

Abstract
Dimers of 2-substituted N,N'-dimethylbenzimidazoline radicals, (2-Y-DMBI)2 {Y = cyclohexyl (Cyc), ferrocenyl (Fc), ruthenocenyl (Rc)} have recently been reported as n-dopants for organic semiconductors. Here their structural and energetic characteristics are reported, along with the mechanisms by which they react with acceptors, A (PCBM, TIPS-pentacene), in solution. X-ray data and DFT both indicate a longer C—C bond for (2-Cyc-DMBI)2 than (2-Fc-DMBI)2, yet DFT and ESR data show that the latter dissociates more readily due to stabilization of the radical by Fc. Depending on the energetics of dimer (D2) dissociation and of D2-to-A electron transfer, D2 reacts with A to form D+ and A•– by either of two mechanisms, differing in whether the first step is endergonic dissociation or endergonic electron transfer. However, the D+/0.5D2 redox potentials – the effective reducing strengths of the dimers – vary little within the series (ca. –1.9 V vs. FeCp2+/0) due to cancelation of trends in the D+/0 potential and D2 dissociation energy. The implications of these findings for use of these dimers as n-dopants, and for future dopant design, are discussed.
Keywords: doping, redox chemistry, structure elucidation, density functional calculations, reaction mechanism
Introduction
Molecular oxidants and reductants have increasingly been used as dopants in organic-semiconductor devices, such as solar cells, light-emitting diodes, field-effect transistors, and thermoelectric devices, in which they can increase the conductivity and/or decrease carrier-injection barriers.[1] They can also be used as surface dopants to modulate the work function (WF) of inorganic electrode materials such as gold[2] and indium tin oxide,[3] and, more recently, have been applied to tuning the WF and conductivity of two-dimensional materials such as graphene[4] and few-layer MoS2.[5] Low ionization energies (IE) enable n-dopants to reduce organic semiconductors with a wide variety of electron affinities (EAs) and to induce large WF changes in electrode materials through simple one-electron transfer, but also preclude dopant air stability. In principle, coupling the electron transfer to a chemical reaction can circumvent this limitation and afford dopants that have high IEs, yet can still act as strong reductants under appropriate conditions. For example, heating pyronin B chloride in vacuum generates a highly reducing species, presumably the corresponding organic radical, which regenerates the stable pyronin B cation on electron transfer to a semiconductor host.[6] Other salts of stable organic cations have also been used as vacuum-processable dopants.[7] Hydride-reduced forms of stable organic cations including leuco crystal violet (LCV) and 2-aryl-2,3-dihydro-1H-benzimidazoles (2-Ar-DMBI-H) have also been used as solution-processable n-dopants for fullerenes;[7b,8] in this case the stable dopant cation is regained by loss of both a hydrogen atom and an electron. Accordingly, hydrogen-reduced byproducts are formed, and the feasibility of doping depends on both the electron- and hydrogen-accepting properties of the host semiconductor.[9] Dimers of highly-reducing neutral species such as those of certain 19-electron organometallic sandwich compounds (e.g., (RhCp2)2 and (RuCp*mes)2, Figure 1)[10] and of certain organic radicals[11] have also been investigated as stable n-dopants. Hydride- and salt-based dopants inevitably form side-products, which, depending on the processing conditions, may be incorporated into the final doped film, in addition to the dopant cation and reduced acceptor; in contrast, the dimeric dopants can potentially undergo clean reaction with acceptors to form stable monomer cations and host anions without formation of byproducts.
Figure 1.
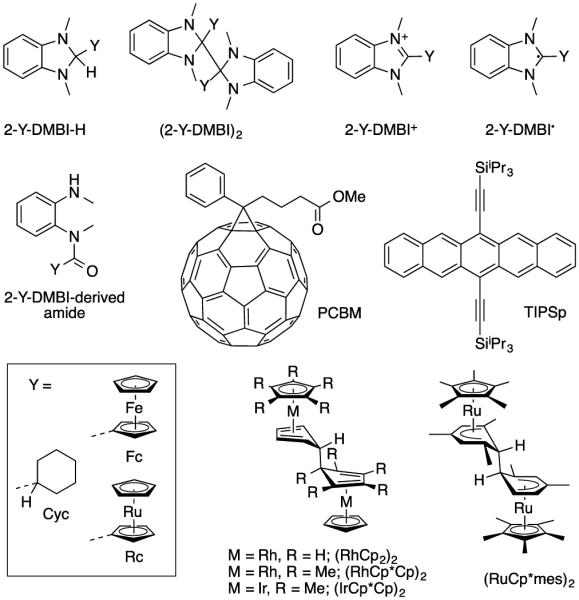
Structures of DMBI derivatives, acceptors (PCBM and TIPSp), and examples of organometallic dimeric dopants discussed in this work.
We have recently reported the use of the dimers formed by benzimidazoline radicals, (2-Y-DMBI)2 {Y = Cyc, Fc, Rc} (Figure 1), as effective n-dopants for organic semiconductors.[12] A room-temperature conductivity of 12.0 S cm–1 was obtained for a film of C60 co-sublimed with (2-Cyc-DMBI)2; this is among the highest values reported for molecularly doped C60. Moreover, spin-coating from solutions containing dopants and either PCBM or P(NDI2OD-T2) (a naphthalene diimide polymer) also gave n-doped films according to UV-photoelectron spectroscopy, photothermal deflection spectroscopy, and conductivity measurements. Solution UV-Vis-NIR experiments indicate that these dimers react several orders-of-magnitude more rapidly with PCBM than do the corresponding Y-DMBI-H compounds, and that they reduce a wider range of acceptors, including 6,13-bis(tri(isopropyl)silylethynyl)pentacene (TIPSp, EA = 3.0 eV).[10a] (2-Fc-DMBI)2 and 2-Fc-DMBI-H have also been recently used to dope the surface of trilayer MoS2, the dimer giving a larger reduction in WF and a larger increase in conductivity.[13]
Knowledge of the mechanism(s) by which these doping reactions proceed, and of the associated thermodynamic and kinetic parameters, is essential to understand the limitations of their doping efficacy, to optimize processing conditions to achieve efficient doping (especially when the dopant and acceptor are mixed in solution prior to film formation), and to develop new dopants that are stronger and/or tolerant of solution processing in air. Two mechanisms can be envisaged for the reaction of dimers with acceptors;[10b] they differ in whether the first step is dimer dissociation (mechanism I, Figure 2) or a dimer-to-acceptor electron transfer (mechanism II) and can be distinguished as shown in Table 1. This contribution reports on the stability of (2-Y-DMBI)2 dimers, their molecular and electronic structure (as determined by X-ray crystallography and density functional theory (DFT) calculations, respectively), energetic parameters related to their reactivity (obtained from electrochemistry, electron-spin resonance (ESR) spectroscopy, and DFT calculations), and the mechanisms of their reactions with acceptors.
Figure 2.
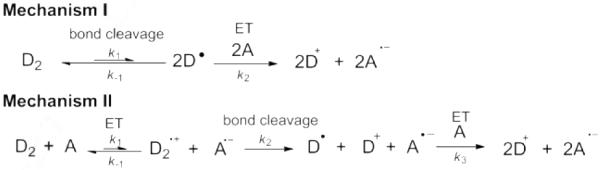
Mechanisms by which dimeric n-dopants (D2) can react with acceptors (A).
Table 1.
Comparison of Mechanisms I and II (Figure 2) for n-Doping with Dimeric Dopants
| Mechanism | Products | Rate-determining step[a] | Rate law[a] | ΔG‡ limited by [a] | Δ S ‡ [a] |
|---|---|---|---|---|---|
| I | D+ A •– ( + A 2–)[b] | D2 dissociation | d[D2]/dt = −k[D2] | ΔGdiss(D2) | >0 |
| II | D+A•– | electron transfer | d[D2]/dt = −k[D2][A] | F{E(D2+/0)–E(A0/−)} | <0 |
Assuming that in each case the first step is rate-determining.
If E(A•–/A2–) is at less reducing potential than Eeff(D2/0.5D•) then A2– can be formed if sufficient D2 is present. Since E(A•–/A2–) is generally significantly more reducing than E(A/A•–), formation of A2– via mechanism II will be much slower than the initial formation of A•–, but at a similar rate if mechanism I is operative.
Results and Discussion
Synthesis and stability of dimers
The dimers were obtained through reduction of salts of the corresponding Y-DMBI+ cations in THF using either 1 wt% Na-Hg or 25 wt% Na-K (CARE – HIGHLY PYROPHORIC). The corresponding amides (Figure 1) were obtained as side products when the Y-DMBI+ salt is poorly soluble in THF. More details of the synthesis and characterization are given in the Supporting Information and experimental details are given in the Supporting Information of Reference [12].
As noted in Reference [12], the dimers are reasonably stable in air as solids, but decompose in solution on exposure to air. In benzene-d6 the air-induced decomposition of (2-Fc-DMBI)2 (Figure S5 of Reference [12]) occurs somewhat more rapidly than some of the dimers of 19-electron sandwich compounds (e.g., compare data for (RhCp2)2 and (RuCp*mes)2 in Table S1 of Reference [10a]). In contrast to the organometallic dimers, which give the corresponding 18-electron monomeric cationic sandwich compounds on exposure of solutions to air, the DMBI dimers are quantitatively converted to the amide species (Figure 1) that are also encountered as side-products in the syntheses (see Supporting Information for characterizing data).
Molecular and electronic structure of the dimers
The dimeric structures of (2-Cyc-DMBI)2 and (2-Fc-DMBI)2 were confirmed by single-crystal X-ray structure determinations (Figure 3, Table S1 in the Supporting Information). CCDC-1048481 ((2-Cyc-DMBI)2) and CCDC-1048482 ((2-Fc-DMBI)2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The asymmetric unit of (2-Cyc-DMBI)2 contains one half molecule, the complete molecule belonging to the point group C2 due to its location on a crystallographic C2 axis. The conformation about the central C—C bond significantly deviates from the perfectly staggered geometries often found for hexasubstituted C—C fragments; the Cyc—C—C—Cyc torsion angle is 140.3° and the angle between the planes of the two DMBI units is 22.7°. The unit cell of (2-Fc-DMBI)2 contains two crystallographically inequivalent molecules, which are nevertheless very similar to one another geometrically. Each of the molecules is located on a crystallographic inversion center and, thus, centrosymmetric (point group Ci), with a perfectly staggered conformation around the central C—C bond; therefore, the Fc–C–C–Fc torsion angle is precisely 180° and the two DMBI units are precisely parallel to one another. The central intermonomer C—C bond lengths (1.640(4) Å for (2-Cyc-DMBI)2; 1.595(5) and 1.601(5) Å for the two independent molecules of (2-Fc-DMBI)2) are longer than those of standard C(sp3)–C(sp3) single bonds (ca. 1.54 Å[14]), but not exceptionally long for hexasubstituted ethanes: for example, values of 1.599(3), 1.635, and 1.636(5) Å have been reported for the central C—C bonds of (FcMe2C)2,[15] (PhEt2C)2,[16] and a Mn(CO)3mes dimer,[17] respectively. The central C—C bonds can also be compared to the tetra- and pentasubstituted ethane moieties in the dimers of 19-electron sandwich compounds (1.553(3)–1.60(3) Å).[18] The differences seen between the experimental bond lengths of (2-Cyc-DMBI)2 and (2-Fc-DMBI)2 are reproduced well in DFT calculations (1.62, 1.58, and 1.58 Å for Y = Cyc, Fc, and Rc dimers, respectively).
Figure 3.
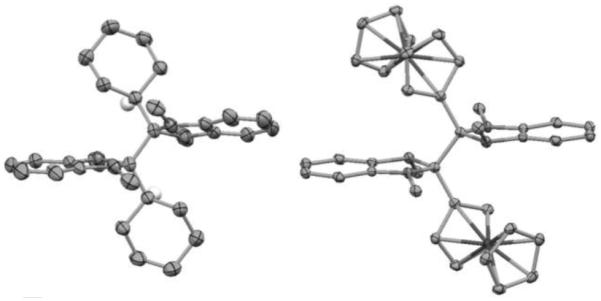
Molecular structures of (2-Cyc-DMBI)2 (left) and (2-Fc-DMBI)2 (right, showing only one of two very similar crystallographically independent molecules) as determined by X-ray crystallography (thermal ellipsoids are shown at the 50% probability level; hydrogen atoms – except for the methine hydrogen atoms of the Cyc groups – are excluded for clarity).
The highest occupied molecular orbitals (HOMOs) obtained from DFT calculations (Figure S8, Supporting Information) are qualitatively similar for all three dimers. In each case the HOMO can be described as arising from an antibonding interaction between the local HOMOs of the two o-phenylenediamine fragments and the σ-orbital associated with the central C—C bond. This pattern is very similar to that seen for dimeric sandwich compounds, where, for example, the HOMO of (RhCp2)2 arises from an anti-bonding interaction between the local HOMOs of two RhCp(butadiene) fragments and the central C—C σ-orbital.[18] Moreover, as in the dimeric sandwich compounds, the destabilization of the HOMO by the interactions with the C—C bond is consistent with the ease of oxidation of the dimers (see values of E(D2+/0) in Table 2), with the irreversibility of these oxidations, and with the lengthening of C—C on oxidation predicted by the DFT calculations (Table 2).[18] There are also additional minor destabilizing contributions from the C—Y σ-orbitals, and, in the case of the Y = Fc and Rc species, small coefficients on the metal centers; these contributions may account for the significant variations between dimers in values of E(D2+/0) (Table 2).
Table 2.
Electrochemical data for dimeric and monomeric 2-Y-DMBI compounds,[a] and adiabatic ionization energies (IEs) and dissociation energies obtained from M06/LANL2DZ/6-31G(d,p) calculations for various 2-Y-DMBI species.
|
E vs. FeCp2
+/0 / V [a] |
IE / eV[d] |
ΔUdiss / kJ mol−1 (eV)[d] |
|||||
|---|---|---|---|---|---|---|---|
| Y | Epa(D2 +/0)[b] | Epc(D+/0)[c] | D2→D2•++e[e] | D•→D++e | 0.5D2→D++e[f] | D2→2D• | D2•+ →D+ + D •[e] |
| Cyc | −0.64 | −2.45 | 5.06 | 3.72 | 4.81 | 210 (2.17) | 81 (0.84) |
| Fc | −0.89 | −2.24 | 4.69 | 3.93 | 4.79 | 165 (1.71) | 91 (0.94) |
| Rc | −0.59 | −2.29 | 4.68 | 3.80 | 4.73 | 181 (1.87) | 97 (1.00) |
In THF / 0.1 M nBu4NPF6.
Peak potential for the irreversible oxidation of D2 measured at 100 mV s−1.
Peak potential for the partially reversible reduction of D+ at 100 mV s−1.
Gas-phase adiabatic IEs and dissociation energies obtained from DFT calculations.
These results should be treated with caution owing to the tendency of DFT to artificially over-delocalize odd-electron systems such as these cations due to self-interaction error[22] Indeed the optimized structures for the D2 •+species are all characterized by spuriously long central C—C bonds (3.23-3.34 Å).
IEeff(0.5D2→D++e) = IE(D) + 0.5ΔUdiss(D2).
Themodynamic parameters relevant to doping
As noted in Table 1 the barriers for the first steps of mechanisms I and II are related to the thermodynamics of dimer dissociation and of dimer-to-acceptor electron-transfer, respectively. Moreover, the overall thermodynamic doping strength depends on both the ease of monomer ionization and the dissociation energetics, and can be expressed as an effective redox potential:
| (1) |
or an effective ionization energy:
| (2) |
Table 2 compares D+/D• and D2•+/D2 redox potentials for the three Y-DMBI systems examined here, along with DFT adiabatic ionization energies (IEs) for monomeric and dimeric species.[19] Both experimental and computational data indicate that the reducing capability of the 2-Y-DMBI• monomers increases in the order Fc < Rc < Cyc. This initially surprised us since the group-8 metallocenes are well-known to stabilize α-carbocations[20] and so might be expected to lead to more reducing 2-Y-DMBI• species than an alkyl group, although their role in stabilizing an aromatic benzimidazole cation is presumably much less significant than in stabilizing, for example, an otherwise unstabilized methylene cation. Evidently the metallocenyl groups stabilize the 2-Y-DMBI• radical monomers relative to their cyclohexyl analogue to a greater extent than they stabilize the 2-Y-DMBI+ cations. Consistent with this, the DFT calculations indicate that, while the spin density of 2-Cyc-DMBI• is almost entirely located in the benzimidazoline rings (primarily at the 2-position), in the metallocenyl species, especially 2-Fc-DMBI•, there is significant spin delocalization onto the Y substituent (Figure 4), consistent with reports for some other molecules in which an organic radical has a group-8 metallocenyl substituent.[21] The electrochemical data also show that there is a large variation in the potential at which the D2 dimers are oxidized, which can be anticipated to lead to rates of reaction via mechanism II increasing in the order Rc < Cyc < Fc.
Figure 4.
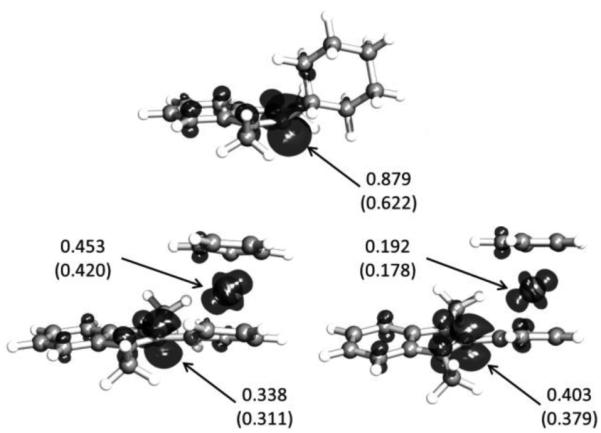
M06/LANL2DZ/6-31G(d,p) DFT-calculated geometries and spin densities (0.05 Å–3 isosurfaces) for 2-Cyc- (top), 2-Fc- (lower left), and 2-Rc-DMBI• (lower right) monomers. Spin fractions in the 2-positions of the imidazoline ring and on the metal atoms, obtained from Mulliken analyses (from natural population analyses in parentheses), are shown.
The DFT values of ΔUdiss for the neutral dimers are also given in Table 2; they vary significantly between the compounds, suggesting that the rate of reaction through mechanism I should increase in the order Cyc < Rc < Fc. The value for (2-Cyc-DMBI)2 is similar to those obtained in the same way for (IrCp*Cp)2 and (RuCp*mes)2 (Figure 1), for which crossover experiments and reactivity studies yielded no evidence for dissociation at room temperature,[10b,18] but the ΔUdiss values for all three DMBI dimers are larger than those for various rhodocene dimers.[18] Within the DMBI dimer series, the values of ΔUdiss do not correlate well with the crystallographic or DFT bond lengths; i.e., (2-Cyc-DMBI)2 has the longest and strongest bond. This is in contrast to the generally observed tendency for C—C bond lengths to decrease linearly with increasing bond strength.[23] However, correlations between bond strength and length are not well-grounded in theory and both organic and inorganic exceptions have been found.[24] Even in systems where the bond length is governed by steric interactions and sharing of electron density in the dimer, the bond strength will depend both on how these factors affect the energy of the dimer and on the stability of the monomer. Indeed the calculated ΔUdiss values decrease with increasing spin delocalization onto the Y substituents (Figure 4). Sterically-induced effects may also destabilize 2-Cyc-DMBI•: in order to avoid close contacts between the CH3 groups and the hydrogen atoms on the 2- and 6-carbons of the cyclohexyl group the geometry around the 2-carbon of the DMBI moiety is significantly distorted from planarity (also shown in Figure 4). A similar lack of correlation between ΔUdiss values and bond lengths was also found for dimeric sandwich compounds, where it was also attributable to variations in the stability of the corresponding monomers.[18]
Experimental evidence for variation in the dissociation energetics of the dimers was obtained from ESR spectroscopy. Solutions of (2-Cyc-DMBI)2 and (2-Rc-DMBI)2 do not show an ESR signal at room temperature, but solutions of (2-Fc-DMBI)2 at comparable concentrations show a structureless ESR signal (g = 2.009), the intensity of which reversibly increases with temperature, and the shape and width of which are similar to those of a spectrum simulated from isotropic contact couplings obtained from DFT calculations on 2-Fc-DMBI• (Figure 5). By comparing the intensity of the ESR signal to that of a standard sample, and measuring ESR intensities over a range of temperatures, we estimated ΔHdiss = +109 kJ mol–1, ΔSdiss = +163 J mol–1 K–1, and ΔGdiss(300 K) = +60 kJ mol–1.
Figure 5.
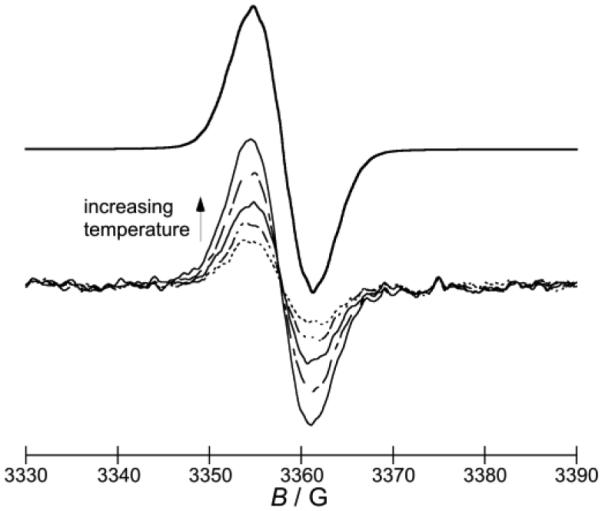
ESR spectra obtained for a solution of (2-Fc-DMBI)2 in chlorobenzene in the range 300-320 K (below), compared to a spectrum simulated with WINSIM[27] based on convolution of coupling constants obtained from M06/LANL2DZ/6-31G(d,p) calculations on 2-Fc-DMBI• with a linewidth of 0.45 G (see Table S2 in the Supporting Information for details).
As noted above, the dopant strengths can be gauged by IEeff (equation 1). These values, based on DFT-calculated quantities, are given in Table 2 and show that all three dimers are expected to exhibit very similar thermodynamic doping abilities: the compound-to-compound variations in IE(D•) and ΔUdiss(D2) effectively cancel one another out, which is not too surprising since, as noted above, the trends in both quantities can be explained in terms of spin delocalization in D•. We have previously observed a similar cancelation when estimating effective dopant strengths for sandwich-compound dimers.[18] The effective IEs estimated here fall within a similar range to those of the dimeric sandwich compounds (4.4-4.8 eV for most compounds examined, 5.4 eV for (RhCp2)2).[18,25] Using Eq 1, E(D+/0), and the ESR-derived value of ΔGdiss(300 K), Eeff for the 2-Fc-DMBI+/0.5(2-Fc-DMBI)2 couple can be estimated to be –1.93 V vs. FeCp2+/0, also similar to values estimated from electrochemical data and DFT calculations for dimeric sandwich compounds (–1.97 to –2.14 V for most examples, –1.72 V for (RhCp2)2).[18] Eeff could not be estimated in the same way for the other two dimers, but the DFT-calculated effective IE values suggest similar values for all three dimers. Thus, the thermodynamic reducing power of all three DMBI dimers is likely similar to that of decamethylcobaltocene (E1/2+/0 = –1.86 V in THF[10a]), which, however, reacts with acceptors by simple one-electron transfer and is much more sensitive to air. The estimated Eeff values are similar to the potentials of the most strongly reducing neutral purely organic compound isolated to date (–1.95 V for the two-electron reduction product of a 2,2'-(pyrrole-2,5-diyl)-bis(4-aminopyridinium) derivative in DMF).[26]
Solution doping reactions
PCBM and TIPSp were used as examples of soluble acceptors (A) with different reduction potentials (E1/20/– = –1.07 and –1.45 V, respectively, vs. FeCp2+/0 in THF) for solution studies of the kinetics of doping reactions. Both are well-established as solution-processable semiconductors and have been used as test systems in our previous n-doping studies;[1d,10a,28] moreover, PCBM•– and TIPSp•– both exhibit distinctive absorption spectra and their salts with organic or organometallic counterions can retain reasonable solubility in the same solvents in which the neutral species are soluble, allowing the kinetics to be monitored using Vis-NIR spectroscopy.[9,10b]
Vis-NIR spectra of all (2-Y-DMBI)2 / A combinations in chlorobenzene revealed the appearance of the characteristic A•– absorptions (Figure 6), while 1H NMR spectra of (2-Y-DMBI)2 / TIPSp mixtures in chlorobenzene-d5 confirmed the formation of the corresponding 2-Y-DMBI+ cations and showed broadening of the TIPSp resonances, consistent with the formation of paramagnetic TIPSp•–; thus, the reactions of (2-Y-DMBI)2 with acceptors form the expected doping products. The rate laws and activation barriers associated with the different (2-Y-DMBI)2 / A reactions in the dark in chlorobenzene were then investigated with Vis-NIR spectroscopy at various relative dimer and acceptor concentrations including pseudo-single-reactant conditions where either the dimer or the acceptor is present in a large excess.
Figure 6.

Representative kinetic data. Left top: UV-vis-NIR spectra of a solution of PCBM (3.7 × 10–4 M) and (2-Cyc-DMBI)2 (1.3 × 10–5 M) in chlorobenzene at various times after mixing. Left bottom: corresponding plot of absorbance at 1030 nm versus time (solid line is fit to a first-order expression). Center top: UV-vis-NIR spectra of a solution of TIPSp (4 × 10–4 M) and (2-Fc-DMBI)2 (4 × 10–4 M) in chlorobenzene. Center bottom: corresponding plots of absorbances at 643, 745, and 810 nm versus time. Right top: Absorbance at 745 nm vs. time for the reaction of TIPSp (7.4 ×10–4 M) and (2-Rc-DMBI)2 (5.3 × 10–5 M) at 55 °C. Right bottom: initial rate of reaction of (2-Rc-DMBI)2 and PCBM divided by the initial concentration of dimer versus the initial concentration of acceptor TIPSp. The linear fits at different temperature with non-zero intercept indicate the rate can be expressed as a sum of two mechanisms, where the intercept is the zero-order rate constant, and slope is the first-order rate constant.
The reactions of PCBM with (2-Cyc-DMBI)2 and (2-Rc-DMBI)2 were found to be first-order in both dimer and fullerene, with the reaction occurring more rapidly in the former case. The reaction was faster still for (2-Fc-DMBI)2 and, even when employing concentrations as low as ca. 10–5 M, the rate law and rate constants could not be reliably determined in an analogous fashion.[29] Thus, the observed rates of reaction with PCBM increase as the ease of dimer oxidation increases. Activation parameters for the Y = Cyc and Rc dimers were obtained from Eyring plots of variable-temperature rate-constant data (Table 3, Figure 7): values of ΔS‡ are negative, as expected for a bimolecular reaction, and values of ΔG‡(300 K) are close to values of ΔGET = F{E(D2+/0)–E(A0/–)}. The rate law, the dependence of rate on Epa(D2+/0), and the activation parameters are entirely consistent with the reduction of PCBM by the Cyc and Rc dimers proceeding by mechanism II, with the first step being rate limiting. Moreover, the reaction of PCBM with (2-Fc-DMBI)2 proceeds much more rapidly than reaction of the same dimer with TIPSp, which is shown below to proceed via mechanism I, at similar concentrations; this suggests (2-Fc-DMBI)2 also reduces PCBM via mechanism II, the faster reaction relative to its Cyc and Rc analogues being consistent with values of Epa(D2+/0).
Table 3.
Summary of Mechanisms and Activation Parameters for Solution Reactions of (2-Y-DMBI)2 with PCBM and TIPSp.
| A | Y | ΔUdiss (ΔGdiss)[a] / kJ mol−1 | ΔGET[b] / kJ mol −1 | Mechanism | ΔH‡ / kJ mol−1 | ΔS‡ / J mol−1 K−1 | ΔG‡ (300) / kJ mol−1 |
|---|---|---|---|---|---|---|---|
| PCBM | Cyc | +210 ([c]) | +41 | II | +29.9 ± 0.3 | −71.2 ± 3.3 | +51.2 ± 1.4 |
| PCBM | Fc | +165 (+60) | +17 | II | [c] | [c] | [c] |
| PCBM | Rc | +181 ([c]) | +46 | II | +45.4 ± 3.5 | −95.4 ± 11.2 | +74.0 ± 6.8 |
| TIPSp | Cyc | +210 ([c]) | +78 | II | +64.2 ± 1.9 | −44.6 ± 5.8 | +77.6 ± 3.6 |
| TIPSp | Fc | +165 (+60) | +54 | I | [c] | [c] | [c] |
| TIPSp | Rc | +181 ([c]) | +82 | I | +118.1 ± 8.7 | +27.8 ± 18.3 | +109.8 ± 14.2 |
| TIPSp | Rc | +181 ([c]) | +82 | II | +69.1 ± 2.7 | −73.4 ± 15.1 | +91.2 ± 7.2 |
Figure 7.
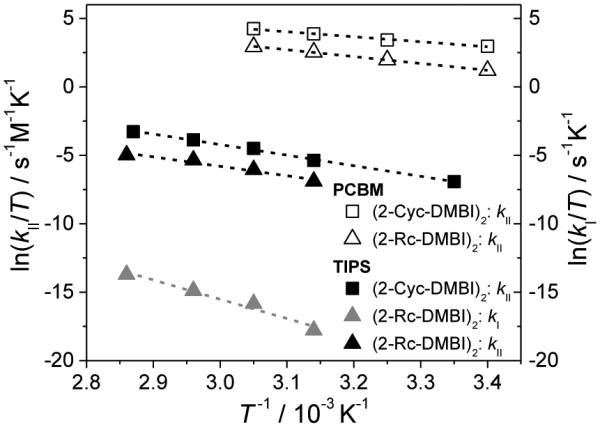
Eyring plots for variable-temperature rate-constant data for PCBM and TIPSp doped with (Y-DMBI)2 derivatives.
The reduction of TIPSp by (2-Cyc-DMBI)2 proceeds considerably more slowly than that of PCBM; indeed at room temperature in the dark the reaction is far from complete after 12 h. However, from reactions at elevated temperatures the rate law was found to be consistent with this reaction also proceeding via mechanism II (with the first step rate-determining) and the value of ΔG‡(300 K) obtained was roughly consistent with the electrochemically estimated value of ΔGET. The slow room-temperature reaction can also be accelerated by exposure to ambient light (Figure S16 in the Supporting Information); as in the previously reported photoinduced reduction of TIPSp using (IrCp*Cp)2, this reaction presumably proceeds by electron transfer from the dimer to photoexcited TIPSp.[18]
The reactions of Fc and Rc dimers with TIPSp are less straightforward. As in the Cyc case, reduction of TIPSp by (2-Rc-DMBI)2 is slow in the dark at room temperature, but can be accelerated in ambient light (Figure S12, Supporting Information). However, rate data acquired at elevated temperature in different concentrations cannot be fit to either of the expected rate laws given in Table 1. Plots of the initial rate divided by the initial concentration of dimer vs. the initial concentration of TIPSp are linear with non-zero intercepts (Figure 6), consistent with a rate law of the type
| (3) |
and, therefore, with both mechanisms I and II contributing to the observed reaction. A rate law of the same form was previously obtained for the reaction of (RhCp*Cp)2 and TIPSp.[10b] A series of such plots at different temperatures was used to extract activation parameters for the two pathways (Table 3, Figure 6). Values of ΔS‡ are positive and negative for pathways I and II, respectively, consistent with expectations. The value of ΔH‡ for the reaction of TIPSp and (2-Rc-DMBI)2 by mechanism I is much larger than the value of ΔHdiss obtained for the Fc compound from ESR data, consistent with the greater bond strength expected for the Rc species. The value of ΔG‡(300) for mechanism II is consistent with the electrochemically estimated value of ΔGET.
At room temperature, (2-Fc-DMBI)2 reduces TIPSp much more rapidly than do the other two dimers at comparable concentrations. In the presence of excess TIPSp the evolution of the TIPSp•– signals is consistent with a reaction first order in dimer. In the presence of excess dimer, [TIPSp•–] linearly approaches a maximum and then linearly decreases at a very similar rate while signals attributable to TIPSp2– appear (Figure S10, Supporting Information).[10b] The independence of the rate on [TIPSp] and the formation of TIPSp2– on a similar time scale are consistent with mechanism I with the first step being rate determining. Reactions at higher temperatures, however, revealed some dependence of rate on [TIPSp], but the data could not be fitted to a combination of mechanisms I and II as was the case for (2-Rc-DMBI)2. At the highest temperatures examined (45 and 55 °C), a plot against [TIPSp] of the initial rate divided by the initial value of [D2]0.5 was found to be linear, consistent with the rate law expected if the second step of mechanism I is rate limiting:
| (4) |
where k2 is the rate constant for electron transfer from the monomer to TIPSp (Figure S11, Supporting Information).[30] This temperature-dependent change in which step is rate-determining precluded determination of activation barriers from the temperature range examined.
The formation of TIPSp2– when sufficient (2-Fc-DMBI)2 is present is also significant from a thermodynamic point of view, suggesting that for the Fc-DMBI+/0.5(2-Fc-DMBI) couple Eeff ≤ E(TIPSp–/2–), i.e. ≤ ca. –1.93 V vs. FeCp2, consistent with the value of –1.93 V obtained from equation 1.[31] Consistent with the observation of TIPSp2–, (2-Fc-DMBI)2 can also reduce 5,11-bis(triethylsilylethynyl)-anthradithiophene (TES-ADT, E0/– = –1.71 V vs. FeCp2+/0 in THF) to its radical anion (Figure S18, Supporting Information).
Discussion
The three (2-Y-DMBI)2 dimers examined here are strong reducing agents with estimated M+/0.5M2 redox potentials of ca. –1.9 V vs. FeCp2+/0; (2-Cyc-DMBI)2 is one of the strongest isolable neutral all-organic reductants isolated to date. These potentials, which gauge the thermodynamic reducing strengths of the dimers, are rather insensitive to the choice of the 2-substituent, Y, because the 2-Y-DMBI• radicals are stabilized by Y in the order Cyc < Rc < Fc, leading to both an increasingly weak bond and an increasingly less reducing monomer. The role of Y in stabilizing the radical monomer also leads to an unusual lack of a negative correlation between the lengths of the central C—C bonds of the dimers and their dissociation energy. The cancelation of trends in dissociation energy and monomer oxidation potential found for the present series suggests that more strongly reducing related compounds could be designed by incorporating features, such as more sterically demanding Y- or N,N'-substituents, that weaken the central C—C bond of the dimer of (2-Cyc-DMBI)2, but do not significantly affect the radical stability and, consequently, the monomer redox potential.
In contrast to the small variation in the estimated thermodynamic electron-donor capabilities, the choice of Y dramatically influence the kinetics of the reaction of the DMBI dimers with acceptors as a consequence of the variation in E(D2•+/0) and ΔUdiss. The Y = Fc derivative has both the weakest bond, due to the role of the substituent in stabilizing the radical monomer, and the most cathodic dimer oxidation potential, and is consequently the most reactive of the species examined, whether reacting with PCBM – a relatively easily reduced acceptor – through an electron-transfer mechanism (II) or with TIPSp – a more challenging acceptor – through a mechanism where dimer dissociation is the first step (I). The Y = Cyc and Rc species also react with PCBM through the electron-transfer mechanism, but successively more slowly, consistent with their increasingly anodic oxidation potentials. Both mechanisms are operative for the reaction of TIPSp with the Y = Rc dimer, which has intermediate bond strength and the most anodic E(D2•+/0), whereas the Y = Cyc dimer, which has the strongest bond and an intermediate E(D2•+/0), reacts with TIPSp only through the electron-transfer mechanism. This suggests that if films of these components are processed rapidly in air, only minimal decomposition of the dimer and minimal reduction (and, therefore, ensuing aerial decomposition) of TIPSp may occur, allowing for subsequent activation of doping through exposure to light. Moreover, reductions using the Y = Cyc dimer are anticipated to be even slower for acceptors with slightly more cathodic reduction potentials. Slower solution reactions may also be advantageous in obtaining uniformly doped films in cases where the DMBI+ salt of the organic semiconductor is poorly soluble and precipitation of the salt occurs on a shorter timescale than film formation. On the other hand, the full thermodynamic reducing strength of the dimers is presumably more reliably exploitable when using the kinetically reactive Fc derivative. This may be advantageous, for example, in ensuring complete reaction in the case of surface doping of materials such as oxides, carbon nanotubes, graphene, and MoS2.
Conclusion
Despite having very different chemical structures, the (2-Y-DMBI)2 dimers show some similarities to the dimers of 19-electron sandwich compounds in the nature of their HOMOs, a lack of negative correlation between bond length and bond strength, and reactvity towards acceptors. The understanding of the thermodynamic and kinetic properties of these dimers gained in this work is important for the selection of a dopant for a particular application, and for the development of new dopants with different combinations of properties. Moreover, in addition to n-doping of organic semiconductors and surface doping of electrode materials, easily handled strong molecular reducing agents that form stable byproducts may be of interest for other applications.[32]
Supplementary Material
Acknowledgements
This research was funded by the National Science Foundation (Materials Network Program, DMR-1209468; MRSEC Program, DMR-0820382; and DMR-1305247), the Office of Naval Research (N00014141-0171), and the National Institutes of Health (through GM065790).
References
- [1].a Lüssem B, Riede M, Leo K. Phys. Status Solidi A. 2013;210:9. [Google Scholar]; b Zhang Y, Zhou H, Seifter J, Ying L, Mikhailovsky A, Heeger AJ, Bazan GC, Nguyen T-Q. Adv. Mater. 2013;25:7038. doi: 10.1002/adma.201302159. [DOI] [PubMed] [Google Scholar]; c Reineke S, Lindner F, Schwartz G, Seidler N, Walzer K, Lussem B, Leo K. Nature. 2009;459:234. doi: 10.1038/nature08003. [DOI] [PubMed] [Google Scholar]; d Naab BD, Himmelberger S, Diao Y, Vandewal K, Wei P, Lussem B, Salleo A, Bao Z. Adv. Mater. 2013;25:4663. doi: 10.1002/adma.201205098. [DOI] [PubMed] [Google Scholar]; e Kim GH, Shao L, Zhang K, Pipe KP. Nat. Mater. 2013;12:719. doi: 10.1038/nmat3635. [DOI] [PubMed] [Google Scholar]
- [2].Bröker B, Blum R-P, Frisch J, Vollmer A, Hofmann OT, Rieger R, Müllen K, Rabe JP, Zojer E, Koch N. Appl. Phys. Lett. 2008;93:243303. [Google Scholar]
- [3].Osikowicz W, Crispin X, Tengstedt C, Lindell L, Kugler T, Salaneck WR. Appl. Phys. Lett. 2004;85:1616. [Google Scholar]
- [4].a Wei P, Liu N, Lee HR, Adijanto E, Ci L, Naab BD, Zhong JQ, Park J, Chen W, Cui Y, Bao Z. Nano Lett. 2013;13:1890. doi: 10.1021/nl303410g. [DOI] [PubMed] [Google Scholar]; b Paniagua SA, Baltazar J, Sojoudi H, Mohapatra SK, Zhang S, Henderson CL, Graham S, Barlow S, Marder SR. Mater. Horiz. 2014;1:111. [Google Scholar]
- [5].Kiriya D, Tosun M, Zhao P, Kang JS, Javey A. J. Am. Chem. Soc. 2014;136:7853. doi: 10.1021/ja5033327. [DOI] [PubMed] [Google Scholar]
- [6].Chan CK, Kim EG, Brédas JL, Kahn A. Adv. Funct. Mater. 2006;16:831. [Google Scholar]
- [7].a Wei P, Menke T, Naab BD, Leo K, Riede M, Bao Z. J. Am. Chem. Soc. 2012;134 doi: 10.1021/ja211382x. [DOI] [PubMed] [Google Scholar]; b Fenghong L, Ansgar W, Martin P, Karl L, Xianjie L. The Journal of Physical Chemistry B. 2004;108 [Google Scholar]
- [8].Wei P, Oh JH, Dong G, Bao Z. J. Am. Chem. Soc. 2010;132 doi: 10.1021/ja103173m. [DOI] [PubMed] [Google Scholar]
- [9].Naab BD, Guo S, Olthof S, Evans EGB, Wei P, Millhauser GL, Kahn A, Barlow S, Marder SR, Bao Z. J. Am. Chem. Soc. 2013;135:15018. doi: 10.1021/ja403906d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a Guo S, Kim SB, Mohapatra SK, Qi Y, Sajoto T, Kahn A, Marder SR, Barlow S. Adv. Mater. 2012;24:699. doi: 10.1002/adma.201103238. [DOI] [PubMed] [Google Scholar]; b Guo S, Mohapatra SK, Romanov A, Timofeeva TV, Hardcastle KI, Yesudas K, Risko C, Brédas J-L, Marder SR, Barlow S. Chem. Eur. J. 2012;18:14760. doi: 10.1002/chem.201202591. [DOI] [PubMed] [Google Scholar]
- [11].a Zeika O, Limmert M, Gruessing A, Lux A, Hartmann H, Werner A. Eur. Pat. Appl. 2007 EP 1837927 A1. [Google Scholar]; b Limmert M, Hartmann H, Zeika O, Werner A, Ammann M. Eur. Pat. Appl. 2007 EP 1837926 A1. [Google Scholar]
- [12].Naab BD, Zhang S, Vandewal K, Salleo A, Barlow S, Marder SR, Bao Z. Adv. Mater. 2014;26:4268. doi: 10.1002/adma.201400668. [DOI] [PubMed] [Google Scholar]
- [13].Tarasov A, Zhang S, Tsai M-Y, Campbell PM, Graham S, Barlow S, Marder SR, Vogel EM. Adv. Mater. 2015;27:1175. doi: 10.1002/adma.201404578. [DOI] [PubMed] [Google Scholar]
- [14].Sutton LE, Mitchell A, Sommerfeld A, Cross L. Tables of interatomic distances and configuration in molecules and ions, Supplement 1956-1959. Chemical Society; London: 1965. [Google Scholar]
- [15].Wadepohl H, Von Der Lieth C-W, Paffen F-J, Pritzkow H. Chem. Ber. 1995;128:317. [Google Scholar]
- [16].Rüchardt C, Beckhaus H-D. Angew. Chem. Int. Ed. in Engl. 1985;24:529. [Google Scholar]
- [17].Gaudet MV, Hanson AW, White PS, Zaworotko MJ. Organometallics. 1989;8:286. [Google Scholar]
- [18].Mohapatra SK, Fonari A, Risko C, Yesudas K, Moudgil K, Delcamp JH, Timofeeva TV, Brédas J-L, Marder S, Barlow S. Chem. Eur. J. 2014 doi: 10.1002/chem.201404007. DOI: 10.1002/chem.201404007. [DOI] [PubMed] [Google Scholar]
- [19]. The energies calculated here are gas-phase IEs; the solid-state IEs that are relevant to determining whether or not a given acceptor can be doped in a film are typically ca. 1 eV lower. However, for a series of similar compounds, the same trends can be expected.
- [20].a Watts WE, Wilkinson G, Stone FGA, Abel EW. Vol. 8. Pergamon; London: 1988. [Google Scholar]; b Gleiter R, Bleiholder C, Rominger F. Organometallics. 2007;26:4850. [Google Scholar]
- [21].a Koivisto BD, Ichimura AS, McDonald R, Lemaire MT, Thompson LK, Hicks RG. J. Am Chem. Soc. 2006;128:690. doi: 10.1021/ja0566331. [DOI] [PubMed] [Google Scholar]; b Sporer C, Heise H, Wurst K, Ruiz-Molina D, Kopacka H, Jaitner P, Köhler F, Novoa JJ, Veciana J. Chem. Eur. J. 2004;10:1355. doi: 10.1002/chem.200305349. [DOI] [PubMed] [Google Scholar]
- [22].Koch WH. A Chemist’s Guide to Density Functional Theory. Wiley & Sons; New York: 2000. M. C. [Google Scholar]
- [23].Zavitsas AA. J. Phys. Chem. A. 2003;107:897. [Google Scholar]
- [24].a Schreiner PR, Chernish LV, Gunchenko PA, Tikhonchuk EY, Hausmann H, Serafin M, Schlecht S, Dahl JEP, Carlson RMK, Fokin AA. Nature. 2011;477:308. doi: 10.1038/nature10367. [DOI] [PubMed] [Google Scholar]; b Kaupp M, Metz B, Stoll H. Angew. Chem. Int. Ed. 2000;39:4607. [PubMed] [Google Scholar]
- [25]. The values of IEeff given in the SI of ref. 18 refer to a THF dielectric. However, the gas-phase values quoted here can easily be obtained from gas-phase values of IE(M•) and ΔUdiss(D2) also given there.
- [26].a Farwaha HS, Bucher G, Murphy JA. Org. Biomol. Chem. 2013;11:8073. doi: 10.1039/c3ob41701h. [DOI] [PubMed] [Google Scholar]; b Converted from the original value quoted vs. SCE using value of +0.45 V for FeCp2+/0 vs. SCE in DMF.
- [27].a Duling DR. J. of Magn. Reson., Series B. 1994;104:105. doi: 10.1006/jmrb.1994.1062. [DOI] [PubMed] [Google Scholar]; b http://www.niehs.nih.gov/research/resources/software/tox-pharm/tools/index.cfm, accessed Apr 2014.
- [28].a Qi Y, Mohapatra SK, Bok Kim S, Barlow S, Marder SR, Kahn A. Appl. Phys. Lett. 2012;100:083305. [Google Scholar]; b TIPSp is generally regarded as a hole-transport material, but can act as electron-transport material when n-doped.
- [29]. Kinetic measurements were carried out by mixing solutions in an inert atmosphere in a glove-box, sealing the cuvette, and then transferring to a spectrometer located outside the glove-box. Rate constants are challenging to determine accurately if the reaction proceeds significantly on the timescale between mixing the solutions and acquiring the first spectrum. Although, in principle, reducing the reagent concentrations can circumvent this problem, at very low concentrations complications can arise due to the sensitivity of the radical anions formed to traces of oxygen, moisture, or other impurities.
- [30]. Equation 4 can be derived by assuming D2 is in pre-equilibrium with D•, but this equation and that given in Table 1 can also be obtained as limits of the general rate equation derived via application of the steady-state approximation (see Supporting Information)
- [31]. It should be noted however, that inferences from reactivity and from electrochemistry are not completely comparable since (i) the electrochemical data for the D+/0 couple and the TIPSp–/2– couple were determined in THF in the presence of electrolyte, whereas the dissociation parameters and the kinetic data were obtained in chlorobenzene, and (ii) any ion pairing between Fc-DMBI+ and TIPSp2– could increase the thermodynamic feasibility of the reaction.
- [32].Connelly NG, Geiger WE. Chem. Rev. 1996;96:877. doi: 10.1021/cr940053x. For example, they may be used to generate of one- or two-electron reduced species such as low-valent organometallics (see. and organic mixed-valence radical anions. [DOI] [PubMed] [Google Scholar]; Hankache J, Wenger OS. Chem. Rev. 2011;111:5138. doi: 10.1021/cr100441k. [DOI] [PubMed] [Google Scholar]; Heckmann A, Lambert C. Angew. Chem. Int. Ed. 2012;51:326. doi: 10.1002/anie.201100944. either for in situ spectroscopic studies or for isolation. Moreover, one- and two-electron inorganic and organic reductants including SmI2. [DOI] [PubMed] [Google Scholar]; Szostak M, Fazakerley NJ, Parmar D, Procter DJ. Chem. Rev. 2014;114:5959. doi: 10.1021/cr400685r. [DOI] [PubMed] [Google Scholar]; Tori M, Sono M. Heterocycles. 2014;89:1369. tetrakis(dimethylamino)ethylene. [Google Scholar]; Médebielle M, Dolbier WR. J. Fluor. Chem. 2008;129:930. and bipyridinylidene species. [Google Scholar]; Murphy JA. J. Org. Chem. 2014;79:3731. doi: 10.1021/jo500071u. are useful reagents in synthetic organic chemistry; the varied kinetic behavior of the present compounds may lead to reactivity that is complementary to that of currently used reductants. However, to gain more widespread acceptance for applications of this type, it may be necessary to devise alternative synthesis that avoid the use of harzardous alkali-metal-based reductants. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.