

Abstract
The cohesin complex holds sister chromatids together until all chromosomes are properly attached to the mitotic spindle. Cleavage of the cohesin subunit Rad21 at the metaphase to anaphase transition allows separation of sister chromatids and is fundamental for the creation of identical daughter cells. Sororin blocks removal of cohesin from chromosomes from S phase until mitosis. In mitosis, Sororin is phosphorylated by Cdk1 releasing it from the cohesin complex. Aurora B phosphorylation of Sororin may play a similar role as Cdk1. Using PhosTag electrophoresis we detect multiple Sororin species suggesting that phosphorylation of Sororin in mitosis is heterogeneous. Mutating the Cdk1 consensus site S21 to alanine eliminates many of the phosphorylated species suggesting that S21 is a key site of phosphorylation in vivo. Inhibiting Aurora B reduces phosphorylation of Sororin in cells, but only if Cdk1 sites are intact suggesting that some phosphorylation events on Sororin may be sequential. Surprisingly, mutating Aurora B consensus sites in Sororin causes it to relocalize to the nucleolus during interphase and blocks its interaction with chromosomes in Aurora B-inhibited cells. These observations indicate that phosphorylation plays unexpected roles in regulating the subcellular localization of Sororin.
Keywords: cohesion, phostag, mitosis, phosphorylation, nucleolus
Introduction
Many intricate control mechanisms ensure that chromosomes are equally segregated to daughter cells during mitosis. Despite these mechanisms, tumor cells commonly show alterations in chromosome number as a consequence of defects in these controls [Barber et al., 2008; Schvartzman et al., 2010]. Changes in gene dosage as a result of chromosome losses or gains may contribute to malignant behavior of cancer cells. During mitosis, the spindle assembly checkpoint is the ultimate arbiter of anaphase onset, and ensures that division only occurs when conditions for equal chromatid segregation are satisfied (i.e. amphitelic attachment of all chromosomes to the spindle) [Kadura and Sazer, 2005; Maresca and Salmon, 2010; Musacchio and Salmon, 2007; Pinsky and Biggins, 2005]. In turn, amphitelic attachment requires that identical sister chromatids are physically held together. The cohesin complex not only fulfills this structural role, but by associating with chromosomes before DNA replication, further ensures that chromatid attachment only occurs between identical DNA molecules generated during S phase. The cohesin ring is composed of the long coiled-coil proteins SMC1 and SMC3 and the kleisin subunit Rad21 [Michaelis et al., 1997]. Additional proteins associate with the ring to regulate loading and unloading onto chromatin. The cohesin complex is loaded onto chromosomes in telophase and associates dynamically with chromatin until S phase. Dynamic association is dependent on the protein WapL, which by binding to Pds5 associated with the ring induces ring opening [Gandhi et al., 2006; Kueng et al., 2006; Shintomi and Hirano, 2009]. Passage of the replication complex through the ring induces acetylation of SMC subunits which in turn recruits the cohesin regulator Sororin [Lafont et al., 2010; Nishiyama et al., 2010; Rankin et al., 2005]. Sororin displaces WapL from Pds5 to stabilize the association of the cohesin complex with newly synthesized sister chromatids [Nishiyama et al., 2010]. In this manner, Sororin is critical to maintain sister chromatid cohesion until anaphase onset when Rad21 is cleaved to allow chromatid segregation.
Phosphorylation of Sororin during mitosis releases the protein from the cohesin complex [Dreier et al., 2011]. Phosphorylation is counteracted at the centromere, allowing a subpopulation of Sororin to persist, presumably to prevent sister chromatids from separating at the centromere until anaphase [Liu et al., 2012]. On the other hand, cohesin along chromosome arms is no longer protected by Sororin, and this, along with phosphorylation of cohesin subunits by Aurora B and Plk1, allows removal of arm cohesin, a process known as the prophase pathway [McGuinness et al., 2005; Sumara et al., 2002; Uhlmann, 2004]. The role of the prophase pathway in mitotic fidelity is not fully understood; however, expression of a Sororin mutant containing alanine substitutions at critical phosphorylation sites delays loss of arm cohesion [Dreier et al., 2011]. Sororin can be phosphorylated by Cdk1 and Aurora B and both kinases appear necessary to displace Sororin from the cohesin complex [Dreier et al., 2011; Nishiyama et al., 2010; Nishiyama et al., 2013].
Here we use PhosTag electrophoresis to characterize phosphorylated species of Sororin. Multiple species of phosphorylated Sororin are observed in mitotic human cells indicating that different subpools with different numbers of phosphate likely co-exist. Mutating S21 to alanine reduces the appearance of multiple phosphorylated species. Inhibiting Aurora B also reduces the number of phosphorylated species detected by PhosTag. By combining an Aurora B inhibitor with Cdk1 phosphorylation-site mutants of Sororin, we obtain evidence that Aurora B may prefer Cdk1-phosphorylated Sororin as a substrate. Finally, mutating Aurora B consensus sites creates a version of Sororin localized to the nucleolus in interphase and unable to associate with mitotic chromosomes upon inhibition of Aurora B. These results highlight several new levels of complexity in the regulation of Sororin by phosphorylation.
Materials and Methods
Cell Line and Culture Conditions
HeLa M cells were grown in a humidified atmosphere containing 10% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM)(Mediatech, Inc.) supplemented with 10% fetal bovine serum (Atlanta Biologicals). All chemicals were obtained from Cayman Chemicals unless otherwise noted. We used nocodazole at 3.3µM, Taxol at 2µM, MG132 at 20µM and ZM447439 (AstraZeneca) at 2.5µM. Transfections were carried out using ExtremeGene (Roche) or polyethyleneimine (Polysciences). A typical experiment in a 6-well plate would include 2–5 µg DNA which was mixed with 50–100 µl of DMEM and 6–15 µl of polyethyleneimine (1mg/ml) followed by a 10 min incubation before adding to cells.
Western Blotting and PhosTag electrophoresis
HeLa M cells were scraped into phosphate buffered saline (PBS) followed by centrifugation (16000 x g, 4°C) for 5 minutes and stored at −80°C. Pellets were lysed with RIPA Buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% NP-40, 1% DOC, 0.1% SDS) supplemented with 1 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin A, 1mM dithiothreitol, 0.1 M phenylmethyl sulfonyl fluoride, 1 mM sodium fluoride and 1 mM sodium vanadate for 20 minutes on ice and centrifuged at maximum speed for 20 minutes at 4°C [Dreier et al., 2011] Protein concentration of each lysate was determined by using the BCA Protein Assay Kit (Pierce). Equal protein amounts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (Millipore) in Transfer Buffer (25mM Tris, 192mM Glycine, 10% Methanol). The membranes were blocked for 1 hour with PBST [1X PBS plus 0.05% Tween 20] containing 5% non-fat dry milk. Membranes were probed with V5 antibodies from Invitrogen (diluted 1:2500) followed by secondary antibodies from Biorad diluted 1:10,000. Antibody binding was detected using the Clarity chemiluminescence substrate (Biorad).
Phosphorylated species of Sororin were separated by adding PhosTag acrylamide (Wako Chemicals) to SDS-PAGE gels. Cells were lysed as described above and equal amounts of protein separated using SDS-PAGE gels containing 7.5% acrylamide, 50µM PhosTag and 100µM MnCl2. After electrophoretic separation, gels were incubated in Transfer Buffer containing 1mM ethylenediamine tetraacetic acid (EDTA) for 10 minutes with agitation. Gels were then agitated in Transfer Buffer without EDTA for an additional 10 minutes. SDS was added to Transfer Buffer to a final concentration of 0.01% during electro-blotting. Electrophoresis and transfer was carried out using the Biorad Mini Protean System which, in both cases was packed in ice while under voltage. All PhosTag experiments were carried out at least twice; duplicate experiments entailed analyzing lysates from a completely different transfection.
In vitro Phosphorylation.
Glutathione-S-transferase (GST)-Sororin fragments were prepared and purified from E.coli as previously described [Dreier et al., 2011]. Full-length Aurora B with an N-terminal HIS-tag was purified from E.coli. Also, a fragment of INCENP (amino acids 822–919) containing the “IN” box with an N-terminal HIS-tag was purified from E.coli. Aurora B/INCENP822–919 was mixed with GST-Sororin in the presence of 32P-γ-ATP (Perkin Elmer) in a buffer containing 50mM Tris pH7.5 and 10mM MgCl2. The final ATP molarity was 60 μM/reaction. The amount of 32P added was 4μCi/reaction, and the specific activity per reaction was 6 Ci/mMols. Reactions were carried out at 37°C for 30 minutes, stopped by boiling for 5 minutes in a buffer containing 2% SDS, 100mM Tris, 0.05% bromophenol blue and 30% glycerol. Proteins were separated by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes and visualized by autoradiography. The same membrane was then analyzed using the Western method with antibodies against GST (Thermo Scientific).
Generation and Analysis of Sororin Point Mutants
QuikChange Multi Site-Directed Mutagenesis (Stratagene) was used to create point mutations in Sororin that was already cloned as a green fluorescent protein (GFP) fusion into pDEST47. Additional V5-tagged fusion constructs were previously described [Dreier et al., 2011]. All constructs were confirmed by sequencing. Imaging studies were carried out with transiently transfected HeLa M cells. Images of live cells were captured at 40X using a wide-field Olympus inverted fluorescence microscope.
Results
Resolution of phosphorylated Sororin species by PhosTag electrophoresis.
We transiently transfected HeLa M cells with V5-tagged Sororin and separated lysates using SDS-polyacrylamide gels containing PhosTag-acrylamide [Kinoshita et al., 2006]. PhosTag is a dinuclear metal complex that binds two cations to create a chemical moiety that serves as a phosphate-binding site. Phostag gels were previously observed to specifically slow phosphorylated proteins [Kinoshita et al., 2006]. Western blotting with V5 antibodies revealed a single major species in cells blocked in S phase with hydroxyurea. In contrast, multiple slowly migrating Sororin species were observed in cells blocked in mitosis with nocodazole (Fig. 1). Using this method, we were able to detect 5–6 mitotic Sororin species. These observations suggest that mitotic cells contain different pools of Sororin either containing different numbers of phosphorylated residues or phosphorylated at different sites. Sororin is phosphorylated in mitosis in a Cdk1-dependent manner and contains 9 residues conforming to the Cdk1 consensus [Dreier et al., 2011]. Phosphoproteome analysis has also provided evidence that all nines sites analyzed in Fig. 1A can be phosphorylated in vivo [Beausoleil et al., 2006; Cantin et al., 2008; Chen et al., 2009; Dephoure et al., 2008; Gnad et al., 2007; Olsen et al., 2006; Van Hoof et al., 2009]. Furthermore, five of these sites (S79, S83, T111, T115 and T159) were directly confirmed to be Cdk1 sites by an in vitro phosphorylation experiment, where human Sororin was incubated with ATP and purified Cdk1, followed by mass spectrometric analysis. [Nishiyama et al., 2013]. In a separate study, S21 was confirmed to be a Cdk1 site on Sororin [Zhang et al., 2011]. Specifically, in vitro kinase assays were performed with recombinant human Sororin and recombinant purified Cdk1 followed by mass spectrometry. The remaining three Cdk1 consensus sites on Sororin are known to be phosphorylated in vivo, however there is no direct evidence that they are phosphorylated by Cdk1 as yet [Beausoleil et al., 2006; Chen et al., 2009].
Figure 1:
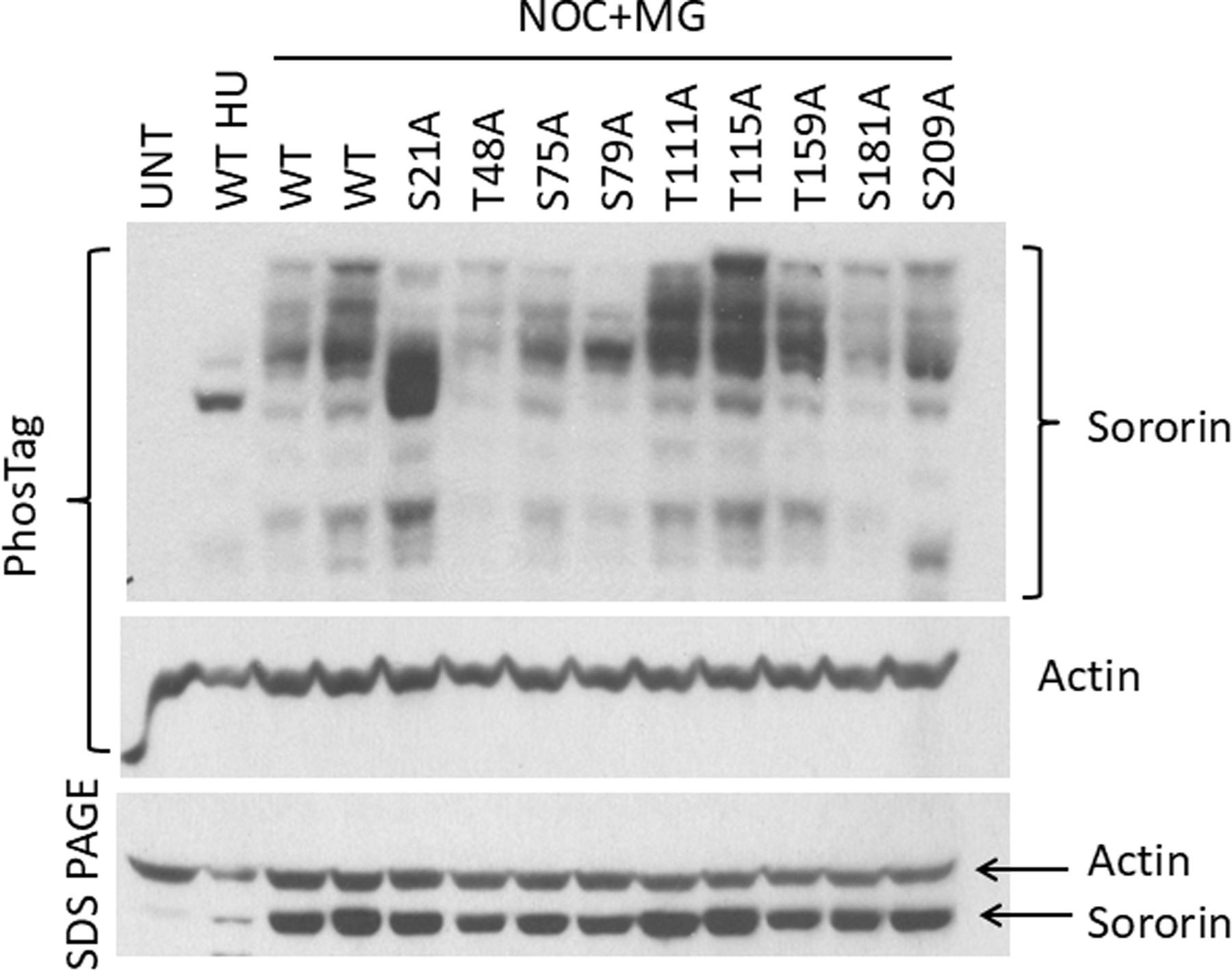
PhosTag analysis of Cdk1 site mutants of Sororin. HeLa M cells transiently transfected with V5-tagged Sororin mutants were either blocked in S phase with hydroxyurea (HU) or in mitosis with nocodazole (NOC) for 18 hours. MG132 (MG) was added for an additional two hours to allow for comparison to inhibitor-treated samples in Figures 3–5. Two separate wild-type (WT)-transfected lysates were run to determine variability. In the top panel, lysates were separated by PhosTag electrophoresis and probed with an antibody to Sororin, followed by stripping and reprobing for Actin. A smaller aliquot of the same lysates was separated by regular SDS-PAGE for comparison. “UNT” : untransfected.
We analyzed PhosTag mobility of Sororin point mutants in which the 9 sites were singly converted to alanine. Multiple Sororin species were observed with each single mutant. Mutating S21 to alanine consistently eliminated many of the slowest migrating bands leaving the bulk of Sororin near the position observed with wild-type Sororin from hydroxyurea-treated S phase cells (Fig. 1). To quantify these results, we divided each lane into 4 sections. Quantitation of signals indicated that ~40% of S21A Sororin was in the section with the fastest migration, indicating a lower amount of phosphorylation (4th section; Fig. S1). In contrast, ~10% of wild type (WT) Sororin was in the fast migrating section. These results indicate an overall loss of phosphorylation in S21A compared to wild-type. Additionally, ~20% of WT Sororin was in the slowest migrating section compared to ~10% of the S21A mutant (1st section Fig. S1). Therefore, S21 is a key site of Sororin phosphorylation in vivo. In some experiments the S75A, S79A and S209A mutants showed subtle reduction of the slowest migrating bands providing evidence that these sites are phosphorylated in vivo (Fig. 1 and unpublished data). The subtle effect of most mutations on Sororin mobility in PhosTag gels may indicate that Sororin is multiply phosphorylated and the effects of single mutations are difficult to detect on this background.
Intersection between Cdk1 and Aurora B phosphorylation of Sororin.
Aurora B is reported to phosphorylate Sororin and we were also able to detect phosphorylation of recombinant Sororin by purified Aurora B [Nishiyama et al., 2013](Fig. 2). Phosphorylation in vitro was lowest using a fragment of Sororin encompassing three Aurora B consensus sites S79, S83, and S145. Next we used PhosTag electrophoresis to characterize the effect of inhibiting Aurora B on Sororin in HeLa cells. Inhibiting Aurora B with ZM447439 reduced the intensity of some of the slowest migrating forms of Sororin detected by PhosTag electrophoresis (Figs. 3, 4, 5). Sororin contains 7 Aurora B consensus sites, one of which (S79) also matches a Cdk1 site. Interestingly, mutating either 8 or 9 Cdk1 sites to alanine produced a Sororin protein that migrated the same after nocodazole treatment compared to hydroxyurea treatment in PhosTag gels (Fig. 3A)(mutations are summarized in Table 2). This suggests that mitotic phosphorylation of Sororin, as detected by PhosTag, is mostly abolished by mutating Cdk1 sites even though 6 of the 7 Aurora B sites are intact. One interpretation of this result is that Aurora B may prefer Sororin as a substrate after it has been phosphorylated at Cdk1 sites. Adding ZM447439 to nocodazole-arrested cells had no effect on the migration of either the 8A or 9A Cdk1 mutants of Sororin, providing additional evidence that these mutants may not be optimal Aurora B substrates.
Figure 2:
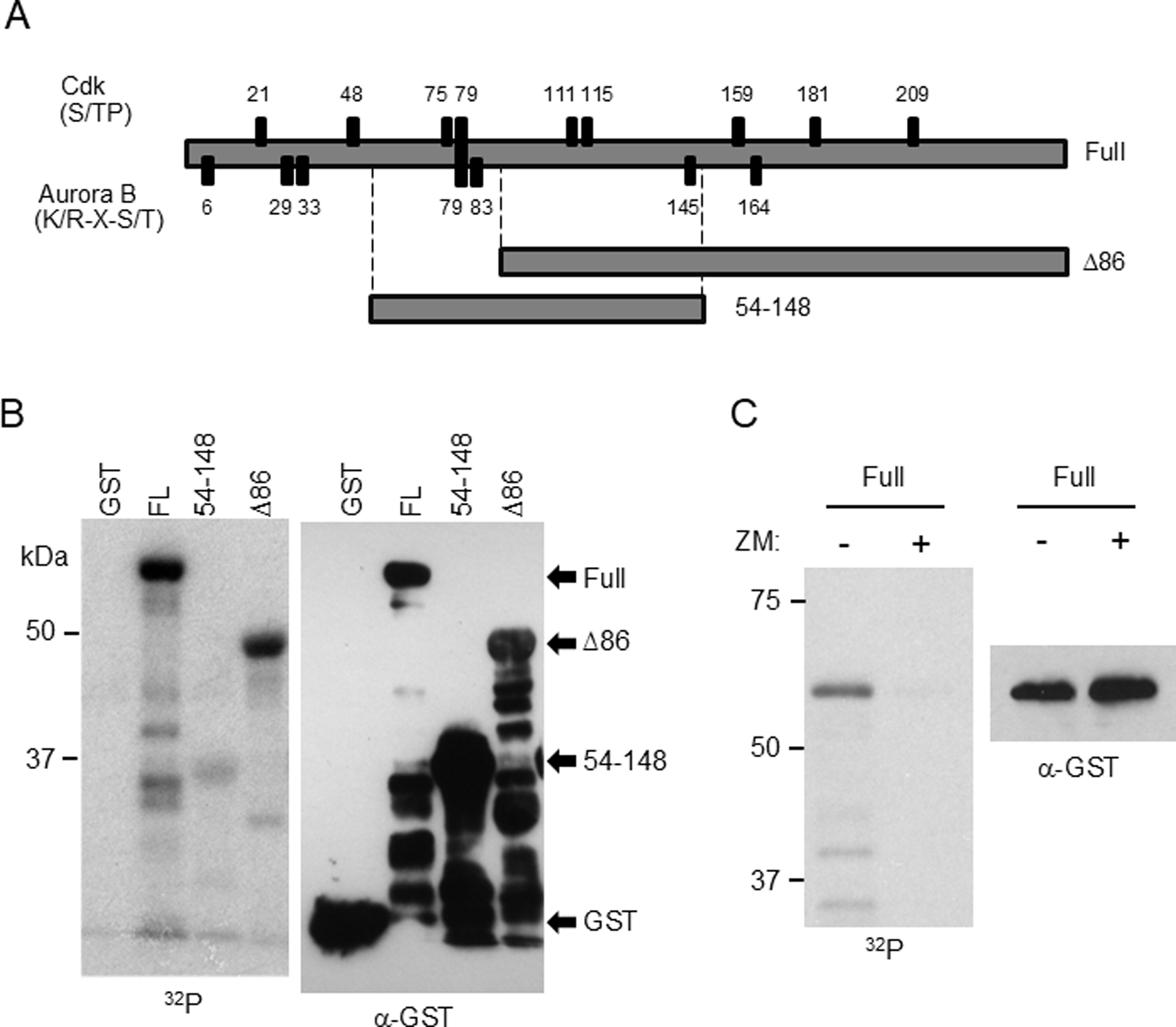
Phosphorylation of Sororin by Aurora B in vitro. GST-tagged Sororin fragments were purified from E. coli and incubated with recombinant Aurora B and the IN-box of INCENP in the presence of 32P-labeled ATP. (A) Schematic showing Cdk and Aurora B consensus sites as well as regions represented in the Sororin fragments used as substrates. (B). Autoradiograph of in vitro-phosphorylated Sororin is shown in the left panel. The same blot, probed with an antibody to GST is shown on the right. (C) ZM447439 added to in vitro reactions to determine specificity.
Figure 3:
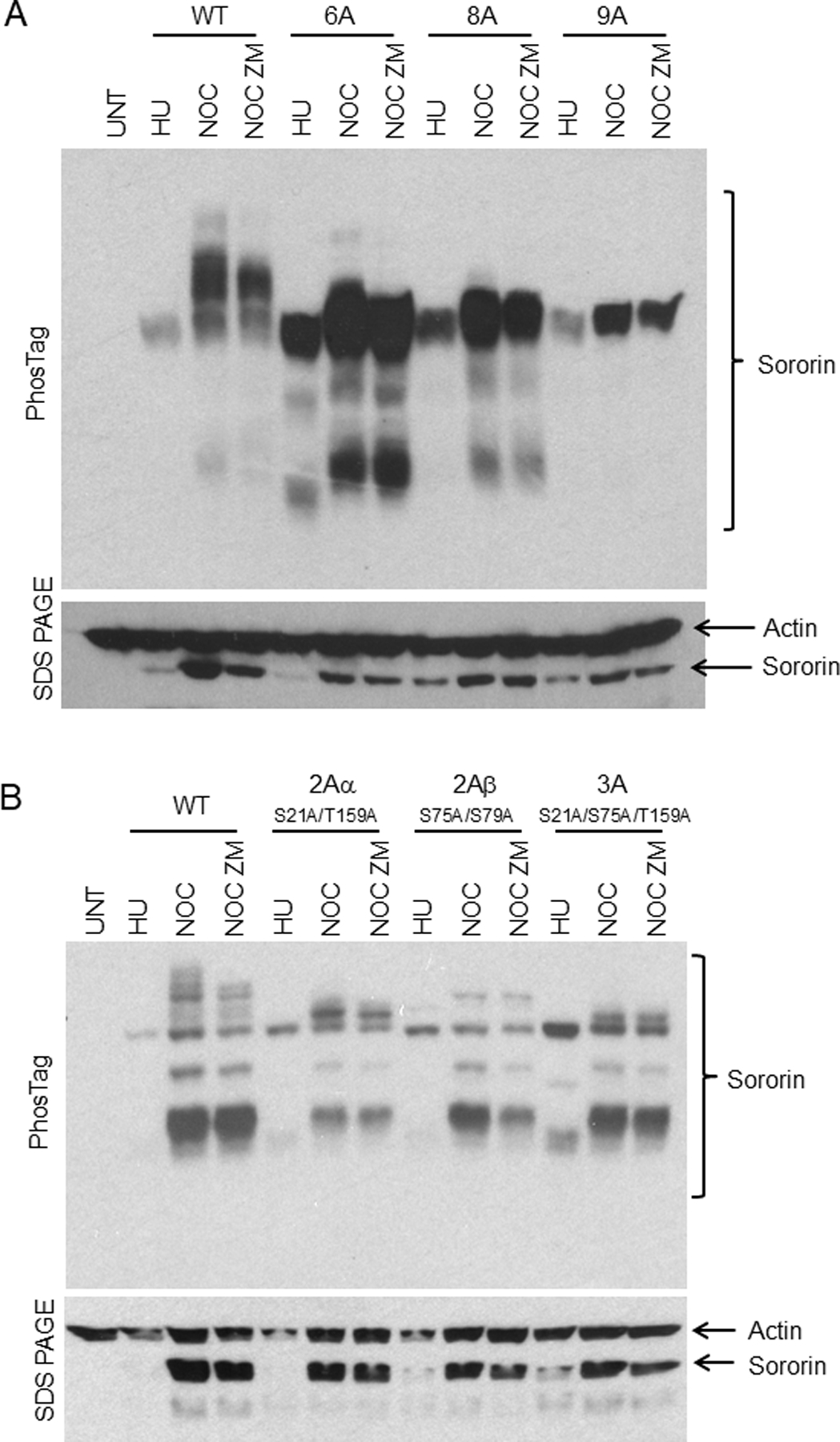
PhosTag analysis of Sororin after inhibition of Aurora B. HeLa M cells were transiently transfected with the indicated compound mutants of Sororin. Cells were treated for 18 hours with either hydroxyurea (HU) to block them in S phase or with nocodazole to block them in mitosis. The nocodazole-treated samples were then treated with MG132 alone (“NOC”) or MG132 + 2.5μM ZM447439 (“NOC ZM”) for 2 hours. ZM447439 is added to inhibit Aurora B, and MG132 is added to prevent mitotic exit when Aurora B is inhibited. In the top panel, lysates were separated by PhosTag electrophoresis before Western analysis. In the bottom panel, an aliquot of the same lysate was separated by standard SDS-PAGE and probed with antibodies to Sororin and Actin for loading. Experiments shown in “A and B” were carried out under the same conditions with different mutants transfected as indicated. “UNT”: untransfected.
Figure 4:
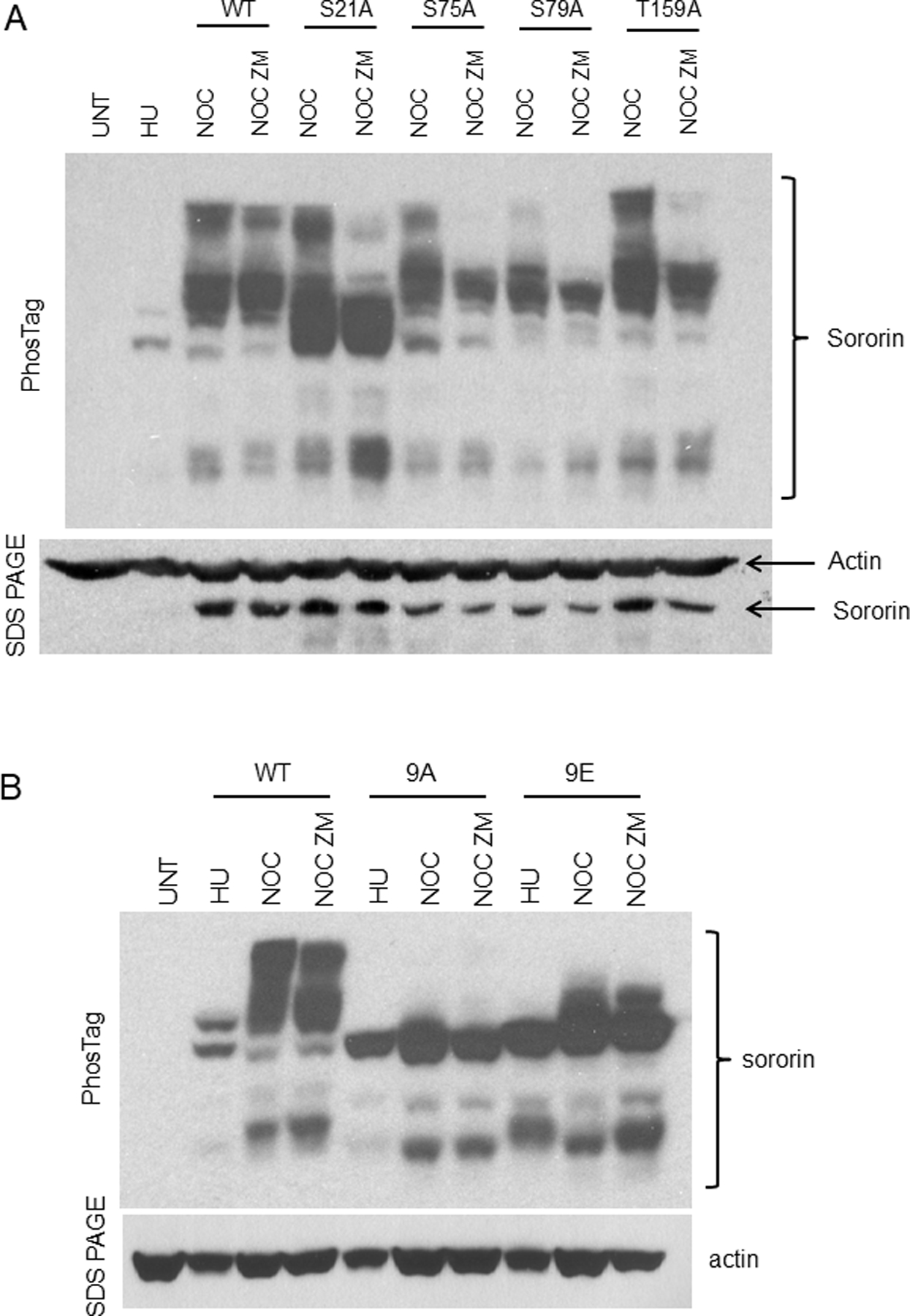
PhosTag analysis of single Cdk1 site mutants of Sororin after inhibition of Aurora B. HeLa M transfection and PhosTag analysis was carried out as described in Figure 3. (A) Single site mutations at Cdk1 consensus sites incorporated into V5-tagged Sororin. (B) Comparison of Sororin-9A and Sororin-9E by PhosTag.
Figure 5:
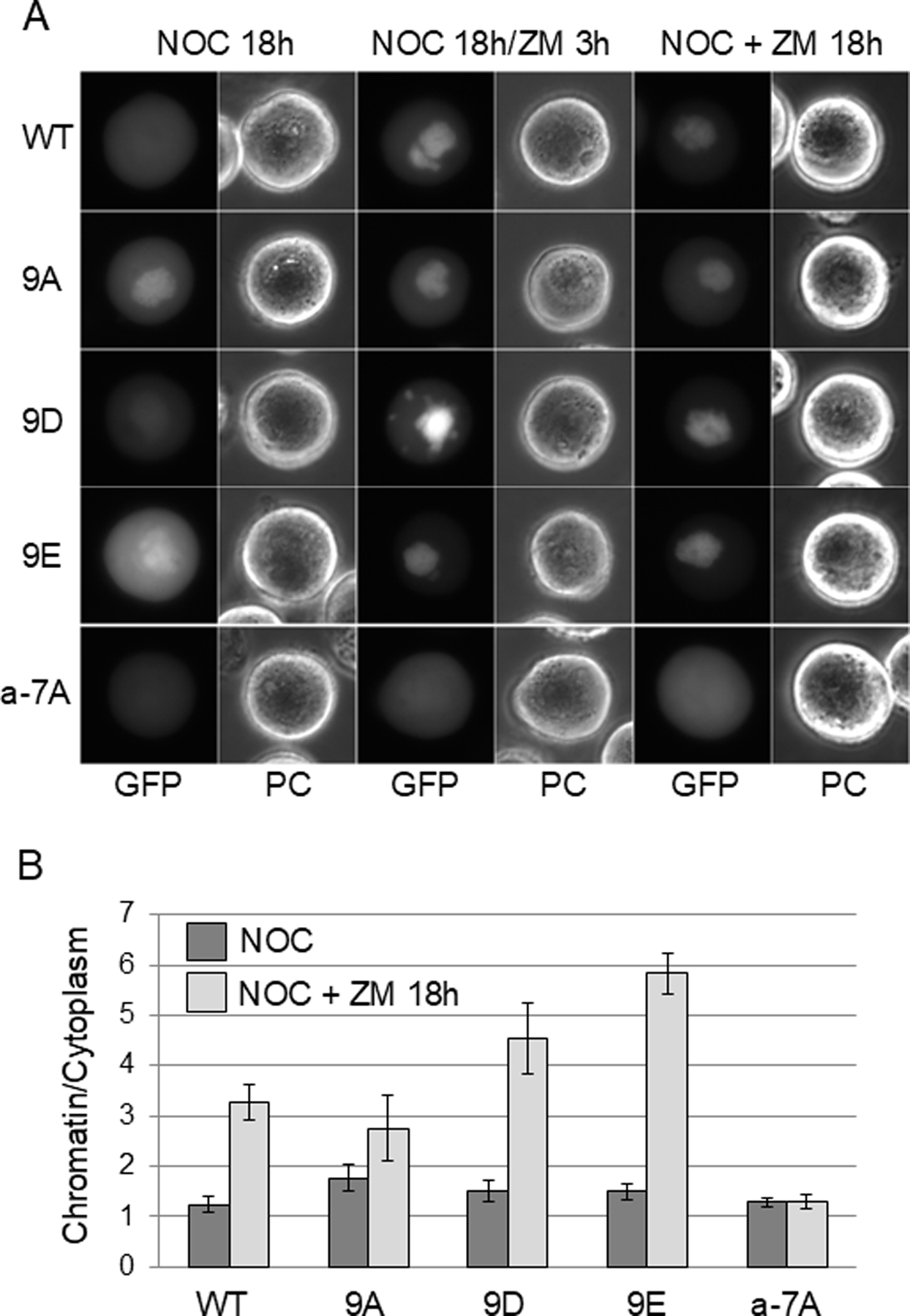
Localization of wild-type and mutant Sororin-GFP in response to Aurora B inhibition. HeLa M cells were transiently transfected with Sororin-GFP. Live cells were imaged by wide-field fluorescence microscopy. (A) Transfected cells were exposed to nocodazole (NOC) for 15 hours followed by ZM447439 (ZM) for an additional 3 hours (NOC 18h/ZM 3h). In the columns on the right, cells were exposed to nocodazole and ZM447439 for 18 hours (NOC + ZM 18h). ZM447439 for either 3 or 18 hours causes association of Sororin with chromatin except in the mutant a-7A which stays dispersed in the mitotic cytoplasm. “PC”: Phase contrast. (B) Quantitation of the chromatin/cytoplasm ratio in images of live cells. Pixel intensity in the central and peripheral regions of the cell were determined and are shown as a ratio. Bars represent standard deviation.
Table 2.
Summary of localization phenotypes of Sororin mutants. ~: partial, nt: not tested. Mutants 9A, 8A, 6A, 3A, 2Aα, 2Aβ were analyzed in a previous study [Dreier et al., 2011].
| WT AA | T | S | S | S | T | S | S | S | T | T | S | T | S | S | S | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cdk1 | Y | Y | Y | Y | Y | Y | Y | Y | Y | on mitotic | ||||||||
| Aurora B | Y | Y | Y | Y | Y | Y | Y | chromatin | ||||||||||
| Position | 6 | 21 | 29 | 33 | 48 | 75 | 79 | 83 | 111 | 115 | 145 | 159 | 164 | 181 | 209 | nucleolar | − ZM | + ZM |
|
|
||||||||||||||||||
| WT | X | X | √ | |||||||||||||||
| 9A | A | A | A | A | A | A | A | A | A | X | √ | √ | ||||||
| 8A | A | A | A | A | A | A | A | A | X | √ | nt | |||||||
| 6A | A | A | A | A | A | A | X | √ | nt | |||||||||
| 3A | A | A | A | X | ~ | nt | ||||||||||||
| 2Aα | A | A | X | ~ | nt | |||||||||||||
| 2Aβ | A | A | X | ~ | nt | |||||||||||||
| a-7A | A | A | A | A | A | A | A | √ | X | X | ||||||||
| a-6A | A | A | A | A | A | A | √ | X | X | |||||||||
| a-5Aα | A | A | A | A | A | √ | X | X | ||||||||||
| a-5Aβ | A | A | A | A | A | √ | X | X | ||||||||||
| a-3Aα | A | A | A | X | X | √ | ||||||||||||
| a-3Aβ | A | A | A | X | X | √ | ||||||||||||
| S79A | A | X | X | √ | ||||||||||||||
| T6A | A | X | X | X | ||||||||||||||
| S29A | A | X | X | X | ||||||||||||||
| S29E | E | X | X | X | ||||||||||||||
To narrow down which Cdk1 sites might contribute to Aurora B phosphorylation, we analyzed additional Sororin mutants. Sororin-6A showed a slight mobility shift in mitosis that could not be detected in every experiment (Fig.3A and unpublished data). The compound mutants 2Aα (S21A;T159A), 2Aβ (S75A; S79A), and 3A (S21A; S75A; T159A) all showed a slight increase in mobility in nocodazole arrested cells, however several phosphorylated bands were missing (Fig. 3B). ZM447439 had minimal effects on the mobility of the 6A, 2Aα, 2Aβ or 3A compound mutants in mitosis. To further narrow down which Cdk1 residues were potentially influencing Aurora B phosphorylation, we tested the effect of ZM447439 on several single point mutants targeting sites mutated in 2Aα, 2Aβ, and 3A. As before, S21A showed a reduction in phosphorylation in nocodazole, but was still reduced further by ZM447439 (Fig. 4A). Similarly, ZM447439 reduced the intensity of phosphorylated species detected by PhosTag in mutants S75A, S79A, and T159A (Fig. 4A). The amount of mobility shift was quantified for the single point mutants. S21A shows ~50% in the fastest migrating section in nocodazole, and ~70% in this section in nocodazole plus ZM447439 (4th section, Fig. S2). Therefore, S21A shows further loss of phosphorylation in the presence of nocodazole and ZM447439, as compared to when in nocodazole alone. S75, S79A and T159A showed ~20% in the fastest migration section of the gel for both nocodazole and nocodazole plus ZM447439, which was similar to wild type Sororin. Therefore, mutating single specific Cdk1 sites does not seem to preclude Aurora B phosphorylation. However combining these Cdk1 mutations in groups of 2, 3, 6, 8 or 9 does appear to abolish Aurora B phosphorylation. Altogether, these results suggest that Aurora B prefers Sororin after it has been multiply phosphorylated at Cdk1 consensus sites.
Next we attempted to test the effect of mimicking Cdk1 phosphorylation of Sororin by converting 9 Cdk1 sites to glutamic acid. Sororin-9E showed a slight mobility shift in nocodazole-treated cells, however ZM447439 had little effect on the mobility of this mutant (Fig. 4B). We interpret this observation as indicating that glutamic acid only partially mimics Cdk1 phosphorylation of Sororin. Further evidence for this interpretation is that Sororin-9E can functionally replace endogenous Sororin knocked-down by shRNA (Table 1). In this experiment, loss of Sororin function causes an increase in mitotic index due to premature loss of cohesion. Phosphorylation of Sororin by Cdk1 releases it from the cohesin complex [Dreier et al., 2011]. If glutamic acid mimics these phosphorylations, Sororin-9E should be unable to associate with the cohesin complex and should not reduce the mitotic arrest associated with Sororin knockdown. Sororin-9E transfected cells had significantly more mitotic cells than those transfected with wild-type Sororin indicating a partial loss of function (Table 1). However, Sororin-9E was clearly able to reduce the mitotic index compared to shRNA alone, suggesting retention of partial function. Conversion of 9 Cdk1 consensus sites to aspartic acid (Sororin-9D) did not hamper its ability to reduce mitotic index suggesting that this negatively charged amino acid, like glutamic acid is a poor mimic of phosphorylation for Sororin (Table 1). We also analyzed the role of the FGF motif in Sororin function in HeLa M cells. This motif is reported to mediate the interaction of Sororin with Pds5 in Xenopus extracts [Nishiyama et al., 2010]. Sororin in which the FGF motif was mutated to AGA could still replace endogenous Sororin and reduce the mitotic index even if present in the 9A background with all Cdk1 sites mutated (Table 1).
Table 1.
Functional analysis of Cdk1 phosphorylation site mutants of Sororin. HeLa M cells co-transfected with a vector expressing an shRNA targeting the 3’UTR of endogenous Sororin and the indicated Sororin cDNAs. A plasmid expressing H2B-GFP was included in all transfections to allow measurement of mitotic index in transfected live cells. Quantitation was carried out in a blinded manner.
| MITOTIC INDEX | ||
|---|---|---|
|
| ||
| PLASMIDS | Experiment 1 Ave ± SD |
Experiment 2 Ave ± SD |
| EMPTY pSUPER | 9.9 ± 1.6 | 9.2 ± 1.9 |
| pSUPER-SOR | 48 ± 3.3 | 48 ± 3.3 |
| pSUPER-SOR + SORWT | 20 ± 1.3 | 27 ± 3.2 |
| pSUPER-SOR + SOR9A | 18 ± 2.9 | 21 ± 7.6 |
| pSUPER-SOR + SOR9D | 17 ± 2.1 | 19 ± 1.3 |
| pSUPER-SOR + SOR9E | 31 ± 1.1 | 28 ± 5.3* |
| pSUPER-SOR + SORAGA-WT | 19 ± 3.5 | 27 ± 4.1 |
| pSUPER-SOR + SORAGA-9A | 29 ± 2.1 | 26 ± 2.0 |
p=0.03 when compared to SORWT (both experiments combined).
Mutating Aurora B sites alters Sororin localization in an unexpected manner.
PhosTag analysis suggests that Aurora B may phosphorylate Sororin in vivo. Aurora B phosphorylation is reported to collaborate with Cdk1 phosphorylation to release Sororin from the cohesin complex in mitosis [Nishiyama et al., 2013]. Therefore, we mutated the 7 Aurora B consensus sites in Sororin in various combinations and analyzed the effect on localization (Table 2). We predicted that like Sororin-9A in which Cdk1 sites are mutated, loss of Aurora B sites might block removal of Sororin from chromatin during mitosis. In contrast, mutating all Aurora B sites created a Sororin mutant (“a-7A”) that was dispersed in the mitotic cytoplasm similarly to the wild-type protein (Fig. 5 – “NOC 18h” samples).
Interestingly, inhibiting Aurora B with ZM447439 caused a major relocalization of wild-type Sororin-GFP from the mitotic cytoplasm onto the chromatin (Fig. 5). This effect was also observed in cells that were not previously arrested in nocodazole (Fig. S3A). On the surface, this observation appears consistent with a role for Aurora B in releasing Sororin from the cohesin complex along with Cdk1. Similarly, ZM447439 induced mutants Sororin-9D and Sororin-9E to relocalize onto mitotic chromatin (Fig. 5). Surprisingly, mutating Aurora B sites on Sororin created a protein (a-7A) that was unable to relocalize onto chromatin in Aurora B inhibited cells (Fig. 5). If Aurora B releases Sororin from chromatin in mitosis by direct phosphorylation, mutating the consensus sites should phenocopy inhibition of the kinase. In contrast, the mutant is resistant to the chromatin-association effect of inhibiting Aurora B.
Next we analyzed additional compound Aurora B consensus site mutants of Sororin. Mutants a-6A, a-5Aα and a-5Aβ showed reduced association with chromatin after ZM447439 treatment similarly to the a-7A mutant (Fig. 6A). However, mutants a-3Aα, a-3Aβ and S79A retained a wild-type ability to localize to chromatin after ZM447439 treatment (Fig. 6B). The main difference between these sets of mutants resides in the N-terminus, specifically residues T6, and S29. Next, we mutated T6 to alanine and S29 to either alanine or glutamic acid. The effect of these mutations on localization after ZM447439 treatment was more subtle than the compound mutants we had analyzed. Mutations at either T6 or S29 did however induce a significant reduction in ZM447439-induced chromatin association compared to the wild-type protein (Fig. 7B). These results indicate that T6, and S29 play an important role in regulating chromatin binding of Sororin in Aurora B-inhibited cells.
Figure 6:
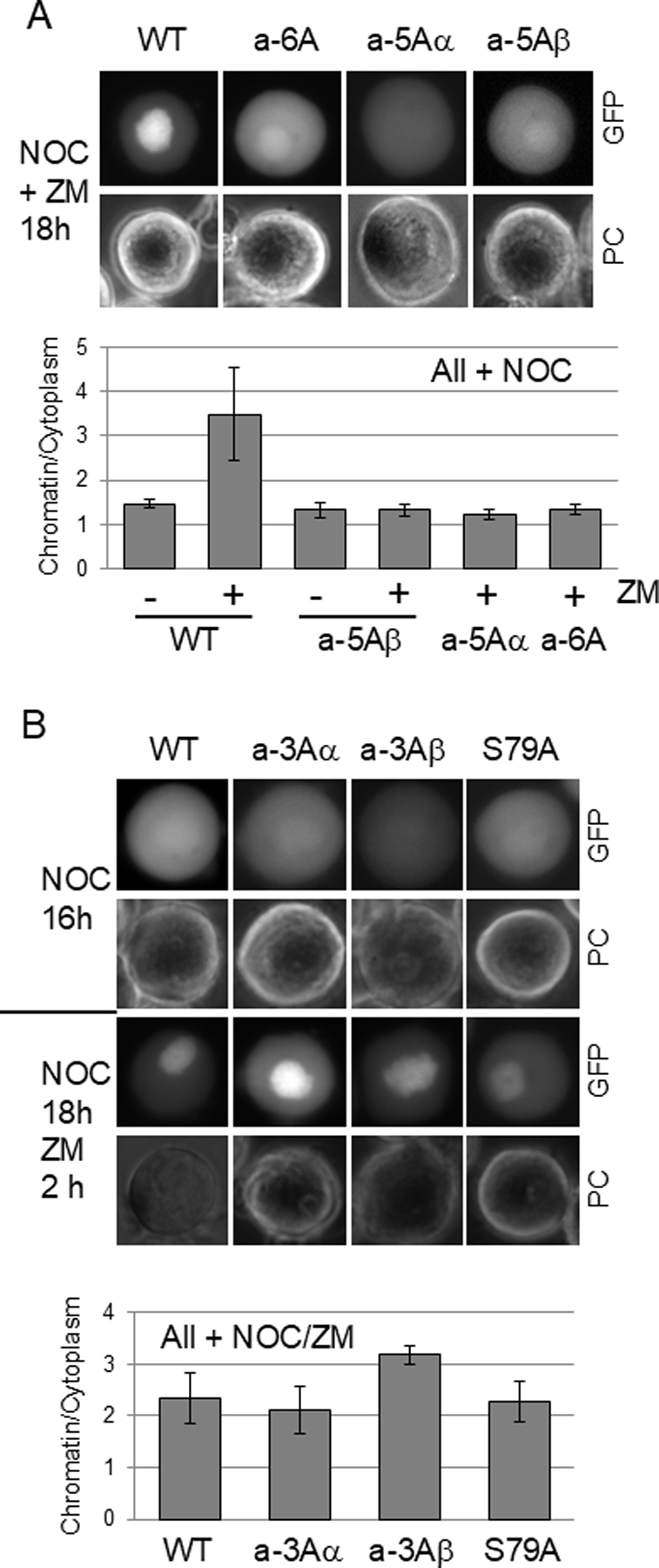
Localization of additional mutants of Sororin after inhibition of Aurora B. HeLa M cells were transfected with mutants of Sororin-GFP in which various Aurora B consensus sites were converted to alanine and then analyzed by fluorescence microscopy. (A) Sororin mutants exposed to nocodazole and ZM447439 for 18 hours. (B) Sororin mutants arrested in mitosis with nocodazole and imaged either before (NOC 16h) or after a 2 hour treatment with ZM447439 (NOC 18h ZM 2h). For both “A” and “B”, the chromatin/cytoplasm expression ratio is shown below each corresponding panel.
Figure 7:
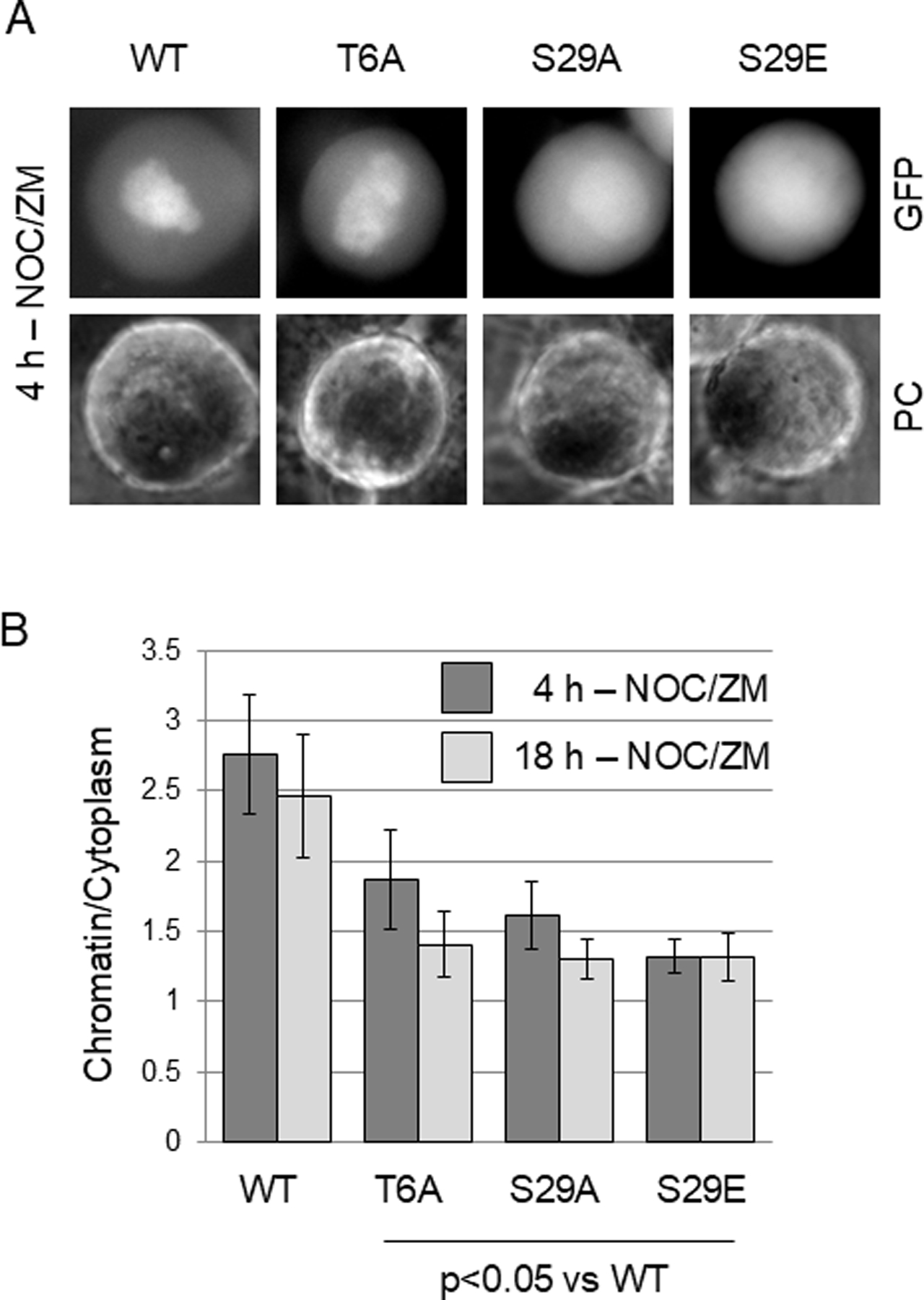
Effect of mutations at T6 and S29 on the localization of Sororin-GFP after Aurora B inhibition. Live HeLa M cells transiently transfected with the indicated mutants were analyzed by wide-field fluorescence microscopy. (A) Representative images of Sororin-GFP transfected cells after inhibition of Aurora B. One day after transfection, nocodazole and ZM447439 were added simultaneously. Images of live cells were captured 4 hours later. (B) Quantitation of chromatin/cytoplasm ratio. One day after transfection, nocodazole and ZM447439 were added simultaneously. Cells were imaged either 4 or 18 hours later.
Further analysis of Aurora-site Sororin mutants revealed one additional unexpected finding. Many of the compound mutants were localized to the nucleolus in interphase unlike wild-type Sororin or any of the Cdk1-site mutants we have previously analyzed (Fig. 8 and S3C). Specifically, mutants a-7A, a-6A, a-5Aα and a-5Aβ were nucleolar in interphase (Figs. 8A and B). Adding ZM447439 did not cause wild-type Sororin to relocalize to the nucleolus suggesting that Aurora B phosphorylation of Sororin and nucleolar localization of mutants are independent effects (Fig. 8B). Adding either nocodazole or nocodazole plus ZM447439 did not appear to alter the nucleolar localization phenotype (Fig. S3B,C). To narrow down the sites responsible for nucleolar localization we analyzed additional Sororin mutants. Mutants a-3Aα, a-3Aβ and S79A were dispersed throughout the interphase nucleus similar to wild-type Sororin (Fig. 8C). In the seven compound Aurora B site mutants, nucleolar localization was correlated with failure to bind to chromatin in ZM447439 treated mitotic cells (Table 2). Interestingly, single point mutants at residues T6 or S29 did not create mutants localized to the nucleolus despite reducing mitotic chromatin binding (Fig. 8D; Table 2). Altogether, these observations suggest that nucleolar localization and failure to bind to chromatin after ZM447439 treatment are regulated by different sets of phosphorylation sites. Also, all mutants that show nucleolar localization have common mutations at T6, S29 and S79 (Table 2). Therefore, we hypothesize that phosphorylation of two or three of these sites in combination is required to remove Sororin from the nucleolus.
Figure 8:
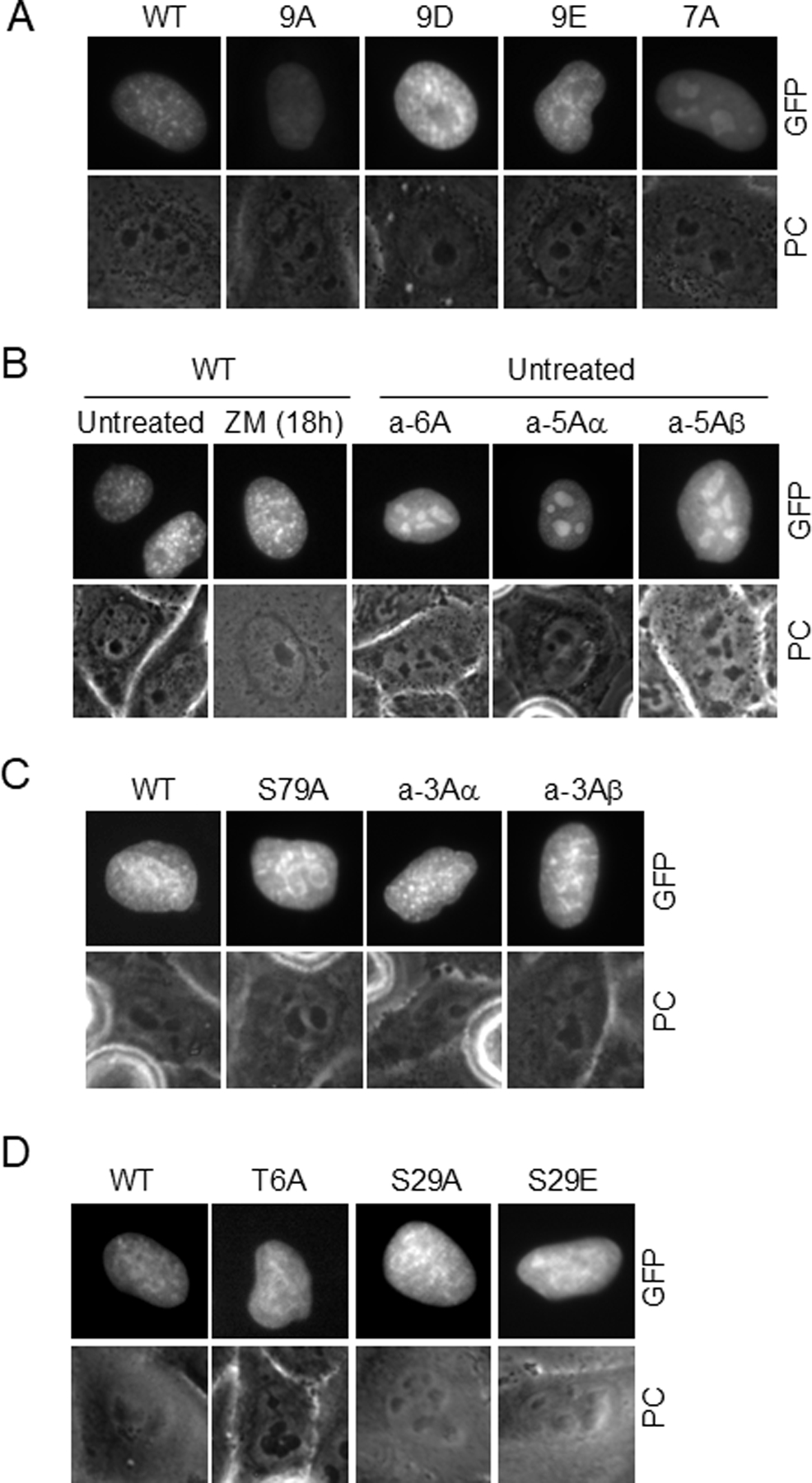
Analysis of phosphorylation sites required to remove Sororin-GFP from the nucleolus. HeLa M cells were transiently transfected with the indicated mutants and live interphase cells imaged by wide-field fluorescence microscopy. (A) Interphase nuclei in untreated cultures indicate dispersed, but punctate staining in WT, 9A, 9D, 9E. Mutant a-7A shows nucleoloar enrichment. (B) Comparison of Sororin mutants bearing either 5 or 6 Aurora consensus site mutants to the wild-type protein. (C) Comparison of wild-type, S79A or two triple Sororin mutants exposed to nocodazole for 18 hours. Cells that had not yet entered mitosis were imaged indicating a wild-type localization in these mutants. Similar results were obtained in untreated cells. (D) Effect of mutations at T6 and S29 on interphase Sororin localization. Cells were untreated at the time of imaging.
Discussion
Sororin stabilizes sister chromatid cohesion after S phase, allowing both chromatids to travel together during prometaphase until anaphase when Rad21 is cleaved. Sororin stabilizes cohesin by binding to Pds5 and displacing WapL, which would otherwise open the hinge of the ring causing release of the cohesin complex [Nishiyama et al., 2010]. During prophase, cohesin is lost from chromosome arms in part because Sororin is phosphorylated during mitosis and unable to bind Pds5 and block cohesin removal [Dreier et al., 2011; Nishiyama et al., 2010; Nishiyama et al., 2013]. Phosphorylation of Sororin is counteracted at the centromere by PP2A presumably to inhibit premature removal of centromeric cohesin [Liu et al., 2012]. Direct phosphorylation of cohesin subunits by Aurora B and Plk1 also contribute to removal of arm cohesin [Uhlmann, 2004]. Dephosphorylation of cohesin near the centromere appears to collaborate with Sororin in maintaining centromeric cohesion until anaphase.
Sororin is phosphorylated by Cdk1 and Aurora B during mitosis to remove Sororin from the cohesin complex [Dreier et al., 2011; Nishiyama et al., 2010; Nishiyama et al., 2013]. Also, Cdk1 phosphorylation of T159 of Sororin creates a Plk1 docking site to facilitate its phosphorylation of the cohesin protein SA2 [Zhang et al., 2011]. Our results indicate several additional layers of complexity in Sororin modification in mitotic cells. First, PhosTag electrophoresis identifies a number of modified forms suggesting that Sororin modification is heterogeneous in mitosis. In other systems, such as Cdk-mediated inactivation of Rb, sequential phosphorylation of multiple residues by different kinases creates subpopulations of modified protein with slightly different activities [Harbour and Dean, 2000]. The fact that the phosphorylation state of Sororin depends on whether it is associated with the centromere or along chromosome arms predicts that different pools of modified Sororin should be detectable in mitosis. Interestingly, phosphorylation of all 9 Cdk1 consensus sites have been detected by in vivo phosphoproteome analysis, yet alanine mutation of most of these sites do not alter the number of shifted bands observed by PhosTag. Most likely, Sororin is phosphorylated on many sites and single mutations have little effect on this background. In contrast, mutation of S21 had a major effect on Sororin migration in PhosTag gels. One interpretation of this finding is that S21 must be phosphorylated to allow other sites to be modified.
Mutation of 9 Cdk1 sites in Sororin created a protein that did not show a mitotic mobility shift upon PhosTag electrophoresis. One possibility is that Cdk1 phosphorylation is a pre-requisite for other phosphorylation events. Mutating either 2 or 3 Cdk1 sites (mutants 2Aα, 2Aβ, 3A) did produce shifted species in PhosTag gels, however these mutants were not affected by inhibiting Aurora B, unlike the wild-type protein. Mutating single Cdk1 sites found in the 2Aα, 2Aβ and 3A mutants produced a Sororin protein that was still affected by Aurora B inhibition. These observations suggest that Aurora B may prefer a multiply Cdk1-phosphorylated Sororin substrate. Future experiments are aimed at testing the effect of sequential phosphorylation of Sororin by Cdk1 and Aurora B using recombinant proteins.
Mutating Cdk1 sites in Sororin to alanine creates a protein that is not released from the cohesin complex in mitosis and remains associated with condensed mitotic chromosomes [Dreier et al., 2011]. Aurora B phosphorylation of Sororin is reported to have a similar effect [Nishiyama et al., 2013]. Consistent with this idea, inhibiting Aurora B with ZM447439 caused relocalization of Sororin from the mitotic cytoplasm to the condensed chromosomes. Surprisingly, mutating 7 Aurora B sites on Sororin to alanine did not create a protein that was associated with chromosomes in contrast to the effect of mutating Cdk1 sites. In addition, ZM447439 was unable to relocalize the Aurora-site mutant of Sororin (a-7A) to chromosomes. These observations argue against a simple situation in which Aurora B and Cdk1 phosphorylation of Sororin releases it from mitotic chromosomes. Several scenarios may explain the effects of ZM447439 and Sororin-a-7A. For example, relocalization of wild-type Sororin to chromosomes after ZM447439 treatment may be due to loss of phosphorylation of a chromosome receptor such as a cohesin protein or histone H3. Serine 10 of histone H3 is a major substrate of Aurora B, and although Sororin is not known to interact with this modified histone, it is intriguing that when associated with chromatin, Sororin-GFP is not found in the inner chromatid region as expected for cohesin binding. Instead, Sororin-9A and wild-type Sororin in ZM447439-treated cells is found wherever DNA is located [Dreier et al., 2011]. It is also possible that Aurora B regulates a direct interaction of Sororin with DNA. Along these lines, Sororin can be isolated from cell lysates using DNA-cellulose, however we did not detect an association between recombinant Sororin and DNA oligonucleotides [Dreier et al., 2011] (unpublished data).
Aurora B phosphorylates Sororin, but we argue that this modification does not simply release the protein from chromatin. Otherwise, mutation of Aurora B sites would create a protein associated with mitotic chromosomes. Our experiments suggest instead that phosphorylation of residues T6, S29 and S79 in some combination releases Sororin from the nucleolus. Also, phosphorylation of T6 and S29 are required for Sororin to bind to chromatin in Aurora B inhibited cells. It is possible that phosphorylation of Aurora B consensus sites is required for proper folding of Sororin. However it is unlikely that a mitotic kinase (Aurora B) is required for folding of a protein required during S phase to stabilize cohesin. Furthermore, wild-type Sororin is not found in the nucleolus during interphase when Aurora B is naturally low, or further inhibited with ZM447439. Another explanation for the behavior of Sororin-a-7A, is that a cell cycle constitutive kinase phosphorylates one of these sites and that this modification releases Sororin from the nucleolus and is required for its association with chromosome receptors in mitosis. Our results would suggest that these receptors are masked by Aurora B-dependent phosphorylation. Regulation of Sororin by phosphorylation appears to be more complicated than simply being a mechanism to release the protein from the cohesin complex. Our observation provide evidence that some residues may set the stage for additional modifications, and that phosphorylation regulates Sororin localization in interphase as well as mitosis.
Supplementary Material
Acknowledgements
We gratefully acknowledge Mike Bekier for preparing recombinant Aurora B/INCENP.
Contract Grant Sponsor: National Institute of General Medical Sciences
Contract Grant Number: R15GM100440-01
ABBREVIATIONS:
- DMEM
Dulbecco’s Modified Eagle’s Medium
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- EDTA
ethylenediamine tetraacetic acid
- GST
Glutathione-S-transferase
- PVDF
polyvinylidene
- GFP
green fluorescent protein
Footnotes
Conflict of Interest
The authors state that they have no conflict of interest to declare.
References
- Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C, Hieter P. 2008. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A 105:3443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. 2006. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24:1285–92. [DOI] [PubMed] [Google Scholar]
- Cantin GT, Yi W, Lu B, Park SK, Xu T, Lee JD, Yates JR 3rd. 2008. Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J Proteome Res 7:1346–51. [DOI] [PubMed] [Google Scholar]
- Chen RQ, Yang QK, Lu BW, Yi W, Cantin G, Chen YL, Fearns C, Yates JR 3rd, Lee JD. 2009. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res 69:2663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. 2008. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A 105:10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier MR, Bekier ME 2nd, Taylor WR. 2011. Regulation of sororin by Cdk1-mediated phosphorylation. Journal of Cell Science 124:2976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R, Gillespie PJ, Hirano T. 2006. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol 16:2406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F, Ren S, Cox J, Olsen JV, Macek B, Oroshi M, Mann M. 2007. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol 8:R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. 2000. Rb function in cell-cycle regulation and apoptosis. Nat. Cell. Biol. 2:E65–7. [DOI] [PubMed] [Google Scholar]
- Kadura S, Sazer S. 2005. SAC-ing mitotic errors: how the spindle assembly checkpoint (SAC) plays defense against chromosome mis-segregation. Cell Motil Cytoskeleton 61:145–60. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5:749–57. [DOI] [PubMed] [Google Scholar]
- Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters JM. 2006. Wapl controls the dynamic association of cohesin with chromatin. Cell 127:955–67. [DOI] [PubMed] [Google Scholar]
- Lafont AL, Song J, Rankin S. 2010. Sororin cooperates with the acetyltransferase Eco2 to ensure DNA replication-dependent sister chromatid cohesion. Proc Natl Acad Sci U S A 107:20364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Rankin S, Yu H. 2012. Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat Cell Biol 15:40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca TJ, Salmon ED. 2010. Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci 123:825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness BE, Hirota T, Kudo NR, Peters JM, Nasmyth K. 2005. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol 3:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. 1997. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91:35–45. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. 2007. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8:379–93. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V, Bando M, Shirahige K, Hyman AA, Mechtler K, Peters JM. 2010. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 143:737–49. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Sykora MM, Huis in ‘t Veld PJ, Mechtler K, Peters JM. 2013. Aurora B and Cdk1 mediate Wapl activation and release of acetylated cohesin from chromosomes by phosphorylating Sororin. Proc Natl Acad Sci U S A 110:13404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–48. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Biggins S. 2005. The spindle checkpoint: tension versus attachment. Trends in Cell Biology 15:486–93. [DOI] [PubMed] [Google Scholar]
- Rankin S, Ayad NG, Kirschner MW. 2005. Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell 18:185–200. [DOI] [PubMed] [Google Scholar]
- Schvartzman JM, Sotillo R, Benezra R. 2010. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10:102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintomi K, Hirano T. 2009. Releasing cohesin from chromosome arms in early mitosis: opposing actions of Wapl-Pds5 and Sgo1. Genes Dev 23:2224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters JM. 2002. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol Cell 9:515–25. [DOI] [PubMed] [Google Scholar]
- Uhlmann F 2004. The mechanism of sister chromatid cohesion. Exp Cell Res 296:80–5. [DOI] [PubMed] [Google Scholar]
- Van Hoof D, Munoz J, Braam SR, Pinkse MW, Linding R, Heck AJ, Mummery CL, Krijgsveld J. 2009. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 5:214–26. [DOI] [PubMed] [Google Scholar]
- Zhang N, Panigrahi AK, Mao Q, Pati D. 2011. Interaction of Sororin protein with polo-like kinase 1 mediates resolution of chromosomal arm cohesion. J Biol Chem 286:41826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.