

ABSTRACT
Influenza virus infections represent a serious threat to human health. Both extrinsic and intrinsic factors determine the severity of influenza. The MX dynamin-like GTPase 1 (Mx1) gene has been shown to confer strong resistance to influenza A virus infections in mice. Most laboratory mouse strains, including C57BL/6J, carry nonsense or deletion mutations in Mx1 and thus a nonfunctional allele, whereas wild-derived mouse strains carry a wild-type Mx1 allele. Congenic C57BL/6J (B6-Mx1r/r) mice expressing a wild-type allele from the A2G mouse strain are highly resistant to influenza A virus infections, to both mono- and polybasic subtypes. Furthermore, in genetic mapping studies, Mx1 was identified as the major locus of resistance to influenza virus infections. Here, we investigated whether the Mx1 protective function is influenced by the genetic background. For this, we generated a congenic mouse strain carrying the A2G wild-type Mx1 resistance allele on a DBA/2J background (D2-Mx1r/r). Most remarkably, congenic D2-Mx1r/r mice expressing a functional Mx1 wild-type allele are still highly susceptible to H1N1 virus. However, pretreatment of D2-Mx1r/r mice with alpha interferon protected them from lethal infections. Our results showed, for the first time, that the presence of an Mx1 wild-type allele from A2G as such does not fully protect mice from lethal influenza A virus infections. These observations are also highly relevant for susceptibility to influenza virus infections in humans.
IMPORTANCE Influenza A virus represents a major health threat to humans. Seasonal influenza epidemics cause high economic loss, morbidity, and deaths each year. Genetic factors of the host strongly influence susceptibility and resistance to virus infections. The Mx1 (MX dynamin-like GTPase 1) gene has been described as a major resistance gene in mice and humans. Most inbred laboratory mouse strains are deficient in Mx1, but congenic B6-Mx1r/r mice that carry the wild-type Mx1 gene from the A2G mouse strain are highly resistant. Here, we show that, very unexpectedly, congenic D2-Mx1r/r mice carrying the wild-type Mx1 gene from the A2G strain are not fully protected against lethal influenza virus infections. These observations demonstrate that the genetic background is very important for the protective function of the Mx1 resistance gene. Our results are also highly relevant for understanding genetic susceptibility to influenza virus infections in humans.
INTRODUCTION
Influenza A virus represents a major health threat to humans. Seasonal influenza epidemics cause high economic loss, morbidity, and deaths each year (1). Annually, about 500 million people are infected by the influenza A virus worldwide, of whom about 500,000 die (1). In recent history, the emergence of new influenza virus subtypes has caused severe pandemics (2–4), the most severe being the Spanish flu pandemic in 1918, which resulted in 30 to 50 million deaths worldwide (5). Furthermore, a new variant of the H1N1 virus, pH1N1, caused a worldwide pandemic in 2009 (6–12). Seasonal influenza A viruses are transmitted from human to human, but bird influenza A viruses may also directly infect humans who have been in close contact with infected birds. There are presently three virus subtypes, H5N1, H9N2, and H7N9, which are circulating in birds and which have the potential to infect humans. Infection with these subtypes may cause severe disease with lethal outcomes (13–17). There is some evidence from animal models that H7N9 virus may be able to spread by contact and air transmission (18, 19), making it a likely candidate for future pandemics in humans. Therefore, it is important to better understand the biological mechanisms that result in severe outcomes after influenza A virus infection.
The course and outcome of an influenza A virus infection are influenced by viral and host factors. Host risk factors, such as obesity or pregnancy, became evident during the 2009 swine flu pandemic (20, 21). Furthermore, genetic factors in humans associated with a higher susceptibility to influenza virus infections and severe disease outcome have been suspected for the 1918 pandemic, as well as H5N1 virus infections in patients (22–24). However, evidence for genetic predisposition in humans is circumstantial (22–24), and the details of the biological mechanisms for health and genetic factors predisposing to severe influenza in humans remain largely unknown (22–27). Recently, the importance of IFITM3 as a crucial factor for host susceptibility has been demonstrated in mice and humans (28).
The mouse is one of the most important mammalian model systems for studying host responses to influenza A virus and for assessing, for example, virus virulence, disease severity, genetic predisposition, immune responses, and vaccine efficacy (reference 29 and references therein). The importance of host factors for host susceptibility and resistance has been demonstrated clearly in animal models. We and others have shown in mouse models that susceptibility of the host to influenza A virus infection strongly depends on the genetic background (30–38).
Also in mice, the MX dynamin-like GTPase 1 (Mx1) gene has been identified as one of the most important influenza virus resistance genes (reviewed in references 39 to 41). Mx1 acts as a cell-autonomous restriction factor against many viral pathogens. Expression of Mx1 is induced by type I or type III interferons (42). Structure analysis of MX1 proteins revealed a globular G domain connected to a stalk region (43). The stalk is able to mediate self-assembly into a ring-like oligomer that is thought to interact directly with viral RNP particles and thereby block replication (43). The amino acid sequence in the L4 loop of the stalk determines specificity against different virus pathogens (44). It has been further suggested that additional cellular host factors may be involved in the antiviral activity of Mx1 (45, 46).
The protective activity of Mx1 against myxoviruses has been originally discovered in A2G mice that carry a wild-type allele (47). However, most laboratory mice are deficient for Mx1 because of deletions or nonsense mutations (48, 49), whereas many wild-derived strains carry a functional Mx1 allele (49, 50). The A2G allele of Mx1 has subsequently been demonstrated to be highly protective against lethal influenza virus infections in various mouse models (51–55). Congenic C57BL/6J.A2G-Mx1r/r (B6-Mx1r/r) mice survive infections with mouse-adapted H1N1 virus (56) and are also resistant to lethal infections with highly virulent polybasic H5N1 virus (54). Furthermore, SPRET/Ei mice, which carry another Mx1 wild-type allele, are strongly protected against influenza virus infections (57). A genetic mapping study in a backcross of (C57BL/6 × SPRET/Ei)F1 × C57BL/6 mice identified Mx1 as the major resistance locus (57). Furthermore, the founder strains of the Collaborative Cross recombinant inbred population (58) carry five different haplotypes in the Mx1 genomic region, two of which (PWK/PhJ and NZO/HILtJ) were highly protective against influenza virus infections (49). A/J, C57BL/6J, 129S1/SvImJ, and NOD/ShiLtJ carry a deletion or stop codon in the Mx1 gene and were highly susceptible (49). A third wild-derived allele was found in CAST/EiJ mice, exhibiting one amino acid difference from the presumed ancestral PWK/PhJ allele. It was expressed after influenza A virus infection but did not protect CAST/EiJ mice from a lethal infection (49). It is yet unclear whether genetic background or the specific Mx1 allele in CAST/EiJ mice is responsible for the susceptible phenotype. In a mapping study using pre-Collaborative Cross mice, Mx1 was found as the strongest resistance quantitative trait locus (QTL), explaining 42% of the variation in body weight loss in this population (49).
We showed previously that in the absence of Mx1, C57BL/6J (B6-Mx1−/−) mice survive infections with a less virulent strain of a mouse-adapted H1N1 (PR8M) virus, whereas DBA/2J (D2-Mx1−/−) mice were highly susceptible (30). On the other hand, Mx1-deficient (B6-Mx1−/−) mice were highly susceptible to the more virulent mouse-adapted H1N1 (PR8) virus (55, 59). However, in the presence of the Mx1 allele from A2G mice, congenic B6-Mx1r/r mice were strongly protected against infections with this virus (55).
To further investigate the role of Mx1 in different genetic backgrounds, we generated a congenic D2(B6).A2G-Mx1r/r (D2-Mx1r/r) mouse line carrying the wild-type Mx1 allele from A2G and challenged these mice with H1N1 (PR8) virus. Most surprisingly, we found that D2-Mx1r/r mice were highly susceptible to PR8 infections even in the presence of the wild-type A2G Mx1 allele.
(Part of this work has been performed as Ph.D. thesis work [D.-L.S.] at the University of Veterinary Medicine, Hannover, Germany.)
MATERIALS AND METHODS
Ethics statement.
All experiments in mice were approved by an external committee according to the national guidelines of the animal welfare law in Germany (BGBl. I S. 1206 and 1313 and BGBl. I S. 1934). The protocol used in these experiments has been reviewed by an ethics committee and approved by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (permit numbers 33.9.42502-04-051/09 and 3392 42502-04-13/1234). No approval was necessary for work with 10-day-old embryonated chicken eggs. Laboratory C57BL/6J (B6-Mx1−/−) and DBA/2J (D2-Mx1−/−) mice carrying mutant Mx1 alleles were purchased from Janvier, France. Mice were maintained under specific-pathogen-free conditions at the animal facilities of the Helmholtz Centre for Infection Research (HZI). Embryonated chicken eggs were purchased from Charles River Laboratories, Germany.
Virus.
Original stocks of mouse-adapted PR8 virus were obtained from Peter Stäheli, University of Freiburg (PR8, A/PuertoRico/8/34 H1N1, Freiburg variant [59, 60]). Mouse-adapted H3N2 virus (A/Hong Kong/1/68 H3N2) was obtained from Otto Haller, University of Freiburg. All viruses were propagated in the chorioallantoic cavity of 10-day-old pathogen-free embryonated chicken eggs, aliquoted, and stored at −80°C.
Mice.
Congenic B6.A2G-Mx1r/r (B6-Mx1r/r) mice carrying a functional A2G Mx1 allele were provided by Peter Stäheli, University of Freiburg, Germany. Congenic D2(B6).A2G-Mx1r/r (D2-Mx1r/r mice) carrying a wild-type Mx1 allele were generated in our laboratory by backcrossing D2-Mx1−/− mice for 10 generations onto B6-Mx1r/r mice. In each generation, the presence of the Mx1 wild-type-containing region on chromosome 16 was confirmed by PCR genotyping.
Genotyping of mice.
For genotyping, genomic DNA was extracted from mouse tails with the DNeasy blood and tissue kit according to the manufacturer's instructions (Qiagen). DNA concentration was quantified with a spectrophotometer (NanoDrop 1000; Thermo Scientific). A total of 100 ng DNA and 10 pmol primer oligonucleotides were used for PCR with LightCycler 480 Probes Master (Roche) according to the manufacturer's instructions. For the PCR genotyping, polymerase was activated at 95°C for 10 min, followed by 40 cycles of a denaturation step at 94°C for 1 min, primer annealing at 61°C for 1 min, and an elongation reaction at 72°C for 1 min. A three-primer PCR strategy was used for Mx1 allele genotyping (Peter Stäheli, personal communication). Primers were designed for sequences flanking the Mx1 locus (exon8 forward, e8fn, 5′-GGAGCTCACCTCCCACATCT-3′; exon8 reverse, e8r, 5′-AGCATGGCTGTGTCACAAGCA-3′; exon12 reverse, e12r, 5′-CGAAGGCAGTTTGGACCATCT-3′). Mice carrying a wild-type Mx1 gene yielded a 950-bp product, whereas mutant Mx1 alleles were detected by the presence of a 1,255-bp product. The correct background in congenic mice after backcrossing was verified by the Mouse Universal Genotyping Array (MUGA). Array processing was performed by Neogen. The analysis demonstrated that 99.02% of the single nucleotide polymorphisms (SNPs) in D2-Mx1r/r mice matched the DBA/2J genotype and carried a 32.73-Mb region from the B6-Mx1r/r mice on chromosome 16, which includes 1.5 Mb of the original A2G region.
Infection of mice.
Female mice at the age of 8 to 12 weeks were anesthetized by intraperitoneal injection of ketamine-xylazine solution in sterile NaCl (100 mg/ml ketamine [WDT, Garbsen, Germany]; 20 mg/ml Xylavet [CP-Pharma, Burgdorf, Germany]) with a dose adjusted to the individual body weight (200 μl/20 g body weight). Infection was performed by intranasal application of virus solution in 20 μl sterile phosphate-buffered saline (PBS). Subsequently, survival and body weight loss were monitored until day 14 postinfection (p.i.). In addition to mice that were found dead, mice with a weight loss of more than 30% of the starting body weight were euthanized and recorded as dead.
RT-PCR for Mx1 transcript analysis.
Reverse transcription-PCR (RT-PCR) was performed to confirm wild-type Mx1 expression in D2-Mx1r/r mice. Mice were anesthetized and infected intranasally with 2 × 103 focus-forming units (FFU) of PR8 in 20 μl PBS. Lungs were prepared, washed in PBS, and stored in 2 ml RNAlater (Qiagen). Subsequently, lungs were homogenized using a PolyTron 2100 homogenizer. Total RNA was prepared using TRIzol chloroform according to the manufacturer's instructions (Invitrogen). One microgram of total RNA was reverse transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen, USA) according to the manufacturer's instructions. Five microliters of cDNA product was amplified with specific primers (e8fn and e12r) to determine expression of the Mx1 wild-type allele. Only D2-Mx1r/r but not D2-Mx1−/− mice yielded a product of 467 bp that is generated from expression of the Mx1 wild-type allele. For quantitative RT-PCR, after alpha interferon (IFN-α) treatment or influenza virus infection, RNA was reverse transcribed using SuperScript III reverse transcriptase. Subsequently, the quantitative PCR was performed in a LightCycler 480 real-time PCR system (Roche) using the SsoFast EvaGreen Supermix kit (Bio-Rad) according to the manufacturer's instructions. Primers for the Mx1 transcript were designed to cross an intron-exon boundary (Mx1EE-F, 5′-CCTGGAGGAGCAGAGTGACAC-3′; Mx1EE-R, 5′-GGTTAATCGGAGAATTTGGCAA-3′). Primers for the Ifnb1 transcript were modified from a previous study by Takaki et al. (61): Ifnb1-F (5′-CCAGCTCCAAGAAAGGACGA-3′) and Ifnb1-R (5′-CGCCCTGTAGGTGAGGTTGAT-3′). Primers for β-actin were used as the housekeeping gene control (Bact2-F, 5′-AGGTGACAGCATTGCTTCTG-3′; Bact2-R, 5′-GCTGCCTCAACACCTCAAC-3′). The specificity of the PCR amplification was assessed by the melting curve at the end of the reaction. Relative expression levels of Mx1 and Ifnb1 were calculated by the threshold cycle (2−ΔΔCT) method (62) and calculated as fold change induction compared to PBS-treated controls.
Determination of infectious viral particles.
For determining viral load in lungs, lungs were prepared and put into 2 ml PBS containing 0.1% bovine serum albumin (BSA). Lung tissue was subsequently homogenized using the PolyTron 2100 homogenizer. Debris was removed by centrifugation, and aliquots were stored at −70°C. Virus titers were determined on MDCK II (Madin-Darby canine kidney II) cells as focus-forming units (FFU) as described previously (59). Briefly, MDCK II cells were seeded in 96-well plates and serial 10-fold dilutions of homogenized lung samples in Dulbecco's modified Eagle's medium (DMEM) containing 5 μg/ml N-acetylated trypsin (NAT; Sigma) were added. After incubation for 24 h at 37°C, cells were washed, fixed with 4% formalin, and permeabilized with quencher buffer (0.5% Triton X-100 with 20 mM glycine in PBS), followed by incubation with a primary anti-influenza virus polyclonal antibody (Virostat) and a secondary horseradish peroxidase (HRP) antibody (KPL). Subsequently, a substrate (True Blue; KPL) was used for immunological staining. Foci were counted and calculated as FFU per lung homogenate. The detection limit of the assay was 80 infectious particles/lung. Thus, for samples where no foci were detected, data points were set to 80 FFU/lung.
Cytokine and chemokine analysis in BAL fluid.
Female B6-Mx1r/r and D2-Mx1r/r mice (five in each group and time point) at the age of 10 to 12 weeks were infected with 2 × 103 FFU of PR8. Control mice were mock infected with PBS. After 3 and 5 days p.i., mice were euthanized by isoflurane. A sterile 22-gauge catheter was inserted into the exposed trachea lumen. By instillation of PBS, a volume of 0.5 ml bronchoalveolar lavage (BAL) fluid per mouse was collected. BAL fluid was stored at −70°C until measurement. Chemokine and cytokine levels of granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), gamma interferon (IFN-γ), interleukin-1α (IL-1α), IL-6, IL-10, IL-17, IP-10, KC, monocyte chemoattractant protein 1 (MCP-1), MIP-1a, RANTES, tumor necrosis factor alpha (TNF-α), and vascular endothelial growth factor (VEGF) were analyzed using the mouse cytokine/chemokine magnetic bead panel Mcytomag-70K from Millipore according to the instruction manual of the manufacturer. Plates were read in the Luminex 100 apparatus.
Interferon pretreatment.
One day prior to influenza virus infection, mice were anesthetized and treated with 50,000 IU of recombinant human alpha interferon B/D (type I interferon [IFN-α], provided by Peter Stäheli, University of Freiburg) in 20 μl of sterile phosphate-buffered saline by intranasal application. The control group received 20 μl of sterile PBS.
Statistical analysis.
Data and statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, California). Results were presented as means ± standard errors of the means (SEM) for body weight change and virus titers. Statistical significance between groups was determined using the Mann-Whitney U test for body weight and virus titers. The log rank test was used to determine significant differences between survival curves. For analysis of BAL fluid proteome data, the Kruskal-Wallis test was used.
RESULTS
D2-Mx1r/r mice are not resistant to lethal H1N1 influenza A virus infections.
The A2G wild-type allele of Mx1 was shown to confer high resistance against many influenza virus subtypes in mice with an A2G, C57BL/6J, or BALB/c genetic background. On the other hand, we found that DBA/2J mice lacking Mx1 were highly susceptible compared to C57BL/6J mice lacking Mx1. Therefore, we wanted to investigate if the wild-type Mx1 allele was also able to protect highly susceptible DBA/2J mice from lethal infection. For this, we generated a congenic DBA/2J(B6).A2G-Mx1r/r (D2-Mx1r/r) mouse strain by backcrossing DBA/2J mice for 10 generations with congenic C57BL/6J.A2G-Mx1r/r (B6-Mx1r/r) mice (received from Peter Stäheli, Freiburg, Germany) that carried the A2G Mx1 wild-type allele. By SNP genotyping (data not shown), we confirmed that the congenic D2-Mx1r/r strain carried a 32.73-Mb region from the B6-Mx1r/r strain on chromosome 16 which includes 1.5 Mb of the original A2G region. Furthermore, the presence of the wild-type allele was confirmed by diagnostic PCR (data not shown). Also, congenic D2-Mx1r/r mice expressed the Mx1 wild-type allele after infection with H1N1 (PR8) by RT-PCR (data not shown).
We then infected D2-Mx1r/r and B6-Mx1r/r mice as well as D2-Mx1−/− and B6-Mx1−/− mice with A/PuertoRico/8/34 (H1N1) virus (59, 60). As described before, B6-Mx1−/− and D2-Mx1−/− mice were highly susceptible to these infections. They rapidly lost body weight and died between days 4 and 8 p.i. (Fig. 1). On the other hand, B6-Mx1r/r mice exhibited only slight body weight losses and survived the infection, confirming previous observations (Fig. 1). Most surprisingly, infected D2-Mx1r/r mice were not protected from lethal infections. They showed severe clinical symptoms and lost body weight similarly to Mx1-deficient DBA/2J mice, and all infected D2-Mx1r/r mice were dead at day 9 p.i. (Fig. 1). Furthermore, D2-Mx1r/r mice produced high levels of chemokines and cytokines in their lungs (Fig. 2), indicating strong inflammatory responses which are associated with high levels of virus replication and a severe course of infection.
FIG 1.
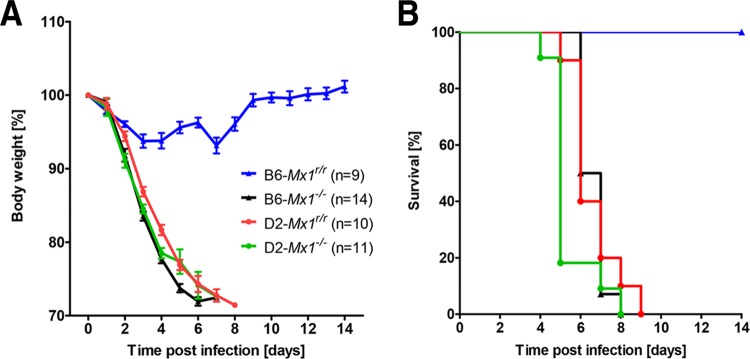
D2-Mx1r/r mice were highly susceptible to H1N1 virus (PR8) infections whereas B6-Mx1r/r mice were resistant. Eight- to 12-week-old female mice (D2-Mx1r/r, B6-Mx1r/r, D2-Mx1−/−, and B6-Mx1−/−) were infected intranasally with 2 × 103 FFU of PR8 (H1N1) influenza A virus. Body weight loss (A) and survival rates (B) were monitored over a period of 14 days. Mice that lost 30% or more of the starting body weight were sacrificed and recorded as dead. Data represent mean percentages of body weight change (± SEM) compared to starting body weight (100%). Differences in body weight loss were significant between D2-Mx1r/r and B6-Mx1r/r mice after day 3 p.i. (P < 0.0001, nonparametric Mann-Whitney test). Survival rates were significantly different between D2-Mx1r/r and B6-Mx1r/r mice (P < 0.0001, log rank Mantel-Cox test). n, number of mice per group.
FIG 2.

Chemokine and cytokine levels in BAL fluid of D2-Mx1r/r mice exhibit stronger inflammatory responses than do those of B6-Mx1−/− mice. Female D2-Mx1r/r (circles) and B6-Mx1r/r (squares) mice were infected with 2 × 103 FFU of PR8F intranasally. Bronchoalveolar lavage (BAL) fluid was collected from mock-infected control mice at day 3 posttreatment and from infected mice at day 3 and day 5 p.i. The concentration of chemokines and cytokines was determined by using the mouse cytokine/chemokine magnetic bead panel (Mcytomag-70K) from Millipore. At each time point, five biological replicates were analyzed.
To confirm that congenic D2-Mx1r/r mice carried a functional Mx1 allele, we outcrossed them to B6-Mx1−/− mice and compared the phenotype of the resulting F1 mice with the phenotype of F1 mice derived from an outcross of B6-Mx1r/r to D2-Mx1−/−. Thus, in the first case, the Mx1 wild-type allele is inherited from the congenic D2-Mx1r/r mice, whereas in the second case, the wild-type allele is derived from the original B6-Mx1r/r congenic strain. After infection with PR8, F1 mice from both crosses showed increased survival compared to D2-Mx1−/− mice (Fig. 3; differences not significant). These observations further demonstrated that the A2G Mx1 allele in D2-Mx1r/r mice is fully functional. D2-Mx1r/r mice showed no difference in upregulation of Mx1 and Ifnb1 genes after infection compared to B6-Mx1r/r mice, demonstrating that the interferon pathway and activation of downstream genes are not affected in D2-Mx1r/r mice (Fig. 4).
FIG 3.
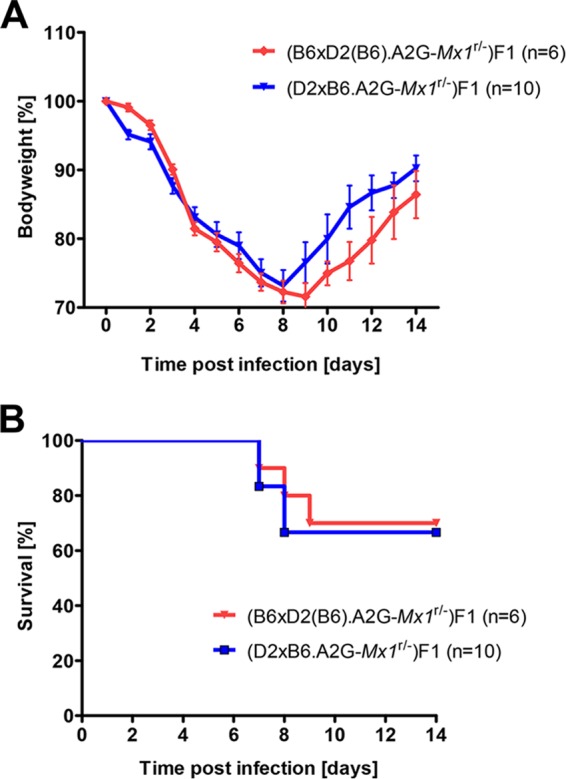
Confirmation of functional wild-type Mx1 in D2-Mx1r/r mice by outcrossing to B6-Mx1−/− mice. Congenic D2-Mx1r/r mice were outcrossed to B6-Mx1−/− mice, and the phenotypes of the resulting F1 mice [(B6 × D2(B6).A2G-Mx1r/−)F1 or reciprocal crosses] were compared to the phenotype of F1 mice derived from an outcross of B6-Mx1r/r to D2-Mx1−/− [(D2 × B6.A2G-Mx1r/−)F1 or reciprocal cross]. F1 mice from both crosses did not show significant differences in body weight loss (A) or survival (B) (Mann-Whitney U test for body weight change analysis and log rank Mantel-Cox test for survival curves). n, number of mice per group.
FIG 4.
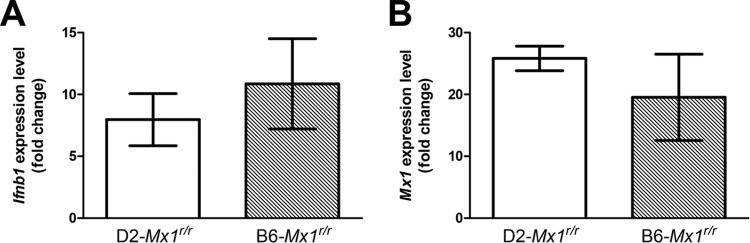
Upregulation of Ifnb1 and Mx1 in congenic D2-Mx1r/r mice after influenza A virus infection. Eight- to 12-week-old female D2-Mx1r/r and B6-Mx1r/r mice were inoculated with 2 × 103 FFU of PR8 H1N1 virus or PBS intranasally. On day 3 postinoculation, lung homogenates were prepared and levels of Ifnb1 (A) and Mx1 (B) mRNA expression were measured by quantitative RT-PCR and compared to PBS-treated controls. Infection with PR8 H1N1 virus induced comparable fold changes of Ifnb1 and Mx1 expression levels in both D2-Mx1r/r and B6-Mx1r/r mice (P = 0.5303 for Ifnb1 and P = 0.4346 for Mx1, two-tailed Student's t test, n = 3).
The protective effect of Mx1 on survival and virus replication is influenced by copy number and genetic background.
We then compared systematically the effect of Mx1r/r copy number and combinations of DBA/2J and C57BL/6J background on survival after PR8 infections (Table 1; Table 2 shows pairwise significance comparisons). The presence of only one instead of two copies of Mx1 increased mortality rates in C57BL/6J mice to 21.5%, and mice died between days 10 and 12 (B6-Mx1r/r versus B6-Mx1r/−, Table 1 and Fig. 5; differences not significant). The increase in mortality was also observed for mice with a hybrid B6 × D2 genetic background [F1(B6 × D2)-Mx1r/r versus F1(B6 × D2)-Mx1r/−, Table 1 and Fig. 5; differences not significant). Mice that were homozygous for the mutant Mx1 allele were most susceptible and succumbed to the infection, independent of their genetic background (D2-Mx1−/− and B6-Mx1−/−; Table 1 and Fig. 1, differences significant). Furthermore, the hybrid B6 × D2 genetic background decreased survival in the presence of either one or two wild-type Mx1 alleles compared to a pure C57BL/6J background [F1(B6 × D2)-Mx1r/− versus B6-Mx1r/−, differences not significant, and F1(B6 × D2)-Mx1r/r versus B6-Mx1r/r, differences significant; Table 1 and Fig. 1 and 5]. Mice with a pure DBA/2J background did not survive infection in the presence or absence of Mx1 (D2-Mx1r/r and D2-Mx1−/−, Table 1 and Fig. 1 and 5, differences not significant).
TABLE 1.
Survival after H1N1 virus infections is influenced by wild-type Mx1 copy number and genetic background
| Mouse strain | Genetic background | Mx1 allele | No. of killed mice/no. of infected mice | Survival proportion (%) |
|---|---|---|---|---|
| B6-Mx1r/r | B6 | r/r | 0/9 | 100 |
| B6-Mx1r/− | B6 | r/− | 3/14 | 78.57 |
| B6-Mx1−/− | B6 | −/− | 14/14 | 0 |
| D2-Mx1r/r | D2 | r/r | 10/10 | 0 |
| D2-Mx1r/− | D2 | r/− | 7/7 | 0 |
| D2-Mx1−/− | D2 | −/− | 11/11 | 0 |
| F1(B6 × D2)-Mx1r/r | B6 × D2 | r/r | 1/14 | 92.83 |
| F1(B6 × D2)-Mx1r/− | B6 × D2 | r/− | 5/16 | 68.75 |
| B6 × F1(B6 × D2)-Mx1r/− | B6 × F1(B6 × D2)a | r/− | 2/11 | 81.82 |
| D2 × F1(B6 × D2)-Mx1r/− | D2 × F1(B6 × D2)a | r/− | 5/8 | 37.50 |
Secondary outcross performed by outcrossing F1(B6 × D2)-Mx1r/r with B6-Mx1−/− or D2-Mx1−/−.
TABLE 2.
Pairwise comparison of survival rates (log rank test)
| Mouse strain | Significance for mouse strain: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| B6-Mx1r/r | B6-Mx1r/− | B6-Mx1−/− | D2-Mx1r/r | D2-Mx1r/− | D2-Mx1−/− | F1(B6 × D2)-Mx1r/r | F1(B6 × D2)-Mx1r/− | B6 × F1(B6 × D2)-Mx1r/− | |
| B6-Mx1r/r | |||||||||
| B6-Mx1r/− | 0.1476 | ||||||||
| B6-Mx1−/− | <0.0001 | <0.0001 | |||||||
| D2-Mx1r/r | <0.0001 | <0.0001 | 0.8207 | ||||||
| D2-Mx1r/− | <0.0001 | <0.0001 | 0.0043 | 0.0288 | |||||
| D2-Mx1−/− | <0.0001 | <0.0001 | 0.0135 | 0.0357 | 0.9404 | ||||
| F1(B6 × D2)-Mx1r/r | 0.4227 | 0.3207 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |||
| F1(B6 × D2)-Mx1r/− | 0.0706 | 0.4231 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.1147 | ||
| B6 × F1(B6 × D2)-Mx1r/− | 0.1900 | 0.9504 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.4187 | 0.4735 | |
| D2 × F1(B6 × D2)-Mx1r/− | 0.0056 | 0.0162 | 0.0003 | 0.0040 | 0.0002 | 0.0003 | 0.0060 | 0.1695 | 0.0645 |
FIG 5.
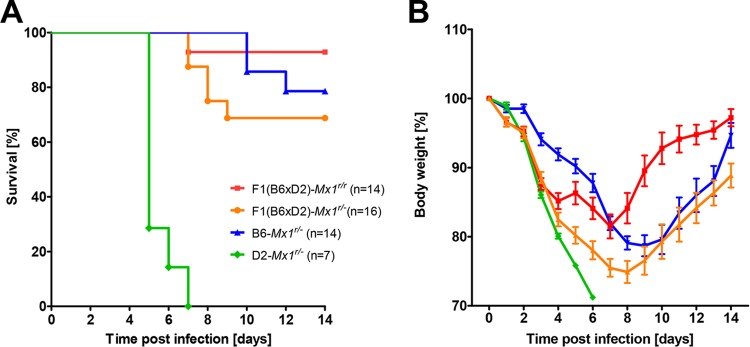
Resistance to lethal H1N1 virus infections is controlled by Mx1 copy number and genetic background. F1 mice of different Mx1 allele combinations and different C57BL/6J and DBA/2J background combinations were tested for susceptibility to PR8 H1N1 virus. Groups of 8- to 12-week-old female mice [F1(B6 × D2)-Mx1r/r, F1(B6 × D2)-Mx1r/−, B6-Mx1r/−, and D2-Mx1r/−] were infected intranasally with 2 × 103 FFU of PR8 virus, and survival was monitored until day 14 p.i. Mice that lost 30% or more of the starting body weight were sacrificed and recorded as dead. For the F1(B6 × D2)-Mx1r/− group, data from reciprocal crosses [(B6 × D2(B6).A2G-Mx1r/−)F1 (n = 6) and (D2 × B6.A2G-Mx1r/−)F1 (n = 10)] were combined. Two copies of the wild-type Mx1 locus increased resistance compared to that with one copy. Introduction of the DBA/2J background in hybrid F1(B6 × D2)-Mx1r/r mice increased susceptibility, and the pure DBA/2J background in Mx1r/r mice increased susceptibility further.
Next, we compared virus replication in the lungs of D2-Mx1r/r mice, B6-Mx1r/r mice, F1 mice expressing one copy of the wild-type Mx1 allele, and D2-Mx1−/− and B6-Mx1−/− mice carrying a mutant allele. After infection with PR8 virus, DBA/2J mice (with or without a functional Mx1 allele) exhibited very high levels of viral load in infected lungs at day 1 p.i. (Fig. 6B and D), whereas infected B6-Mx1r/r mice rapidly reduced viral titers in lungs at day 3 p.i. (Fig. 6A). Most interestingly, F1(B6 × D2)-Mx1r/− mice initially exhibited high viral loads in infected lungs but reduced viral titers in their lungs at day 3 p.i., which further decreased until day 5 p.i. (Fig. 6E). Thus, the Mx1 restrictive function on viral replication requires a hybrid or pure C57BL/6J background.
FIG 6.

Restriction of virus replication is determined by the presence of a functional Mx1 allele and genetic background. Eight- to 12-week-old female B6-Mx1r/r (A), D2-Mx1r/r (B), B6-Mx1−/− (C), D2-Mx1−/− (D), and F1(B6 × D2)-Mx1r/− (E) mice were infected intranasally with 2 × 103 FFU of PR8 virus. Infectious virus particles in lung homogenates were determined by focus-forming assay at days 1, 3, and 5 p.i. Viral loads on day 1 p.i. were significantly different between infected mice that carried a DBA/2J genetic background and those that carried a C57BL/6J genetic background (B6-Mx1r/r compared to D2-Mx1r/r using Mann-Whitney test, P = 0.0022), and only mice carrying both a functional Mx1 allele and a C57BL/6J genetic background reduced viral loads from day 1 to day 3 p.i. D2-Mx1r/r, B6-Mx1r/r, and B6-Mx1−/− mice, n = 6 per time point; D2-Mx1−/− mice, n = 5 per time point; F1(B6 × D2)-Mx1r/− mice, n = 3 per time point.
D2-Mx1r/r mice are partially resistant to low-dose infection with H1N1 and to infections with H3N2 influenza A virus.
In addition, we studied pathology in D2-Mx1r/r mice after infection with a low dose of H1N1 virus (10 FFU). In this case, D2-Mx1r/r showed significantly less body weight loss and almost all infected mice survived compared to D2-Mx1−/− infected mice that rapidly lost body weight and all died (Fig. 7A). Also, D2-Mx1r/r mice were partially protected against infections with the H3N2 virus subtype, which was evident by a shift in the survival curve in D2-Mx1r/r compared to D2-Mx1−/− mice (Fig. 7B). However, overall survival rates were much lower than those of B6-Mx1r/r mice, which all survived an infection with H3N2 (Fig. 7B). Of note, F1(B6 × D2)-Mx1r/− mice with a hybrid C57BL/6J genetic background were also fully protected against mortality from H3N2 infections (Fig. 7B).
FIG 7.

D2-Mx1r/r mice were protected against infections with low-dose H1N1 (PR8) virus and partially protected against infections with H3N2 virus. Eight- to 12-week-old female mice [B6-Mx1r/r, D2-Mx1r/r, F1(B6 × D2)-Mx1r/−, B6-Mx1−/−, and D2-Mx1−/−] were infected intranasally with 10 FFU of PR8 H1N1 virus (A) or with 2 × 103 FFU of H3N2 virus (B). Survival rates were monitored over a period of 14 days p.i. Mice that lost 30% or more of the starting body weight were sacrificed and recorded as dead. Almost all infected D2-Mx1r/r mice survived infection with low-dose PR8 virus. D2-Mx1r/r mice were partially protected against H3N2 infections compared to B6-Mx1−/− (log rank Mantel-Cox test, P = 0.0065). Also, all F1(B6 × D2)-Mx1r/− mice with a hybrid C57BL/6J genetic background survived the H3N2 infections.
D2-Mx1r/r mice are resistant to H1N1 influenza A virus after interferon pretreatment.
Mx1 is one of the main interferon response genes and can be induced by exogenous treatment with interferon (55). Therefore, we investigated the pathology in D2-Mx1r/r mice after pretreatment with IFN-α 1 day before infection. We first confirmed that IFN-α pretreatment induces Mx1 expression in both D2-Mx1r/r and B6-Mx1r/r mice (Fig. 8). Most remarkable, all pretreated D2-Mx1r/r mice survived an infection with PR8 virus, whereas all PBS mock-treated mice lost body weight and died (Fig. 9A). Furthermore, D2-Mx1r/r mice pretreated with IFN-α exhibited lower viral loads than did mock-treated animals at day 1 p.i. (Fig. 9B). In immunohistochemical staining, we did not observe viral antigen at the day 3 p.i. point in IFN-α-pretreated mice compared to a wide spread of virus in D2-Mx1r/r mice that were pretreated with PBS (data not shown). When D2-Mx1−/− or B6-Mx1−/− mice were pretreated with IFN-α prior to infection, all infected mice died and no difference from mock-treated mice was observed, demonstrating that survival depends on the presence of a functional Mx1 allele (data not shown).
FIG 8.
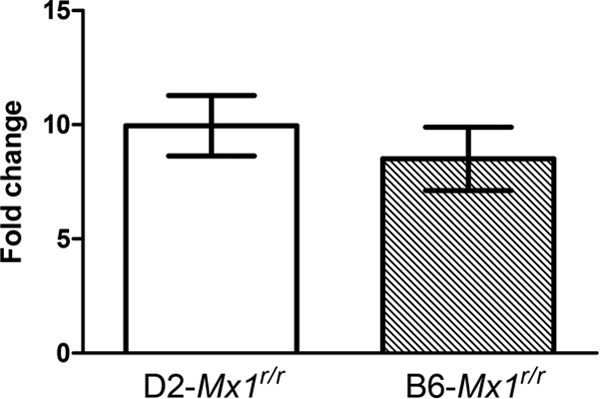
Upregulation of Mx1 in congenic D2-Mx1r/r mouse strains after IFN-α treatment. D2-Mx1r/r and B6-Mx1r/r mice were treated with 50,000 IU of recombinant IFN-α intranasally. Levels of Mx1 mRNA expression were measured by quantitative RT-PCR and compared to those in PBS-treated controls. Treatment with IFN-α induces comparable fold increases of Mx1 expression in both D2-Mx1r/r and B6-Mx1r/r mice. No difference was detected between B6-Mx1r/r and D2-Mx1r/r mice (P = 0.4918, two-tailed Student's t test, n = 3).
FIG 9.

Alpha interferon pretreatment rescues D2-Mx1r/r mice. Eight- to 11-week-old female D2-Mx1r/r mice were pretreated with 1 μg recombinant human alpha interferon B/D (IFN-α) intranasally 1 day prior to infection, and PBS was given as a mock control. All mice were subsequently infected intranasally with 2 × 103 FFU of PR8 virus. (A) Survival rate was monitored for 14 days p.i. Mice that lost 30% or more of the starting body weight were sacrificed and recorded as dead. IFN-α-pretreated mice showed higher survival rates than did PBS-treated controls (log rank survival, P = 0.0019, n = 5 mice per group). (B) Virus particles in lung homogenates from IFN-α-pretreated and PBS-treated mice were determined in a focus-forming assay on day 1 p.i. Virus titers were significantly different between IFN-α- and PBS-pretreated groups (**, P = 0.01, Mann-Whitney U test). n = 5 mice per group.
DISCUSSION
Most laboratory mouse strains, including C57BL/6J, are deficient in Mx1 and susceptible to H1N1 (mouse-adapted PR8) infections. However, the presence of a wild-type Mx1 allele makes B6-Mx1r/r resistant to H1N1 infections (55). Furthermore, many studies have shown that Mx1 is a strong genetic resistance factor controlling influenza virus replication and protecting the host from severe pathology and mortality (51–55). These studies combined suggested that the Mx1 allele from A2G mice is able to protect from lethal infections independently of the genetic background.
Most surprisingly, we found that congenic D2-Mx1r/r mice that carry the Mx1 wild-type allele still exhibited an equally highly susceptible phenotype as did D2-Mx1−/− mice after infection with H1N1 virus (PR8). All infected D2-Mx1r/r mice rapidly lost body weight and died. We confirmed in D2-Mx1r/r infected mice that the wild-type allele in D2-Mx1r/r was expressed after infection. Furthermore, (B6 × D2(B6).A2G-Mx1r/−)F1 mice which received the Mx1 allele from the congenic D2-Mx1r/r strain were as resistant to PR8 infections as F1 mice that were generated by crossing B6-Mx1r/r to D2-Mx1−/− mice. These experiments confirmed that D2-Mx1r/r mice carried a fully functional protective Mx1 allele.
In agreement with previously published data (55), we also observed that B6-Mx1r/r mice were resistant to mouse-adapted PR8 (H1N1) virus infections whereas Mx1-deficient B6-Mx1−/− mice succumbed to the infection. Previously, the 50% lethal dose (LD50) after PR8 infection had been determined for B6-Mx1r/r mice at 6.7 × 106 infectious particles (56), for B6-Mx1−/− mice at 20 to 32 infectious particles (38), and for D2-Mx1−/− at 2 to 3 infectious particles (38). In our studies, D2-Mx1r/r mice survived an infection with a low dose (10 FFU) of PR8 virus. However, they succumbed to an infection dose of 2 × 103 infectious particles. We decided to not further determine the exact LD50 for D2-Mx1r/r mice with respect to the 3Rs (reduction, refinement, and replacement) of animal ethics, because it would not add any more highly relevant information. Thus, in conclusion, the LD50 for B6-Mx1−/− mice is about 5 orders of magnitude lower than that for B6-Mx1r/r mice whereas the LD50 for D2-Mx1−/− mice is only about 1 to 2 orders of magnitude lower than that for D2-Mx1r/r mice. D2-Mx1r/r mice are 3 to 5 orders of magnitude more susceptible than B6-Mx1r/r mice.
Thus, our results show for the first time that the presence of the A2G Mx1 allele, which is able to rescue A2G and congenic C57BL/6J mice from lethal influenza A virus infections, does not exert its protective function in a DBA/2J genetic background. These observations suggest that additional genetic factors are required for the protective Mx1 functions or that the DBA/2J background is highly permissive to infections and that expression of Mx1 comes too late.
Recently, the wild-derived mouse strain CAST/EiJ was found to be highly susceptible to H1N1 virus infections, although these mice express a full-length Mx1 allele with only one amino acid difference from the ancestral PWK/PhJ allele (49). However, it is yet unclear if the high susceptibility in CAST/EiJ mice is caused by the genetic background or the polymorphism in the Mx1 allele.
Furthermore, we systematically tested different background combinations and Mx1 allele combinations to determine how resistance and susceptibility were influenced by wild-type Mx1 and combinations of C57BL/6J and DBA/2J backgrounds. We observed that changing the genetic background from a pure DBA/2J to a hybrid B6 × D2 and then to a pure C57BL/6J background incrementally increased survival. Thus, for protective nonlethal outcome of an H1N1 virus infection, at least one C57BL/6J genome had to be present. In the presence of a C57BL/6J genome, an increase from one to two copies of the wild-type Mx1 allele also increased survival. These observations suggest that the Mx1 gene and presumed resistance factors from C57BL/6J act in an additive fashion. Furthermore, we found that the presence of a DBA/2J background in pure DBA/2J or in hybrid B6 × D2 mice resulted in high viral loads in the lung at day 1 p.i., regardless of whether the wild-type Mx1 allele was present or not. However, in mice with a C57BL/6J or hybrid B6 × D2 background, the presence of the Mx1 wild-type allele always resulted in a reduction of virus lung titers on day 3 p.i. The absence of the wild-type Mx1 allele resulted in very high titers in a DBA/2J background, which were not reduced at day 3, and lower initial virus titers in mice with a C57BL/6J background, which increased until day 3 p.i.
These observations suggest that a pure DBA/2J background renders the host highly permissive to an early rapid viral replication whereas the presence of at least one copy of the wild-type Mx1 gene results in reduction of virus replication on day 3 p.i. The most likely explanation for the high susceptibility of D2-Mx1r/r mice is therefore that Mx1 is induced too late after infection with H1N1 virus (PR8) to exert its protective functions.
Mx1 is upregulated after infection in both DBA/2J and C57BL/6J mice with higher levels in DBA/2J mice (our unpublished data). Furthermore, it has been shown previously that pretreatment of B6-Mx1r/r mice with IFN-α can rescue mice from an otherwise lethal infection with a very highly virulent H1N1 virus (hvH1N1) (55). We therefore investigated whether IFN-α pretreatment may have a beneficial effect in D2-Mx1r/r mice. Indeed, pretreated D2-Mx1r/r mice survived lethal infections with PR8 virus. Thus, our results showed that wild-type Mx1 is able to protect DBA/2J mice when it is already present at the time of infection. These results further support the hypothesis that in nontreated DBA/2J mice viral replication during the first 2 days is very rapid and expression of Mx1 is too late to restrict the massive viral replication and prevent severe tissue damage and subsequent death.
Mx1 expression in D2-Mx1r/r mice was investigated only at the RNA level. We can thus not exclude the possibility that protein half-lives may be different in different genetic backgrounds. However, our experiments with IFN pretreatment suggest that this possibility is very unlikely, because in this case, D2-Mx1r/r mice would not be protected.
D2-Mx1r/r mice showed a lower mortality rate when infected with 2 × 103 FFU of H3N2 virus. In this case, we speculate that H3N2 virus may exhibit a lower replication rate very early after infection or that H3N2 virus does not suppress induction of Mx1 as efficiently as PR8 and that induction of Mx1 protein is early or strong enough to partially rescue infected D2-Mx1r/r mice.
Humans, as a species, carry genes that are functional orthologs of Mx1, named MX1 and MX2 (39). Thus, the differences in susceptibility and resistance to influenza A virus in humans that are attributed to genetic factors (24, 63) are most likely not caused by the presence or absence of MX1. Therefore, our observations which demonstrate that the genetic background may render an individual highly susceptible, even in the presence of a functional MX1 resistance gene, are also highly relevant for understanding genetic susceptibility to influenza virus infections in humans.
In summary, our results show that, in contrast to studies that were performed previously, the wild-type influenza virus resistance Mx1 gene (derived from A2G) does not necessarily result in high resistance to lethal influenza A virus infections. Rather, the protective effect of Mx1 depends strongly on the genetic background, the virulence of the virus, and the kinetics of Mx1 induction.
ACKNOWLEDGMENTS
We thank the animal caretakers at the Central Animal Facilities at the HZI for maintaining the mice for this study and Christin Fricke and Karin Lammert for excellent technical assistance. We thank Claudia Pommerenke for her support in analyzing the genotyping data for congenic D2-Mx1r/r mice. Original stocks of influenza A viruses were obtained from Peter Stäheli (University of Freiburg) and Otto Haller (University of Freiburg). Congenic B6-Mx1r/r mice, the protocol for genotyping Mx1 congenic mice, and recombinant human alpha interferon B/D were obtained from Peter Stäheli. We thank Blair Prochnow for critical reading of the manuscript.
This work was supported by intramural grants from the Helmholtz Association (Program Infection and Immunity) and a research grant, FluResearchNet (no. 01KI07137), from the German Ministry of Education and Research to K.S. D.-L.S. received partial financial support from a Studying Abroad Scholarship from the Ministry of Education, Taiwan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Fauci AS. 2006. Seasonal and pandemic influenza preparedness: science and countermeasures. J Infect Dis 194(Suppl 2):S73–S76. doi: 10.1086/507550. [DOI] [PubMed] [Google Scholar]
- 2.Klenk HD, Garten W, Matrosovich M. 2011. Molecular mechanisms of interspecies transmission and pathogenicity of influenza viruses: lessons from the 2009 pandemic. Bioessays 33:180–188. doi: 10.1002/bies.201000118. [DOI] [PubMed] [Google Scholar]
- 3.Kilbourne ED. 2006. Influenza pandemics of the 20th century. Emerg Infect Dis 12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell CJ, Webster RG. 2005. The genesis of a pandemic influenza virus. Cell 123:368–371. doi: 10.1016/j.cell.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Johnson NP, Mueller J. 2002. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull Hist Med 76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 6.Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki-Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman-Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maines TR, Jayaraman A, Belser JA, Wadford DA, Pappas C, Zeng H, Gustin KM, Pearce MB, Viswanathan K, Shriver ZH, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM. 2009. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraser C, Donnelly CA, Cauchemez S, Hanage WP, Van Kerkhove MD, Hollingsworth TD, Griffin J, Baggaley RF, Jenkins HE, Lyons EJ, Jombart T, Hinsley WR, Grassly NC, Balloux F, Ghani AC, Ferguson NM, Rambaut A, Pybus OG, Lopez-Gatell H, Alpuche-Aranda CM, Chapela IB, Zavala EP, Guevara DM, Checchi F, Garcia E, Hugonnet S, Roth C. 2009. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 324:1557–1561. doi: 10.1126/science.1176062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, Lopez-Gatell H, Olivera H, Lopez I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov AI, Cox NJ. 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munster VJ, de Wit E, van den Brand JM, Herfst S, Schrauwen EJ, Bestebroer TM, van de Vijver D, Boucher CA, Koopmans M, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Fouchier RA. 2009. Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science 325:481–483. doi: 10.1126/science.1177127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang TT, Palese P. 2009. Unraveling the mystery of swine influenza virus. Cell 137:983–985. doi: 10.1016/j.cell.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 13.Gambotto A, Barratt-Boyes SM, de Jong MD, Neumann G, Kawaoka Y. 2008. Human infection with highly pathogenic H5N1 influenza virus. Lancet 371:1464–1475. doi: 10.1016/S0140-6736(08)60627-3. [DOI] [PubMed] [Google Scholar]
- 14.Krug RM. 2006. Virology. Clues to the virulence of H5N1 viruses in humans. Science 311:1562–1563. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe T, Kiso M, Fukuyama S, Nakajima N, Imai M, Yamada S, Murakami S, Yamayoshi S, Iwatsuki-Horimoto K, Sakoda Y, Takashita E, McBride R, Noda T, Hatta M, Imai H, Zhao D, Kishida N, Shirakura M, de Vries RP, Shichinohe S, Okamatsu M, Tamura T, Tomita Y, Fujimoto N, Goto K, Katsura H, Kawakami E, Ishikawa I, Watanabe S, Ito M, Sakai-Tagawa Y, Sugita Y, Uraki R, Yamaji R, Eisfeld AJ, Zhong G, Fan S, Ping J, Maher EA, Hanson A, Uchida Y, Saito T, Ozawa M, Neumann G, Kida H, Odagiri T, Paulson JC, Hasegawa H, Tashiro M, Kawaoka Y. 2013. Characterization of H7N9 influenza A viruses isolated from humans. Nature 501:551–555. doi: 10.1038/nature12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morens DM, Taubenberger JK, Fauci AS. 2013. H7N9 avian influenza A virus and the perpetual challenge of potential human pandemicity. mBio 4(4):e00445-13. doi: 10.1128/mBio.00445-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. 2013. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H, Wang D, Kelvin DJ, Li L, Zheng Z, Yoon SW, Wong SS, Farooqui A, Wang J, Banner D, Chen R, Zheng R, Zhou J, Zhang Y, Hong W, Dong W, Cai Q, Roehrl MH, Huang SS, Kelvin AA, Yao T, Zhou B, Chen X, Leung GM, Poon LL, Webster RG, Webby RJ, Peiris JS, Guan Y, Shu Y. 2013. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science 341:183–186. doi: 10.1126/science.1239844. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. 2013. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 341:410–414. doi: 10.1126/science.1240532. [DOI] [PubMed] [Google Scholar]
- 20.Scriven J, McEwen R, Mistry S, Green C, Osman H, Bailey M, Ellis C. 2009. Swine flu: a Birmingham experience. Clin Med 9:534–538. doi: 10.7861/clinmedicine.9-6-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yates L, Pierce M, Stephens S, Mill A, Spark P, Kurinczuk J, Valappil M, Brocklehurst P, Thomas S, Knight M. 2010. Influenza A/H1N1v in pregnancy: an investigation of the characteristics and management of affected women and the relationship to pregnancy outcomes for mother and infant. Health Technol Assess 14:109–182. doi: 10.3310/hta14340-02. [DOI] [PubMed] [Google Scholar]
- 22.Albright FS, Orlando P, Pavia AT, Jackson GG, Cannon Albright LA. 2008. Evidence for a heritable predisposition to death due to influenza. J Infect Dis 197:18–24. doi: 10.1086/524064. [DOI] [PubMed] [Google Scholar]
- 23.Gottfredsson M, Halldorsson BV, Jonsson S, Kristjansson M, Kristjansson K, Kristinsson KG, Love A, Blondal T, Viboud C, Thorvaldsson S, Helgason A, Gulcher JR, Stefansson K, Jonsdottir I. 2008. Lessons from the past: familial aggregation analysis of fatal pandemic influenza (Spanish flu) in Iceland in 1918. Proc Natl Acad Sci U S A 105:1303–1308. doi: 10.1073/pnas.0707659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horby P, Sudoyo H, Viprakasit V, Fox A, Thai PQ, Yu H, Davila S, Hibberd M, Dunstan SJ, Monteerarat Y, Farrar JJ, Marzuki S, Hien NT. 2010. What is the evidence of a role for host genetics in susceptibility to influenza A/H5N1? Epidemiol Infect 138:1550–1558. doi: 10.1017/S0950268810000518. [DOI] [PubMed] [Google Scholar]
- 25.Mancuso P. 2013. Obesity and respiratory infections: does excess adiposity weigh down host defense? Pulm Pharmacol Ther 26:412–419. doi: 10.1016/j.pupt.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlsson EA, Marcelin G, Webby RJ, Schultz-Cherry S. 2012. Review on the impact of pregnancy and obesity on influenza virus infection. Influenza Other Respir Viruses 6:449–460. doi: 10.1111/j.1750-2659.2012.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Brien KB, Vogel P, Duan S, Govorkova EA, Webby RJ, McCullers JA, Schultz-Cherry S. 2012. Impaired wound healing predisposes obese mice to severe influenza virus infection. J Infect Dis 205:252–261. doi: 10.1093/infdis/jir729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, Wise HM, Kane L, Goulding D, Digard P, Anttila V, Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, Everingham K, Dawson H, Hope D, Ramsay P, Walsh TS, Campbell A, Kerr S, Harrison D, Rowan K, Addison J, Donald N, Galt S, Noble D, Taylor J, Webster N, Taylor I, Aldridge J, Dornan R, Richard C, Gilmour D, Simmons R, White R, Jardine C, Williams D, Booth M, Quasim T, Watson V, Henry P, Munro F, Bell L, Ruddy J, Cole S, Southward J, Allcoat P, Gray S, McDougall M, Matheson J, Whiteside J, Alcorn D, Rooney K, Sundaram R, Imrie G, Bruce J, McGuigan K, Moultrie S, Cairns C, Grant J, Hughes M, Murdoch C, Davidson A, Harris G, Paterson R, Wallis C, Binning S, Pollock M, Antonelli J, Duncan A, Gibson J, McCulloch C, Murphy L, Haley C, Faulkner G, Freeman T, Baillie JK, Chaussabel D, Adamson WE, Carman WF, Thompson C, Zambon MC, Aylin P, Ashby D, Barclay WS, Brett SJ, Cookson WO, Drumright LN, Elderfield RA, Garcia-Alvarez L, Gazzard BG, Griffiths MJ, Habibi MS, Hansel TT, Herberg JA, Holmes AH, Hussell T, Johnston SL, Kon OM, Levin M, Moffatt MF, Nadel S, Openshaw PJ, Warner JO, Aston SJ, Hay A, McCauley J, O'Garra A, Banchereau J, Hayward A, Kellam P, Simmonds P, McNamara PS, Semple MG, Smyth RL, Nguyen-Van-Tam JS, Ho LP, McMichael AJ, Dougan G, Brass AL. 2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–523. doi: 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilk E, Schughart K. 2012. The mouse as model system to study host-pathogen interactions in influenza A infections. Curr Protoc Mouse Biol 2:177–205. doi: 10.1002/9780470942390.mo110173. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava B, Blazejewska P, Hessmann M, Bruder D, Geffers R, Mauel S, Gruber AD, Schughart K. 2009. Host genetic background strongly influences the response to influenza a virus infections. PLoS One 4:e4857. doi: 10.1371/journal.pone.0004857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trammell RA, Toth LA. 2008. Genetic susceptibility and resistance to influenza infection and disease in humans and mice. Expert Rev Mol Diagn 8:515–529. doi: 10.1586/14737159.8.4.515. [DOI] [PubMed] [Google Scholar]
- 32.Ding M, Lu L, Toth LA. 2008. Gene expression in lung and basal forebrain during influenza infection in mice. Genes Brain Behav 7:173–183. doi: 10.1111/j.1601-183X.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 33.Boon AC, deBeauchamp J, Hollmann A, Luke J, Kotb M, Rowe S, Finkelstein D, Neale G, Lu L, Williams RW, Webby RJ. 2009. Host genetic variation affects resistance to infection with a highly pathogenic H5N1 influenza A virus in mice. J Virol 83:10417–10426. doi: 10.1128/JVI.00514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boon AC, Debeauchamp J, Krauss S, Rubrum A, Webb AD, Webster RG, McElhaney J, Webby RJ. 2010. Cross-reactive neutralizing antibodies directed against pandemic H1N1 2009 virus are protective in a highly sensitive DBA/2 influenza mouse model. J Virol 84:7662–7667. doi: 10.1128/JVI.02444-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otte A, Sauter M, Alleva L, Baumgarte S, Klingel K, Gabriel G. 2011. Differential host determinants contribute to the pathogenesis of 2009 pandemic H1N1 and human H5N1 influenza A viruses in experimental mouse models. Am J Pathol 179:230–239. doi: 10.1016/j.ajpath.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boon AC, Finkelstein D, Zheng M, Liao G, Allard J, Klumpp K, Webster R, Peltz G, Webby RJ. 2011. H5N1 influenza virus pathogenesis in genetically diverse mice is mediated at the level of viral load. mBio 2(5):e00171-11. doi: 10.1128/mBio.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trammell RA, Liberati TA, Toth LA. 2012. Host genetic background and the innate inflammatory response of lung to influenza virus. Microbes Infect 14:50–58. doi: 10.1016/j.micinf.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Pica N, Iyer A, Ramos I, Bouvier NM, Fernandez-Sesma A, Garcia-Sastre A, Lowen AC, Palese P, Steel J. 2011. The DBA.2 mouse is susceptible to disease following infection with a broad, but limited, range of influenza A and B viruses. J Virol 85:12825–12829. doi: 10.1128/JVI.05930-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haller O, Staeheli P, Kochs G. 2007. Interferon-induced Mx proteins in antiviral host defense. Biochimie 89:812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Haller O, Staeheli P, Kochs G. 2009. Protective role of interferon-induced Mx GTPases against influenza viruses. Rev Sci Tech 28:219–231. [DOI] [PubMed] [Google Scholar]
- 41.Haller O. 1981. Inborn resistance of mice to orthomyxoviruses. Curr Top Microbiol Immunol 92:25–52. [DOI] [PubMed] [Google Scholar]
- 42.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol 81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, Kochs G, Daumke O. 2011. Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35:514–525. doi: 10.1016/j.immuni.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 44.Patzina C, Haller O, Kochs G. 2014. Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J Biol Chem 289:6020–6027. doi: 10.1074/jbc.M113.543892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wisskirchen C, Ludersdorfer TH, Muller DA, Moritz E, Pavlovic J. 2011. The cellular RNA helicase UAP56 is required for prevention of double-stranded RNA formation during influenza A virus infection. J Virol 85:8646–8655. doi: 10.1128/JVI.02559-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wisskirchen C, Ludersdorfer TH, Muller DA, Moritz E, Pavlovic J. 2011. Interferon-induced antiviral protein MxA interacts with the cellular RNA helicases UAP56 and URH49. J Biol Chem 286:34743–34751. doi: 10.1074/jbc.M111.251843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindenmann J, Lance CA, Hobson D. 1963. The resistance of A2G mice to myxoviruses. J Immunol 90:942. [PubMed] [Google Scholar]
- 48.Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O. 1988. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol 8:4518–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, Bradel-Tretheway B, Bryan JT, Buus RJ, Gralinski LE, Haagmans BL, McMillan L, Miller DR, Rosenzweig E, Valdar W, Wang J, Churchill GA, Threadgill DW, McWeeney SK, Katze MG, Pardo-Manuel de Villena F, Baric RS, Heise MT. 2013. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog 9:e1003196. doi: 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin HK, Yamashita T, Ochiai K, Haller O, Watanabe T. 1998. Characterization and expression of the Mx1 gene in wild mouse species. Biochem Genet 36:311–322. doi: 10.1023/A:1018741312058. [DOI] [PubMed] [Google Scholar]
- 51.Cilloniz C, Pantin-Jackwood MJ, Ni C, Carter VS, Korth MJ, Swayne DE, Tumpey TM, Katze MG. 2012. Molecular signatures associated with Mx1-mediated resistance to highly pathogenic influenza virus infection: mechanisms of survival. J Virol 86:2437–2446. doi: 10.1128/JVI.06156-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hodgson NR, Bohnet SG, Majde JA, Krueger JM. 2012. Influenza virus pathophysiology and brain invasion in mice with functional and dysfunctional Mx1 genes. Brain Behav Immun 26:83–89. doi: 10.1016/j.bbi.2011.07.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moritoh K, Yamauchi H, Asano A, Yoshii K, Kariwa H, Takashima I, Isoda N, Sakoda Y, Kida H, Sasaki N, Agui T. 2009. Generation of congenic mouse strains by introducing the virus-resistant genes, Mx1 and Oas1b, of feral mouse-derived inbred strain MSM/Ms into the common strain C57BL/6J. Jpn J Vet Res 57:89–99. [PubMed] [Google Scholar]
- 54.Tumpey TM, Szretter KJ, Van Hoeven N, Katz JM, Kochs G, Haller O, Garcia-Sastre A, Staeheli P. 2007. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol 81:10818–10821. doi: 10.1128/JVI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grimm D, Staeheli P, Hufbauer M, Koerner I, Martínez-Sobrido L, Solorzano A, García-Sastre A, Haller O, Kochs G. 2007. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc Natl Acad Sci U S A 104:6806–6811. doi: 10.1073/pnas.0701849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P. 2007. Protective role of beta interferon in host defense against influenza A virus. J Virol 81:2025–2030. doi: 10.1128/JVI.01718-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanlaere I, Vanderrijst A, Guenet JL, De Filette M, Libert C. 2008. Mx1 causes resistance against influenza A viruses in the Mus spretus-derived inbred mouse strain SPRET/Ei. Cytokine 42:62–70. doi: 10.1016/j.cyto.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 58.Collaborative Cross Consortium. 2012. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 190:389–401. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blazejewska P, Koscinski L, Viegas N, Anhlan D, Ludwig S, Schughart K. 2011. Pathogenicity of different PR8 influenza A virus variants in mice is determined by both viral and host factors. Virology 412:36–45. doi: 10.1016/j.virol.2010.12.047. [DOI] [PubMed] [Google Scholar]
- 60.Liedmann S, Hrincius ER, Anhlan D, McCullers JA, Ludwig S, Ehrhardt C. 2014. New virulence determinants contribute to the enhanced immune response and reduced virulence of an influenza A virus A/PR8/34 variant. J Infect Dis 209:532–541. doi: 10.1093/infdis/jit463. [DOI] [PubMed] [Google Scholar]
- 61.Takaki H, Takeda M, Tahara M, Shingai M, Oshiumi H, Matsumoto M, Seya T. 2013. The MyD88 pathway in plasmacytoid and CD4+ dendritic cells primarily triggers type I IFN production against measles virus in a mouse infection model. J Immunol 191:4740–4747. doi: 10.4049/jimmunol.1301744. [DOI] [PubMed] [Google Scholar]
- 62.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 63.Horby P, Nguyen NY, Dunstan SJ, Baillie JK. 2012. The role of host genetics in susceptibility to influenza: a systematic review. PLoS One 7:e33180. doi: 10.1371/journal.pone.0033180. [DOI] [PMC free article] [PubMed] [Google Scholar]