

Abstract
The [4+2] cycloaddition of 3-alkoxyfurans with N-substituted maleimides provides the first general route for preparing endo-cantharimides. Unlike the corresponding reaction with 3H furans, the reaction can tolerate a broad range of 2-substitued furans including alkyl, aromatic, and heteroaromatic groups. The cycloaddition products were converted into a range of cantharimide products with promising lead-like properties for medicinal chemistry programs. Furthermore, the electron-rich furans are shown to react with a variety of alternative dienophiles to generate 7-oxabicyclo[2.2.1]heptane derivatives under mild conditions. DFT calculations have been performed to rationalize the activation effect of the 3-alkoxy group on a furan Diels–Alder reaction.
Keywords: cantharimides, cycloadditions, dienes, furans, phthalimide
Introduction
To access new areas of chemical space, medicinal chemistry programs are increasingly focusing on fragments and scaffolds with rigid 3D structures that contain a significant proportion of sp3 carbon atoms.[1] This in turn presents a considerable synthetic challenge as these molecules are generally not straightforward to synthesize, and late-stage derivatization is often far from trivial. Further challenges reside in the control of relative and absolute stereochemistry due to the presence of numerous chiral centres. Current structural scaffolds of interest include strained small-ring molecules (cyclopropanes, oxetanes, azetidines),[2] as well as fused (dihydrobenzofurans, indolines, tetrahydroquinolines)[3] and bridged bicylic and polycyclic compounds (bicyclopentanes, cubanes, etc).[4] Natural products have also traditionally provided chemists with inspiration, as they include bioactive molecules with complex 3D architectures.[5] Many of these compounds, however, have high molecular weights or are too structurally complex to be suitable for use as scaffolds for medicinal chemistry applications. Nevertheless, smaller natural products contain ring systems that are potentially ideal scaffolds for use in medicinal chemistry, provided that efficient synthetic routes can be developed with appropriate functional groups at positions on the central core.
The exo-cantharimide skeleton (Figure 1, derived from cantharidin, a natural product secreted by many species of blister beetle with well-established cytotoxic activity)[6] has been exploited in a wide range of molecules with useful biological properties. The motif is present in several cytotoxic compounds,[7] antiplasmodial agents,[8] androgen receptor antagonists[9] and in a positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4).[10] More generally, the 7-oxabicyclo[2.2.1]heptyl skeleton is found in a number of other important natural products[11]–[13] and it has proved to be a valuable intermediate for synthetic chemists.[14]–[17] The properties of the exo-cantharimide skeleton have been extensively explored with a range of N-substituted derivatives showing useful biological properties. However, there are few methods for the introduction of substituents around the 7-oxabicyclo[2.2.1]heptyl ring system.[18] Furthermore, the corresponding endo-cantharimide scaffold has rarely been reported at all.[19]
Figure 1.
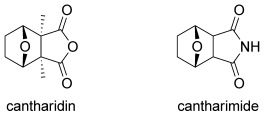
Natural product cantharidin and cantharimide.
The exo-cantharimide skeleton is typically prepared by the [4+2] cycloaddition of furans and maleic anhydride, followed by alkene reduction and condensation with an amine (Scheme 1).[20] A curious feature of the cycloaddition reaction is the high stereoselectivity for the exo diastereomer observed, believed to be the result of a highly reversible cycloaddition process which is operating under thermodynamic control.[21], [22] It is possible to access the corresponding endo-cantharimide by a Diels–Alder reaction of furan with maleimide;[23] however, experimental and computational studies have shown that this reaction is under thermodynamic control with the exo-cantharimide being the thermodynamic product.[24] As a consequence, the endo-adduct of maleimide and furan is known to rapidly isomerize either in hot solvent or when exposed to visible light, which impedes both the isolation and application of these compounds.[25] Another serious limitation of furan Diels–Alder reactions is that any deactivating substituents on the furan have a profound effect on the equilibrium position of the cyclization. For example, there are no reported examples of the [4+2] cycloaddition of 2-aryl or 2-heteroaryl furans with dienophiles of any type.
Scheme 1.
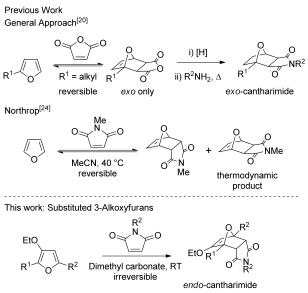
Synthetic approaches to cantharimides.
There is a long tradition of activating dienes for Diels–Alder reactions through the use of electron-donating substituents, which are known to reduce the activation energy for the cycloaddition reaction.[26] However, this is generally a kinetic effect and reducing the kinetic barriers to a thermodynamically controlled reaction would only increase the rate at which isomerization occurs. To access stable endo-cantharimides it is therefore necessary to develop reactions with a significantly improved thermodynamic driving force.[24]
We have recently developed a straightforward approach to 2-substituted-3-alkoxyfurans by gold-catalysed solvolytic cyclisation of suitably functionalised propargylic alcohols (Scheme 2).[27] Preliminary studies indicated that 3-alkoxyfurans underwent rapid and endo-selective reactions with N-methylmaleimide to generate kinetically stable cantharimide products. The distinct 3D structure of the endo-cantharimide motif, coupled with its physical properties, should make it a valuable new scaffold for medicinal chemistry applications. Such an approach should enable control of substituents at a variety of positions on the tricyclic ring system.
Scheme 2.

Gold-catalysed synthesis of 3-alkoxyfurans 2 from propargylic alcohols 1.[27]
Results and Discussion
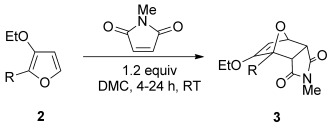
The reaction of 3-ethoxyfuran 2 a with 1.2 equivalents of N-methylmaleimide proceed in a variety of solvents at room temperature to give cantharimide 3 a in near quantitative yield (Table 1, entries 1 to 4). Crucially the cantharimide was formed with a clear preference for the endo diastereomer and the two isomers could be readily separated by flash column chromatography. The identity of the solvent had little impact on yield or diastereoselectivity, so dimethyl carbonate (DMC) was selected on the grounds of its excellent environmental profile.[28] The reaction could also be scaled up to use 1 g of furan 2 a, giving cantharimide 3 a in 95 % yield (entry 5, endo/exo ratio of 75:25). A purified sample of endo-3 a was treated under the same reaction conditions and no isomerization was observed, suggesting the reaction proceeds under kinetic control. However, it was possible to increase the proportion of exo-3 a by heating the reaction at 80 °C for 16 h (entry 6). The cyclization was equally effective when 3-methoxyfuran 2 b was used as a diene, giving the corresponding adduct in excellent yield as an 80:20 mixture of endo and exo diastereomers (entry 7).
Table 1.
[4+2] cycloaddition of 3-alkoxyfurans 2 with N-methylmaleimide.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Solvent[a] | T [°C] | Reaction t [h] | Product | Yield [%] | endo/exo[b] |
| 1 | Et | Et2O | 25 | 4 | 3 a | 98[c] | 65:35 |
| 2 | Et | PhMe | 25 | 4 | 3 a | 100[c] | 65:35 |
| 3 | Et | EtOH | 25 | 4 | 3 a | 100[c] | 70:30 |
| 4 | Et | DMC | 25 | 4 | 3 a | 93[d] | 70:30 |
| 5[e] | Et | DMC | 25 | 4 | 3 a | 95[d] | 75:25 |
| 6 | Et | DMC | 80 | 16 | 3 a | 93[d] | 55:45 |
| 7 | Me | DMC | 25 | 4 | 3 b | 89[d] | 80:20 |
DMC refers to dimethyl carbonate.
Determined by analysis of the crude 1H NMR spectrum.
Yield determined by 1H NMR spectroscopy using pentachlorobenzene as an internal standard.
Isolated yield.
Reaction conducted with 1.0 g of furan 2 a.
These reaction conditions were applied to a wide range of 3-ethoxyfurans with different substituents at the 2-position, with the results summarized in Table 2. The reaction tolerated furans with primary and secondary aliphatic substituents (Table 2, entries 2 and 3). It was also possible to incorporate a tert-butoxycarbonyl (N-Boc) piperidine, as shown in entry 4. The reaction was very effective with an aromatic group at the 2-position, giving the first reported examples of 4-arylcantharimides (entries 5 to 10). The reaction of 2-phenylfuran 2 f gave an 80:20 mixture of endo and exo diastereomers in good yield. This reaction could also be conducted on a 1.0 g scale, giving the two diastereomers 3 f in a combined yield of 86 %, and with complete isomeric separation following chromatography on silica gel. The relative stereochemistry of the two diastereomers was confirmed by X-ray crystallography (Figure 2).
Table 2.
[4+2] cycloaddition of 3-alkoxyfurans 2 with N-methylmaleimide.
 | |||
|---|---|---|---|
| Entry | R | Isolated yield [%][a] | endo/exo[b] |
| 1 |  |
93 | 70:30 |
| 2 |  |
95 | 85:15 |
| 3 |  |
90 | 80:20 |
| 4 |  |
85 | 75:25 |
| 5 |  |
86 (86)[c] | 80:20 |
| 6 |  |
75 | 80:20 |
| 7 |  |
78 | 80:20 |
| 8 |  |
95 | 80:20 |
| 9 |  |
84 | 75:25 |
| 10 |  |
86 | 80:20 |
| 11 | 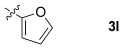 |
85 | 90:10 |
| 12 |  |
96 | 70:30 |
| 13 |  |
92 | 70:30 |
Combined isolated yield of endo-3 and exo-3.
Determined by analysis of the 1H NMR spectrum of the crude product.
Reaction conducted with 1.0 g of furan 2 f.
Figure 2.
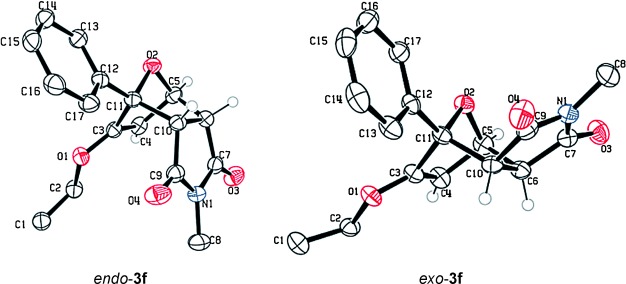
Crystal structures of cantharimides 3 f. Ellipsoids are shown at the 50 % probability level. Only hydrogen atoms belonging to the cyclic core are shown for clarity.[29]
The reaction was tolerant of electron-poor aromatic substituents (Table 2, entries 6 and 9), an electron-rich aromatic substituent (entry 8) and an aryl bromide substituent (entry 7). It was also possible to use a sterically encumbered 2-tolyl substituent to give cantharimide 3 k in 86 % yield. Furthermore, the reaction was effective when the 3-alkoxyfuran possessed a heteroaromatic substituent, as can be seen in entries 11 to 13 (85–96 % yields). The chemoselective reaction of bis-furan 2 l with N-methylmaleimide to give exclusively the enol ether adduct is an interesting demonstration of the high reactivity of the 3-alkoxyfuran unit in a [4+2] cycloaddition reaction. It was also possible to functionalize a 3-alkoxyfuran at the 5-position prior to the cycloaddition reaction, in order to introduce a substituent at the 7-position of the endo-cantharimide scaffold (Scheme 3).
Scheme 3.
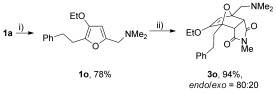
Synthesis of a 7-substituted endo-cantharimide: i) (H2C—NMe2)I (2 equiv), MeCN, 16 h, RT; ii) N-methylmaleimide (1.2 equiv), DMC, 24 h, RT.
The cycloaddition of 3-alkoxyfuran 2 a was effective with a number of alternative N-substituted maleimides, as illustrated in Table 3.[30] Sterically more challenging N-substituents could be incorporated in high yield and without an extended reaction time.
Table 3.
[4+2] cycloaddition of 3-alkoxyfuran 2 a with maleimides 4.
 | |||
|---|---|---|---|
| Entry | R | Yield5[%] | endo/exo[a] |
| 1 | Ph 4 a | 94 | 65:35 |
| 2 | 4-MeC6H4 4 b | 83 | 55:35 |
| 3 | c-Pr 4 c | 87 | 60:40 |
Determined by analysis of the 1H NMR spectrum of the crude product.
Additionally, it was possible to combine the gold-mediated furan synthesis with the cycloaddition reaction in a single step (Scheme 4, conditions i). Treating propargylic alcohol 1 a with gold catalyst and N-methylmaleimide gave diethyl acetal 6 in good yield. It appeared that the gold catalyst was responsible for the in situ conversion of enol ether 3 a into the corresponding diethyl acetal, as the interconversion can be avoided by poisoning the catalyst with 2.5 mol % PPh3 prior to addition of the N-methylmaleimide, to give enol ether 3 a (endo/exo ratio of 70:30). Treatment of a sample of enol ether 3 a with catalytic PPh3AuNTf2 in ethanol was also observed to result in formation of acetal 6.
Scheme 4.
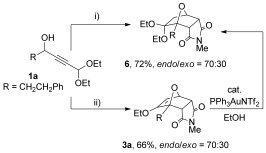
One-pot cantharimide synthesis from propargylic alcohol 1 a. i) N-methylmaleimide, 2 mol % PPh3AuNTf2, EtOH; ii) 2 mol % PPh3AuNTf2, EtOH then 2.5 mol % PPh3 then N-methylmaleimide.
Transformation of cycloaddition products
The endo-cantharimides contain an enol ether moiety, which can be readily transformed into a variety of functional groups (Scheme 5). For example, enol ether endo-3 f can by hydrogenated to generate ether 7 with complete diastereocontrol.[31] The enol ether also underwent hydroboration and oxidation to give alcohol 8, with complete regio- and stereocontrol. Enol ether endo-3 f could be hydrolysed to give ketone 9 in good yield by passing it through a strong cation exchange (SCX-2) cartridge.[32] Treating ketone 9 with NaBH4 afforded alcohol 10, again with high stereocontrol.
Scheme 5.
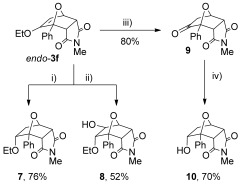
Functional-group interconversion of enol ether endo-3 f: i) H2, 10 % Pd/C; ii) 9-BBN then H2O2/NaOH; iii) SCX-2 cartridge; iv) NaBH4, MeOH. 9-BBN=9-borabicyclo[3.3.1]nonane.
The acid-mediated aromatization of 7-oxabicyclo[2.2.1]heptane derivatives has been previously applied to the synthesis of aromatic rings, and this approach could be used to prepare substituted phthalimide 11.[33] The one-pot cantharimide synthesis described in Scheme 4 was used to convert alcohol 1 f into the crude cantharimide, which could be converted into phthalimide 11 by acid-mediated ring-opening and aromatization (Scheme 6).
Scheme 6.
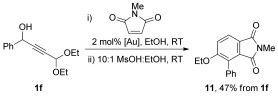
Synthesis of substituted phthalimide 11 by acid-catalysed aromatisation of cantharimide intermediates.
Physicochemical properties
An important challenge for drug development is the generation of novel heterocyclic building blocks with suitable properties for use in screening and medicinal chemistry programs.[34] The cantharimides accessed using this methodology have appropriate physicochemical properties for lead-like compounds, including lipophilicity,[35] molecular weight and polar surface area[36] (Figure 3). Another attractive feature of these scaffolds is the high proportion of sp3-hybridized carbon atoms, which is typically associated with improved protein binding selectivity and frequency, better solubility and a reduced chance of off-target effects.[37] Indeed, cantharimides 7, 10, endo-3 f and exo-3 f were screened against the hERG receptor (IC50>50 μm) and the aryl hydrocarbon receptor (EC50>100 μm), which are responsible for common off target effects, and no affinity was observed. In addition the in vitro clearance of alcohol 10 in the presence of human microsomes was determined and only a low level of turnover was observed (<0.53 mL min−1 g−1).[38]
Figure 3.
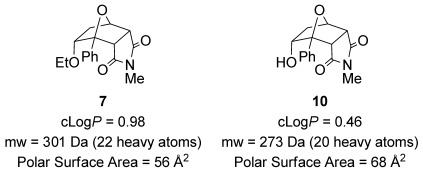
Physicochemical properties of endo-cantharimides.[39]
[4+2] cycloadditions with other dienophiles
The [4+2] cycloaddition of furans with maleate esters is known but was reported to require either forcing pressure[40] or high catalyst loadings of a Lewis acid.[41] In contrast, the catalyst-free reaction of dimethyl maleate 12 a and furan 2 a proceeded at room temperature to give adduct 13 a in a good yield and with excellent endo selectivity (Table 4, entry 1). The reactions of dimethyl and diethyl fumarate (12 b and 12 c) with furan 2 a proceeded more rapidly, giving the corresponding adducts in 77–89 % yield after 4 h (entries 2 and 3). There is a clear selectivity in both examples for the product which possessed exo stereochemistry with respect to the 3-position (3-exo-13).[42] Heating furan 2 a with ethyl vinyl ketone at 80 °C for 16 h, followed by hydrolysis of the enol ether on an SCX-2 cartridge, gave diketone 13 d with high regiocontrol (95:5), although as a 60:40 mixture of endo/exo isomers.
Table 4.
[4+2] cycloaddition of 3-alkoxyfuran 2 a with different dienophiles.
 | |||||
|---|---|---|---|---|---|
| Entry | Dienophile12 | Reaction t [h] | Product13 | Isolated yield [%] | Product ratio[a] |
| 1 |  |
72 |  |
70 | 12:1 |
| 2 | 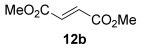 |
4 |  |
77 | 15:85 |
| 3 |  |
4 | 89 | 15:85 | |
| 4[b,c] | 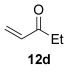 |
16 | 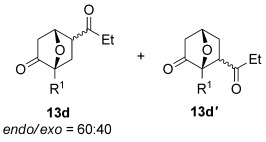 |
60 | 95:5 |
| 5[c,d] |  |
6 |  |
89 | 95: 5 |
Determined by analysis of the 1H NMR spectrum of the crude product.
Reaction conducted at 80 °C.
Crude product flushed through a SCX-2 cartridge.
Reaction conducted with 2 mol % HfCl4.[41]
The catalyst-free reaction of furan 2 a with ethyl acrylate 12 e was relatively slow at room temperature, with <100 % conversion after 24 h. However, it was possible to accelerate the reaction through the use of 2 mol % HfCl4, giving the ketone 13 e in 89 % yield and with good regiocontrol (95:5) after 6 h at room temperature (Table 4, entry 5). The catalyst loading for this reaction is much lower than the high (sometimes stoichiometric) loading reported for the Lewis acid-catalyzed reactions of 3H furans and acrylates.[41], [43]
Computational study
The reactions of five 3-alkoxyfurans and N-methylmaleimide were explored with the M06-2X exchange–correlation function of Truhlar et al.,[44] a density functional that has been successfully used to model the reaction and activation energies of different cycloaddition processes.[45] 2-Substituted-3-methoxyfurans were chosen as suitable models for our 3-alkoxyfurans and these were compared to the corresponding 3H furans.
The 3-alkoxy group has a dramatic effect on the thermodynamics of the cycloaddition reaction, as is evident in Table 5. All five reactions of 3-alkoxyfurans have a clear thermodynamic driving force for the formation of both endo- and exo-addition products (Figure 5) and the data is consistent with a reaction that is likely to be kinetically controlled. In contrast, the values of ΔG for the corresponding reactions of 3H furans are all greater by 24–34 kJ mol−1 (Figure 4). This effect is most significant when the 2-substituent is aromatic, as this results in a value of ΔG close to zero for the furans 14 h–j. As expected, the 3-alkoxy group also has a significant effect on the free energy of activation for the cycloaddition reaction, with the kinetic barrier reduced by 11–23 kJ mol−1. The effect of solvation on these reactions was also considered but was found to have little effect (see Supporting Information).
Table 5.
Calculated ΔG and ΔG≠ for the reactions of furans 14 and N-methylmaleimide 15.[a]
 | ||||||
|---|---|---|---|---|---|---|
| 14 | R | X | endo | exo | ||
| ΔG | ΔG≠ | ΔG | ΔG≠ | |||
| 14 a | Me | OMe | −41.5 | 81.1 | −47.7 | 82.1 |
| 14 b | cPr | OMe | −44.1 | 75.0 | −53.2 | 78.6 |
| 14 c | 4-MeOC6H4 | OMe | −32.5 | 76.8 | −30.1 | 85.3 |
| 14 d | Ph | OMe | −34.0 | 83.6 | −28.2 | 91.4 |
| 14 e | 4-F3CC6H4 | OMe | −25.3 | 85.9 | −23.8 | 96.8 |
| 14 f | Me | H | −11.8 | 97.9 | −16.8 | 96.2 |
| 14 g | cPr | H | −12.5 | 92.2 | −10.3 | 92.8 |
| 14 h | 4-MeOC6H4 | H | −6.1 | 95.4 | −3.6 | 98.8 |
| 14 i | Ph | H | −1.7 | 101.1 | 0.7 | 105.5 |
| 14 j | 4-F3CC6H4 | H | −0.9 | 101.6 | 0.9 | 108.3 |
All values in kJ mol−1. All data is calculated for species in the gas phase.
Figure 5.
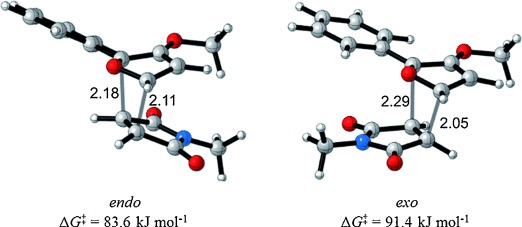
M06-2X/6-31G(d)-optimized endo and exo transition states for the reaction of furan 14 d and N-methylmaleimide 15.[46] Distances in Å.
Figure 4.
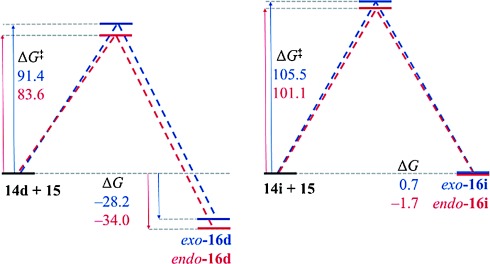
Calculated ΔG and ΔG≠ values for the reactions of 2-phenylfurans 14 d and 14 i and N-methylmaleimide 15 in kJ mol−1.
The reversibility of most furan Diels–Alder reactions has been attributed to the loss of aromatic stabilization upon formation of an adduct, which results in a facile retro-cycloaddition.[22] In order to examine the effect of a 3-methoxy group on this phenomenon, thermodynamic cycles involving the partial hydrogenation of 3-methoxyfuran 17 a and furan 20 a to the corresponding 2,5-dihydrofurans 18 a and 21 a were considered (Scheme 7). It is notable that the free energy of hydrogenation for furan 20 a was 25.9 kJ mol−1 greater than for 3-methoxyfuran 17 a. The corresponding reaction free energies for cyclopentadienes 17 b and 20 b were also calculated but no significant difference was observed. The implications of these calculations are that 1) the difference in behaviour between 3H and 3-methoxy furans in cycloaddition reactions can be attributed to differences associated with loss of aromaticity rather than with C—C bond formation and 2) a 3-methoxy group can reduce the energetic penalty associated with the loss of aromaticity upon the Diels–Alder reaction of a furan, increasing the thermodynamic stability of the cycloaddition product.
Scheme 7.
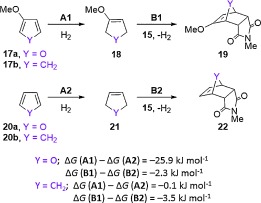
Thermodynamic cycle involving the hydrogenation of dienes 17 and 20.
Conclusions
We have demonstrated that 3-alkoxyfurans are excellent dienes for [4+2] cycloadditions with a wide variety of maleimides and other dienophiles. This methodology significantly expands the nature of cantharimides that can be readily prepared with high endo selectivity. The reaction tolerates alkyl, aryl and heteroaryl substituents and the enol ether cycloaddition product can be transformed into a diverse collection of drug-like compounds. Finally, DFT calculations have confirmed that a 3-alkoxy group has a significant effect on both the thermodynamic driving-force and the activation energy of the Diels–Alder reaction of 2-substituted furans with N-methylmaleimides. The former effect can potentially be attributed to the 3-alkoxygroup leading to a reduced energetic penalty associated with the loss of furan aromaticity that occurs during the cycloaddition reaction.
Experimental Section
General cycloaddition procedure
A solution of the maleimide (1.2 equiv) in dimethyl carbonate (3.6 m) was added to a stirring solution of 3-alkoxyfuran (1.0 equiv) in dimethyl carbonate (1.5 m) at room temperature and the reaction stirred at room temperature for 4–24 h. The reaction was then diluted with ethyl acetate and loaded onto an aminopropyl cartridge. After 5 min the cartridge was then flushed with ethyl acetate and the solvent removed in vacuo to give the crude cycloaddition product.
Experimental procedures, 1H and 13C NMR spectra, characterization data of all compounds, compound screening data, details of computational studies including energy minimized geometries and XRD crystallography files are available in the Supporting Information.
Acknowledgments
This work was supported by GlaxoSmithKline and the Engineering and Physical Sciences Research Council (EPSRC Industrial CASE Award) and the UCL Ph.D. program in Drug Discovery.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201406286.
References
- [1].pp. 6752–6756.
- [1a].Lovering F, Bikker J, Humblet C. J. Med. Chem. 52 doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- [1b].Reymond JL, van Deursen R, Blum LC, Ruddigkeit L. Med. Chem. Commun. 2009;1 [Google Scholar]
- [1c].Blum LC, Reymond JL. J. Am. Chem. Soc. 2010;131 doi: 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- [1d].Ritchie TJ, MacDonald SJF. Drug Discovery Today. 2009;14 doi: 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- [2].Cyclopropanes: pp. 1603–1624.
- [2a].Gnad F, Reiser O. Chem. Rev. 103 doi: 10.1021/cr010015v. [DOI] [PubMed] [Google Scholar]
- [2b].Ishikawa S, Sheppard TD, D′Oyley JM, Kamimura A, Motherwell WB. Angew. Chem. Int. Ed. 2003;52 doi: 10.1002/anie.201304720. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 Oxetanes. [Google Scholar]
- [2c].Burkhard JA, Wuitschik G, Rogers-Evans M, Müller K, Carreira EM. Angew. Chem. Int. Ed. 2013;49 doi: 10.1002/anie.200907155. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010;122 [Google Scholar]
- [2d].Davis OA, Bull JA. Angew. Chem. Int. Ed. 2010;53 doi: 10.1002/anie.201408928. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 Azetidines. [Google Scholar]
- [2e].Wuitschik G, Rogers-Evans M, Buckl A, Bernasconi M, Märki M, Godel T, Fischer H, Wagner B, Parrilla I, Schuler F, Schneider J, Alker A, Schweizer WB, Müller K, Carreira EM. Angew. Chem. Int. Ed. 2014;47 doi: 10.1002/anie.200800450. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008;120 [Google Scholar]
- [3].pp. 3975–3984.
- [3a].Liu D, Zhao G, Xiang L. Eur. J. Org. Chem [Google Scholar]
- [3b].Bertolini F, Pineschi M. Org. Prep. Proced. Int. 2009;41 [Google Scholar]
- [4].pp. 3414–3424.
- [4a].Stepan AF, Subramanyam C, Efremov IV, Dutra JK, O’Sullivan TJ, DiRico KJ, McDonald WS, Won A, Dorff PH, Nolan CE, Becker SL, Pustilnik LR, Riddell DR, Kauffman GW, Kormos BL, Zhang L, Lu Y, Capetta SH, Green ME, Karki K, Sibley E, Atchison KP, Hallgren AJ, Oborski CE, Robshaw AE, Sneed B, O’Donnell CJ. J. Med. Chem. 55 [Google Scholar]
- [4b].Wlochal J, Davies RDM, Burton J. Org. Lett. 2012;16 doi: 10.1021/ol501750k. [DOI] [PubMed] [Google Scholar]
- [5].Newman DJ, Cragg GM. J. Nat. Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- [6].pp. 1687–1690.
- [6a].McCluskey A, Bowyer MC, Collins E, Sim ATR, Sakoff JA, Baldwin ML. Bioorg. Med. Chem. Lett. 10 doi: 10.1016/s0960-894x(00)00323-1. [DOI] [PubMed] [Google Scholar]
- [6b].McCluskey A, Ackland SP, Bowyer MC, Baldwin ML, Garner J, Walkom CC, Sakoff JA. Bioorg. Chem. 2000;31 doi: 10.1016/s0045-2068(02)00524-2. [DOI] [PubMed] [Google Scholar]
- [7].pp. 1155–1159.
- [7a].Kok SHL, Chui CH, Lam WS, Chen J, Lau FY, Wong RSM, Cheng GYM, Lai PBS, Leung TWT, Yu MWY, Tanga JCO, Chan ASC. Bioorg. Med. Chem. Lett. 17 [Google Scholar]
- [7b].Lin LH, Huang H-S, Lin CC, Lee L-W, Lin PY. Chem. Pharm. Bull. 2007;52 doi: 10.1248/cpb.52.855. [DOI] [PubMed] [Google Scholar]
- [7c].Attar RM, Jure-Kunkel M, Balog A, Cvijic ME, Dell-John J, Rizzo CA, Schweizer L, Spires TE, Platero JS, Obermeier M, Shan W, Salvati ME, Foster WR, Dinchuk J, Chen SJ, Vite G, Kramer R, Gottardis MM. Cancer Res. 2004;69 doi: 10.1158/0008-5472.CAN-09-1111. [DOI] [PubMed] [Google Scholar]
- [7d].Shimizu T, Iizuka M, Matsukura H, Hashizume D, Sodeoka M. Chem. Asian J. 2009;7 doi: 10.1002/asia.201200077. [DOI] [PubMed] [Google Scholar]
- [7e].McCluskey A, Walkom C, Bowyer MC, Ackland SP, Gardiner E, Sakoff JA. Bioorg. Med. Chem. Lett. 2012;11 doi: 10.1016/s0960-894x(01)00594-7. [DOI] [PubMed] [Google Scholar]
- [7f].Campbell BE, Tarleton M, Gordon CP, Sakoff JA, Gilbert J, McCluskey A, Gasser RB. Bioorg. Med. Chem. Lett. 2001;21 doi: 10.1016/j.bmcl.2011.04.031. [DOI] [PubMed] [Google Scholar]
- [8].Bajsa J, McCluskey A, Gordon CP, Stewart SG, Hill TA, Sahu R, Duke SO, Tekwani BL. Bioorg. Med. Chem. Lett. 2010;20:6688–6695. doi: 10.1016/j.bmcl.2010.09.004. [DOI] [PubMed] [Google Scholar]
- [9].Salvati ME, Balog A, Shan W, Rampulla R, Giese S, Mitt T, Furch JA, Vite GD, Attar RM, Jure-Kunkel M, Geng J, Rizzo CA, Gottardis MM, Krystek SR, Gougoutas J, Galella MA, Obermeier M, Furad A, Chandrasena G. Bioorg. Med. Chem. Lett. 2008;18:1910–1915. doi: 10.1016/j.bmcl.2008.02.006. [DOI] [PubMed] [Google Scholar]
- [10].Jones CK, Engers DW, Thompson AD, Field JR, Blobaum AL, Lindsley SR, Zhou Y, Gogliotti RD, Jadhav S, Zamorano R, Bogenpohl J, Smith Y, Morrison R, Daniels JS, Weaver CD, Conn PJ, Lindsley CW, Niswender CM, Hopkins CR. J. Med. Chem. 2011;54:7639–7647. doi: 10.1021/jm200956q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].pp. 469–472.
- [11a].Tori M, Torii T, Tachibana K, Yamada S, Tsuyuki T, Takahashi T. Bull. Chem. Soc. Jpn. 50 [Google Scholar]
- [11b].Yamano Y, Ito M, Wada A. Org. Biomol. Chem. 1977;6 doi: 10.1039/b807482h. [DOI] [PubMed] [Google Scholar]
- [11c].Lee CL, Chiang LC, Cheng LH, Liaw CC, El-Razek MHA, Chang FR, Wu YC. J. Nat. Prod. 2008;72 doi: 10.1021/np900158f. [DOI] [PubMed] [Google Scholar]
- [11d].Kusano A, Shibano M, Kussano G, Miyase T. Chem. Pharm. Bull. 2009;44 doi: 10.1248/cpb.44.2078. [DOI] [PubMed] [Google Scholar]
- [13].Lin YT, Lin FY, Isobe M. Org. Lett. 2014;16:5948–5951. doi: 10.1021/ol5029755. [DOI] [PubMed] [Google Scholar]
- [14].pp. 173–185.
- [14a].Vogel P, Fattori D, Gasparini F, Le Drian C. Synlett [Google Scholar]
- [14b].Hudlicky T, Entwistle DA, Pitzer KK, Thorpe AJ. Chem. Rev. 1996;96 doi: 10.1021/cr9403300. [DOI] [PubMed] [Google Scholar]
- [15].Grieco PA, Zelle RE, Lis R, Finn J. J. Am. Chem. Soc. 1983;105:1403–1404. doi: 10.1021/ja00279a041. [DOI] [PubMed] [Google Scholar]
- [16].Sim JY, Hwang GS, Kim KH, Ko EM, Ryu DH. Chem. Commun. 2007:5064–5065. doi: 10.1039/b710537a. [DOI] [PubMed] [Google Scholar]
- [17].pp. 3783–3800.
- [17a].Shoji M, Hayashi Y. Eur. J. Org. Chem [Google Scholar]
- [17b].Shoji M. Bull. Chem. Soc. Jpn. 2007;80 [Google Scholar]
- [18].pp. 2685–2688.
- [18a].Göksu G, Gül M, Öcal N, Kaufmann DE. Tetrahedron Lett. 49 [Google Scholar]
- [18b].Deng LP, Liu FM, Wang HY. J. Heterocycl. Chem. 2008;42 [Google Scholar]
- [19].pp. 2955–2964.
- [19a].Jarosz S, Mach M, Szewczyk K, Skóra S, Ciunik Z. Eur. J. Org. Chem [Google Scholar]
- [19b].Howell SJ, Spencer N, Philp D. Org. Lett. 2002;4 doi: 10.1021/ol017044c. [DOI] [PubMed] [Google Scholar]
- [20].Grogan GH, Rice LM. J. Med. Chem. 1963;6:802–805. doi: 10.1021/jm00342a042. [DOI] [PubMed] [Google Scholar]
- [21].Tobia D, Harrison R, Phillips B, White TL, DiMare M, Rickborn B. J. Org. Chem. 1993;58:6701–6706. [Google Scholar]
- [22].Rulísek L, Šebek P, Havlas Z, Hrabal R, Čapek P, Svatos A. J. Org. Chem. 2005;70:6295–6302. doi: 10.1021/jo050759z. [DOI] [PubMed] [Google Scholar]
- [23].Goh YW, Pool BR, White JM. J. Org. Chem. 2008;73:151–156. doi: 10.1021/jo7018575. [DOI] [PubMed] [Google Scholar]
- [24].Boutelle RC, Northrop BH. J. Org. Chem. 2011;76:7994–8002. doi: 10.1021/jo201606z. [DOI] [PubMed] [Google Scholar]
- [25].Kwart H, Burchuk I. J. Am. Chem. Soc. 1952;74:3094–3097. [Google Scholar]
- [26].pp. 7807–7808.
- [26a].Danishefsky S, Kitahara T. J. Am. Chem. Soc. 96 doi: 10.1021/ja00426a066. [DOI] [PubMed] [Google Scholar]
- [26b].Kozmin SA, Rawal VH. J. Org. Chem. 1974;62 [Google Scholar]
- [26c].Holmes JM, Albert AL, Gravel M. J. Org. Chem. 1997;74 doi: 10.1021/jo901347u. [DOI] [PubMed] [Google Scholar]
- [26d].Zhou S, Sánchez-Larios E, Gravel M. J. Org. Chem. 2009;77 doi: 10.1021/jo202655h. [DOI] [PubMed] [Google Scholar]
- [26e].Choi J, Park H, Yoo HJ, Kim S, Sorensen EJ, Lee C. J. Am. Chem. Soc. 2012;136 doi: 10.1021/ja505721v. [DOI] [PubMed] [Google Scholar]
- [27].Pennell MN, Foster RW, Turner PG, Hailes HC, Tame CJ, Sheppard TD. Chem. Commun. 2014;50:1302–1304. doi: 10.1039/c3cc48290a. [DOI] [PubMed] [Google Scholar]
- [28].pp. 854–862.
- [28a].Henderson RK, Jiménez-González C, Constable DJC, Alston SR, Inglis GGA, Fisher G, Sherwood J, Binks SP, Curzons AD. Green Chem. 13 [Google Scholar]
- [28b].Laird T. Org. Process Res. Dev. 2011;16 [Google Scholar]
- [29].CCDC 1035038 and 1035039 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via. http://www.ccdc.cam.ac.uk/data_request/cif.
- [30].Borikar SP, Paul V, Puranik VG, Sathe VT, Lagunas-Rivera S, Ordónez M. Synthesis. 2011;10:1595–1598. [Google Scholar]
- [31].Wang H, Houk K. Chem. Sci. 2014;5:462–470. doi: 10.1039/C3SC52538D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Biotage ISOLUTE SCX-2 solid phase extraction column
- [33].pp. 10267–10271.
- [33a].Ram RN, Kumar N. Tetrahedron. 64 [Google Scholar]
- [33b].Medimagh R, Marque S, Prim D, Marrot J, Chatti S. Org. Lett. 2008;11 doi: 10.1021/ol9003965. [DOI] [PubMed] [Google Scholar]
- [33c].Romero M, Renard P, Caignard DH, Atassi G, Solans X, Constans P, Bailly C, Pujol MD. J. Med. Chem. 2009;50 doi: 10.1021/jm061184g. [DOI] [PubMed] [Google Scholar]
- [33d].Ball M, Boyd A, Churchill G, Cuthbert M, Drew M, Fielding M, Ford G, Frodsham L, Golden M, Leslie K, Lyons S, McKeever-Abbas B, Stark A, Tomlin P. Org. Process Res. Dev. 2007;16 [Google Scholar]
- [33e].Peter A, Singaram B. Tetrahedron Lett. 2012;23 [Google Scholar]
- [33f].Sarang PS, Yadav AA, Patil PS, Krishna UM, Trivedi GK, Salunkhe MM. Synthesis. 1982 [Google Scholar]
- [33g].Mahmoud E, Watson DA, Lobo RF. Green Chem. 2014;16 [Google Scholar]
- [34].pp. 3743–3748.
- [34a].Teague SJ, Davis AM, Leeson PD, Oprea T. Angew. Chem. Int. Ed. 38 doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999;111 [Google Scholar]
- [34b].Nadin A, Hattotuwagama C, Churcher I. Angew. Chem. Int. Ed. 1999;51 doi: 10.1002/anie.201105840. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [35].pp. 881–890.
- [35a].Leeson PD, Springthorpe B. Nat. Rev. Drug Discovery. 6 doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- [35b].Gleeson P, Bravi G, Modi S, Lowe D. Bioorg. Med. Chem. 2007;17 doi: 10.1016/j.bmc.2009.07.002. [DOI] [PubMed] [Google Scholar]
- [35c].Hughes JD, Blagg J, Price DA, Bailey S, DeCrescenzo GA, Devraj RV, Ellsworth E, Fobian YM, Gibbs ME, Gilles RW, Greene N, Huang E, Krieger-Burke T, Loesel J, Wager T, Whiteley L, Zhang Y. Bioorg. Med. Chem. Lett. 2009;18 doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- [36].Linnankoski J, Mäkelä JM, Ranta VP, Urtti A, Yliperttula M. J. Med. Chem. 2006;49:3674–3681. doi: 10.1021/jm051231p. [DOI] [PubMed] [Google Scholar]
- [37].Clemons PA, Bodycombe NE, Carrinski HA, Wilson JA, Shamji AF, Wagner BK, Koehler AN, Schreiber SL. Proc. Natl. Acad. Sci. USA. 2010;107:18787–18792. doi: 10.1073/pnas.1012741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].IVC testing performed by Cyprotex, 15 Beech Lane, Macclesfield, Cheshire, SK10 2DR, UK
- [39].Polar Surface Area and.clog P were calculated using ChemBioDraw Ultra 14.0, CambridgeSoft
- [40].pp. 496–499.
- [40a].Kotsuki H, Nishizawa H, Ochi M, Matsuoka K. Bull. Chem. Soc. Jpn. 55 [Google Scholar]
- [40b].McCluskey A, Keane MA, Walkom CC, Bowyer MC, Sim ATR, Young DJ, Sakoff JA. Bioorg. Med. Chem. Lett. 1982;12 [Google Scholar]
- [41].Hayashi Y, Nakamura M, Nakao S, Inoue T, Shoji M. Angew. Chem. Int. Ed. 41:4079–4082. doi: 10.1002/1521-3773(20021104)41:21<4079::AID-ANIE4079>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002;114 [Google Scholar]
- [42].Kassick AJ, Jiang J, Bunda J, Wilson D, Bao J, Lu H, Lin P, Ball RG, Doss GA, Tong X, Tsao KLC, Wang H, Chicchi G, Karanam B, Tschirret-Guth R, Samuel K, Hora DF, Kumar S, Madeira M, Eng W, Hargreaves R, Purcell M, Gantert L, Cook J, DeVita RJ, Mills SG. J. Med. Chem. 2013;56:5940–5948. doi: 10.1021/jm400751p. [DOI] [PubMed] [Google Scholar]
- [43].pp. 3339–3340.
- [43a].Kotsuki H, Asao K, Ohnishi H. Bull. Chem. Soc. Jpn. 57 [Google Scholar]
- [43b].Moore JA, Partain EM. J. Org. Chem. 1984;48 [Google Scholar]
- [43c].Brion F. Tetrahedron Lett. 1983;23 [Google Scholar]
- [44].Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- [45].Paton RS, Kim S, Ross AG, Danishefsky SJ, Houk KN. Angew. Chem. Int. Ed. 50:10366–10368. doi: 10.1002/anie.201103998. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123 [Google Scholar]
- [46].CYLview, 1.0b; Legault, C. Y., Université de Sherbrooke, 2009. http://www.cylview.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.