

Abstract

Herein we describe the design and synthesis of a series of pyridopyrazine-1,6-dione γ-secretase modulators (GSMs) for Alzheimer’s disease (AD) that achieve good alignment of potency, metabolic stability, and low MDR efflux ratios, while also maintaining favorable physicochemical properties. Specifically, incorporation of fluorine enabled design of metabolically less liable lipophilic alkyl substituents to increase potency without compromising the sp3-character. The lead compound 21 (PF-06442609) displayed a favorable rodent pharmacokinetic profile, and robust reductions of brain Aβ42 and Aβ40 were observed in a guinea pig time-course experiment.
Keywords: Alzheimer’s disease, gamma secretase modulators, fluorine, lipophilic metabolism efficiency, LipMetE
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that is characterized by gradual loss of memory, impaired speech and motor functions, and is ultimately fatal. An estimated 5.2 million people currently suffer from AD in the US alone, and it is a major unmet medical need given the lack of disease modifying therapies to slow the progression of the disease.1 Accumulation of brain amyloid plaques is a characteristic feature of AD pathology. These plaques consist primarily of the neurotoxic peptide amyloid β42 (Aβ42), which is formed via sequential cleavage of the amyloid precursor protein (APP) by β-secretase (BACE) and γ-secretase.2 Development of compounds that inhibit or modulate these enzymes has therefore become a major focus in the search for disease modifying drugs.
γ-Secretase inhibitors (GSIs) have progressed to phase II and phase III clinical trials and demonstrated robust reduction of Aβ42 in human cerebrospinal fluid (CSF).3 However, several adverse events were observed, such as gastrointestinal toxicity, increased occurrence of skin cancer, and negative effects on cognition.4,5 These findings may in part be attributed to inhibition of Notch signaling, which is critical for cell differentiation.6 It has also been hypothesized that accumulation of the C99 BACE cleavage product of APP could be a contributing factor to the cognitive decline.7 In contrast to GSIs, γ-secretase modulators (GSMs) do not inhibit the proteolytic activity of γ-secretase, but rather shift the cleavage site of APP to reduce formation of Aβ42 and Aβ40, in favor of the shorter, less toxic species Aβ38 and/or Aβ37.8,9 Development of GSMs has therefore emerged as a promising approach for the treatment of AD. γ-Secretase is an intramembrane-cleaving aspartyl protease and is composed of at least four subunits; presenilin, nicastrin, Aph1, and Pen2.10 Through the use of photoaffinity probes, we and others have demonstrated that several classes of GSMs bind to allosteric sites on presenilin that are distinct from the binding sites of GSIs.11,12
Examination of the patent literature points to significant challenges in identifying potent GSMs within favorable CNS physicochemical property space.13 We have previously disclosed an amide series (generically represented by 2) of GSMs using E2012 (1) as a scaffold (Scheme 1).14−16 However, efforts to align potency (IC50 < 100 nM) with adequate physicochemical properties and acceptable absorption, distribution, metabolism, and excretion (ADME) attributes were unfruitful in this series. Furthermore, stronger inhibition of cytochrome P450 enzymes (CYP450) was noted with more potent analogues. Our focus was therefore shifted toward designing novel, conformationally restricted cores with increased polarity.17,18 As previously published, merging the hydroxy-substituted bicyclic core of 3 with pyridyl amide 2 afforded a novel pyridopyrazine-1,6-dione heterocyclic scaffold (i.e., 4 and 5; Scheme 1) that maintained the key polar interactions required for potency but was devoid of liabilities associated with the phenol.17
Scheme 1. Evolution of the Pyridopyrazine-1,6-dione Series.

Development of robust, parallel enabled chemistry allowed for rapid optimization to afford pyridone 4,17,19 which has moderate Aβ42-lowering activity in CHO APP cells (Aβ42 IC50 = 101 nM) and favorable physicochemical properties: cLogP = 3.3,20 central nervous system multiple parameter optimization score (CNS MPO) = 4.6,21 and LipE = 3.9.22 Compound 4 displayed dose-dependent reduction of brain Aβ42 in a guinea pig model, but additional gains in potency and in vivo efficacy were ultimately needed. Ongoing parallel chemistry efforts delivered naphthyl analogue 5, which displayed excellent in vitro activity while maintaining good properties (Aβ42 IC50 = 15 nM; cLogP = 2.85). However, despite excellent human liver microsomal stability, this compound exhibited high turnover in rat liver microsomes (HLM CLint,app = 8.1 mL/min/kg; RLM CLint,app = 469 mL/min/kg), which complicates in vivo efficacy and safety studies in rodents.23,24 Herein, we describe our efforts to optimize the pyridopyrazine-1,6-dione series to combine potency under 10 nM with good ADME profile and favorable physicochemical properties. The strategic use of fluorine played a key role in achieving these objectives.
Through parallel medicinal chemistry using a previously developed protocol (Scheme 2), we sought to identify heterocyclic replacements for the naphthyl group of 5.17,25 Several interesting compounds emerged such as thiazole 9 and isothiazole 10, which maintained moderate Aβ42-lowering activity (Table 1). However, consistent with previous observations, increased polarity in the right-hand section of the molecule came at the expense of high MDR efflux ratio (MDR ER).26 To address this issue, we explored replacing the methyl group of 9 with an electron-withdrawing trifluoromethyl group to reduce the electron density of the heterocycle. This modification also had the potential to improve potency since we had previously observed a dramatic improvement in Aβ42-lowering activity by replacing a methyl group with a trifluoromethyl substituent in this region of the molecule.17
Scheme 2. Synthesis of 9–13 and 15–21.

Table 1. Activity of 9–14.


Aβ42 IC50 values were obtained in a whole cell assay using CHO APPwt cells. Aβ42 IC50 values are the geometric mean of at least two experiments.
MDR efflux ratio using a MDR1/MDCK assay utilizing MDCK cells transfected with the gene that encodes human P-glycoprotein.26
Human liver microsome-derived scaled intrinsic clearance.23
The resulting analogue 11 achieved both design objectives; not only was the Aβ42 IC50 improved from 302 to 18 nM, but the MDR ER was reduced from 4.9 to 2.5 while maintaining a cLogP value below 3.0. Similarly, library hit 12 incorporating a difluoromethyl-substituted isoxazole exhibited a promising in vitro profile. Using the same tactic of introducing a trifluoromethyl group, potency was improved from 72 to 17 nM (compound 13), and the MDR ER was reduced from 3.7 to 2.1 while maintaining good stability in human liver microsomes (CLint,app = 20.0 mL/min/kg). Finally, we explored a small set of targets designed specifically as naphthyl replacements. These efforts resulted in benzothiazole 14, which had potency approaching that of 5 while maintaining good ADME and properties (Aβ42 IC50 = 25 nM, MDR ER = 2.1, and cLogP = 3.0).
The lead compounds were then evaluated in vivo to examine their pharmacokinetic (PK) profile. However, compounds 13 and 14 were found to have inadequate oral exposure when dosed in rat at 5 mg/kg even though RLM stability had been improved relative to 5 (5, 13, and 14 had RLM CLint,app = 469, 89.5, and 87.0 mL/min/kg, respectively). One concern with these compounds was the presence of four aromatic rings resulting in reduced sp3-character.27 The design strategy was therefore shifted to improve this parameter while maintaining good alignment of potency, physicochemical properties, and ADME profile. Re-examination of the library data revealed compounds with simple alkyl substituents in the ortho-position that maintained reasonable activity and that could serve as starting points for further optimization. Two of these included t-butyl analogue 15 and cyclobutyl compound 16, which had Aβ42 IC50 values of 128 and 73 nM, respectively (Table 2). Metabolic stability was poor, which may be expected given that the aliphatic substituents are susceptible to oxidative metabolism.
Table 2. Activity of 15–20.


Aβ42 IC50 values were obtained in a whole cell assay using CHO APPwt cells. Aβ42 IC50 values are the geometric mean of at least two experiments.
MDR efflux ratio using a MDR1/MDCK assay utilizing MDCK cells transfected with the gene that encodes human P-glycoprotein.26
Human liver microsome-derived scaled intrinsic clearance.23
Lipophilic efficiency.22
Lipophilic metabolism efficiency.28
We hypothesized that clearance could be improved by introducing fluorine atoms to block potential sites of oxidative metabolism. Furthermore, incorporation of fluorine into this region would likely improve potency based on previous structure–activity relationship (SAR) observations.17 Toward this end, difluoro-substitution was installed on the cyclobutyl group of 16, as well as an additional fluorine in the para-position of the phenol since we have previously shown that blocking this metabolic soft spot can lead to an improvement in both HLM and RLM stability.17 The resultant compound 17 did indeed have significantly better metabolic stability (HLM CLint,app = 17.3 vs 93.8 mL/min/kg for 16), but in vitro Aβ42-lowering activity was only marginally improved from 73 to 59 nM.
To increase potency and metabolic stability of 15, one of the methyl groups of the t-butyl substituent was replaced with a trifluoromethyl group to afford 18. The resultant 2,2-dimethyltrifluoroethyl moiety has previously been reported in the literature as a t-butyl replacement with improved metabolic stability.29 As in the case of 17, a fluorine atom was also introduced in the para-position of the right-hand aromatic ring to block this metabolically labile site. The resulting analogue 18 represented a significant advancement in that Aβ42-lowering activity was enhanced from 128 to 30 nM, and HLM stability was improved from 95.7 to 23.7 mL/min/kg while also maintaining low MDR ER and acceptable lipophilicity. The synthetic route to this custom phenol allowed ready access to the corresponding monomethyl analogues (i.e., 19 and 20; see Supporting Information). Interestingly, removing one of the methyl groups of 18 followed by separation of enantiomers resulted in a further gain in potency and LipE. Compound 19 had an Aβ42 IC50 value of 20 nM, whereas the enantiomer 20 was >4-fold less active. Metabolic stability was improved further, which is consistent with a reduction in cLogP. As shown in Table 2, fluorine appears to play a key role in improving the potency of 15 to ultimately deliver 19; however, in the absence of an X-ray structure of γ-secretase, the nature of the specific molecular interactions remains unknown.
While compounds 11, 13, and 14 (Table 1) relied on a fourth aromatic ring to achieve good potency, incorporation of fluorine allowed successful design of potent GSMs such as 19 (Table 2) that have reduced aromatic ring-count and increased sp3-character.27 Specifically, incorporation of fluorine enabled the creation of lipophilic aryl-substituents with improved metabolic stability. Lipophilic metabolism efficiency (LipMetE) is a useful parameter to gauge metabolic stability at a given LogD (eq 1).28 Higher LipMetE is preferable because it indicates that metabolic stability can be achieved within a wider LogD range. This is particularly relevant when pursuing an intramembrane target like γ-secretase, which may require higher lipophilicity to achieve sufficient potency and in vivo efficacy. As shown in Table 2, optimization of the ortho-alkyl substituent and incorporation of fluorine at specific sites led to significant improvements in metabolic stability while keeping lipophilicity relatively unchanged. This is reflected in an increase in LipMetE as exemplified by compounds 15 and 18, which have LipMetE values of 0.9 and 1.8, respectively.28
| 1 |
While optimizing the terminal aryl ring we also explored introduction of conformational control elements on the methylene linker to orient the aryl ether into the putative hydrophobic pocket of the binding site. A methyl scan revealed that potency could be improved about 3-fold by introducing an S-methyl substituent as shown in 21 (Table 3; the corresponding R-enantiomer was 6-fold less active than 21, not shown). Lead GSM 21 (PF-06442609)30 represented a major milestone in that single-digit potency (Aβ42 IC50 = 6 nM) was successfully aligned with excellent microsomal stability, good passive permeability,31 and low MDR efflux ratio (HLM CLint,app = 12.7 mL/min/kg; RRCK Papp,A→B = 16.0 × 10–6 cm/s;31 MDR ER = 1.4) while maintaining favorable physicochemical properties (cLogP = 3.1; SFLogD = 3.4;32 CNS MPO = 4.0). This is further highlighted by the fact that 21 has one of the highest LipE and LipMetE values of the compounds explored in this series (LipE = 4.8; LipMetE = 2.0). In addition to good HLM stability, 21 also had significantly improved stability in rat liver microsomes as compared to initial lead 5 (RLM CLint,app = 69.6 vs 469 mL/min/kg for 21 and 5, respectively), which contributed to acceptable rat pharmacokinetic parameters (CL = 34.1 mL/min/kg; F = 51%; Table 3). In addition, 21 achieved good brain penetration in rat as indicated by a brain to plasma ratio (B/P) of 0.8 and an unbound brain to unbound plasma ratio (Cb,u/Cp,u) of 0.4. Finally, GSM 21 exhibited only moderate hERG activity (IC50 = 9.2 μM),33 and it was devoid of Notch inhibition (NICD IC50 > 50 μM),17 which is consistent with the desired profile of a GSM.
Table 3. Pharmacokinetic Parameters.

| In Vitro Potency/Selectivity | |
| Aβ42 IC50 | 6 nM |
| NICD IC50 | >10 μM |
| Physicochemical Properties | |
| cLogP/SFLogD | 3.1/3.4 |
| LipE/LipMetE | 4.8/2.0 |
| CNS MPO | 4.0 |
| solubility (pH 6.5)a | 101 μM |
| In Vitro ADME | |
| HLM CLint,app | 12.7 mL/min/kg |
| RLM CLint,app | 69.6 mL/min/kg |
| RRCK Papp,A→B | 16.0 × 10–6 cm/s |
| MDR ER | 1.4 |
| Rat PK | |
| B/Pb | 0.8 |
| Cb,u/Cp,ub,c | 0.4 |
| CLd | 34.1 mL/min/kg |
| T1/2d | 1.15 h |
| Fe | 51% |
Kinetic solubility was measured at Analiza, Inc.35
Determined from 10 mg/kg oral dose, 1 h time point.
Plasma and brain free fractions of 21 in rat were 2.7% and 1.3%, respectively.
Determined from 1 mg/kg intravenous dose.
Calculated using the exposure from 1 mg/kg intravenous dose and 5 mg/kg oral dose.
In vivo efficacy of 21 was evaluated in a guinea pig time-course experiment. Compound 21 was administered orally at a dose of 60 mg/kg, and Aβ42, Aβ40, Aβ38, and Aβ-total were measured along with compound exposure at time-points ranging from 1 to 24 h. Sustained reductions of both Aβ42 and Aβ40 were achieved, while Aβ-total remained relatively unchanged (Figure 1). A 41% reduction of brain Aβ42 was observed at the 3 h time-point for the 60 mg/kg dose, whereas Aβ38 was increased 46% in accordance with the typical profile of a GSM. The corresponding unbound brain and plasma exposures at the 3 h time-point were 225 ± 45 and 680 ± 152 nM, respectively (see Supporting Information for further details on efficacy and exposure data).
Figure 1.
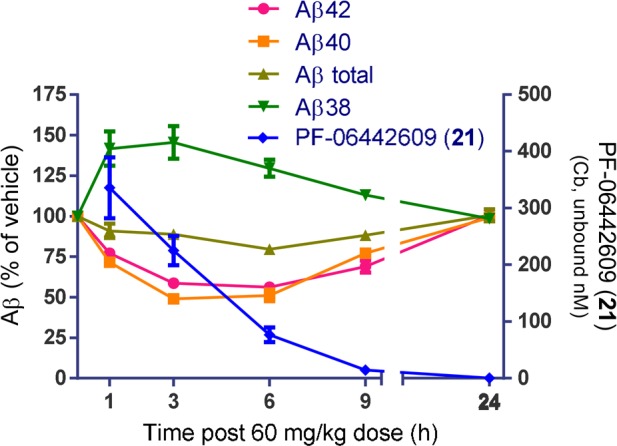
Guinea pig time course study.
While the initial synthesis of 21 (Scheme 2, using 7) was adequate for producing milligram quantities for in vitro screening, it was not suitable for large scale synthesis to support in vivo studies. Steric bulk imparted by the 1,1,1-trifluoropropan-2-yl substituent resulted in a sluggish chloride displacement reaction that was accompanied by formation of a side product resulting from elimination of 7 rather than alkylation. This prompted our investigation into an alternative disconnection giving rise to amine 27 and lactone 29 (Scheme 3). This strategy offered a more convergent approach, allowing for C–O bond formation between phenol 25 and the readily available (S)-N-Boc-alaninol. The final step in generating 21 was envisioned as a lactone-to-lactam conversion utilizing this chiral amine.
Scheme 3. Synthesis of 21.

Reagents and conditions: (a) 2-bromo-3,3,3-trifluoroprop-1-ene, (PPh3)2PdCl2, K2CO3, THF, H2O, rt, 70%; (b) BBr3, CH2Cl2, −78 °C to rt, 74%; (c) 75 psi H2, [RuCl(p-cymene)(S)-Segphos)]Cl, EtOH, 50 °C, 72%, 96% ee; (d) (S)-N-(t-butoxycarbonyl)alaninol methanesulfonate, Cs2CO3, DMF, 60 °C; (e) TFA, CH2Cl2, rt, then fumaric acid in MeOH, 78% (2 steps from 26); (f) 1,2-dibromoethane, Cs2CO3, DMF, 90 °C, 76%; (g) 27, DABAL-Me3, THF, 70 °C, 72%; (h) TFAA (3.5 equiv), DBU (8 equiv), CH3CN, 0 °C to rt, 85%.
Our synthesis of the requisite phenol 25 began with Suzuki coupling of commercially available boronic acid 22 with 2-bromo-3,3,3-trifluoropropene to afford the styrene derivative 23. Demethylation of the methoxy group using BBr3 afforded phenol 24, and asymmetric catalytic hydrogenation with ([RuCl(p-cymene)(S)-Segphos)]Cl) delivered the chiral phenol 25 in excellent ee (96% ee; 72% yield).34 Alkylation of 25 with (S)-N-Boc-alaninol methanesulfonate, followed by Boc deprotection and isolation of the amine as a solid fumarate salt completed the synthesis of amine 27.
The highly crystalline lactone 29 was prepared from the known pyridone carboxylic acid 28(17) via bis-alkylation with 1,2-dibromoethane. The amide 30 could then be obtained through simple amidation with 27 in hot methanol. However, to achieve acceptable conversion it was necessary to use superstoichiometric quantities (>3 equiv) of the precious amine 27, which was undesirable for large scale synthesis. Instead, the amidation was more efficiently carried out using the Lewis acidic DABAL-Me3 reagent to afford 30 in 72% yield. Lactam formation to generate the final target 21 was subsequently achieved in a one pot procedure through in situ generation of a mixed anhydride with trifluoroacetic anhydride followed by intramolecular alkylation of the amide by treatment with DBU.
We have described the design strategies and synthetic efforts leading to advanced GSM 21 using pyridopyrazine-1,6-dione leads 4 and 5 as starting points. Strategic use of fluorine played a key role in aligning potency and ADME properties such as reducing the MDR efflux ratio for GSMs incorporating a heterocyclic d-ring. Specific introduction of fluorine also enabled creation of metabolically less liable lipophilic aryl substituents leading to superior in vitro potency and HLM stability while maintaining good physicochemical properties and increased sp3-character. This is reflected in improved LipE relative to 4: compounds 4 and 21 have whole-cell LipE values of 3.9 and 4.8, respectively. LipMetE was utilized to gauge progress with respect to metabolic stability by normalizing for changes in lipophilicity. Compound 21 had a LipMetE value of 2.0, which was the highest in the series and compares favorably to the initial t-butyl substituted library hit 15 (LipMetE = 0.9). Finally, 21 demonstrated good rodent brain penetration and oral bioavailability as well as robust in vivo Aβ42- and Aβ40-lowering in guinea pig. Additional efforts focused on further improving in vivo efficacy and reducing the efficacious plasma concentration will be disclosed in a future publication.
Acknowledgments
We thank Stacey Becker, Emily Miller, Michael Marconi, Emily Sylvain, and Karin Wallace for their contributions to the in vivo studies. We also thank the Pfizer ADME technology group for generating the in vitro pharmacokinetic data to support the SAR efforts.
Supporting Information Available
Experimental data for compound synthesis and characterization, assay protocols, in vivo efficacy, and exposure data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ 1723 Dove Cottage Drive, Charlotte, North Carolina 28226, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Thies W.; Bleiler L. Alzheimer’s disease facts and figures. Alzheimers Dement. 2013, 9, 208–245. [DOI] [PubMed] [Google Scholar]
- Karran E.; Mercken M.; De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discovery 2011, 10, 698–712. [DOI] [PubMed] [Google Scholar]
- Bateman R. J.; Siemers E. R.; Mawuenyega K. G.; Wen G.; Browning K. R.; Sigurdson W. C.; Yarasheski K. E.; Friedrich S. W.; Demattos R. B.; May P. C.; Paul S. M.; Holtzman D. M. A γ-secretase inhibitor decreases amyloid-β production in the central nervous system. Ann. Neurol. 2009, 66, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Lessons from a failed γ-secretase Alzheimer trial. Cell 2014, 159, 721–726. [DOI] [PubMed] [Google Scholar]
- Coric V.; van Dyck C. H.; Salloway S.; Andreasen N.; Brody M.; Richter R. W.; Soininen H.; Thein S.; Shiovitz T.; Pilcher G.; Colby S.; Rollin L.; Dockens R.; Pachai C.; Portelius E.; Andreasson U.; Blennow K.; Soares H.; Albright C.; Feldman H. H.; Berman R. M. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [DOI] [PubMed] [Google Scholar]
- Haapasalo A.; Kovacs D. M. The many substrates of presenilin/γ-secretase. J. Alzheimer’s Dis. 2011, 25, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani Y.; Yarimizu J.; Saita K.; Uchino H.; Akashiba H.; Shitaka Y.; Ni K.; Matsuoka N. Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 2012, 32, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S.; Eriksen J. L.; Das P.; Sagi S. A.; Wang R.; Pietrzik C. U.; Findlay K. A.; Smith T. E.; Murphy M. P.; Bulter T.; Kang D. E.; Marquez-Sterling N.; Golde T. E.; Koo E. H. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [DOI] [PubMed] [Google Scholar]
- Kounnas M. Z.; Danks A. M.; Cheng S.; Tyree C.; Ackerman E.; Zhang X.; Ahn K.; Nguyen P.; Comer D.; Mao L.; Yu C.; Pleynet D.; Digregorio P. J.; Velicelebi G.; Stauderman K. A.; Comer W. T.; Mobley W. C.; Li Y. M.; Sisodia S. S.; Tanzi R. E.; Wagner S. L. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron 2010, 67, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B.; Iwatsubo T.; Wolfe M. S. Presenilins and γ-Secretase: Structure, Function, and Role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozdnyakov N.; Murrey H. E.; Crump C. J.; Pettersson M.; Ballard T. E.; am Ende C. W.; Ahn K.; Li Y.-M.; Bales K. R.; Johnson D. S. γ-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. J. Biol. Chem. 2013, 288, 9710–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump C. J.; Johnson D. S.; Li Y.-M. Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry 2013, 52, 3197–3216. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M.; Stepan A. F.; Kauffman G. W.; Johnson D. S. Novel γ-secretase modulators for the treatment of Alzheimer’s disease: a review focusing on patents from 2010 to 2012. Expert. Opin. Ther. Pat. 2013, 23, 1349–1366. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Johnson D. S.; Subramanyam C.; Bales K. R.; am Ende C. W.; Fish B. A.; Green M. E.; Kauffman G. W.; Lira R.; Mullins P. B.; Navaratnam T.; Sakya S. M.; Stiff C. M.; Tran T. P.; Vetelino B. C.; Xie L.; Zhang L.; Pustilnik L. R.; Wood K. M.; O’Donnell C. J. Design and synthesis of dihydrobenzofuran amides as orally bioavailable, centrally active γ-secretase modulators. Bioorg. Med. Chem. Lett. 2012, 22, 2906–2911. [DOI] [PubMed] [Google Scholar]
- For related approaches, seeHall A.; Patel T. R. γ-Secretase modulators: current status and future directions. Prog. Med. Chem. 2014, 53, 101–145. and references therein. [DOI] [PubMed] [Google Scholar]
- Gijsen H. J.; Mercken M. γ-Secretase modulators: can we combine potency with safety?. Int. J. Alzheimer’s Dis. 2012, 2012, 295207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M.; Johnson D. S.; Subramanyam C.; Bales K. R.; am Ende C. W.; Fish B. A.; Green M. E.; Kauffman G. W.; Mullins P. B.; Navaratnam T.; Sakya S. M.; Stiff C. M.; Tran T. P.; Xie L.; Zhang L.; Pustilnik L. R.; Vetelino B. C.; Wood K. M.; Pozdnyakov N.; Verhoest P. R.; O’Donnell C. J. Design, synthesis, and pharmacological evaluation of a novel series of pyridopyrazine-1,6-dione γ-secretase modulators. J. Med. Chem. 2014, 57, 1046–1062. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Johnson D. S.; Humphrey J. M.; am Ende C. W.; Evrard E.; Efremov I.; Kauffman G. W.; Stepan A. F.; Stiff C. M.; Xie L.; Bales K. R.; Hajos-Korcsok E.; Murrey H. E.; Pustilnik L. R.; Steyn S. J.; Wood K. M.; Verhoest P. R. Discovery of indole-derived pyridopyrazine-1,6-dione γ-secretase modulators that target presenilin. Bioorg. Med. Chem. Lett. 2015, 25, 908–913. [DOI] [PubMed] [Google Scholar]
- Tran T. P.; Mullins P. B.; am Ende C. W.; Pettersson M. Synthesis of pyridopyrazine-1,6-diones from 6-hydroxypicolinic acids via a one-pot coupling/cyclization reaction. Org. Lett. 2013, 15, 642–645. [DOI] [PubMed] [Google Scholar]
- Values for cLogP were calculated using the BIOBYTE (www.biobyte.com) program cLogP, version 4.3.
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LipE = −log(EC50) – LogD where EC50 is in molar units and LogD was experimentally determined using shake flask LogD.32 For additional detail, seeRyckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: a lipophilic efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. [DOI] [PubMed] [Google Scholar]
- Assay method adapted from published protocols:Riley R. J.; McGinnity D. F.; Austin R. P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug. Metab. Dispos. 2005, 33, 1304–1311. [DOI] [PubMed] [Google Scholar]
- Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug. Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- Compound 14 was not prepared via the alkylation route in Scheme 2, but rather using the HATU-mediated coupling/cyclization as previously published: See Pfizer WO 2012/131539.
- Feng B.; Mills J. B.; Davidson R. E.; Mireles R. J.; Janiszewski J. S.; Troutman M. D.; de Morais S. M. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug. Metab. Dispos. 2008, 36, 268–275. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- LipMetE was calculated using measured shake flask LogD32 and unbound intrinsic clearance (CLint,u) derived from the intrinsic apparent clearance (CLint,app) and calculated human liver microsomal binding values. For additional details seeStepan A. F.; Kauffman G. W.; Keefer C. E.; Verhoest P. R.; Edwards M. Evaluating the differences in cycloalkyl ether metabolism using the design parameter ″lipophilic metabolism efficiency″ (LipMetE) and a matched molecular pairs analysis. J. Med. Chem. 2013, 56, 6985–6990. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Shishido Y. Synthesis of aromatic compounds containing a 1,1-dialkyl-2-trifluoromethyl group, a bioisostere of the tert-alkyl moiety. Bioorg. Med. Chem. Lett. 2007, 17, 6079–6085. [DOI] [PubMed] [Google Scholar]
- Compound 21, also known as PF-06442609, is now commercially available from Sigma Aldrich; catalog #PZ0258.
- Callegari E.; Malhotra B.; Bungay P. J.; Webster R.; Fenner K. S.; Kempshall S.; LaPerle J. L.; Michel M. C.; Kay G. G. A comprehensive non-clinical evaluation of the CNS penetration potential of antimuscarinic agents for the treatment of overactive bladder. Br. J. Clin. Pharmacol. 2011, 72, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shake flask LogD. Assay method adapted from published protocol:Hay T.; Jones R.; Beaumont K.; Kemp M. Modulation of the partition coefficient between octanol and buffer at pH 7.4 and pKa to achieve the optimum balance of blood clearance and volume of distribution for a series of tetrahydropyran histamine type 3 receptor antagonists. Drug Metab. Dispos. 2009, 37, 1864–1870. [DOI] [PubMed] [Google Scholar]
- Kutchinsky J.; Friis S.; Asmild M.; Taboryski R.; Pedersen S.; Vestergaard R. K.; Jacobsen R. B.; Krzywkowski K.; Schroder R. L.; Ljungstrom T.; Helix N.; Sorensen C. B.; Bech M.; Willumsen N. J. Characterization of potassium channel modulators with QPatch automated patch-clamp technology: system characteristics and performance. Assay Drug Dev. Technol. 2003, 1, 685–693. [DOI] [PubMed] [Google Scholar]
- RuCl(p-cymene)(R)-dm-segphos)Cl, CAS [944451–30–3].
- Solubility was measured by Analiza, Inc., Cleveland, OH (http://analiza.com/physchem/logd.html#qf).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.