

Summary
ADAM metallopeptidase domain 17 (ADAM17) is responsible for processing large numbers of proteins. Recently, 1 family involving 2 patients with a homozygous mutation in ADAM17 were described, presenting with skin lesions and diarrhea. In this report, we describe a second family confirming the existence of this syndrome. The proband presented with severe diarrhea, skin rash, and recurrent sepsis, eventually leading to her death at the age of 10 months. We performed exome sequencing and detailed pathological and immunological investigations. We identified a novel homozygous frameshift mutation in ADAM17 (NM_003183.4:c.308dupA) leading to a premature stop codon. CD4+ and CD8+ T-cell stimulation assays showed severely diminished tumor necrosis factor–α and interleukin-2 production. Skin biopsies indicated a focal neutrophilic infiltrate and spongiotic dermatitis. Interestingly, the patient developed unexplained systolic hypertension and nonspecific hepatitis with apoptosis. This report provides evidence for an important role of ADAM17 in human immunological response and underscores its multiorgan involvement.
Keywords: ADAM17, Exome sequencing, Congenital enteropathy, Immunodeficiency, Inflammation
1. Introduction
ADAM metallopeptidase domain 17 (ADAM17) is a sheddase belonging to the ADAM family of disintegrins and metalloproteases. Because of its wide variety of ligands such as epidermal growth factor receptor ligands, tumor necrosis factor (TNF)–α, and angiotensin I converting enzyme 2, ADAM17 is crucially involved in various pathological conditions including cancer, inflammation, neurodegeneration, and fibrosis. More than 70 different substrates for ADAM17 have been reported [1]. Recently, 2 siblings were described with a homozygous loss of function mutation in the gene encoding for ADAM17 [2]. Whereas 1 sibling died at the age of 12 years, the other sibling presented in adulthood and experienced repeated skin infections only. Mechanistic data and assessment of different organ systems potentially affected by a loss of ADAM17 function were limited in this report. Here we report a second family with a novel mutation in the ADAM17 gene confirming the existence of this new rare syndrome and provide evidence for a multiorgan involvement.
2. Clinical history
2.1. General history and infectious manifestions
The examined family consisted of 5 family members with the proband being the third child, a girl, of healthy Armenian parents. There was no history of consanguinity. The pregnancy was uneventful except for intrauterine diagnosed oligohydramnios and growth restriction. After a full-term vaginal delivery, birth weight was 2790 g (20th–50th percentile). At birth, the main clinical feature of the infant was its skin that was covered by a collodion membrane, which coincided with diffuse erythema. Except for an ear tag, no other dysmorphic features were present. Apart from the affected child, the siblings and parents were examined clinically by a staff geneticist for dysmorphic features, which were not found. She was discharged soon after birth but readmitted 13 days after birth because of persistent skin rash, diarrhea, and weight loss. From the first hospital admission until her death, there were numerous (>12) episodes with fever and irritability and, for some episodes, clinical signs of sepsis with tachycardia and cold peripheries. Febrile episodes were accompanied by significant elevations of blood inflammatory markers, for which antibiotic therapy was empirically started. C-reactive protein was intermittently elevated with a peak level of 72 mg/ mL; in-between levels were normal (<5 mg/mL). Leukocytes were elevated most of the time, with levels around 15 × 109/L and a peak level of 34 × 109/L. Blood cultures taken during these incidents were only positive on 3 of these episodes. Enterogenic bacteria, Acinetobacter spp (2×), and Escherichia coli (1×) were cultured. After initiation of selective intestinal decontamination, that is, reducing the concentration of potentially pathogenic bacteria and fungi using tobramycin, colistin, and amphotericin B, there were no more septic/ bacteremic events. After prolonged hospitalization in different hospitals, the patient ultimately developed respiratory insufficiency related to a respiratory syncytial virus and died at the age of 10 months because of refractory hypoxia. Autopsy was not granted by the family.
2.2. Cutaneous involvement
Tender skin lesions were seen within 7 days after birth. The skin felt dry, rough, and scaly, reminiscent of atopic dermatitis/ichthyosis vulgaris (Fig. 1). There were diffuse generalized pustular rash and erythema with yellow crusted scales, similar to those seen in seborrheic dermatitis, over the scalp and face (periocular, cheek, chin, jaw, upper lip) and in the folds of axillae and groins. The clinical picture was suggestive of acrodermatitis enterohepathica but failed to respond to zinc supplementation. Multiple bacterial swabs of the skin grew Staphylococcus aureus. Hair on scalp and eyebrows was present at birth but shed soon thereafter. Hair growth returned at the age of 6 months. The nails were not affected. The severity of skin abnormalities was episodic in nature and appeared to coincide with the severity of intestinal symptoms. Dermatological treatments included corticosteroids, antimycotics/antibiotics, and plain ointments, with some clinical response. The skin infections responded best to systemic broad-spectrum antibiotics.
Fig. 1.
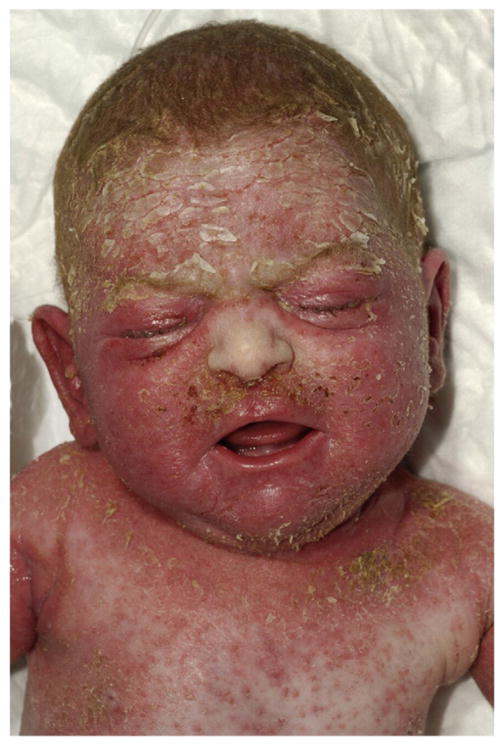
Dermatological features at 3 weeks of age. Erythema of the face and flexures, axillary maceration, widespread pustules in the chest, and seborrhoeic scaling of the scalp and forehead can be seen.
2.3. Gastrointestinal and hepatic manifestations and growth
Severe watery diarrhea developed in the first week of life. Diarrhea did not completely disappear after discontinuation of feeding, suggesting a combination of osmotic and secretory diarrhea. Feeding in the form of an elemental amino acid–based formula was restarted but did not prevent novel episodes of loose to watery stools. Mucus was also noticed frequently, but there was never any rectal bleeding. Fecal-reducing substances were episodically elevated, associated with mildly decreased fecal pH suggesting carbohydrate malabsorption. She was on full parenteral nutrition for periods of less than 1.5 weeks and on partial parenteral nutrition for about 6 months. Elevation of transaminases developed soon after the start of parenteral nutrition. Increased levels varied over time and did not clearly correlate with clinical signs of sepsis or increased C-reactive protein concentrations. Maximum levels were aspartate aminotransferase 396 U/L and alanine transaminase 426 U/L, which improved after switching from Intralipid (Fresenius Kabi, Bad Homburg, Germany) to Omegaven (Fresenius Kabi) in the parenteral nutrition but did not completely resolve. While being on full enteral nutrition with a high energy intake (145 kcal/[kg d]), there was only a modest weight gain. Weight and height remained below −2 SD of the World Health Organization reference curve.
2.4. Renal and cardiac manifestations
Systolic blood pressure became consistently elevated at the age of 6 months (mean, 127 mm Hg), whereas diastolic blood pressure was normal (mean diastolic, 56 mm Hg). Cardiac evaluation revealed a mild pulmonary valve stenosis and small atrial septum defect but no other structural abnormalities. The cardiac findings were not accompanied by clinical symptoms such as shortness of breath. Hypertension was refractory to amlodipine but responded well to oral labetolol (mean, 104/68 mm Hg with a maximal dose of 3.5 mg/[kg d]). Ultrasound revealed relatively large kidneys (right kidney, 6.6 × 3.4 × 3.2 cm; left kidney, 6.4 × 3.5 × 3.2 cm; both >95th percentile) with increased cortical echogenicity. There was no conclusive biochemical evidence for an activated renin-angiotensin-aldosterone system. The patient showed moderate proteinuria with persistently elevated excretion of β2-microglobulin. Glomerular filtration rate and tubular handling of phosphate and sodium revealed no abnormalities.
3. Materials and methods
We obtained written informed consent from both parents for participation in this study. The study was performed according to the Declaration of Helsinki protocols and was approved by the UCLA institutional review board. Data containing patient information were extracted from patient’s medical records. Blood samples for specific immunological and genetic analyses were collected at the age of 7 months from the affected girl and her parents. Esophagogastroduodenoscopy and coloscopy were performed at the age of 3 and 6 months. A skin biopsy was performed at the age of 5 months; and a liver biopsy, at the age of 6 months. No other biopsies were taken. Biopsy material was collected during these procedures for basic histology and immunohistochemistry on paraffin-embedded tissues and for electron microscopy using standard methods.
Genomic DNA was isolated from peripheral blood lymphocytes according to standard procedures. Microarray analysis was performed using an Affymetrix GeneChip 260K Nsp1 (Santa Clara, CA) genotyping array according to the manufacturer’s instructions. Genomic DNA was exome enriched (Agilent SureSelect XT Human All Exon 50 Mb, Santa Clara, CA), sequenced (Illumina GA 2000, San Diego, CA), and mapped (Novocraft Novoalign, Petaling Jaya, Malaysia) according to the manufacturers’ instructions, yielding 100 × 100 paired end reads at a mean depth of 58×, with 86% of targeted bases having at least 20× coverage. Variants were identified as previously described [3].
The cytokine pattern of T-cell subsets was determined using fluorescent cell barcoding as recently described by our group [4]. Heparinized blood was stimulated and incubated within 3 hours after collection. Results were expressed as the percentage of cytokine-producing CD69+ cells within the total CD4+ or CD8+ T-cell population. Values were corrected for unstimulated cultures. For the cytokine expression studies, results were expressed as the average with the 10th and 90th percentiles. As these data are based on the report of a single patient, no statistical analyses were performed.
4. Results
4.1. Genetic analyses
Using Sanger sequencing, no pathogenic mutations could be identified in the coding regions of the genes SLC39A4 (acrodermatitis enterohepatica), SPINK5 (Netherton syndrome), and MYO5B (myosin 5B, microvillus inclusion disease). Microarray analysis showed a normal female profile (arr [1–22,X]x2). The SNP array did show 7 large stretches of copy number–neutral homozygosity on chromosomes 2, 4, 6, 8, 10, 11, and X. Whole-exome sequencing was subsequently performed. After filtering 70 324 raw variants by consequence rank, allele frequency, and controls, 151 variants remained. Among these 151 candidate variants, one was a homozygous frameshift mutation in ADAM17 (NM_003183.4:c.308dupA). The mutation was in a 5-Mb homozygosity block. This novel mutation was predicted to introduce a frame shift and a premature stop codon (p.Asn103LysfsTer20) and expected to result in expression of a severely truncated protein or in an absence of protein as a consequence of nonsense-mediated mRNA decay. Homozygosity for the ADAM17 mutation was only seen in the affected proband and not in the siblings, and heterozygosity was confirmed in both parents using Sanger sequencing.
4.2. Histology
Skin biopsies were obtained from the erosive axillary and the scaly, dry abdominal skin. At the time of the biopsy, there were clinical signs of systemic infection with fever and elevated concentrations of C-reactive protein. Biopsies were taken 4 days after start of broad-spectrum antibiotics (meropenem and vancomycin), when the skin lesions were starting to improve. Skin samples revealed features of parakeratosis with focal neutrophilic granulocytes and spongiotic dermatitis with exocytosis of lymphocytes (Fig. 2A). No epidermolytic keratosis, acantholysis, or epidermolysis was present. The hair shafts appeared normal. Result of periodic acid–Schiff staining was negative. Immunohistochemistry on routine paraffin sections showed normal expression of lymphoepithelial Kazal type–related inhibitor (Lekti) in the epidermis, ruling out Netherton disease. Immunofluorescence staining for ADAM17 antibody was scheduled, but the patient died before we could obtain a skin sample for frozen sections. Electron microscopy revealed spongiosis without additional diagnostic features (data not shown).
Fig. 2.
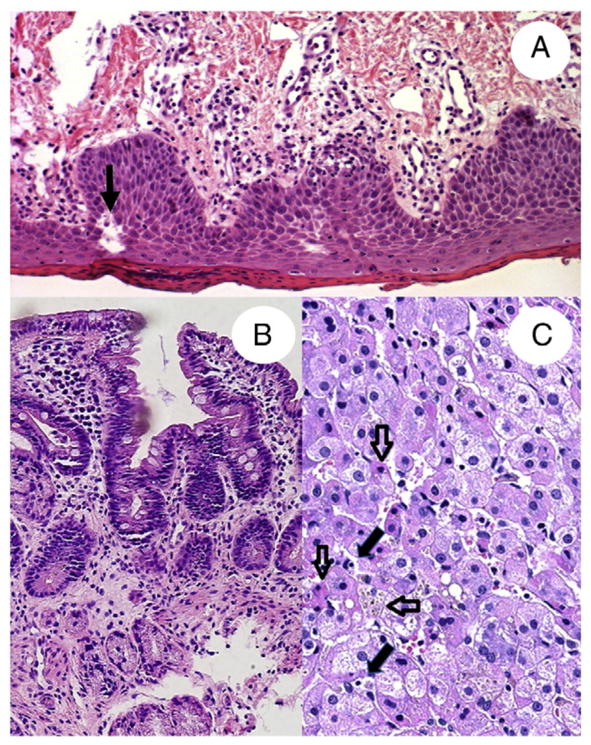
A, Hematoxylin and eosin stain of a skin biopsy taken from an affected scaly area on the abdomen revealing marked parakeratosis, neutrophils, and spongiosis (arrow). B, Hematoxylin and eosin stain of a duodenal biopsy obtained at 6 months of age. Duodenal biopsy shows focal villous atrophy and minimal crypt hyperplasia. C, Hematoxylin and eosin stain of a liver biopsy showing mild lymphocytic infiltrate (filled arrows), hepatocellular cholestasis (horizontal open arrow), and foci of apoptotic activity (vertical open arrows).
A duodenal biopsy at 3 months of age showed patchy mostly mild focal villous atrophy and crypt hyperplasia without intraepithelial lymphocytosis at 6 months (Fig. 2B). Periodic acid–Schiff staining showed focal thinning of glycocalix on the surface of enterocytes. Although we did not observe apoptosis, we did see some scattered mitosis in the crypts. In all gastrointestinal compartments, there was a slight patchy increase in lymphoplasmocytic inflammation in combination with the presence of eosinophils. There were no other clear abnormal histological findings in the esophagus, stomach, or colon. Electron microscopy of intestinal biopsies showed a relatively normal brush border microvilli on the enterocytes and well-preserved Paneth cells and endocrine cells (data not shown). Result of immunostaining for cytomegalovirus was negative. In summary, the intestinal histological changes were minimal and nonspecific and do not explain well the clinical intestinal phenotype.
Evaluation of the liver revealed a mild intralobular lymphocytic infiltrate without interface hepatitis (Fig. 2C). In some portal areas, there was mild ductular reaction in the absence of inflammation. There was no evidence of fibrosis. There was evidence of hepatocelular cholestasis and scattered foci with apoptotic bodies, without a zonal distribution. The presence of this apoptotic activity was confirmed by caspase 3 staining (data not shown). At the time of the liver biopsy, transaminases were elevated with an aspartate aminotransferase of 160 U/L, alanine transaminase of 218 U/L, and γ-glutamyl transpeptidase of 97 U/L.
4.3. Immunological function
Immunocompetence of the patient was investigated in a number of ways. She had elevated white blood cell counts (15–20 × 109/L) most of the time, with normal differentiation most of the time. Phenotyping of her lymphocytes (including Tregs) were normal for her age; and all T cells were autologous, thereby excluding a severe combined immunodeficiency or Omenn syndrome. We assessed the function of her T cells by measuring T-cell activation and intracellular cytokine production upon stimulation (Table and Fig. 3). The results showed normal T-cell activation (CD69 positivity) upon stimulation with phorbol myristate acetate (PMA), phytohemagglutinin (PHA), and staphylococcal enterotoxin B (SEB) in CD4 and especially CD8 cells. Intracellular TNF-α production was diminished, especially in the CD8+ T cells. Moreover, intracellular interleukin (IL)-2 production was impaired as well, whereas interferon (IFN)-γ production was normal. Polyfunctional analyses were not performed because there were hardly any cells with double-positive expression of cytokines.
Table.
Frequencies of cytokine-producing T lymphocytes upon in vitro stimulation in the patient compared to healthy controls
| Stimulation | Cytokine/activation marker | % Cytokine-producing cells/CD69+ cells | |||
|---|---|---|---|---|---|
|
| |||||
| Within CD3+CD4+ T cells | Within CD3+CD8+ T cells | ||||
|
|
|
||||
| Patient | Healthy controls | Patient | Healthy controls | ||
| PMA | TNF-α | 10.5 a | 53.0 (36.9–69.6) | 3.4 a | 14.1 (7.3–22.0) |
| IL-2 | 0.0 a | 15.4 (3.1–22.8) | 0.05 a | 2.8 (0.6–5.8) | |
| IFN-γ | 5.4 | 2.2 (0.9–3.3) | 8.3 | 6.9 (2.5–15.5) | |
| CD69 | 90.4 | 100 (100–100) | 94.7 | 100 (100–100) | |
| PHA | TNF-α | 3.5 | 8.2 (2.9–12.5) | 0.4 a | 4.6 (0.8–9.5 |
| IL-2 | 0.6 a | 1.8 (0.1–3.2) | 0.04 a | 0.3 (0.1–0.6) | |
| IFN-γ | 2.0 | 1.0 (0.3–1.7) | 1.6 | 2.4 (0.5–4.0) | |
| CD69 | 15.6 a | 35.2 (23.6–50.4) | 21.4 a | 44.4 (27.0–61.6) | |
| SEB | TNF-α | 1.8 | 3.8 (1.5–6.8) | 0.02 a | 1.0 (0.3–1.9) |
| IL-2 | 0.4 a | 1.4 (0.3–3.8) | 0.05 a | 0.2 (0.1–0.5) | |
| IFN-γ | 0.7 | 0.3 (0–1.9) | 0.2 | 0.7 (0.1–1.6) | |
| CD69 | 7.0 | 9.7 (3.4–15.6) | 9.2 | 11.9 (6.6–17.5) | |
NOTE. PBMCs were in vitro stimulated with PMA, PHA, or SEB; and frequencies of activated (CD69+) and TNF-α–, IFN-γ–, and IL-2–producing T lymphocytes within the CD3+CD4+ T cells and CD3+CD8+ T cells were determined. Healthy controls (n = 20) of the same age as the patient (age 6 months) were used. Average frequencies (10th and 90th percentile) are denoted for the healthy controls.
Abbreviations: PMA, phorbol myristate acetate; PHA, phytohemagglutinin; SEB, staphylococcal enterotoxin B.
Patient results below the 10th percentile of the healthy controls.
Fig. 3.

Representative flow cytometry analysis of TNF-α production in the CD3+CD8+ T lymphocytes of the patient (A and B) and a healthy control (C). PBMCs were in vitro stimulated with medium only (A) or PHA (B and C) and stained for CD69 and TNF-α expression. Results show a higher percentage of TNF-α–producing cells in the healthy control subject (2.5%) compared to the patient (0.4%) upon stimulation with PHA.
5. Discussion
To our knowledge, this is the second report describing phenotypic and genetic characteristics of a disorder associated with loss of ADAM17. Our report provides new information on additional clinical and pathological changes observed in patients with ADAM17 mutation. In the previously described siblings with an ADAM17 mutation, extracellular production of TNF-α was diminished upon stimulation of peripheral blood mononuclear cells (PBMCs). We confirmed this finding and provide additional evidence that this is probably due to an intracellular pathway defect, whereby diminished IL-2 production plays a pivotal role. IL-2 is known to be cardinal in the development and function of lymphocytes and hence inflammation. In addition, our data suggest an upregulation of IFN-γ production. A recent observation that inhibition of ADAM17 causes an activation of IFN-γ via IL-1β [5] is consistent with our findings. The immunological defects are therefore likely to lead to an increased susceptibility to severe bacterial infections and enhanced inflammatory state as seen in our patient.
Sheddase enzymes cleave and thereby release many membrane-bound substrates, including desmogleins and desmocollins from the cell surface. Perturbance of the skin barrier leads to severe dermatitis such as in atopic dermatitis associated by loss of filaggrin [6], Netherton disease due to secondary desmoglein 1 and desmocollin 1 cleavage [7], and sinobronchial allergic mycosis syndrome due to primary desmoglein 1 mutations [8]. sinobronchial allergic mycosis syndrome is also associated with metabolic wasting as also found in the patient reported here.
The etiology of primarily systolic hypertension in our case remains elusive. We were unable to find a clear relation between the hypertension and the mutation, in part because the patient died before we could exclude all other causes. Although we did not find evidence for a dysregulated renin-angiotensin-aldosterone system, it is tempting to speculate that the elevated blood pressure is secondary to an imbalance between angiotensin-converting enzyme I and II. The activation of the latter may be reduced secondary to a reduced or abolished sheddase activity of ADAM17. It is unclear whether the prolonged hypertension had any longer-term effect on cardiac function or morphology, as the patients passed away before we could evaluate this. One of the previously described patients was found to have a mild cardiomyopathy [2].
The liver histology and electron microscopy findings were not specific for parenteral nutrition–induced cholestasis, although it could have contributed to the liver pathology. We did not observe steatosis or macrophage aggregates or fibrosis, which can be found in total parenteral nutrition-induced liver injury. In addition, transaminases remained elevated even after total parenteral nutrition had been discontinued. Sepsis can also be associated with abnormal liver biochemistry; but in our patient, we did not observe a correlation between transaminase levels and signs of sepsis or increases in levels of C-reactive protein. Multiple studies have indicated that ADAM17 is involved in hepatocyte apoptosis. ADAM17-mediated shedding of epidermal growth factor receptor and TNF receptor has been shown to inhibit apoptotic signals in a hepatitis model in mice [9]. In hypoxia-exposed liver cell lines, downregulation of ADAM17 led to upregulation of caspase-3 [10], a critical regulator of apoptosis, which is consistent with our observation of high caspase-3 expression in hepatocytes in the affected patient. It has been shown that ADAM17 inhibition stimulates caspase-1 expression in isolated PMBCs [5] and that caspase-1 is strongly involved in cell death receptor FAS-mediated apoptosis [11]. Finally, trials with inhibitors of ADAM17 have shown indications of liver toxicity [12]. It is of interest that there was no mention of hepatic involvement in the 2 siblings described in the earlier report by Blaydon et al [2].
Our report underscores the broad regulatory role of ADAM17 in human biological processes. Blaydon et al [2] postulated that, in humans, compensatory mechanisms might exist to limit the severity of the phenotype. However, our patient illustrates that disease severity is variable and can lead to very early mortality. Although several reports have indicated the potential benefits of ADAM17–blocking agents for the treatment of inflammatory conditions or certain types of cancer, the severity and extensiveness of the phenotype associated with loss of ADAM17 function might make this a less attractive strategy.
Acknowledgments
We thank Dr Annechien Lambeck, medical immunologist, for excellent technical support.
Footnotes
Disclosures: The authors declare that they have no conflicts of interest. This work was supported by grants from the National Institute of Diabetes, Digestive and Kidney Diseases, Bethesda, MD (DK083762) and California Institute of Regenerative Medicine, San Francisco, CA (RT2-01985).
References
- 1.Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011;32:380–7. doi: 10.1016/j.it.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Blaydon DC, Biancheri P, Di WL, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–8. doi: 10.1056/NEJMoa1100721. [DOI] [PubMed] [Google Scholar]
- 3.Yourshaw M, Taylor P, Rao AR, Martín MG, Nelson SF. Rich annotation of DNA sequencing variants by leveraging the Ensembl Variant Effect Predictor with plugins. Brief Bioinform. doi: 10.1093/bib/bbu008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stam J, Abdulahad W, Huitema MG, et al. Fluorescent cell barcoding as a tool to assess the age-related development of intracellular cytokine production in small amounts of blood from infants. PLoS One. 2011;6:e25690. doi: 10.1371/journal.pone.0025690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma M, Mohapatra J, Acharya A, Deshpande SS, Chatterjee A, Jain MR. Blockade of tumor necrosis factor–alpha converting enzyme (TACE) enhances IL-1beta and IFN-gamma via caspase-1 activation: a probable cause for loss of efficacy of TACE inhibitors in humans? Eur J Pharmacol. 2013;701:106–13. doi: 10.1016/j.ejphar.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Palmer CN, Irvine AD, Terron-Kwiatkowski A, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 7.Descargues P, Deraison C, Prost C, et al. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J Invest Dermatol. 2006;126:1622–32. doi: 10.1038/sj.jid.5700284. [DOI] [PubMed] [Google Scholar]
- 8.Samuelov L, Sarig O, Harmon RM, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45:1244–8. doi: 10.1038/ng.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murthy A, Defamie V, Smookler DS, et al. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Invest. 2010;120:2731–44. doi: 10.1172/JCI42686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang XJ, Feng CW, Li M. ADAM17 mediates hypoxia-induced drug resistance in hepatocellular carcinoma cells through activation of EGFR/PI3K/Akt pathway. Mol Cell Biochem. 2013;380:57–66. doi: 10.1007/s11010-013-1657-z. [DOI] [PubMed] [Google Scholar]
- 11.Los M, Van de Craen M, Penning LC, et al. Requirement of an ICE/CED-3 protease for Fas/APO-1–mediated apoptosis. Nature. 1995;375:81–3. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 12.Moss ML, Sklair-Tavron L, Nudelman R. Drug insight: tumor necrosis factor–converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2008;4:300–9. doi: 10.1038/ncprheum0797. [DOI] [PubMed] [Google Scholar]