

Abstract
Purpose
We evaluated the role of a poly(ethylene glycol) (PEG) surface coating to increase residence times and alter the cellular fate of nano- and microparticles delivered to the lung.
Methods
Three sizes of PRINT hydrogel particles (80×320 nm, 1.5 and 6 um donuts) with and without a surface PEG coating were instilled in the airways of C57/b6 mice. At time points of 1, 7, and 28 days, BALF and whole lungs were evaluated for the inflammatory cytokine Il-6 and chemokine MIP-2, histopathology, cellular populations of macrophages, dendritic cells (DCs), and granulocytes, and particulate uptake within these cells through flow cytometry, ELISAs, and fluorescent imaging.
Results
Particles of all sizes and surface chemistries were readily observed in the lung with minimal inflammatory response at all time points. Surface modification with PEGylation was found to significantly increase lung residence times and homogeneous lung distribution, delaying macrophage clearance of all sizes, with the largest increase in residence time observed for 80×320 nm particles. Additionally, it was observed that DCs were recruited to the airway following administration of unPEGylated particles and preferentially associated with these particles.
Conclusions
Pulmonary drug delivery vehicles designed with a PEG surface coating can be used to delay particle uptake and promote cell-specific targeting of therapeutics.
Keywords: pulmonary drug delivery, PEGylation, nanoparticle, microparticle
Introduction
Drug delivery via the pulmonary route offers an alternative course of administration with tremendous therapeutic benefits (1). Respiratory drug delivery can target local lung diseases, such as asthma, COPD, lung cancer, and can also be used to delivery therapeutics systemically, including delivery of insulin for diabetes management, or antigens for vaccination (2–5). This drug delivery can be achieved through a variety of methods, ranging from traditional nebulizers and inhaler devices to more recent developments in dry powder (DPI) therapeutics, offering benefits of patient compliance and ease of administration (6, 7).
There remains a significant unmet need in the delivery of respiratory drugs to target or de-target cells in the lung depending on the desired site of action of the therapeutic. Alveolar macrophages (AM or MΦ) often present a significant barrier to efficient pulmonary drug delivery applications because of i) their sequestration and undesirable clearance of certain deposited drug particles in the lung whose site of action involves non-macrophage targets, like aerosolized therapeutics targeted to the alveoli and epithelial cells (8, 9) or ii) by their protection and harboring of disease targets, such as infectious bacteria like tuberculosis, which makes it difficult to treat effectively (10). Because particles with geometric diameters of 2–3 microns have been shown to be preferentially taken up by AMs (11), large, porous particles (LPPs) (12) and swellable microparticles (13) were developed to deposit in the alveolar airspace and successfully avoid macrophage uptake due to their large (> 5 μm) geometric size. Alternatively, it has been suggested that nanoparticles can avoid the rigorous AM clearance faced by microparticles, as nanoparticles are not as rapidly internalized by AMs (6). Any such therapeutic carriers which could avoid macrophage uptake offer the potential to provide sustained controlled release to the airspace and possibly into systemic circulation. While this detargeting of AMs holds clear therapeutic potential, specific targeting of these and other lung cells may hold alternative therapeutic advantages. For example, dendritic cell (DC) uptake of particles containing an antigen may be advantageous for vaccine applications or immunomodulation therapies (14–19). Physicochemical particle properties targeting macrophages have also been explored for enhancing host defense and regulating immune responses, and may be required for successful treatment of tuberculosis (10, 20).
The addition of a poly(ethylene glycol) (PEG) surface coating to nanoparticles has been studied extensively for intravenous (IV) administration of drug carriers, as the increase in surface hydrophilicity can prevent protein opsonization, which in turn limits cellular uptake and extends circulation times (21–24). Given the overwhelming evidence of PEGylation in avoiding macrophage uptake in IV applications, PEGylation has also been investigated in the context of pulmonary drug delivery (25). PEGylation has been successfully used to extend the lung half-life of antibodies, small molecule drugs, and other proteins in the lung (25–28). Various particles containing PEG in their formulation have been evaluated for pulmonary delivery as novel carriers, such as liposomes, polymeric micelles, and dendrimers (25), while others have been further optimized for therapeutic effects, including gene delivery, vaccines, and lung cancer (25, 29–33). PEGylation has been studied on LPPs and swellable polymeric particles made of PEG, with emphasis placed on the final geometric size accounting for the increase in macrophage avoidance and sustained release (13, 34). Finally, PEG coatings have also been studied in the lung in the context of mucocilliary transport, as a high density PEG coating can promote mucus penetration of nano- and micro-particles in the lung (35, 36). Alternative coatings have been explored in the lung, as a particle coating of phospholipid 1,2-dipalmitoylphosphatidylcholine (DPPC) was shown to reduce uptake in alveolar macrophages (37). Despite this extensive history of PEG in the lung, there has been surprisingly limited work done to explore the in vivo cellular fate and inflammatory response to micro- and nano-particles coated with PEG. Indeed, to our knowledge, there has not been a study to date which directly evaluates the role of a PEG surface coating on lung particle residence times.
To determine if PEGylation is a feasible approach for extending the residence time of a particle in the lung, we utilized the Particle Replication in Non-wetting Templates (PRINT) method, which allows for the precise control over many particle characteristics such as size, shape, composition, porosity, modulus, and surface functionality (38). PRINT particles have been previously demonstrated for pulmonary applications (5, 39, 40) and PRINT nanoparticles have also been successfully functionalized with a high density PEG coating (21). In this study, non-spherical polymeric hydrogel particles ranging from 80 nm to 6 μm in diameter were fabricated using PRINT and functionalized with a PEG coating. These particles were then instilled into the lungs of mice and particle uptake and residence time in the lung was determined by flow cytometry and confocal microscopy, with additional evaluation of the induction of inflammatory cytokines and recruitment of inflammatory cells.
Materials and Methods
Animals
All studies were conducted in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee (IACUC) at UNC. All animals were maintained in pathogen-free facilities at UNC and were between 8 and 15 weeks of age. C57BL/6 were obtained from Jackson Laboratories.
Materials
Solvents and buffers of reagent grade and cell media were obtained by Fisher Scientific. PRINT 80 × 320nm, 1.5μm and 6μm donut molds were provided by Liquidia Technologies. Pre-particle reagents of 2-aminoethylmethacrylate (AEM), poly(ethylene glycol)700 diacrylate (PEG700DA) and diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) were obtained from Sigma; tetra(ethylene glycol) monoacrylate (HP4A) was synthesized in house via previously described methods (41). Particle dye maleimide-Dylight 650 was obtained from Fisher. Lipopolysaccharide (LPS) was obtained from Sigma Aldrich. Methoxy-PEG5k-SCM was purchased from Creative PEGworks.
Fabrication of PEGylated Hydrogel PRINT Particles
Particles were composed of 10% PEG700DA (cross-linker) (Sigma), 67% hydroxyl-tetraethylene glycol monoacrylate (HP4A, monomer), 20% 2-aminoethyl methacrylate hydrochloride (AEM), 2% fluorescent dye (Dylight 650 Maleimide, Thermo Fisher), and 1% 2,4,6-Trimethylbenzoyl-diphenyl-phosphineoxide (TPO, photoinitiator) (Sigma) by weight. This pre-particle solution was filled into a Fluorocur mold of the intended geometric shape and cross-linked in a UV curing chamber (λ=365 nm). Particles were then transferred from the filled mold onto a polyvinyl alcohol (PVOH) harvest layer. Water was used to dissolve away the harvesting layer and free particles were collected. PEG5k-succinimidyl carboxymethyl ester (PEG5k-SCM) was reacted with the amine functional handles resulting in a layer of PEG on the particle surface (21). Both PEGylated and unPEGylated particles were then succinylated to quench any remaining free amine groups.
Particle Characterization
Geometric particle dimensions were measured by scanning electron microscopy (SEM) analysis; samples were sputter-coated with 1–5 nm of Au/Pd (Cressington Scientific Instruments) and imaged by using Hitachi model S-4700. Particle zeta potential was measured using a Zetasizer Nano ZS (Malvern Instruments, Ltd.).
Fluorescent Microscopy of In vitro Macrophage Uptake
MH-S, a mouse alveolar macrophage cell line, (ATCC) was used for particle uptake experiments. Cells were plated at 4 × 104 cells/well in 8-well chamber slides (LabTek) 48 hours before particle addition. Cells were dosed with 25 μg particles in media consisting of high glucose Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) and 10% fetal bovine serum (FBS). Particles were incubated on the cells for 4 hours at 37°C with 5% CO2. After incubation, cells were washed with Dulbecco’s Phosphate Buffer Saline (PBS) solution and fixed with 4% paraformaldehyde (PFA) solution. Staining of actin (Alexa Fluor 555, Invitrogen) and DAPI (Vectashield, Vector Labs) of fixed cells was then performed. Fluorescent imaging of stained cells was performed on a Zeiss 710 laser scanning confocal microscope (Zeiss).
In vivo Lung Instillation
To assess PRINT particle uptake in the lungs of mice, 8–10 week old female C57Bl/6 mice (Jackson Laboratories) were anesthetized with isofluorane and 50 μL of a particle solution was orotracheally administered. The anesthetized animal was suspended via the incisors in a near vertical position. The tongue was retracted to prevent swallowing and the particle suspension was administrated to the mouth. To complete the dose and ensure uniform instillation, the mouse was then monitored for five to ten breaths in this vertical position, with the nose gently occluded to ensure aspiration. Particles were dosed at 50 μg per mouse in PBS solution. Administration of 50 μL PBS or 20 μg/50 μL lipopolysaccharide (LPS) served as negative and positive controls, respectively. At timepoints of one, seven, and twenty-eight days post-particle dose, mice were euthanized and broncheoalveolar lavage fluid (BALF) was collected and the whole lung was excised.
Tissue and Cell Preparation
BAL was performed by inserting a cannula in an incision in the trachea and flushing the lungs with 1 mL HBSS. For collection of BALF cells, three sequential 1 mL washes were performed to ensure collection of all BALF cells; for protein analysis of the BALF, animals were lavaged with a single 1 mL wash to ensure a detectable concentration. BALF cells were obtained by centrifugation, separating BALF cells from supernatant. For single cell whole lung suspensions, lungs were resected, physically agitated and digested in 5 mg/mL Collagenase D, 20 units/mL DNase in HBSS with 2% FBS for 1 hr at 37°C. These cells were then passed through a 70 μm sieve. For histology sectioning, lungs were inflated through the cannulae with a mixture of 1:1 PBS:OCT using a gravity inflation; inflated lungs were embedded in OCT (Tissue-Tek) and flash frozen using an isopentane, dry ice slurry.
Flow Cytometry Analysis
Cells collected from BALF and single cell suspensions collected from homogenized whole lung were stained with a panel of fluorescent antibodies for flow cytometric analysis of particle uptake and cell populations. The following antibodies were used for staining (BioLegend): CD45-PacBlue, CD11c-PE, MHCII-PE-Cy7, and Ly6-G-AF700. Cells were fixed with 4% PFA in PBS after staining and kept at 4°C until analysis. Flow cytometry data was collected on the LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
To correct for slight differences in fluorescence between particle types, relative particle fluorescence was independently determined by using a SpectraMax M5 plate-reader. Particles containing DyLight650 were incubated at 50 μg/mL in PBS and fluorescent intensity of each particle size was measured at 672 nm following excitation at 652 nm. Fluorescence values from all particle sizes were then normalized to that of the 80×320 particles and this ratio was used to adjust median fluorescence intensity (MFI) flow cytometry results.
Cytokine Analysis
Enzyme-linked immuno assay (ELISA) kits for interleukin 6 (IL-6, BD Biosciences) and macrophage inflammatory protein 2 (MIP-2, R & D Systems) were used to measure protein release in BALF supernatant and performed following manufacturer’s instructions.
Fluorescent Lung Tissue Staining
Frozen lung was sliced into 7 μm thick sections, fixed with acetone, and fluorescently stained for phalloidin (Alexa Fluor 555 Phalloidin, Molecular Probes), CD11c (AF888 anti-mouse CD11c, BioLegend), and cell nuclei (DAPI, Thermo Scientific). Images were obtained on the Zeiss 710 confocal microscope.
Results
Fabrication and Characterization of PEGylated Hydrogel PRINT Particles
Three different particle shapes, 80×320 nm, 1.5 μm donuts, and 6 μm donuts composed of PEG700-DA and with amine-functional handles were fabricated using the PRINT method. SEM images show the monodispersity of the fabricated hydrogel particles (Figure 1). Methoxy-PEG5k-SCM was then conjugated to these particles (designated 80×320P, 1.5P, 6P) to provide a stealthing PEG layer on the surface. PEGylated and unPEGylated particles were indistinguishable by SEM. Any remaining free amine groups were quenched with succinic anhydride. Particles that were not PEGylated, (designated 80×320U, 1.5U, 6U), were also quenched with succinic acid so that all particle groups would have a comparable negative surface charge. The particles were then characterized by surface area, volume, and surface charge (Table 1).
Figure 1.

SEM images of PRINT hydrogel particles. (A) 80×320 nm, (B) 1.5 μm donut, (C) 6 μm donut.
Table 1.
Particle characterization of surface area (SA), volume (Vol), and charge. Values from a representative batch of particles.
| SA (μm2) | Vol (μm3) | Zeta Potential (mV) | ||
|---|---|---|---|---|
|
| ||||
| Particle | UnPEGylated | PEGylated | ||
|
| ||||
| 80×320 nm | 0.097 | .0018 | −34.7 | −28.5 |
| 1.5 μm Donut | 5.655 | 0.79 | −25.3 | −32.3 |
| 6 μm Donut | 88.593 | 40.68 | −5.6 | −31.3 |
PEGylation of particles significantly reduces uptake by macrophages in vitro
To confirm the surface modification of the particles with a PEG layer, particles with and without PEG surface modification were incubated in vitro with MH-S mouse alveolar macrophages at a dose of 25 μg / 40k cells. Cells were fixed and stained for fluorescent confocal microscopy 4 hours after particle dose. As seen in the confocal images (Figure 2), PEGylation significantly reduced particle uptake in macrophages for all particle sizes tested. As expected, the macrophages most readily internalized the unPEGylated 1.5 μm donuts compared to the other two particle sizes, as particles with geometric diameters of 1–5 microns have been shown to be preferentially taken up by alveolar macrophages (11, 42).
Figure 2.

Particles dosed to MH-S cells at 25 μg/40 k cells and imaged 4 hours post-dose. Particles (red), Nuclei (blue), Actin (green). Representative images from n=3 independent experiments.
Particle effects on cell populations and cytokine/chemokine response in the lung
Relative cell populations of macrophages (MΦ), dendritic cells (DCs), and granulocytes in the whole lung and bronchoalveolar lavage fluid (BALF) were analyzed by flow cytometry to determine the effect of particle administration on cell recruitment in the lung (Figure 3, representative cell gating in S. Figure 1A). Macrophages (CD45+CD11c+MHCIIlo) constituted the major cell type in the BALF, ranging between 85–99% of the total (Figure 3A). DC (CD45+CD11c+MHCIIhi) percentages of total cells ranged from 1–5% (Figure 3B). Granulocyte (CD45+CD11c−Ly6G+) populations were negligible (Figure 3C) in the saline and particle-treated mice. The macrophage population was observed to change based on the size and surface modification of the particle dosed. One day after particle instillation, it was observed that in the 80×320 nm and 1.5 μm donut treated mice, the unPEGylated particles had a mean macrophage population that was 5% and 10% lower, respectively, than their PEGylated counterparts while there was no statistical difference between the 6 μm donuts of different surface modifications. This trend continues out to day seven for the 80×320 nm sized particles. The LPS-treated mice had a significant influx of granulocytes in the BALF at 24 hours post-dose, such that the relative number of macrophages was decreased by 2-fold.
Figure 3.

Relative populations of macrophages, dendritic cells (DC) and granulocytes in (A–C) BALF and (D–F) whole lung as determined by flow cytometry analysis (BALF representative gating in Supplemental Figure 1, lung in Supplemental Figure 2). Saline and LPS cell populations analyzed only at 1 and 7 days post-dose. Average values shown (n=4 animals) +/− standard deviation. Analysis by two-way ANOVA with Tukey multiple comparisons test; * p < 0.05, ** p < 0.001, *** p < 0.0001.
Interestingly, although the percentage of DCs in the BALF was low; there was a surface modification effect observed on the BALF DC population at 24 hours post-dose that compliments the macrophage population trends. More specifically, the percentage of DCs was 2.5 and 3.8 fold lower in the mice treated with the PEGylated particles verses the unPEGylated particles of the 80×320 nm and 1.5 μm donut shapes, respectively. This difference only occurs at the day one time-point, as no other significant differences in the DC cell populations between particle-treated mice occurs in the BALF collected on days seven and twenty-eight. The 80×320P and 1.5P DC populations were similar to that of saline treated mice (~1%), however, there was a significant increase in BALF DC populations of the other particle-treated mice compared to the saline control groups. The 80×320U, 1.5U, 6U, and 6P particles had about a 4-fold increase in the DC population. Combined with the macrophage population changes, this suggests an influx of DCs to the lung following particle administration of small, unPEGylated particles and both functionalities of 6 μm particles, which further suggests that both surface modification and particle size may play a role in the appearance of DCs in the BALF.
In the whole lung, macrophages, dendritic cells, and granulocytes typically make up less than 20% of the cell population as endothelial, epithelial, and interstitial cells comprise a greater majority of lung cells (Figure 3D–F, representative gating in S. Figure 1A). In these experiments, the macrophage, DC, and granulocyte populations ranged from 5–20%, 5–15%, and 1–6% of the total whole lung cell population remaining after a BAL, respectively. Interestingly, the surface modification effect seen on the DCs in the BALF is not replicated in the whole lung. After 24 hours, the particle-dosed mice had 50% fewer macrophages in the lung than the saline-dosed mice which may indicate macrophage recruitment from the interstitium to the airspace to assist in particle clearance. Similar to the BALF, LPS-treated mice had a significant influx of granulocytes in the lung and the relative number of macrophages in the lung tissue was decreased by 8-fold. Slight increases in the granulocyte population in both BALF and lung was observed with the 6 μm particles at one day post-dose, but reverted back to saline levels at later time points. No other statistical difference between cell populations of the particle-treated mice was observed at one, seven, or twenty-eight days in the whole lung.
Lung architecture and cell infiltration was also analyzed by lung histology (Figure 4). Cell infiltration was noticeable only in the unPEGylated 6 μm donuts one day post dose. All other particles doses at one, seven, and twenty-eight days showed no change in lung architecture or infiltration of granulocytes to the lung, corresponding to flow cytometry results.
Figure 4.

H&E stained lung tissue 24 hours after particle dose. (Scale bar = 200 μm, 2x magnification) Representative images from n=4 animals.
Previous studies have shown that PRINT PEG hydrogel particles do not induce an inflammatory response of TNF-α, IL-1β, and IL-6 production in the lung out to 14 days (40). In this work, IL-6 and MIP-2, a mouse macrophage inflammatory protein plays a key role in neutrophil recruitment, was analyzed in BALF collected at one, seven, and twenty-eight days after particle administration. By ELISA, detectable levels of IL-6 and MIP-2 were only seen one day post particle instillation (Figure 5).
Figure 5.
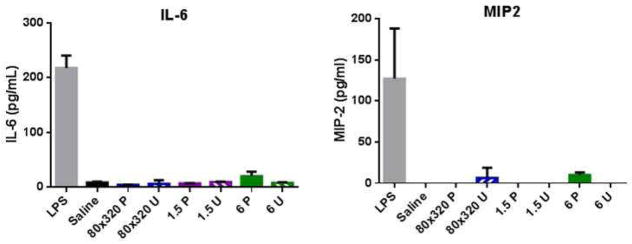
IL-6 cytokine and MIP-2 chemokine levels measured from BALF 24 hours post-instillation. Average values shown (n=4 animals) +/− standard deviation.
Surface modification of particles affects particle retention time and cellular uptake in the lung
To determine particle size and surface modification effects on particle retention time and cellular uptake in the lung, flow cytometry was run on collected BALF and whole lung cells. Using the same flow cytometry gating as shown in S. Figure 1B, macrophage, DC and granulocyte populations were again identified and evaluated for particles containing Dylight 650 dye, thus allowing uptake in specific cell populations to be determined by flow cytometry analysis. The percentage of particle positive cells within a population was determined for BALF macrophages (Figure 6A), lung macrophages (Figure 6D), and lung DCs (Figure 6G). From the particle positive gate, the median fluorescence intensity (MFI), or the average amount of fluorescence per cell, was also determined. This value is a measure of the volume of particles taken up by each particle positive cell type (Figure 6B, E, and H) and has been normalized based on the relative fluorescence intensity of the particles. In addition, the total fluorescence was calculated by multiplying the number of positive particle cells by the MFI in order to give an overall view of the total volume of particles internalized (Figures 6C, F, and I).
Figure 6.

Flow cytometry analysis of particle uptake, median fluorescence intensity (MFI) of particle positive cells, and total fluorescence. (A–C) BALF macrophage, (D–F) lung macrophage and (G–I) lung DCs at 1, 7 and 28 days post-instillation. Average values shown (n=4 animals) +/− standard deviation. Two-way ANOVA with Tukey multiple comparisons test; * p < 0.05, ** p < 0.001, *** p < 0.0001).
Unlike what was seen in vitro, macrophages in the BALF were non-discriminating of surface modification for the smaller 1.5 μm donuts and 80×320 nm particles at one day post instillation with over 80% of macrophages having taken up particles. Yet, at day 28, the PEGylated 80×320 nm particles were present in 60% of BALF macrophages versus only 20% for their unPEGylated counterparts, indicating a time-dependent surface modification response to particle uptake and/or clearance. With the larger 6 μm donuts, the macrophages had preferential uptake of the unPEGylated particles at one and seven days post-dose with no uptake seen at day 28. Because particles were dosed by mass and not by number, the reduced percentage of particle positive cells for the 6 μm donut instilled mice may be due to a reduced surface area coverage of the particles in the lung. In addition, the significantly larger volume of the 6 μm donut may also play a role in reduced uptake by macrophages.
The MFI data normalizes the volume of particles taken up by each particle positive cell. In BALF macrophages (Figure 6B), the MFI of the cells that were associated with the 6 μm donuts was 2–3 times greater than that of the other shapes at one day post-dose, indicating a larger mass of particles per cell. After one day, there was a 1.6-fold reduction in MFI of the 80×320P particle positive macrophages compared to that of the 80×320U and a 3-fold reduction in MFI of the 1.5 P particles compared to the 1.5 U particles after 1 day. This indicates that a greater number of unPEGylated particles were initially taken up by macrophages, thus, surface modification may affect the propensity and degree of particle uptake in vivo. At day twenty-eight, the MFI levels of all particles were extremely low, yet, as mentioned previously, over 60% of macrophages in the BALF were particle positive for PEGylated 80×320 nm particles. We hypothesize that the PEG layer provides a delay in macrophage uptake, continuing through day 28. This may indicate that macrophage clearance and/or turnover rates can be affected by surface modification as well, with the addition of a PEG layer being able to prolong particle residence time in the lung.
The total fluorescence data for BALF macrophages (Figure 6C), shows a clear pattern in the effects of surface modification on particle uptake. At 24 hours post-dose, there is a 1.5, 3.4, and 3.7 fold difference between surface modified and unmodified 80×320 nm, 1.5 μm donut, and 6 μm donut particles, respectively, indicating that PEGylation of particles can significantly reduce uptake by macrophages in the lung of both nanoparticles and microparticles, with perhaps a stronger influence on microparticles. Interestingly, at day seven, the total fluorescence of the 80×320P particles remains at similar levels to those at day one which suggests slower clearance of macrophages with these particles and/or continued uptake of free particles residing in the alveolar space. This hypothesis is supported by the fact that the 80×320U particles have a 3.2 fold reduction in total fluorescence between days one and seven, implying clearance of particle loaded macrophages from the BALF and/or fewer free particles for further uptake by alveolar macrophages.
In macrophages from the lung tissue (Figure 6D), the trends in percent particle positive cells are identical to that of the BALF macrophages. Surface modification again does not play a role in the number of particle positive cells one day after instillation with 80×320 nm and 1.5 μm donut particles. However, unlike in the BALF, the 1.5 μm donut particles had 20% less particle positive cells than the 80×320 nm particles. This indicates a higher preference of the 80×320 nm particles to lung-residing macrophages compared to the other two particle sizes. Interestingly, the percentage of 80×320 nm positive cells increases from day one to day seven, suggesting that there may be free particles residing in the alveolar space which are not immediately internalized by cells in the lung. The unPEGylated 6 μm donuts have over a 5-fold increase in particle positive cells versus the PEGylated particles at both one and seven days post-instillation. In both the BALF and the lung, surface modification on larger micron-sized (6 μm) particles has a greater effect on uptake at early time points than modifications on smaller (80×320 nm and 1.5 μm) particles.
The MFI data for the lung macrophage population (Figure 6E) is quite different from that of the BALF. While the fluorescence intensity of the 80×320 nm particles and 1.5 μm donuts at day one are similar in intensity to that of the BALF, the 6P and 6U particles in the lung tissue had a 25% and 98% reduction in MFI, respectively. This indicates that macrophages in the BALF are associated with a greater number of 6 μm particles compared to macrophages in the lung tissue, and that the unPEGylated particles are more quickly cleared from the lung cells. In furthering the evidence of 80×320P particles having the ability to be retained in the lung both free and in macrophages, the MFI value of these particles increases 2-fold from day one to day seven. One would expect that if only the macrophage clearance rate was reduced, that the MFI levels would have a slower rate of decay, but would not increase over time. Because the percent particle positive cells and the MFI increase over time, this indicates that additional particles may be continually taken up by macrophages in the lung, and are retained within the lung tissue, rather than being cleared to the BALF macrophages. At twenty-eight days post-instillation, all particle MFIs are significantly reduced and no difference is seen between particle types.
While the total fluorescence data from macrophages in the BALF indicated a preference for unPEGylated particles at one day post-dose, the total fluorescence data from macrophages in the lung (Figure 6F) shows no effect of surface modification on the total volume of particles internalized. For the longer seven and twenty-eight day time points, the trends hold from the MFI data, indicating an increase in particle uptake of 80×320 P particles from day one to day seven and then a clearance of particle-loaded macrophages by day twenty-eight.
Unlike the macrophages in both the BALF and the lung, DCs in the lung showed a preference for unPEGylated particles one day post-dose (Figure 6G). Particle uptake of unPEGylated particles increased by 2.0, 4.2, and 1.6-fold in 80×320 nm, 1.5 μm, and 6 μm particles, respectively compared to their PEGylated counterparts. At days seven and twenty-eight, surface modification effects are no longer seen. Interestingly, although there was a higher percentage of particle positive cells with the 80×320 U particles, the MFI data indicates that DCs in the lung have a much higher preference for 1.5 μm donuts, and in particular, the unPEGylated 1.5 μm donuts (Figure 6H). One caveat for the MFI analysis is demonstrated with the 6 μm donut particles at days seven and twenty-eight. Because MFI analyzes the median fluorescence intensity of only particle positive cells, and because of the extremely low percentage of particle positive DCs containing 6 μm donuts (all less than 0.2%), the MFI may not be truly representative of the population and inferences of the data may not be relevant. The vertical axis of the total fluorescence graph of lung DCs (Figure 6I) is an order of magnitude lower than that of the BALF and lung macrophages, but it follows the trend of the MFI data, indicating a strong preference for unPEGylated 1.5 μm particles at one day post-dose. At day seven, the total fluorescence equalizes among the 80×320 nm and 1.5 μm particles which suggests that there may be a homeostatic effect in regulating the volume of particles within DCs in the lung.
These results indicate that there are significant differences in particle uptake and clearance among difference resident lung cell populations. In addition, surface modification, and in this case, PEGylation, of both nano- and micron-sized particles affects particle uptake and retention in the lung. PEGylated 80×320 nm particle uptake in BALF and lung macrophages is seen out to twenty-eight days, suggesting a slower clearance rate of these cells and/or continued uptake of free particles in the alveolar region.
Particle Distribution in the Lung Tissue
To observe the distribution of the particles within the lung tissue, sections of frozen lung tissue were stained for actin, cell nuclei, and CD11c followed by analysis via fluorescent microscopy (Figure 7). As expected, more 80×320 nm particles were observed in the lung tissue due to the number dosed as compared to the micron-sized particles. Interestingly, the PEGylated 80×320nm particles were shown to have a more homogeneous deposition throughout the lung tissue. Because of this higher distribution in the lung epithelium, the retention time of these particles may be extended, supporting the flow cytometry data which shows that PEGylated 80×320 nm particles can remain in the lung out to twenty-eight days. The unPEGylated 80×320 nm particles were shown to be less evenly distributed in the lung and were often seen in larger aggregates than the unPEGylated particles. More free PEGylated 1.5 μm particles were also observed compared to their unPEGylated counterparts. There were no differences in particle distribution in the lung with the 6 μm particles.
Figure 7.

Particle distribution in the lung 1 day post-instillation. Particles (red), Actin (gray), Nuclei (blue), CD11c (green). Representative images from n=4 animals.
Discussion
The ease of access, large absorptive surface area, and reduced enzymatic activity of the lung make it an attractive target for both local and systemic drug delivery, yet drug carriers must circumvent the lung’s highly efficient particle clearance mechanisms in order to successfully deliver their payload. In addition, the drug delivery vehicles must be inert, preferably not contributing to an adverse inflammatory response. While there are a multitude of studies in environmental toxicology analyzing the retention and inflammatory effects of polystyrene, pollutant and industrial (diesel exhaust, dust, carbon fiber) particle burdens in the lung (43, 44), there is a growing interest in studying the effect of lung drug carriers, especially of a controlled design (13, 32, 40). In this study, we observed the effects of size and surface modification on polymeric particles on particle retention and potential inflammatory consequences in the lung. The findings from these studies may help articulate important particle parameters towards designing more efficient pulmonary drug delivery vehicles. Here, we utilized PRINT technology to fabricate monodisperse, cross-linked hydrogel particles of nanometer and micrometer sizes that were further modified by adding an additional PEG layer to the particle surface.
The orientation of the PEG layer on the particle surface is known to directly impact the particle’s interaction with various cells and proteins. In particular, a “brush” conformation, where the PEG chains are roughly perpendicular to the particle surface, is considered a requirement to hinder cellular internalization (21, 45). The method used to functionalize the particles studied here has been previously established in our lab to ensure a brush conformation on 80×320 nm hydrogel particles of the same chemistry (21). While PEG density was not explicitly evaluated for the other two sizes used in this study, we assume that all particle sizes also have a brush conformation. PEG conformation is dictated by the relationship of Flory radius and the pinning distance of PEG chains on the particle surface (46). The calculated Flory radius, as calculated in ref. (21), remains the same regardless of particle size, as the PEG molecular weight and solvent conditions remain unchanged. This leaves potential changes in the pinning distance between PEG chains. This pinning distance is based on the frequency of reactive groups and the particle surface area. As identical particle chemistry is used for all sizes, the frequency of the reactive groups on the particle surface is assumed to be equivalent, ensuring a PEG brush conformation.
Our results support the assumed brush PEG conformation on all particle sizes. The in vitro results in MH-S cells showed dramatic reduction in internalization over the 4 hr time point and all three PEGylated particles showed decreased cellular uptake and increased lung residence time in vivo compared to their unPEGylated counterparts. To our knowledge, this is one of the first reports of PEGylated micron-sized particles avoiding biological clearance mechanisms, as particles of this size range can only feasibly be delivered via inhalation. Indeed, all three sizes of particles in this study were identified to have aerodynamic diameters under 5 microns to facilitate aerosol delivery, regardless of their geometric size (39). Given this large spectrum of sizes, we were originally unsure if a 5k molecular weight PEG would be sufficient to provide the observed adequate stealthing properties to micron-sized particles. There is evidence in the literature to suggest that PEG molecular weight and pinning density can be further optimized within the brush conformation to have enhanced reduction of cellular uptake (47). Future work exploring the role of PEG molecular weight and pinning density to enhance the observed PEGylation effects on micron-sized particles, especially in the lung, is needed and currently ongoing in our group.
In the studies presented here, we evaluated the three sizes of particles dosed at equivalent mass. We chose this dosing strategy to evaluate particle size as a formulation strategy for delivering an equivalent mass of a potential therapeutic agent. Thus, the flow cytometry results of percent positive cells would correspond to the number cells directly interacting with the therapeutic, the MFI would correspond to an approximation for the total dose per cell, and the total fluorescence would correspond to the total dose contained in the collected cellular compartment. Given the large difference in geometric size and thus volume between the particle set, considerable differences in particle number exists between the sizes. Dosing by equivalent particle number would increase the frequency of micron-sized particles in the cellular compartment and may impact the observed inflammatory response. Thus, some of the results presented here are likely an artifact of the differences in particle number dosed. Previous work from our group has shown potential benefit of dosing by an equivalent mass of therapeutic but different particle number through varied drug loadings within particles to increase therapeutic efficacy (48). However, in this work, we assume that the same mass of therapeutic would be loaded into each particle size in order to directly compare the benefit of each size and surface chemistry.
We were especially interested in making these comparisons within key lung populations following pulmonary delivery by utilizing flow cytometry to determine the cellular fate of the particles. Flow cytometry is an especially attractive approach for evaluating particle formulations and is finding increased application in drug delivery applications, especially toward targeting specific cell types. The lung represents a challenging organ in phenotypic identification of the cells identified here, as many subtypes of these cells will express varying amounts of markers that are traditionally used to distinguish cells in other organs. For example, AMs express high levels of CD11c, traditionally considered a DC marker, and a subset of DCs (CD11b+ DCs) can express F4/80, traditionally considered a macrophage marker (19). The gating scheme chosen to broadly identify DCs and macrophages is shown in Supplemental Figure 1 and uses major histocompatibility complex (MHC) II expression to distinguish macrophages and DCs, which assumes a non-inflamed lung environment. This is supported by previous work in our group and the cytokine and histology results in Figures 4 and 5. We are currently exploring more complex flow cytometry strategies to evaluate particle association in some of these critical macrophage and DC subtypes, which may have further application in vaccine and cancer therapeutics.
For therapeutic applications targeting sentinel dendritic cells, our flow cytometry findings show that lung DCs have a significant preference for unPEGylated 1.5 μm particles. Here, increasing the overall lung resident time by avoiding macrophage uptake did not enhance uptake in DCs. Although there are a higher percentage of particle positive DCs in the lung with the 80×320U particles, the total volume of internalized particles is over 3-fold greater for the 1.5U particles, indicating a clear preference for this size larger particle. From a more physiological view, we show that the DC population that migrates out to the airway surface can potentially be modulated by the surface modification of the particle; unPEGylated 80×320 nm and 1.5 μm particles were shown to double the DC population in the BALF one day post-dose.
Interestingly, although the in vitro macrophage uptake studies showed that PEGylated particles of all three particle sizes tested had a significant decrease in number of cells with particle positive cells compared to their unPEGylated counterparts, only the 6 μm donut particles followed this trend in BALF and lung macrophages with a 2-fold and 5-fold decrease in particle positive cells, respectively. This indicates that PEGylated 6 μm donuts may be promising drug carriers as well for disease targets where de-targeting macrophage uptake is preferred. These particles have a large geometric size to avoid macrophage uptake, which is further enhanced with a PEG surface coating. Taking into account total fluorescence, or overall amount of particles internalized, there was indeed a difference due to surface modification of all particle sizes in the BALF indicating the ability to reduce mass of particles taken up by immune cells in the lung via surface PEGylation.
Our results show that these particles can reside in the lung out to twenty-eight days without causing an inflammatory response. This supports previous work from our group, which found that these particles did not activate the inflammasome following pulmonary delivery (40). In this work, we also measured the BALF for MIP-2, a chemokine responsible for neutrophil recruitment. These cytokine and chemokine data correspond to the immune cell recruitment seen in the flow cytometry analysis of the cell populations in the BALF: both the 80×320U and the 6U dosed mice had slightly elevated granulocyte levels at day one compared to saline. Interestingly, histopathology on these samples shows no significant neutrophil recruitment to the lungs. Other protein analysis of the potential acute inflammatory response to these particles, including analysis of lung-specific molecules such as surfactant proteins A and D (SP-A and SP-D respectively) or total mucin content, may lend increase insight to the cellular recruitment process (49–51). Importantly, no long term inflammatory response was observed at the 28 day time point.
At this long time point, the 80×320 nm and 1.5 μm particles were still observed in BALF and lung cells, which may make them a prime candidate for sustained release of drug for diseases targeting macrophages in the lung, such as tuberculosis. Additionally, the PEGylated 80×320 nm actually show an increase in uptake from day one to day seven, indicating that there are likely free particles residing in the alveolar airspace and/or epithelial tissue. This is supported by fluorescent microscopy of the lung tissue which shows the particles significantly distributed throughout the epithelial cells of the lung. In contrast, the unPEGylated particles were more likely to be internalized by cells, explaining the punctate distribution in the lung that implies cellular internalization. The PEGylated particles were not as prone to uptake thus increasing retention and duration on the lung surface. This increased homogeneity in lung distribution may have also resulted from PEGylated particles ability to better interact with the mucus and surfactants within the airway. This may make these particles ideal for gene delivery to treat cystic fibrosis and local delivery of therapeutics to airway cells to treat chronic obstructive pulmonary disease (COPD).
Conclusions
In this work, the role of PEGylation to increase residence times and alter the cellular fate of nano- and microparticles delivered to lung was evaluated. Not only are these particles non-inflammatory, but they were found to be retained in the lung for at least a month, with potential to serve as drug depots. Surface modification with PEGylation was found to significantly affect uptake kinetics by key cell populations in the lung, thus allowing for cell-specific targeted delivery of therapeutics. Particularly, PEGylation was found to increase lung residence times of all sizes of particles tested here, with the largest increase in residence time observed for smaller 80×320 nanometer particles. Additionally it was observed that DCs preferred smaller unPEGylated particles. By tuning the particle size and surface chemistry, design rules for targeted cellular pulmonary delivery are beginning to emerge. Applying the knowledge from these fundamental biological studies of particle interactions in the lung can lead to the design of more efficient drug carriers for a variety of pulmonary delivery applications.
Supplementary Material
Representative flow cytometric analysis of BALF and Lung cells. (A) Gating scheme used to define three major lung cell populations: Granulocytes CD45+CD11c-Ly6G+, Macrophages (Mφs) CD45+CD11c+MHCIIlo, Dendritic cells (DCs) CD45+CD11c+MHCIIhi. (B) Representative gating of particle positive populations of Granulocytes, AMs and DCs. Blue curves representative data from 80×320 day 1 BALF. Red curves represent fluorescence minus one (FMO) controls used to set gates. Black curves represent saline controls.
{kind=link}
Acknowledgments
We thank R. Roberts, K. Reuter, J. Perry, S. Tian, J. P.Y. Ting, A. Pandya, B. Udis, N. Fisher, S. Coquery, L.Wai, and, J. Weaver for useful discussions and technical assistance. We acknowledge Liquidia Technologies for providing PRINT molds, and the core facilities at UNC, including the Nucleic Acids Core Facility, CHANL imaging facility, the LCCC Histopathology Core, the Histology Facility of the Department of Cell and Molecular Physiology, the Department of Microbiology and Immunology Flow Cytometry Core Facility, and DLAM facility. This work was funded in part by the NIH Pioneer Award to J.M.D. (1DP1OD006432), DTRA award (HDTRA1-13-1-0045), and the NSF Graduate Research Fellowship, as well as NCI Center Core Support Grant CA016086.
Abbreviations
- AEM
2-aminoethylmethacrylate
- AM
Alveolar macrophages
- BALF
Broncheoalveolar lavage fluid
- COPD
Chronic Obstructive Pulmonary Disease
- DC
Dendritic cells
- Dpi
Dry powder inhaler
- DPPC
1,2-dipalmitoylphosphatidylcholine
- ELISA
enzyme-linked immuno assay
- HP4A
tetra(ethylene glycol) monoacrylate
- IACUC
Institutional Animal Care and Use Committee
- IV
Intravenous
- LPPs
Large porous particles
- LPS
Lipopolysaccharide
- MΦ
macrophages
- PEG
Poly(ethylene glycol)
- PEG700DA
poly(ethylene glycol)700
Particle replication in non-wetting templates
- SEM
scanning electron microscopy
- TPO
diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide
Footnotes
Disclosures
J.M.D. is a founder and maintains a financial interest in Liquidia Technologies.
References
- 1.Pattonand JS, Byron PR. Inhaling medicines: delivering drugs to the body through the lungs. Nature reviews Drug discovery. 2007;6:67–74. doi: 10.1038/nrd2153. [DOI] [PubMed] [Google Scholar]
- 2.Pulliam B, Sung JC, Edwards DA. Design of nanoparticle-based dry powder pulmonary vaccines. Expert Opin Drug Deliv. 2007;4:651–663. doi: 10.1517/17425247.4.6.651. [DOI] [PubMed] [Google Scholar]
- 3.Chow AH, Tong HH, Chattopadhyay P, Shekunov BY. Particle engineering for pulmonary drug delivery. Pharmaceutical research. 2007;24:411–437. doi: 10.1007/s11095-006-9174-3. [DOI] [PubMed] [Google Scholar]
- 4.Azarmi S, Roa WH, Lobenberg R. Targeted delivery of nanoparticles for the treatment of lung diseases. Advanced drug delivery reviews. 2008;60:863–875. doi: 10.1016/j.addr.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Garcia A, Mack P, Williams S, Fromen C, Shen T, Tully J, Pillai J, Kuehl P, Napier M, Desimone JM, Maynor BW. Microfabricated engineered particle systems for respiratory drug delivery and other pharmaceutical applications. Journal of drug delivery. 2012;2012:941243. doi: 10.1155/2012/941243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mansour HM, Rhee Y, Wu X. Nanomedicine in pulmonary delivery. Int J Nanomedicine. 2009;4:299–319. doi: 10.2147/ijn.s4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newmanand WW, Busse SP. Evolution of dry powder inhaler design, formulation, and performance. Repiratory Medicine. 2002;96:293–304. doi: 10.1053/rmed.2001.1276. [DOI] [PubMed] [Google Scholar]
- 8.Lombry C, Edwards DA, Preat V, Vanbever R. Alveolar macrophages are a primary barrier to pulmonary absorption of macromolecules. American journal of physiology Lung cellular and molecular physiology. 2004;286:L1002–1008. doi: 10.1152/ajplung.00260.2003. [DOI] [PubMed] [Google Scholar]
- 9.Lehnert BE. Pulmonary and thoracic macrophage subpopulations and clearance of particles from the lung. Environ Health Perspect. 1992;97:17–46. doi: 10.1289/ehp.929717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma R, Saxena D, Dwivedi AK, Misra A. Inhalable microparticles containing drug combinations to target alveolar macrophages for treatment of pulmonary tuberculosis. Pharm Res. 2001;18:1405–1410. doi: 10.1023/a:1012296604685. [DOI] [PubMed] [Google Scholar]
- 11.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharmaceutical research. 2008;25:1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, Mintzes J, Deaver D, Lotan N, Langer R. Large Porous Particles for Pulmonary Drug Delivery. Science. 1997;276:1868–1971. doi: 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- 13.El-Sherbiny IM, McGill S, Smyth HD. Swellable microparticles as carriers for sustained pulmonary drug delivery. Journal of pharmaceutical sciences. 2010;99:2343–2356. doi: 10.1002/jps.22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nembrini C, Stano A, Dane KY, Ballester M, van der Vlies AJ, Marsland BJ, Swartz MA, Hubbell JA. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E989–997. doi: 10.1073/pnas.1104264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinman RM. Dendritic cells: versatile controllers of the immune system. Nature medicine. 2007;13:1155–1159. doi: 10.1038/nm1643. [DOI] [PubMed] [Google Scholar]
- 17.Naito T, Suda T, Suzuki K, Nakamura Y, Inui N, Sato J, Chida K, Nakamura H. Lung dendritic cells have a potent capability to induce production of immunoglobulin A. American journal of respiratory cell and molecular biology. 2008;38:161–167. doi: 10.1165/rcmb.2007-0237OC. [DOI] [PubMed] [Google Scholar]
- 18.Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462:449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- 19.Guilliams M, Lambrecht BN, Hammad H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal immunology. 2013;6:464–473. doi: 10.1038/mi.2013.14. [DOI] [PubMed] [Google Scholar]
- 20.Ahsan F, Rivas IP, Khan MA, Torres Suarez AI. Targeting to macrophages: role of physicochemical properties of particulate carriers--liposomes and microspheres--on the phagocytosis by macrophages. Journal of controlled release : official journal of the Controlled Release Society. 2002;79:29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- 21.Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, Napier M, Bear JE, DeSimone JM. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano letters. 2012;12:5304–5310. doi: 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liand L, Huang SD. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochimica et biophysica acta. 2009;1788:2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owens DE, 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 25.Muralidharan P, Mallory E, Malapit M, Hayes D, Jr, Mansour HM. Inhalable PEGylated Phospholipid Nanocarriers and PEGylated Therapeutics for Respiratory Delivery as Aerosolized Colloidal Dispersions and Dry Powder Inhalers. Pharmaceutics. 2014;6:333–353. doi: 10.3390/pharmaceutics6020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bayard FJ, Thielemans W, Pritchard DI, Paine SW, Young SS, Backman P, Ewing P, Bosquillon C. Polyethylene glycol-drug ester conjugates for prolonged retention of small inhaled drugs in the lung. Journal of controlled release : official journal of the Controlled Release Society. 2013;171:234–240. doi: 10.1016/j.jconrel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 27.Koussoroplis SJ, Paulissen G, Tyteca D, Goldansaz H, Todoroff J, Barilly C, Uyttenhove C, Van Snick J, Cataldo D, Vanbever R. PEGylation of antibody fragments greatly increases their local residence time following delivery to the respiratory tract. Journal of controlled release : official journal of the Controlled Release Society. 2014;187:91–100. doi: 10.1016/j.jconrel.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Youn YS, Kwon MJ, Na DH, Chae SY, Lee S, Lee KC. Improved intrapulmonary delivery of site-specific PEGylated salmon calcitonin: optimization by PEG size selection. Journal of controlled release : official journal of the Controlled Release Society. 2008;125:68–75. doi: 10.1016/j.jconrel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Merkel OM, Beyerle A, Librizzi D, Pfestroff A, Behr TM, Sproat B, Barth PJ, Kissel T. Nonviral siRNA Delivery to the Lungs: Investigation of PEG-PEI Polyplexes and Their In Vivo Performance. Molecular pharmaceutics. 2009;6:1246–1260. doi: 10.1021/mp900107v. [DOI] [PubMed] [Google Scholar]
- 30.Meenach SA, Anderson KW, Zach Hilt J, McGarry RC, Mansour HM. Characterization and aerosol dispersion performance of advanced spray-dried chemotherapeutic PEGylated phospholipid particles for dry powder inhalation delivery in lung cancer. Eur J Pharm Sci. 2013;49:699–711. doi: 10.1016/j.ejps.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel B, Gupta V, Ahsan F. PEG-PLGA based large porous particles for pulmonary delivery of a highly soluble drug, low molecular weight heparin. Journal of controlled release : official journal of the Controlled Release Society. 2012;162:310–320. doi: 10.1016/j.jconrel.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Fromen CA, Robbins GR, Shen TW, Kai MP, Ting JPY, DeSimone JM. Controlled analysis of nanoparticle charge on mucosal and systemic antibody responses following pulmonary immunization. Proceedings of the National Academy of Sciences. 2014;2014:22923. doi: 10.1073/pnas.1422923112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ibricevic A, Guntsen SP, Zhang K, Shrestha R, Liu Y, Sun JY, Welch MJ, Wooley KL, Brody SL. PEGylation of cationic, shell-crosslinked-knedel-like nanoparticles modulates inflammation and enhances cellular uptake in the lung. Nanomedicine : nanotechnology, biology, and medicine. 2013;9:912–922. doi: 10.1016/j.nano.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu J, Fiegel J, Krauland E, Hanes J. New polymeric carriers for controlled drug delivery following inhalation or injection. Biomaterials. 2002;23:4425–4433. doi: 10.1016/s0142-9612(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 35.Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang YY, Cone R, Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suk JS, Kim AJ, Trehan K, Schneider CS, Cebotaru L, Woodward OM, Boylan NJ, Boyle MP, Lai SK, Guggino WB, Hanes J. Lung gene therapy with highly compacted DNA nanoparticles that overcome the mucus barrier. Journal of controlled release : official journal of the Controlled Release Society. 2014;178:8–17. doi: 10.1016/j.jconrel.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evora C, Soriano I, Rogers RA, Shakesheff KN, Hanes J, Langer R. Relating the phagocytosis of microparticles by alveolar macrophages to surface chemistry: the effect of 1,2-dipalmitoylphosphatidylcholine. Journal of controlled release : official journal of the Controlled Release Society. 1998;51:143–152. doi: 10.1016/s0168-3659(97)00149-1. [DOI] [PubMed] [Google Scholar]
- 38.Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. Direct fabrication and harvesting of monodisperse shape-specific nanobiomaterials. Journal of the American Chemical Society. 2005;127:10069–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- 39.Fromen CA, Shen TW, Larus AE, Mack P, Maynor BW, Luft JC, DeSimone JM. Synthesis and characterization of monodisperse uniformly shaped respirable aerosols. AIChE Journal. 2013;59:3184–3194. [Google Scholar]
- 40.Roberts RA, Shen T, Allen IC, Hasan W, DeSimone JM, Ting JP. Analysis of the murine immune response to pulmonary delivery of precisely fabricated nano- and microscale particles. PloS one. 2013;8:e62115. doi: 10.1371/journal.pone.0062115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guzman J, Iglesias MT, Riande E. Synthesis and polymerization of acrylic monomers with hydrophilic long side groups. Polymer. 1997;38:5227–5232. [Google Scholar]
- 42.Tabataand Y, Ikada Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials. 1988;9:356–362. doi: 10.1016/0142-9612(88)90033-6. [DOI] [PubMed] [Google Scholar]
- 43.Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, Frew A. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. American journal of respiratory and critical care medicine. 1999;159:702–709. doi: 10.1164/ajrccm.159.3.9709083. [DOI] [PubMed] [Google Scholar]
- 44.Oberdörster G, Oberdörster E, Oberdörster J. Nanotoxicology: An Emerging Discipline Evolving from Studies of Ultrafine Particles. Environmental Health Perspectives. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Molecular pharmaceutics. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Gennes PG. Polymers at an interface: a simplified view. Advances in Colloid and Interface Science. 1987;27:189–209. [Google Scholar]
- 47.Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Müller RH. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids and Surfaces B: Biointerfaces. 2000;18:301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 48.Chu KS, Schorzman AN, Finniss MC, Bowerman CJ, Peng L, Luft JC, Madden AJ, Wang AZ, Zamboni WC, DeSimone JM. Nanoparticle drug loading as a design parameter to improve docetaxel pharmacokinetics and efficacy. Biomaterials. 2013;34:8424–8429. doi: 10.1016/j.biomaterials.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright JR. Immunoregulatory functions of surfactant proteins. Nature reviews Immunology. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 50.Haczku A. Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J Allergy Clin Immunol. 2008;122:861–879. doi: 10.1016/j.jaci.2008.10.014. quiz 880–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young HW, Sun CX, Evans CM, Dickey BF, Blackburn MR. A3 adenosine receptor signaling contributes to airway mucin secretion after allergen challenge. American journal of respiratory cell and molecular biology. 2006;35:549–558. doi: 10.1165/rcmb.2006-0060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative flow cytometric analysis of BALF and Lung cells. (A) Gating scheme used to define three major lung cell populations: Granulocytes CD45+CD11c-Ly6G+, Macrophages (Mφs) CD45+CD11c+MHCIIlo, Dendritic cells (DCs) CD45+CD11c+MHCIIhi. (B) Representative gating of particle positive populations of Granulocytes, AMs and DCs. Blue curves representative data from 80×320 day 1 BALF. Red curves represent fluorescence minus one (FMO) controls used to set gates. Black curves represent saline controls.