

Abstract
The psychostimulant methamphetamine (METH) is highly addictive and neurotoxic to dopamine terminals. METH toxicity has been suggested to be due to the release and accumulation of dopamine in the cytosol of these terminals. The vesicular monoamine transporter 2 (VMAT2; SLC18A2) is a critical mediator of dopamine handling. Mice overexpressing VMAT2 (VMAT2-HI) have an increased vesicular capacity to store dopamine, thus augmenting striatal dopamine levels and dopamine release in the striatum. Based on the altered compartmentalization of intracellular dopamine in the VMAT2-HI mice, we assessed whether enhanced vesicular function was capable of reducing METH-induced damage to the striatal dopamine system. While wildtype mice show significant losses in striatal levels of the dopamine transporter (65% loss) and tyrosine hydroxylase (46% loss) following a 4 × 10 mg/kg METH dosing regimen, VMAT2-HI mice were protected from this damage. VMAT2-HI mice were also spared from the inflammatory response that follows METH treatment, showing an increase in astroglial markers that was approximately one-third of that of wildtype animals (117% vs 36% increase in GFAP, wildtype vs VMAT2-HI). Further analysis also showed that elevated VMAT2 level does not alter the ability of METH to increase core body temperature, a mechanism integral to the toxicity of the drug. Finally, the VMAT2-HI mice showed no difference from wildtype littermates on both METH-induced conditioned place preference and in METH-induced locomotor activity (1 mg/kg METH). These results demonstrate that elevated VMAT2 protects against METH toxicity without enhancing the rewarding effects of the drug. Since the VMAT2-HI mice are protected from METH despite higher basal dopamine levels, this study suggests that METH toxicity depends more on the proper compartmentalization of synaptic dopamine than on the absolute amount of dopamine in the brain.

Keywords: Methamphetamine, VMAT2, vesicle, neurodegeneration, inflammation, dopamine
The addictive psychostimulant methamphetamine (METH) is severely neurotoxic to the dopamine system, selectively targeting dopaminergic projections, depleting striatal dopamine levels, and initiating a large neuroinflammatory cascade.1–5 METH induces a massive efflux of intracellular dopamine into the extracellular space by reducing the pH gradient across the vesicular membrane, emptying vesicular dopamine stores, and reversing plasmalemmal dopamine transporter (DAT) function.6–8 In this way, METH causes a disruption of dopamine compartmentalization at the synaptic terminal. While the mechanism of METH toxicity remains unclear, the accumulation of cytosolic dopamine following METH treatment is thought to be central to the neurotoxicity of the compound.4,9,10 Cytosolic dopamine, when not sequestered into the vesicular compartment, is vulnerable to oxidation and conversion to neurotoxic species, including the formation of dopamine-quinones and cysteinyl adducts.9,11 Additionally, METH promotes dopamine synthesis via upregulation of tyrosine hydroxylase (TH) function12 and opposes dopamine degradation via reductions in monoamine oxidase (MAO) activity.13 These mechanisms elevate cytosolic dopamine levels and potentially exacerbate METH toxicity. Changes to core body temperature can also modify METH toxicity, independent of dopamine content in the striatum.14 Additional proposed mechanisms for METH toxicity have included free radical accumulation and oxidative stress,4,15 altered ion gradients,16 and mitochondrial dysfunction.17 METH toxicity is indicated by losses of the dopamine terminal markers DAT and TH in the striatum.1,5 This damage to dopamine terminals is also accompanied by extensive neuroinflammation through the activation of both astrocytes and microglia, which contribute to neurodegeneration over prolonged periods.2,18–21
The vesicular monoamine transporter 2 (VMAT2, SLC18A2) is a critical mediator of dopamine dynamics in the neuronal terminal. VMAT2 is an H+-ATPase antiporter, which uses the vesicular electrochemical gradient to drive the packaging of cytosolic monoamines (dopamine, serotonin, norepinephrine, histamine) into small synaptic and dense core vesicles.22–25 By preventing the accumulation of dopamine in the neuronal cytosol, VMAT2 also counters intracellular dopamine toxicity.11,26,27 While genetic knockout of VMAT2 in mice is lethal,28–30 mice with a 95% reduction in VMAT2 survive and display both lower monoamine levels and progressive neurodegeneration with aging.31,32 These VMAT2-deficient mice show increased dopamine turnover and increased cysteinyl-DOPA and DOPAC adducts, suggesting that improper dopamine storage leads to an accumulation of potentially neurotoxic byproducts. Most recently, reduced vesicular storage has been linked to dysfunction in the dopamine system in humans. A familial VMAT2 mutation was shown to dramatically reduce vesicular transport and cause an infantile parkinsonism condition.33 Large reductions in VMAT2-mediated transport have also been identified in post mortem Parkinson’s disease brains beyond what could be explained by terminal loss alone.34 These data suggest that manipulation of synaptic terminal dopamine handling promotes nigrostriatal neurodegeneration in human populations.
While the detrimental effects of reduced VMAT2 function have been examined in the studies above, an in vivo model of permanently increased vesicular storage has only recently become available using BAC-transgenic VMAT2-overexpressing (VMAT2-HI) mice.35 As a result of their elevated VMAT2 expression, VMAT2-HI mice display increased vesicular capacity, increased striatal dopamine content and release, and increased locomotor activity.35 VMAT2-HI mice are also protected from the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) since its metabolite, MPP+, is sequestered by VMAT2. VMAT2 is also a known target of METH, and METH treatment reduces VMAT2-mediated vesicular uptake.8,36 Mice with reductions in VMAT2 are more vulnerable to METH toxicity, as measured by exacerbated dopamine terminal degeneration, dopamine depletion, and neuroinflammatory responses.37,38 Based on this work, it was predicted that elevated vesicular function would oppose the neurotoxic and neuroinflammatory consequences of METH. To explore the additional benefits of permanently elevated VMAT2, this study examined the effects of increased vesicular capacity on in vivo METH toxicity in the VMAT2-HI mice.
RESULTS
Increased VMAT2 Protects against Loss of Dopamine Neuron Terminal Markers in the Striatum
Male wildtype and VMAT2-HI mice were treated with four injections (s.c.) of either saline (0.9%) or methamphetamine (free base, 10 mg/kg w/v) every 2 h and sacrificed 48 h after the final injection. METH toxicity preferentially targets striatal dopamine terminals. Following this 4 × 10 mg/kg METH dosing regimen, wildtype mice showed dramatic losses in the striatal dopamine terminal markers, DAT and TH, as measured by immunoblotting and immunohistochemistry (Figure 1A–D). VMAT2-HI mice showed significant protection from this DAT and TH loss following METH treatment. Elevated VMAT2 levels were confirmed in the VMAT2-HI mice and also protected from loss after METH (Figure 1E,F). At higher magnifications, the preservation of striatal TH innervation in METH-treated VMAT2-HI mice was dramatic (Figure 2). In keeping with the typical terminal-targeting of METH toxicity, this 4 × 10 mg/kg METH dose did not induce any cell body loss in the substantia nigra pars compacta as measured by stereological counts of TH-positive neurons (Figure 3).
Figure 1.
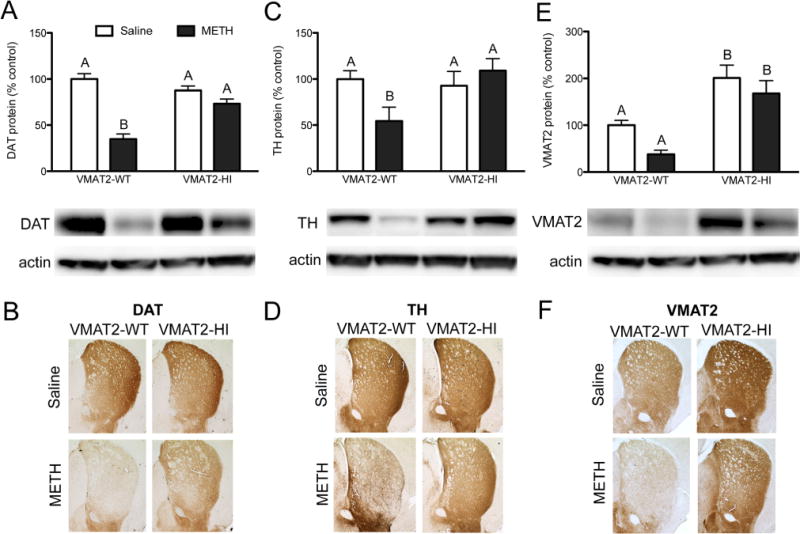
Increased VMAT2 protects against TH and DAT loss in the striatum. Male wildtype and VMAT2-HI mice were treated with four injections of either saline or 10 mg/kg methamphetamine every 2 h and sacrificed 48 h after the final injection. (A–D) Wildtype animals show significant losses in DAT and TH levels. VMAT2-HI mice are significantly protected from losses in both DAT (p < 0.01) and TH (p < 0.05) following METH. (E,F) Wildtype mice also show a trend toward VMAT2 protein loss following METH, while VMAT2-HI mice maintain a VMAT2 level greater than wildtype even following METH. Data are presented as percent of saline-treated wildtype mice. Different letters at the tops of bars indicate differences of at least p < 0.05 as determined by a two-way ANOVA with Bonferroni posthoc tests (n = 6).
Figure 2.

Increased VMAT2 protects against TH+ fiber denervation in the striatum. VMAT2-HI mice are protected from the loss of TH+ fibers in the striatum. Representative images of dorsolateral striatum pictured with cortex on the right side of each image. Scale bar = 200 μm.
Figure 3.
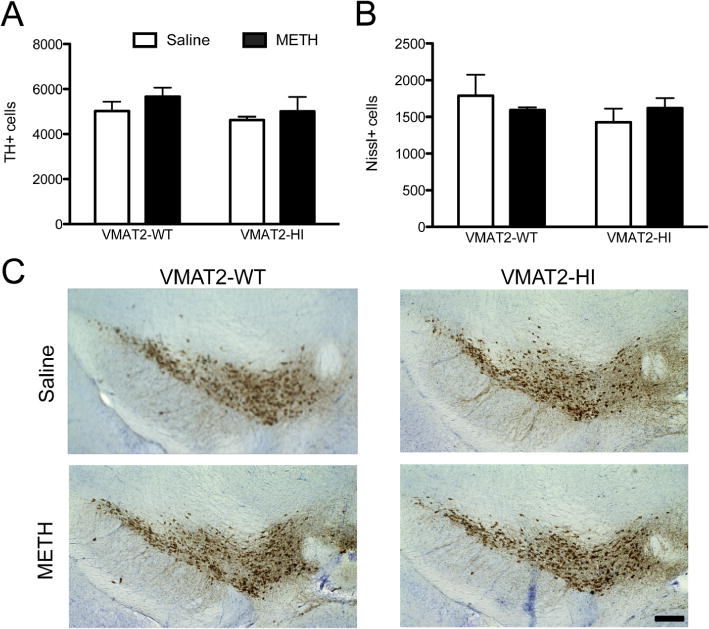
METH treatment did not induce SNpc cell body loss. (A,B) There was no difference in TH+ cells (p > 0.05) or Nissl+ cells (p > 0.05) between the genotypes following a 4 × 10 mg/kg METH dose (n = 6). (C) Representative images of TH staining of the midbrain with and without METH treatment.
Increased VMAT2 Protects against Gliosis in the Striatum
METH is known to induce a large inflammatory response, which can be assessed by glial markers in the striatum.2 Wildtype mice showed a significant increase in glial fibrillary acidic protein (GFAP) expression both by immunoblotting and immunohistochemistry (Figure 4A,B). VMAT2-HI mice were protected from this astrocyte response as indicated by a significantly smaller increase in GFAP levels. Similarly, wildtype mice showed substantial activation of microglia in response to METH as measured by isolectin B4 (IB4) staining (Figure 4C,D). VMAT2-HI mice showed less amoeboid microglia morphology following METH treatment when compared to wildtype animals, indicating reduced activation of striatal microglia.
Figure 4.

Increased VMAT2 protects against gliosis in the striatum. (A,B) VMAT2-HI mice show a significantly smaller increase in astrogliosis as indicated by GFAP expression (n = 6). Different letters above the bars indicate difference of p < 0.05. Data are presented as percent of saline-treated wildtype mice. (C,D) VMAT2-HI mice show less ramified microglia as shown by IB4 staining. Representative images of dorsolateral striatum pictured with corpus callosum in the upper right corner of each image. Scale bar = 200 μm.
Preferential Targeting of the Striosomes Following METH Treatment
METH treatment causes preferential loss of dopaminergic markers like DAT and TH in the striosomes of the striatum, rather than the surrounding matrix.39 Immunohistochemical analysis also showed a loss of dopaminergic terminals in the striosomes following a 4 × 10 mg/kg METH dose in both genotypes as shown by striatal patches with reduced DAT immunoreactivity (Figure 5B). At a lower 4 × 5 mg/kg METH dose, wildtype animals still showed a striosomal loss of DAT (Figure 5A). However, the VMAT2-HI animals were completely spared from this preferential striosome loss at this lower METH dose.
Figure 5.

Preferential targeting of the striosomes following METH treatment. (A,B) Selective loss of striosomal DAT was shown in both wildtype and VMAT2-HI mice. However, this loss was not seen until a higher 4 × 10 mg/kg METH dose in the VMAT2-HI animals (A). (C) Representative tracing of DAT loss in striosomes of wildtype METH-treated striatum following a 4 × 5 mg/kg METH dose.
Increased VMAT2 Level Does Not Alter the Hyperthermic Response Following METH Treatment
Binge METH treatment induces a significant increase in core body temperature that is critical to the neurotoxic effects of the drug.40 Core temperatures were taken at baseline and 1 h after each METH injection in both genotypes. Both wildtype and VMAT2-HI mice showed significant increases in core body temperatures following METH treatment (Figure 6). However, the elevation in core temperature following METH treatment was the same between the genotypes. In addition, when wildtype and VMAT2-HI animals were treated with a lower 4 × 5 mg/kg METH dose that did not cause hyperthermia, VMAT2-HI mice were still protected from terminal degeneration and inflammatory markers (Supporting Information Figure 1).
Figure 6.

Increased VMAT2 level does not alter the hyperthermic response following METH treatment. Core temperatures taken 1 h post each METH injection (injections indicated by arrows). While there was a significant increase in core temperature in both genotypes, there was no difference between the hyperthermic profiles between wildtype and VMAT2-HI mice (p > 0.05) (n = 12).
Increased VMAT2 Does Not Change METH-Induced Conditioned Place Preference
Due to the reinforcing properties of METH, it was important to examine the effects of elevated VMAT2 and the associated increased dopamine output on METH-induced conditioned place preference behavior. A 1 mg/kg methamphetamine conditioning paradigm was used since it is a standard dose used in place preference behaviors.41 Both wildtype and VMAT2-HI mice developed a preference for the METH-paired side of the test chamber following 1 mg/kg METH conditioning sessions (Figure 7A). VMAT2-HI mice showed no difference in time spent on the METH-paired side compared to their wildtype littermates. Furthermore, 1 mg/kg METH increased locomotor activity to the same level in both wildtype and VMAT2-HI mice, despite a greater baseline activity level in the VMAT2-HI mice (Figure 7B).
Figure 7.

Increased VMAT2 does not change METH-induced conditioned place preference or METH-stimulated locomotor activity. Both genotypes show a preference at 1 mg/kg METH (n = 9). However, there was no difference between genotypes on time spent in the METH-paired side of the chamber on test day. Similarly, wildtype and VMAT2-HI mice show no difference in locomotor activity when the genotypes were treated with METH. Different letters at the tops of the bars indicate difference of p < 0.01.
DISCUSSION
Elevated VMAT2 Protects against METH Toxicity
Both in vitro and in vivo evidence shows that VMAT2 function acts as a neuroprotective mechanism in dopamine neurons.35,38,42–44 Reduced VMAT2 levels increase cytosolic dopamine metabolism and cause both progressive dopaminergic loss and an exaggerated response to a toxic insult.29,31,37,38,45 Due to the increased vesicular capacity in the VMAT2-HI mice, it was predicted that these mice would have a reduced cytosolic dopamine burden when challenged with METH, thus protecting the midbrain dopamine pathway. This study shows that the VMAT2-HI mice are spared from dopaminergic terminal loss by immunochemical techniques at two different METH doses (Figure 1 and Supporting Information Figure 1). Furthermore, there is a preferential targeting of the striosomes for degeneration, as compared to the surrounding striatal matrix (Figure 5). Striosomes are characterized by lower levels of superoxide dismutase 2 (SOD2), which reduces reactive oxygen species,46 and also increased vascularization that may increase exposure to the drug.47 Both of these factors may contribute to the elevated METH toxicity in these areas. It appears that elevated VMAT2 levels do not alter the striosome-targeting aspect of METH toxicity since the VMAT2-HI mice still showed a preferential loss of terminal markers in the striosomes, just at a higher METH dose (Figure 5). These results suggest that the neuroprotection in the VMAT2-HI mice represents a shifting of the severity of METH toxicity and not an alteration of the mechanism of this toxicity.
Elevated VMAT2 Reduces the Neuroinflammatory Response to METH
The VMAT2-HI mice showed a reduction in neuroinflammatory markers following METH treatment, which was reflected by less astrogliosis than wildtype animals (Figure 4). Both genotypes showed increased microglial expression in METH-treated mice using isolectin B4 staining. However, it appears that IB4 staining in METH-treated wildtype mice showed amoeboid morphological changes indicating activated microglia, whereas VMAT2-HI mice had less of this dysmorphic microglial staining. Interestingly, the VMAT2-HI mice still had an inflammatory response to METH despite an almost complete protection from dopamine terminal damage after METH. Though it remains unclear as to what mechanisms induce the neuroinflammatory cascade following METH treatment, extracellular dopamine has been implicated in this response. Inflammatory markers have been reported to precede terminal damage,2 which suggests that neuroinflammation is due to the acute effects of METH, presumably via extracellular dopamine release. METH treatment of DAT-knockout mice does not cause dopamine efflux, and these DAT-knockout mice also have no METH-induced glial activation.48 These findings also suggest that the inflammatory cascade is dependent on the efflux of dopamine. Based on the behavioral effects of METH (Figure 7), it is clear that both genotypes experience dopamine efflux after METH, which likely accounts for the initiation of the inflammatory response. The smaller magnitude of the neuroinflammatory response in the VMAT2-HI mice may be due to a more efficient repackaging of displaced dopamine into vesicles compared to wildtype mice. However, VMAT2-HI mice have also been shown to have greater baseline dopamine levels,35 which has the potential to exacerbate an inflammatory response in this scenario. Microdialysis experiments would be needed to examine METH-stimulated dopamine efflux and uptake between the genotypes. It is more likely that the protection from the neuroinflammatory response in the VMAT2-HI mice is occurring at a later time point and is due to the survival of more dopamine terminals after METH. The preservation of terminal integrity in the VMAT2-HI mice would result in fewer cues (e.g., cytokines, chemokines) to signal neuroinflammation. Further studies on the time course of VMAT2’s neuroprotective effect and the release of these cues would be needed to address such questions.
Elevated VMAT2 Does Not Change the Hyperthermic Response to METH
METH administration raises core body temperature, a mechanism integral to the toxicity of the drug.14,40,49,50 Even in the absence of dopamine, elevations in body temperature following METH treatment are still able to induce dopaminergic degeneration.14 Pharmacologically lowered core temperature is also neuroprotective against METH. This protection has been shown with a variety of compounds that change dopamine signaling through dopamine receptor antagonism and TH inhibition.51 Based on decades of work on the hypothermic effects of the VMAT2 inhibitor reserpine,52 it was necessary to show that the VMAT2-HI mice have no alterations to their core temperatures either at baseline or following METH. Wildtype and VMAT2-HI mice showed identical baseline temperatures and hyperthermic responses following METH treatment (Figure 6). These results eliminate the potential confound that the neuroprotection seen in the VMAT2-HI mice is due to an altered response in core temperature.
Elevated VMAT2 Level Does Not Increase The Rewarding Potential For Meth
The data presented here suggest that elevated VMAT2 level or function would be beneficial in protecting against a dopaminergic toxicant. However, the elevated dopamine tissue content and stimulated dopamine release in the VMAT2-HI mice could also cause negative behavioral effects in the presence of a psychostimulant like METH.35 Surprisingly, the VMAT2-HI mice showed no increase in conditioned place preference for 1 mg/kg METH (Figure 7a), a dose shown to induce conditioned place preference and locomotor sensitization in mice.53,54 Additionally, the VMAT2-HI mice showed the same level of enhanced locomotor activity following this METH dose, even though they have elevated activity at baseline (Figure 7B), as previously published.35 These results suggest that vesicle-based approaches aimed at reducing intracellular toxicity may not negatively impact other dopamine-mediated behaviors.
VMAT2-HI Mice Suggest That Dopamine Compartmentalization Is Integral to METH Neurotoxicity
While some may assume that the elevated striatal dopamine levels in the VMAT2-HI mice would actually be detrimental in a METH model, these data suggest that the majority of this dopamine is distributed into the vesicles, thus preserving terminal integrity. The reduced METH toxicity in the VMAT2-HI mice complements the “weak base” mechanism of METH action, which suggests that amphetamines act as a base once inside the synaptic vesicle, reducing the vesicular pH gradient and depleting vesicular contents.55,56 One can imagine various ways that the increased vesicular function in the VMAT2-HI mice may protect a dopamine neuron from METH. First, increased VMAT2 levels may maintain vesicular filling in the presence of METH and counteract the METH-induced vesicular depletion of dopamine. This would keep more dopamine sequestered into vesicles and less potentially toxic dopamine byproducts in the cytosol. This protection would occur acutely after METH treatment. Indeed, the 4 × 10 mg/kg METH dosing paradigm has been shown previously to rapidly decrease vesicular [3H]-dopamine uptake in purified synaptic vesicles from rats.36 Alternatively, increased VMAT2 levels could cause a faster recovery of the pH gradient following the disappearance of METH. METH reduces vesicular uptake as soon as 1 h after binge dosing and maintains this up to 24 h later,36 suggesting that enhanced vesicular storage may positively impact dopamine distribution even long after METH administration has ended. In this way, elevated VMAT2 levels may allow for a more efficient restoration of vesicular dopamine contents in the VMAT2-HI mice. Both of these proposed mechanisms of protection depends on the distribution of cytosolic dopamine throughout the neuron.
Cytosolic Dopamine Accumulation and METH Toxicity
The focus on cytosolic dopamine accumulation as the source for METH-induced dopaminergic toxicity in these experiments was based on findings from mice with reduced VMAT2 expression37 and in vitro measures of vesicular function.55 The current findings also complement previous work done via VMAT2 overexpression in PC12 cells64 and on the effects of the pituitary adenylyl cyclase-activating polypeptide (PACAP38) in mice,65 both of which suggested that manipulation of VMAT2 level can alter dopamine storage and ameliorate aspects of METH toxicity.
Dopamine molecules left unpackaged in the neuronal cytosol can create neurotoxic byproducts through both oxidative and metabolic processes. Cytosolic dopamine can be converted to form reactive oxygen species, such as hydroxyl radicals, superoxide, and hydrogen peroxide.11 Oxidized dopamine can form both dopamine-quinones and cysteinyl adducts that are capable of altering intracellular protein function, including inhibiting DAT and TH,57–59 activating microglia,3 and potentially contributing to dopaminergic neurodegeneration.9 Dopamine can also be metabolized to the catecholaldehyde, 3,4-dihydroxyphenylacetaldehyde (DOPAL),60 which can itself induce quinone and hydroxyl radical formation, protein cross-linking, and oligomerization of potentially hazardous proteins such as α-synuclein.26,61–63 The current study cannot provide a definitive explanation for the neuroprotection afforded by VMAT2 without a quantification of the cytosolic dopamine levels in the VMAT2-HI mice. Future experiments should examine cytosolic dopamine concentration via intracellular patch electrochemistry.66–68 More indirectly, one could also assess dopamine turnover as described by the DOPAC/DA ratio. Due to uncertainties of the timeline of VMAT2’s protective effects post METH insult, a single time point may not reflect the dynamic nature of the toxic response. A full time course experiment would be needed to describe the neurochemical changes behind this protection.
Elevated VMAT2 Levels and Other Potential Mechanisms of METH Toxicity
It is possible that the neuroprotection via increased VMAT2 is not directly dependent upon dopamine handling at all. Although the current study eliminated the possibility that core temperature was modifying neuronal vulnerability, multiple alternative hypotheses remain to explain METH toxicity. With the elaborate arborization and large number of synapses made by striatal dopamine neurons, it has been suggested that these cells carry a substantial bioenergetic burden.69 If increased VMAT2 more efficiently allows for the packaging of transmitter and toxic molecules, it is possible that the oxidative stress and energy consumption of these synapses may also be reduced.4,15 Additionally, it is also possible that VMAT2 function could also contribute to ameliorate other deficits caused by METH including ion gradients,16 mitochondrial dysfunction,17,70 and excitotoxicity via glutamate release onto dopamine neurons,71 though these mechanisms have yet to be entirely characterized.
VMAT2 Inhibitors as a Treatment for Addiction
Recent work has suggested that the inhibition of VMAT2 may be a therapeutic treatment to reduce psychostimulant abuse.72,73 Lobeline, an inhibitor of VMAT2 at the tetrabenazine binding site, decreases amphetamine-evoked dopamine output, but not electrically stimulated output, suggesting that lobeline provides a selective inhibition of amphetamine effects. It has been suggested that this reduction occurs via increased dopamine metabolism and reduced dopamine available for reverse transport through the DAT.74 Thus, by taking a VMAT2 inhibitor, dopamine-releasing drugs such as amphetamine have less of a rewarding or euphoric effect, providing a potential therapeutic use in addiction. However, long-term reductions in VMAT2 function through pharmacological (e.g., reserpine and tetrabenazine) and genetic (e.g., VMAT2-deficient mice) manipulations have been shown to increase vulnerability to neurodegeneration, reduce locomotion, and dramatically alter affective behaviors.31,37,38,75–77 Based on these findings and those from the current study, a targeted VMAT2-inhibitor should be pursued cautiously and, perhaps, for only brief treatment periods.
Neuroprotection via Elevated VMAT2 Function
The data presented here suggest that a positive modulator of VMAT2 may be highly beneficial in preventing cellular toxicity in dopamine neurons. Additionally, these findings suggest that elevated VMAT2 function does not enhance the rewarding effects of METH, a critical distinction that could have confounded the therapeutic potential of a positive VMAT2 modulator. VMAT2 modulation may be especially relevant to dopamine neurons, which are often considered to be vulnerable to insult as seen in a variety of genetic and toxicant models of dopamine dysfunction.42,78 Evidence also suggests that METH toxicity may elevate the risk of dopamine system dysfunction based on emerging studies in human populations. For example, METH and amphetamine users have up to a 3-fold increase in the likelihood of developing Parkinson’s disease throughout their lifetime.79,80 Thus, in hypodopaminergic states like Parkinson’s disease or the METH-treated brain, increasing vesicle function would provide two therapeutic benefits: restoration and enhancement of dopamine signaling and protection against neurotoxicity.
METHODS
Animals
VMAT2-HI mice were created by BAC-transgenic overexpression of the Slc18a2 gene as previously published.35 VMAT2-HI mice were bred onto a Charles River C57BL/6 background. Male mice of 6–8 months of age were used for all studies. Twelve animals were used per group, with half of each group going to either immunoblotting or immunohistochemistry. Mice received food and water ad libitum on a 12:12 light cycle. All procedures were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Emory University.
Methamphetamine Injection Schedule
(+)-Methamphetamine HCl (Sigma, St Louis, MO) was dissolved in 0.9% saline and administered subcutaneously. Mice were given a neurotoxic regimen of four doses of 10 mg/kg (free base) METH, 2 h apart, and killed by rapid decapitation 48 h after the last dose. A 5 mg/kg methamphetamine dose was used for the low dose experiment in Supporting Information Figure 1. Tissue for immunoblotting was flash frozen in liquid nitrogen. Tissue for immunohistochemical analysis was drop-fixed in 4% paraformaldehyde.
Core Body Temperature
Core body temperature was taken 30 min before the first injection and 1 h after the three subsequent injections of saline or METH. Temperature was taken rectally by using a digital thermometer (VWR International, Westchester, PA) lubricated with 100% petroleum jelly.
Antibodies
Rabbit polyclonal anti-VMAT2 serum was raised against a peptide in the C-terminal region of mouse VMAT2 (CTQNNVQPYPVGDDEESESD).35 Rat anti-DAT and rabbit anti-TH were purchased from Millipore. Mouse anti-GFAP was purchased from Cell Signaling Technologies. Mouse anti-β-actin was purchased from Sigma. All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories.
Western Blotting
Western blots were performed as previously described.31 Primary antibodies used were polyclonal rabbit anti-VMAT2 serum (1:20 000), rat anti-DAT (1:5000), rabbit anti-TH (1:1000), mouse anti-GFAP (1:5000), and mouse anti-β-actin (1:5,000). The appropriate HRP-linked secondary antibodies (1:5000) were used. Analysis was calibrated to coblotted dilutional standards of pooled sample from all VMAT2-HI samples.
Immunohistochemistry
Immunohistochemistry was performed as previously described.31 Primary antibodies used were polyclonal rabbit anti-VMAT2 serum (1:50 000), rat anti-DAT (1:1000), mouse anti-GFAP (1:1000), or rabbit anti-TH (1:1000). The appropriate biotinylated secondary antibodies (1:200) were used. Nissl stain was performed for stereological analysis by a 3 min Cresyl Violet dip of mounted sections prior to dehydration, xylene clearing, and coverslipping. All images were acquired with NeuroLucida (MicroBright-Field).
Isolectin B4
For microglial visualization, sections were incubated with biotinylated isolectin B4 (1:250; Invitrogen) overnight at room temperature and then incubated 1 h in avidin–biotin–HRP conjugate solution (Vectastain ABC kit, Vector Laboratories) as previously reported.37 Visualization was performed using DAB as above for 25 min at room temperature.
Stereological Analysis
Stereological sampling was performed using the Stereo Investigator software as previously described, and the number of neurons in the SNpc was estimated using the optical fractionator method in StereoInvestigator (MicroBrightField).32,35 Parameters, cell type definition, and counting intervals were also the same as previously described. Gundersen’s coefficient of error was held to 0.1 for all values.
Conditioned Place Preference (CPP)
Place conditioning was performed as previously published.53 Three compartment CPP chambers (San Diego Instruments, La Jolla, CA) were used for training and testing. The test pairs drug administration with one chamber of a two-chamber arena. In preconditioning (day 1), mice were placed into the open center chamber of the apparatus and allowed to freely explore the apparatus for 30 min. Time spent in each side chamber was recorded. Animals were assigned to METH or saline groups such that side-bias was eliminated. During training (days 2–4), all mice received a saline injection in the morning and were confined to their designated chamber. Six hours later, mice were treated with either METH (1 mg/kg, i.p.) or saline and confined to the opposite side chamber for 30 min. On test day (day 5), mice were placed into the open center chamber and allowed to freely explore the apparatus for 30 min. Time spent in each side chamber was recorded.
Statistical Analysis
All data were analyzed in GraphPad Prism 5. Data were analyzed using two-way ANOVA (with treatment and genotype as factors) with Bonferroni posthoc tests. Outliers were defined by Grubbs’ test for outliers (α = 0.05). All errors shown are SEM.
Supplementary Material
Acknowledgments
We thank Dr. Alison Bernstein for her work on the VMAT2 antiserum. We also thank the Emory University Rodent Behavioral Core for their assistance with the CPP behavior.
Funding
This work was supported by The National Institutes of Health Grants ES023839, P30ES019776, F31NS084739, F31DA037652, T32ES012870, and P50NS071669, The Canadian Institutes of Health Research Grant 210296 and The Lewis Dickey Memorial Fund.
Footnotes
Supporting Information
Figure showing that elevated VMAT2 is also neuroprotective at a lower METH dose. This material is available free of charge via the Internet at http://pubs.acs.org/
Author Contributions
Participated in research design: K.M.L., K.A.S., T.S.G., A.S., and G.W.M. Conducted experiments: K.M.L., K.A.S., A.R.D., and M.W. Performed data analysis: K.M.L., K.A.S., and A.R.D. Wrote or contributed to the writing of the manuscript: K.M.L. and G.W.M.
Notes
The authors declare no competing financial interest.
References
- 1.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 2.LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Ann N Y Acad Sci. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- 4.Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ricaurte GA, Seiden LS, Schuster CR. Further evidence that amphetamines produce long-lasting dopamine neurochemical deficits by destroying dopamine nerve fibers. Brain Res. 1984;303:359–364. doi: 10.1016/0006-8993(84)91221-6. [DOI] [PubMed] [Google Scholar]
- 6.Sulzer D, et al. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peter D, Jimenez J, Liu Y, Kim J, Edwards RH. The chromaffin granule and synaptic vesicle amine transporters differ in substrate recognition and sensitivity to inhibitors. J Biol Chem. 1994;269:7231–7237. [PubMed] [Google Scholar]
- 8.Torres B, Ruoho AE. N-terminus regulation of VMAT2 mediates methamphetamine-stimulated efflux. Neuroscience. 2014;259:194–202. doi: 10.1016/j.neuroscience.2013.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: Evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lotharius J, O’Malley KL. Role of mitochondrial dysfunction and dopamine-dependent oxidative stress in amphetamine-induced toxicity. Ann Neurol. 2001;49:79–89. doi: 10.1002/1531-8249(200101)49:1<79::aid-ana11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 11.Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: A review. Neurotox Res. 1999;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- 12.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-Induced Degeneration of Dopaminergic Neurons Involves Autophagy and Upregulation of Dopamine Synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osamu S, Hideki H, Minoru A, Masakazu O, Yoshinao K. Inhibition of monoamine oxidase by d-methamphetamine. Biochem Pharmacol. 1980;29:2071–2073. doi: 10.1016/0006-2952(80)90493-1. [DOI] [PubMed] [Google Scholar]
- 14.Yuan J, et al. Dopamine is not essential for the development of methamphetamine-induced neurotoxicity. J Neurochem. 2010;114:1135–1142. doi: 10.1111/j.1471-4159.2010.06839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- 16.Callahan BT, Cord BJ, Yuan J, McCann UD, Ricaurte GA. Inhibitors of Na+/H+ and Na+/Ca2+ exchange potentiate methamphetamine-induced dopamine neurotoxicity: possible role of ionic dysregulation in methamphetamine neurotoxicity. J Neurochem. 2001;77:1348–1362. doi: 10.1046/j.1471-4159.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- 17.Brown JM, Yamamoto BK. Effects of amphetamines on mitochondrial function: role of free radicals and oxidative stress. Pharmacol Ther. 2003;99:45–53. doi: 10.1016/s0163-7258(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 18.O’Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- 19.Hess A, Desiderio C, McAuliffe WG. Acute neuropathological changes in the caudate nucleus caused by MPTP and methamphetamine: immunohistochemical studies. J Neurocytol. 1990;19:338–342. doi: 10.1007/BF01188403. [DOI] [PubMed] [Google Scholar]
- 20.Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- 21.Guilarte TR, Nihei MK, McGlothan JL, Howard AS. Methamphetamine-induced deficits of brain monoaminergic neuronal markers: distal axotomy or neuronal plasticity. Neuroscience. 2003;122:499–513. doi: 10.1016/s0306-4522(03)00476-7. [DOI] [PubMed] [Google Scholar]
- 22.Erickson JD, Eiden LE, Hoffman BJ. Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1992;89:10993–10997. doi: 10.1073/pnas.89.22.10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson JD, Eiden LE. Functional identification and molecular cloning of a human brain vesicle monoamine transporter. J Neurochem. 1993;61:2314–2317. doi: 10.1111/j.1471-4159.1993.tb07476.x. [DOI] [PubMed] [Google Scholar]
- 24.Eiden LE, Weihe E. VMAT2: A dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse. Ann N Y Acad Sci. 2011;1216:86–98. doi: 10.1111/j.1749-6632.2010.05906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, et al. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–51. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein DS, et al. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem. 2013;126:591–603. doi: 10.1111/jnc.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alter SP, Lenzi GM, Bernstein AI, Miller GW. Vesicular integrity in Parkinson’s disease. Curr Neurol Neurosci Rep. 2013;13:362. doi: 10.1007/s11910-013-0362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang YM, et al. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi N, et al. VMAT2 knockout mice: Heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci U S A. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fon EA, et al. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 31.Caudle WM, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor TN, Alter SP, Wang M, Goldstein DS, Miller GW. Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology. 2014;76:97–105. doi: 10.1016/j.neuropharm.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine-serotonin vesicular transport disease and its treatment. New Engl J Med. 2013:543–550. doi: 10.1056/NEJMoa1207281. [DOI] [PubMed] [Google Scholar]
- 34.Pifl C, et al. Is Parkinson’s Disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci. 2014;34:8210–8218. doi: 10.1523/JNEUROSCI.5456-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lohr KM, et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc Natl Acad Sci U S A. 2014;111:9977–9982. doi: 10.1073/pnas.1402134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ugarte Y, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE. Methamphetamine rapidly decreases mouse vesicular dopamine uptake: Role of hyperthermia and dopamine D2 receptors. Eur J Pharmacol. 2003;472:165–171. doi: 10.1016/s0014-2999(03)01911-3. [DOI] [PubMed] [Google Scholar]
- 37.Guillot TS, et al. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106:2205–2217. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fumagalli F, et al. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Granado N, et al. Selective vulnerability in striosomes and in the nigrostriatal dopaminergic pathway after methamphetamine administration: Early loss of TH in striosomes after methamphetamine. Neurotoxic Res. 2010;18:48–58. doi: 10.1007/s12640-009-9106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller DB, O’Callaghan JP. Environment-, drug-and stress-induced alterations in body temperature affect the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- 41.Cunningham CL, Noble D. Methamphetamine-induced conditioned place preference or aversion depending on dose and presence of drug. Ann N Y Acad Sci. 1992;654:431–433. doi: 10.1111/j.1749-6632.1992.tb25989.x. [DOI] [PubMed] [Google Scholar]
- 42.Guillot TS, Miller GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol. 2009;2:149–170. doi: 10.1007/s12035-009-8059-y. [DOI] [PubMed] [Google Scholar]
- 43.Caudle WM, Colebrooke RE, Emson PC, Miller GW. Altered vesicular dopamine storage in Parkinson’s disease: a premature demise. Trends Neurosci. 2008;31:303–308. doi: 10.1016/j.tins.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 44.Gainetdinov RR, et al. Increased MPTP Neurotoxicity in Vesicular Monoamine Transporter 2 Heterozygote Knockout Mice. J Neurochem. 1998;70:1973–1978. doi: 10.1046/j.1471-4159.1998.70051973.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang YM, et al. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 46.Medina L, Figueredo-Cardenas G, Reiner A. Differential abundance of superoxide dismutase in interneurons versus projection neurons and in matrix versus striosome neurons in monkey striatum. Brain Res. 1996;708:59–70. doi: 10.1016/0006-8993(95)01320-2. [DOI] [PubMed] [Google Scholar]
- 47.Breuer O, Lawhorn C, Miller T, Smith DM, Brown LL. Functional architecture of the mammalian striatum: Mouse vascular and striosome organization and their anatomic relationships. Neurosci Lett. 2005;385:198–203. doi: 10.1016/j.neulet.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 48.Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: Evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowyer JF, et al. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- 50.Bowyer JF, et al. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 51.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 52.Lessin AW, Parkes MW. The hypothermic and sedative action of reserpine in the mouse. J Pharm Pharmacol. 1957;9:657–662. doi: 10.1111/j.2042-7158.1957.tb12321.x. [DOI] [PubMed] [Google Scholar]
- 53.Thiriet N, et al. Environmental enrichment does not reduce the rewarding and neurotoxic effects of methamphetamine. Neurotoxic Res. 2011;19:172–182. doi: 10.1007/s12640-010-9158-2. [DOI] [PubMed] [Google Scholar]
- 54.Shimosato K, Ohkuma S. Simultaneous Monitoring of Conditioned Place Preference and Locomotor Sensitization Following Repeated Administration of Cocaine and Methamphetamine. Pharmacol, Biochem Behav. 2000;66:285–292. doi: 10.1016/s0091-3057(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 55.Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- 56.Sulzer D, et al. Weak base model of amphetamine action. Ann N Y Acad Sci. 1992;654:525–528. doi: 10.1111/j.1749-6632.1992.tb26020.x. [DOI] [PubMed] [Google Scholar]
- 57.Whitehead RE, Ferrer JV, Javitch JA, Justice JB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]
- 58.Park SU, Ferrer JV, Javitch JA, Kuhn DM. Peroxynitrite inactivates the human dopamine transporter by modification of cysteine 342: Potential mechanism of neurotoxicity in dopamine neurons. J Neurosci. 2002;22:4399–4405. doi: 10.1523/JNEUROSCI.22-11-04399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuhn DM, Arthur RE, Thomas DM, Elferink LA. Tyrosine Hydroxylase Is Inactivated by Catechol-Quinones and Converted to a Redox-Cycling Quinoprotein. J Neurochem. 2001;73:1309–1317. doi: 10.1046/j.1471-4159.1999.0731309.x. [DOI] [PubMed] [Google Scholar]
- 60.Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- 61.Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: Implications for Parkinson’s disease pathogenesis. Brain Res. 2003;989:205–213. doi: 10.1016/s0006-8993(03)03354-7. [DOI] [PubMed] [Google Scholar]
- 62.Burke WJ, et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115:193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- 63.Anderson DG, Mariappan SVS, Buettner GR, Doorn JA. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J Biol Chem. 2011;286:26978–26986. doi: 10.1074/jbc.M111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vergo S, Johansen JL, Leist M, Lotharius J. Vesicular monoamine transporter 2 regulates the sensitivity of rat dopaminergic neurons to disturbed cytosolic dopamine levels. Brain Res. 2007;1185:18–32. doi: 10.1016/j.brainres.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 65.Guillot TS, et al. PACAP38 increases vesicular monoamine transporter 2 (VMAT2) expression and attenuates methamphetamine toxicity. Neuropeptides. 2008;42:423–434. doi: 10.1016/j.npep.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mosharov EV, Gong LW, Khanna B, Sulzer D, Lindau M. Intracellular patch electrochemistry: regulation of cytosolic catecholamines in chromaffin cells. J Neurosci. 2003;23:5835–5845. doi: 10.1523/JNEUROSCI.23-13-05835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mosharov EV, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mosharov EV, et al. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci. 2006;26:9304–9311. doi: 10.1523/JNEUROSCI.0519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27:1478–1483. doi: 10.1002/mds.25135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burrows KB, Gudelsky G, Yamamoto BK. Rapid and transient inhibition of mitochondrial function following methamphetamine or 3,4-methylenedioxymethamphetamine administration. Eur J Pharmacol. 2000;398:11–18. doi: 10.1016/s0014-2999(00)00264-8. [DOI] [PubMed] [Google Scholar]
- 71.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dwoskin LP, Crooks PA. A novel mechanism of action and potential use for lobeline as a treatment for psychostimulant abuse. Biochem Pharmacol. 2002;63:89–98. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- 73.Nickell JR, et al. Lobelane inhibits methamphetamine-evoked dopamine release via inhibition of the vesicular monoamine transporter-2. J Pharmacol Exp Ther. 2010;332:612–621. doi: 10.1124/jpet.109.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller DK, et al. Lobeline Inhibits the Neurochemical and Behavioral Effects of Amphetamine. J Pharmacol Exp Ther. 2001;296:1023–1034. [PubMed] [Google Scholar]
- 75.Taylor TN, et al. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci. 2009;29:8103–8113. doi: 10.1523/JNEUROSCI.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Freis ED. Mental depression in hypertensive patients treated for long periods with large doses of reserpine. New Engl J Med. 1954;251:1006–1008. doi: 10.1056/NEJM195412162512504. [DOI] [PubMed] [Google Scholar]
- 77.Pettibone DJ, Totaro JA, Pflueger AB. Tetrabenazine-induced depletion of brain monoamines: Characterization and interaction with selected antidepressants. Eur J Pharmacol. 1984;102:425–430. doi: 10.1016/0014-2999(84)90562-4. [DOI] [PubMed] [Google Scholar]
- 78.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 79.Curtin K. Methamphetamine/amphetamine abuse and risk of Parkinson’s disease in Utah: A population-based assessment. Drug Alcohol Depend. 2015;146:30–38. doi: 10.1016/j.drugalcdep.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Callaghan RC, Cunningham JK, Sykes J, Kish SJ. Increased risk of Parkinson’s disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 2012;120:35–40. doi: 10.1016/j.drugalcdep.2011.06.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.