and compensated for by the activity(ies) of another domain(s). Our experiments, however, suggest that at sufficiently high concentrations of starter unit, the KS can recognize the SNAC thioester as an adequate mimic of the normal ACP-delivered starter unit to be loaded directly. Additionally, the possibility remains that even in the absence of a covalent bond between the SAT domain and a starter unit, non-covalent binding may increase effective concentrations sufficiently for efficient biosynthesis. Although an active SAT domain is required for starter unit transfer within the HexA/HexB•PksA complex(22), under the super-physiological conditions utilized in this in vitro study, the SAT appears to be bypassed by some starter units and directly loaded on to the KS. Such relatively facile utilization of non-native starter units is fortuitous but useful from the perspective of synthetic biology and rationally-directed biosynthesis.
Unlike other SAT domains that scavenge acetyl-CoA from their cellular environment, PksA receives its hexanoyl starter unit directly in complex with Hex A/B and thus may have less pressure to be discriminating in its starter unit selection. Analogous observations have been made in the case of the HR-PKS•NR-PKS pair Hpm3 and Hpm8, where a SNAC mimic of the starter unit can rescue production in the absence of an active SAT domain(25). Based on the limited data from these two examples, it can be anticipated that when SAT domains exist in FAS•NR-PKS or HR-PKS•NR-PKS complexes, their primary function is interprotein transfer of a relatively large and/or complex starter unit and they are likely to be additionally flexible in the modified primers they accept and/or can be bypassed to directly load the KS. Diversity of production is then determined by the tolerance of the KS and PT to provide full polyketide chain elongation and correct cyclization register. At present no X-ray structure of a SAT domain has been achieved, but its availability would open the door to mutagenesis experiments to remodel its binding site with the goal of efficient uptake of altered starter units and transfer to sustain directed biosynthesis in a physiological context.
METHODS
Reagents
All chemicals were purchased from Sigma Aldrich unless otherwise noted.
Cloning
Cloning details for all constructs used in this study are provided in the Supporting Information. All plasmids used are summarized in Table S1, and primers are given in Table S2.
Protein Expression and Purification
All proteins were heterologously expressed in E. coli BL21(DE3) and isolated as described previously(27), with the exception that Co2+-TALON resin (GoldBio) was used in place of Ni2+-NTA resin. Expression of soluble pentadomain PksA SAT-KS-MAT-PT-ACP was achieved by an additional one-hour cold shock in ice water prior to induction with 1 mM IPTG. Following purification, proteins were dialyzed against assay buffer (100 mM potassium phosphate pH 7, 10% glycerol) and optionally frozen at −80 °C prior to use. If necessary, proteins were concentrated using 10K Molecular Weight Amicon Ultra Centrifugation Filters (Millipore). Protein concentration was measured in triplicate using the Bradford assay (Bio-Rad) and bovine serum albumin as a standard (New England Biolabs).
In vitro assays
Purified SAT-KS-MAT tridomain (10 μM) and PT-ACP didomain (10 μM) were combined with 1 mM malonyl-SNAC in assay buffer (100 mM potassium phosphate, pH 7.0, 10% glycerol, 1 mM TCEP). Reactions were initiated by addition of 1 mM acyl-SNAC starter unit and allowed to run for 4 h at room temperature. The 500 μL reactions were quenched with 10 μL hydrochloric acid and extracted three times with 500 μL ethyl acetate. Extractions were pooled and evaporated to dryness using a Speed-Vac. Enzymatic products were resuspended in 300 μL 40% aqueous acetonitrile for HPLC analysis.
HPLC and UPLC-ESI-MS analysis of enzymatic products
100 μL injections of enzymatic products were analyzed over a Prodigy ODS3 analytical column (4.6 × 250 mm, 5 μ, Phenomenex) by a gradient HPLC method using an Agilent 1200 instrument equipped with an autosampler. Bisolvent method: linear gradient of 20% acetonitrile/80% water to 90% acetonitrile over 35 min, hold 90% acetonitrile for 10 min, re-equilibrate to 20% acetonitrile over 5 min followed by a 10 min hold at 20% acetonitrile. UV-vis spectra were recorded over a range of 200–800 nm, and chromatograms recorded at 280 nm. High-resolution mass data were obtained by UPLC-ESI-MS on a Waters Acquity/Xeno-G2 in positive ion mode.
Synthesis of acyl-SNAC substrates
Malonyl-SNAC was produced using the malonyl-CoA synthetase MatB from Rhizobium leguminosarum and HPLC purified as described previously(26, 31). The synthesis of all acyl-SNAC starter units used in this study is described in the Supporting Information.
Supplementary Material
Acknowledgments
We thank Dr. J. W. Scheerer (present address Department of Chemistry, College of William & Mary) for exploratory experiments, the National Institutes of Health for financial support (ES001670) and A. G. Newman and P. A. Storm for helpful conversations and guidance.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
References
- 1.Cronan JE, Thomas J. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol. 2009;459:395–433. doi: 10.1016/S0076-6879(09)04617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 3.Crawford JM, Townsend CA. New insights into the formation of fungal aromatic polyketides. Nat Rev Microbiol. 2010;8:879–889. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirschning A, Hahn F. Merging chemical synthesis and biosynthesis: a new chapter in the total synthesis of natural products and natural product libraries. Angew Chem Int Ed Engl. 2012;51:4012–4022. doi: 10.1002/anie.201107386. [DOI] [PubMed] [Google Scholar]
- 5.Moore BS, Hertweck C. Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Natural Product Reports. 2002;19:70–99. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- 6.Goss RJ, Shankar S, Fayad AA. The generation of “unnatural” products: synthetic biology meets synthetic chemistry. Nat Prod Rep. 2012;29:870–889. doi: 10.1039/c2np00001f. [DOI] [PubMed] [Google Scholar]
- 7.Weissman KJ. Mutasynthesis – uniting chemistry and genetics for drug discovery. Trends Biotechnol. 2007;25:139–142. doi: 10.1016/j.tibtech.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 9.Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, Kushnir S, Ismail S, Fansa EK, Wittinghofer A, Schulz F, Sanchez-Garcia E. Predicted incorporation of non-native substrates by a polyketide synthase yields bioactive natural product derivatives. Chembiochem. 2014;15:1991–1997. doi: 10.1002/cbic.201402206. [DOI] [PubMed] [Google Scholar]
- 10.Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MC. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341:1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 12.Udwary DW, Merski M, Townsend CA. A method for prediction of the locations of linker regions within large multifunctional proteins, and application to a type I polyketide synthase. Journal of Molecular Biology. 2002;323:585–598. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumann J, Hertweck C. Advances in cloning, functional analysis and heterologous expression of fungal polyketide synthase genes. J Biotechnol. 2006;124:690–703. doi: 10.1016/j.jbiotec.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 14.Cox RJ. Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 15.Crawford JM, Dancy BC, Hill EA, Udwary DW, Townsend CA. Identification of a starter unit acyl-carrier protein transacylase domain in an iterative type I polyketide synthase. Proc Natl Acad Sci U S A. 2006;103:16728–16733. doi: 10.1073/pnas.0604112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford JM, Korman TP, Labonte JW, Vagstad AL, Hill EA, Kamari-Bidkorpeh O, Tsai SC, Townsend CA. Structural basis for biosynthetic programming of fungal aromatic polyketide cyclization. Nature. 2009;461:1139–1143. doi: 10.1038/nature08475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Image II, Xu W, Image I, Tang Y, Image I. Classification, prediction, and verification of the regioselectivity of fungal polyketide synthase product template domains. J Biol Chem. 2010;285:22764–22773. doi: 10.1074/jbc.M110.128504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vagstad AL, Newman AG, Storm PA, Belecki K, Crawford JM, Townsend CA. Combinatorial domain swaps provide insights into the rules of fungal polyketide synthase programming and the rational synthesis of non-native aromatic products. Angew Chem Int Ed Engl. 2013;52:1718–1721. doi: 10.1002/anie.201208550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman AG, Vagstad AL, Storm PA, Townsend CA. Systematic domain swaps of iterative, nonreducing polyketide synthases provide a mechanistic understanding and rationale for catalytic reprogramming. J Am Chem Soc. 2014;136:7348–7362. doi: 10.1021/ja5007299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang PK, Cary JW, Yu J, Bhatnagar D, Cleveland TE. The Aspergillus parasiticus polyketide synthase gene pksA, a homolog of Aspergillus nidulans wA, is required for aflatoxin B1 biosynthesis. Mol Gen Genet. 1995;248:270–277. doi: 10.1007/BF02191593. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe CM, Townsend CA. Initial characterization of a type I fatty acid synthase and polyketide synthase multienzyme complex NorS in the biosynthesis of aflatoxin B(1) Chem Biol. 2002;9:981–988. doi: 10.1016/s1074-5521(02)00213-2. [DOI] [PubMed] [Google Scholar]
- 22.Foulke-Abel J, Townsend CA. Demonstration of starter unit interprotein transfer from a fatty acid synthase to a multidomain, nonreducing polyketide synthase. Chembiochem. 2012;13:1880–1884. doi: 10.1002/cbic.201200267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, Smith LH, Cox RJ, Beltran-Alvarez P, Arthur CJ, Simpson FRST. Catalytic relationships between type I and type II iterative polyketide synthases: The Aspergillus parasiticus norsolorinic acid synthase. Chembiochem. 2006;7:1951–1958. doi: 10.1002/cbic.200600341. [DOI] [PubMed] [Google Scholar]
- 24.Crawford JM, Vagstad AL, Ehrlich KC, Townsend CA. Starter unit specificity directs genome mining of polyketide synthase pathways in fungi. Bioorg Chem. 2008;36:16–22. doi: 10.1016/j.bioorg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Qiao K, Gao Z, Meehan MJ, Li JW, Zhao X, Dorrestein PC, Vederas JC, Tang Y. Enzymatic synthesis of resorcylic acid lactones by cooperation of fungal iterative polyketide synthases involved in hypothemycin biosynthesis. J Am Chem Soc. 2010;132:4530–4531. doi: 10.1021/ja100060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vagstad AL, Bumpus SB, Belecki K, Kelleher NL, Townsend CA. Interrogation of global active site occupancy of a fungal iterative polyketide synthase reveals strategies for maintaining biosynthetic fidelity. J Am Chem Soc. 2012;134:6865–6877. doi: 10.1021/ja3016389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crawford JM, Thomas PM, Scheerer JR, Vagstad AL, Kelleher NL, Townsend CA. Deconstruction of iterative multidomain polyketide synthase function. Science. 2008;320:243–246. doi: 10.1126/science.1154711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H, Zhan J, Watanabe K, Xie X, Tang Y. A polyketide macrolactone synthase from the filamentous fungus Gibberella zeae. Proc Natl Acad Sci U S A. 2008;105:6249–6254. doi: 10.1073/pnas.0800657105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Awakawa T, Yokota K, Funa N, Doi F, Mori N, Watanabe H, Horinouchi S. Physically discrete beta-lactamase-type thioesterase catalyzes product release in atrochrysone synthesis by iterative type I polyketide synthase. Chem Biol. 2009;16:613–623. doi: 10.1016/j.chembiol.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 31.An JH, Kim YS. A gene cluster encoding malonyl-CoA decarboxylase (MatA), malonyl-CoA synthetase (MatB) and a putative dicarboxylate carrier protein (MatC) in Rhizobium trifolii–cloning, sequencing, and expression of the enzymes in Escherichia coli. Eur J Biochem. 1998;257:395–402. doi: 10.1046/j.1432-1327.1998.2570395.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.