

Abstract
Objective:
De novo SCN2A mutations have recently been associated with severe infantile-onset epilepsies. Herein, we define the phenotypic spectrum of SCN2A encephalopathy.
Methods:
Twelve patients with an SCN2A epileptic encephalopathy underwent electroclinical phenotyping.
Results:
Patients were aged 0.7 to 22 years; 3 were deceased. Seizures commenced on day 1–4 in 8, week 2–6 in 2, and after 1 year in 2. Characteristic features included clusters of brief focal seizures with multiple hourly (9 patients), multiple daily (2), or multiple weekly (1) seizures, peaking at maximal frequency within 3 months of onset. Multifocal interictal epileptiform discharges were seen in all. Three of 12 patients had infantile spasms. The epileptic syndrome at presentation was epilepsy of infancy with migrating focal seizures (EIMFS) in 7 and Ohtahara syndrome in 2. Nine patients had improved seizure control with sodium channel blockers including supratherapeutic or high therapeutic phenytoin levels in 5. Eight had severe to profound developmental impairment. Other features included movement disorders (10), axial hypotonia (11) with intermittent or persistent appendicular spasticity, early handedness, and severe gastrointestinal symptoms. Mutations arose de novo in 11 patients; paternal DNA was unavailable in one.
Conclusions:
Review of our 12 and 34 other reported cases of SCN2A encephalopathy suggests 3 phenotypes: neonatal-infantile–onset groups with severe and intermediate outcomes, and a childhood-onset group. Here, we show that SCN2A is the second most common cause of EIMFS and, importantly, does not always have a poor developmental outcome. Sodium channel blockers, particularly phenytoin, may improve seizure control.
SCN2A encodes the major α subunit (Nav1.2) of voltage-gated sodium channels in excitatory neurons.1 We first identified inherited mutations in SCN2A in the self-limited autosomal dominant epilepsy syndrome, benign familial neonatal infantile seizures (BFNIS).2,3
De novo SCN2A mutations have increasingly been recognized to cause severe disorders, reported in 39 patients with epileptic encephalopathies (EEs)4–20 and 5 patients with intellectual disability without epilepsy.21–23 Cohorts with prototypic EEs, Ohtahara and West syndrome, due to SCN2A mutations are described. To date, only single cases of other syndromes are reported, including epilepsy of infancy with migrating focal seizures (EIMFS), which begins before 6 months of age and is characterized by multiple types of focal seizures that “migrate” from one hemisphere to another, usually with severe developmental impairment.
We sought to delineate the phenotypic spectrum of SCN2A encephalopathy, and show that SCN2A is an important cause of EIMFS.
METHODS
Eleven patients were identified from 580 patients with heterogeneous EEs, disorders characterized by refractory seizures, abundant epileptiform activity, and developmental arrest or regression.
We used 2 approaches to identify mutations: targeted sequencing in 568 patients with molecular inversion probes (MIPs),9 and whole-exome sequencing (WES) in 20 patients (8 also had MIPs). WES was performed using the Illumina (San Diego, CA) Genome Analyzer IIx massively parallel sequencing system; alignment and variant calling were performed using standard methods.24 If nonsynonymous, frameshift, and splice-site variants were absent in control populations, segregation analysis was performed. Variants were considered pathogenic if they arose de novo, were inherited from a mosaic parent, or segregated with the disorder. A mosaic mutation was confirmed by TOPO TA cloning using standard methods (Invitrogen, Carlsbad, CA). No patients with pathogenic SCN2A mutations in this cohort were excluded from this study and no patient had more than one pathogenic variant. WES looked for potentially pathogenic mutations in all annotated genes including movement disorder genes. Nonepilepsy genes were not included in the MIPs gene panel. A twelfth patient had an SCN2A mutation identified on clinical epilepsy gene panel testing.
Clinical records were reviewed for epilepsy and developmental history and other features. Pediatric epileptologist K.B.H. reviewed 74 EEGs in 11 patients (1–17 EEGs per patient, median 6), recorded between 3 days and 17 years, for background activity, interictal epileptiform discharges, ictal rhythms, and video of seizures and nonepileptic episodes. EIMFS was defined as onset before 6 months of multiple types of focal seizures, with clinical or EEG evidence of “migration” of the seizure from one hemisphere to the other. Ohtahara syndrome was diagnosed in the presence of neonatal-onset tonic seizures and a burst-suppression EEG. West syndrome was defined as infantile spasms with hypsarrhythmia on EEG. Pediatric neuroradiologist S.M. reviewed 16 MRI brain scans from 10 patients, at ages 3 days to 17 years. Patient 5 had 4 and patient 10 had 2 MRI brain scans for which only reports were available.
Standard protocol approvals, registrations, and patient consents.
The ethics committees of the Austin and Royal Children's Hospitals in Melbourne approved this study.
RESULTS
Of our 12 patients aged 1 to 22 years (median 4 years, 5 male), 3 were deceased (table). No patient had a similarly affected family member, but 5 had a first- or second-degree relative with febrile seizures or mild epilepsy. Eleven were born at term, and one at 36 + 5 weeks. None had hypoxic-ischemic encephalopathy.
Table.
Clinical features of our patients with SCN2A mutations

SCN2A mutations.
Missense mutations were identified in 11 patients and a frameshift mutation in one (figure 1). Missense mutations disrupted highly conserved nucleotides and all were predicted to be probably damaging. Five mutations (patients 2, 3, 4, 9, 11) have been reported with limited clinical information.9 Seven patients have novel mutations. A recurrent mutation (p.Arg1882Gln) occurred in patients 3 and 10.
Figure 1. Location of mutations in SCN2A.
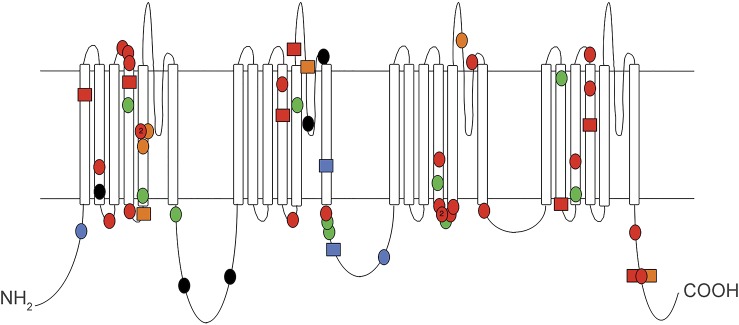
Mutations are denoted as follows: green = benign familial neonatal infantile seizures; orange = intermediate neonatal-infantile phenotype; red = severe neonatal-infantile phenotype; blue = childhood phenotype; black = intellectual disability without epilepsy; squares = cases in this study; circles = other published cases; 2 (in circles) = mutations at the same residue to 2 different amino acids.
Eleven patients had de novo mutations. In the twelfth, the variant was not maternally inherited; paternal DNA was unavailable.
Patient 5 was mosaic with 7 of 41 colonies (17%) containing the mutant allele only, correlating with 34% of cells containing a heterozygous SCN2A mutation.
Epilepsy.
Seizures commenced at age 1 to 4 days in 8 cases, and 10 days, 6 weeks, 14 months, and 17 years in the remaining 4 (median 2 days). Abnormal antenatal movements were present in 4, absent in 4, and unknown in 4.
Nine had an identifiable epilepsy syndrome: EIMFS in 7 and Ohtahara syndrome in 2. One patient with seizure onset at 1 day and the older patients with seizure onset at 14 months and 17 years did not fit a recognizable epilepsy syndrome. No patient presented with West syndrome.
Each patient had multiple types of focal seizures. Semiologies included asymmetric tonic seizures with versive features, hemiclonic, asymmetric bilateral clonic, and hypomotor with prominent autonomic features. Seizures typically lasted <2 minutes and clustered in 11 patients. Rare prolonged seizures occurred in 5 patients. Maximal seizure frequency was multiple hourly in 9, multiple daily in 2, and multiple weekly in one and occurred within 3 months of seizure onset in 10. Epileptic spasms in infancy occurred in 3 patients. Video-EEG recordings corroborated the reported seizure semiologies. Myoclonus was precipitated by vigabatrin and lamotrigine in one patient. Other patients did not have myoclonic, absence, or atonic seizures.
Seizures ceased at age 3 to 6 months in 4, recurring in 2 at 2 years. Nine patients had ongoing multiple weekly to yearly seizure clusters at most recent review or death. Exacerbations were seen with intercurrent illness (afebrile and febrile) in 5 and ambient heat in one. Status epilepticus, comprising clusters of very brief seizures without recovery between seizures, occurred in 3 patients.
EEG.
Interictal EEG background was persistently normal in 3 patients. Two had a burst-suppression pattern in the neonatal period and a slow and disorganized background when older. The remaining 7 patients had a continuous, but usually slow and disorganized, awake background. Occasional EEGs showed more organized background activity, typically during times of lower seizure frequency or at an older age. Sleep was recorded in 6 of these 7 cases, and 4 often showed a discontinuous pattern. Background activity was transiently or persistently asymmetric in 5 of 12 patients.
Multifocal interictal epileptiform discharges were seen in all patients, and almost all EEGs (figure 2), occurring frequently and often continuously when seizures were frequent. Discharges were polymorphic, most frequently sharp and sharp-slow wave complexes. Brief bursts of poorly formed focal fast activity, spikes, and polyspikes were also seen. Two patients (one 16 months, one >2 years) had centrotemporal spikes, typical of benign epilepsy with centrotemporal spikes. None had generalized spike-wave activity or typical hypsarrhythmia. Only 6 EEGs (one each in 6 patients) did not show epileptiform activity: when seizures were controlled in 4, in a sedated sleep EEG in one, and during a chest infection with respiratory failure in one. In that patient, vigabatrin had been recently introduced, and clonazepam and levetiracetam doses were reduced. Despite the improved EEG, frequent seizures continued.
Figure 2. Interictal and ictal EEG features in SCN2A encephalopathy.

(A) Interictal EEG in sleep in patient 7 (severe neonatal-infantile phenotype) at age 3 months showing disorganized background with absence of sleep spindles, and frequent multifocal polymorphic interictal epileptiform discharges. (B) Interictal EEG in sleep in patient 9 (intermediate neonatal-infantile phenotype) at age 3 months showing normal background activity including sleep spindles, with frequent multifocal polymorphic interictal epileptiform discharges. (C) Focal seizure in patient 5 at age 4 months arising from the left frontal region in C.a, with intraictal activation of a contralateral ictal rhythm of different frequency beginning in C.b, which recruits over the right parieto-occipital region in C.c. Both rhythms terminate in C.d.
A median of 10 focal seizures (range 3–83) was recorded in 9 patients, with 1 to 5 independent foci of ictal onset per patient (≥2 foci in 8 patients). Seven patients displayed intraictal activation, with an independent remote ictal rhythm developing. Ictal recordings included spiking rhythms and low-voltage fast activity. Clinical features correlated with the ictal focus (e.g., contralateral motor features with central ictal rhythm). One patient had seizures recorded at presentation at 14 months, but these EEGs were not available. Two patients did not have seizures recorded.
Episodes consistent with epileptic spasms were recorded in 3 patients in infancy. Two patients had midline sharp-slow wave complexes typical of spasms but, in the other, no EEG change was seen. One other patient had a cluster of episodes clinically consistent with asymmetric tonic seizures, which ceased after phenytoin loading, without change on EEG.
Antiepileptic drugs.
Nine patients received 1 to 3 antiepileptic medications at recent review or death. Two seizure-free patients and one with ongoing seizures were on no treatment. The median number of antiepileptic drugs tried was 6 (range 4–11).
Eight patients were treated with phenytoin. Five showed a marked seizure improvement, producing seizure cessation or near cessation from multiple daily to multiple hourly attacks. Four patients required supratherapeutic phenytoin levels of >80 (some >100 μmol/L), above the therapeutic range of 40 to 80 μmol/L. Seizure exacerbations occurred with phenytoin levels below the patient's effective range in 4 of 5. No patient had seizure exacerbation with phenytoin. Two of 5 patients showed a minor overall improvement with phenytoin, becoming more alert and interactive, although major developmental gains were not seen. Conversely, one patient had an exacerbation of their movement disorder, one a reduction in alertness despite improved seizures, and 2 had gingival hypertrophy. Phenytoin was not ceased in these patients, although the dose was reduced in one. One was later successfully weaned from all antiepileptic drugs, and 4 remain on phenytoin at 4.2 to 15.5 mg/kg/d.
Six patients had seizure improvement with other sodium channel blockers (carbamazepine, oxcarbazepine, lamotrigine, topiramate). The 3 patients with spasms improved on high-dose prednisolone.
Apart from the emergence of myoclonus with vigabatrin and lamotrigine in one patient, no other antiepileptic drug exacerbated seizures.
Development.
Development was normal before seizures in 3 patients, abnormal in one, and not assessable in 8 with seizure onset at <1 week of age. The patient with seizure onset at 17 years had normal development until 15 months when severe regression occurred.
Periods of developmental plateauing and regression occurred during times of high seizure frequency in all patients. Eight had severe to profound intellectual disability or developmental delay. Five could walk independently and 6 were unable to sit. Eight patients had good visual interaction. Hearing was normal. Three patients had autistic features.
Of 7 patients with EIMFS, developmental outcome was normal in one and one had mild language delay, and the remaining 5 had moderate to profound intellectual disability.
Other clinical features.
Additional neurologic features included movement disorders in 10 patients, axial hypotonia in 11, spasticity in 8, and early handedness in 8. Movement disorders included dystonia, chorea, dyskinesia, and stereotypies. Dystonia required intrathecal baclofen in 2 patients. Extreme irritability was seen in 5, auditory startle in 5, strabismus in 4, an unusual shuffling gait in 2, acquired microcephaly in 2, and optic atrophy in one. Episodic ataxia occurred in one patient (not drug-related), episodic hemiparesis in one (not postictal), and episodic leg jerking of unknown nature in one.
Nine patients had severe gastrointestinal symptoms, including constipation, gastroesophageal reflux, or chronic diarrhea. Three patients required supplemental feeding via nasogastric or gastrostomy tube. Respiratory symptoms occurred in 4 patients (episodic hyperventilation in 3, breath-holding spells in one, and hiccoughs in one). One patient's episodic hyperventilation was accompanied by tachycardia and temperature instability, suggestive of autonomic dysfunction. Dysmorphic features were not noted.
Brain imaging.
All 10 patients with scans available for review had abnormalities on brain MRI (table, figure 3). Eight patients had T2 hyperintensities, in white matter in 8, basal ganglia in 5, thalamus in 4, and brainstem in 5. White matter T2 hyperintensities were seen on only one scan in 3 of 4 patients who had sequential imaging: on the first scan (10–60 days) but not on follow-up imaging (20 days to 22 months) in 2, and on the second scan (81 days) but not the first (9 days) in the other. Delayed myelination was noted in 2 patients, reduced white matter volume in 4, T1 hyperintensities in the basal ganglia in 3, diffusion restriction in the cerebellum in one, and moderate cerebellar and cerebral atrophy in one. Six patients had elongated superior temporal sulci, 4 had small anterior temporal lobes, one had small frontal lobes, and 2 had underopercularization of the sylvian fissure.
Figure 3. MRI brain scans of patients with SCN2A encephalopathy.

(A) T2-weighted axial MRI of patient 6 at age 3 days showing T2 hyperintensity in the white matter (arrow). (B) T2-weighted sagittal MRI of patient 8 at age 18 days showing T2 hyperintensity in the pons (arrow). (C) T2-weighted axial MRI of patient 9 at age 60 days showing T2 hyperintensity in the white matter posteriorly (arrow). (D) T2-weighted axial MRI of patient 9 at age 22 months showing normal white matter including normal terminal zones of myelination and resolution of the white matter signal abnormality.
MRI brain scans were reported as normal in patients 5 and 10. No patient had imaging features of hypoxic-ischemic injury or malformations.
Death.
Sudden unexpected death occurred in patient 3 and patient 12 in sleep at age 21 months and 20 years, respectively. Patient 7 died in the context of refractory seizures and extreme autonomic dysfunction at 22 months.
Postmortem analysis in patient 3 revealed the cause of death to be pneumonia. Histopathologic examination of the brain revealed reduced total brain weight, marked cortical dyslamination of temporal lobes, cortical gliosis most prominent in the temporal lobes and around the interhemispheric fissure, and bilateral hippocampal CA1 neuronal loss, gliosis, and astrocytosis.
Patient 3 had commenced phenytoin a month before death, with a marked seizure improvement. Weekly ECGs during this period showed no cardiac rhythm abnormality. As the postmortem phenytoin level was subtherapeutic at 23 μmol/L, phenytoin toxicity was unlikely to have contributed to death.
DISCUSSION
Our study expands the phenotypic spectrum of SCN2A encephalopathy and emphasizes SCN2A as a major gene for EIMFS.
Seven patients with SCN2A mutations had EIMFS, constituting 26% (7/27) of our total cohort of patients with EIMFS and rendering SCN2A the second most commonly mutated gene for EIMFS, after KCNT1.25,26 Phenotypic features that distinguish SCN2A-associated EIMFS from KCNT1-associated EIMFS25 include severe movement disorders and response to phenytoin in some patients. Two patients showed a considerably better outcome than is usual for EIMFS. To date, there has only been a single case of SCN2A causing EIMFS who also had a paternally inherited 15q11.2 deletion.14 In contrast to previous series, only 2 of our cases had Ohtahara syndrome and none had West syndrome, as hypsarrhythmia was not present in those with infantile spasms.6,8,10–13,15
Despite variability in age at onset and severity of outcome, there are common features in SCN2A encephalopathy. Our patients presented at a median of 2 days with multiple types of brief focal seizures that cluster, reaching maximal frequency of multiple hourly seizures within 3 months of onset. Episodes consistent with infantile spasms or tonic seizures occurred in some, sometimes without an EEG correlate, raising the possibility that these are paroxysmal movement disorders mimicking seizures in some patients. Multifocal epileptiform discharges are usual. Movement disorders, axial hypotonia, and gastrointestinal problems are frequent.
A marked improvement in seizures with high therapeutic or supratherapeutic phenytoin was seen in 5 patients, effecting seizure control following periods of multiple hourly seizures, with seizures recurring when phenytoin levels lowered. Some improvement in seizure control was also noted with other sodium channel blocking drugs. While the observation of seizure improvement is based only on retrospective clinical review, this finding reinforces previous observations of drug response: withdrawal of phenytoin provoking status epilepticus in one case, status treated with fosphenytoin in another, 2 seizure-free on lamotrigine, and one with seizure control on lidocaine.6,12,14 Phenytoin did not, however, significantly improve development in the patients with seizure improvement and was not associated with clear seizure improvement in all patients, although seizures were not exacerbated. The reasons for these differential effects within and between patients are unclear and warrant further study.
Movement disorders appear to be a key feature of SCN2A encephalopathy. Severe and polymorphic movement disorders including dystonia and chorea occurred in most patients with neonatal-onset seizures and poor developmental outcome. Movement disorders in patients with neonatal-onset seizures and better developmental outcome were milder, consisting of dyskinesia that resolved in infancy. Patients with childhood-onset seizures developed prominent stereotypies. Movement disorders are common in other genetic EEs. The movement disorders in SCN2A encephalopathy overlap those seen in other conditions such as SCN8A encephalopathy,27 in contrast to other conditions such as STXBP1 encephalopathy, in which a distinctive movement disorder, a “figure-of-eight” head stereotypy, can provide a diagnostic clue.28
Three of our patients and 3 previously reported patients have died (6/46, 13%), emphasizing the extreme severity of this condition. This mortality rate is comparable to Dravet syndrome, another sodium channelopathy due to SCN1A mutations, which is approximately 15% by age 20 years.29 Two of our patients died unexpectedly in sleep. Sudden unexpected death in epilepsy (SUDEP) is recognized in other sodium channelopathies including Dravet syndrome and 2 cases of SCN8A encephalopathy.27,30,31 Currently, numbers of patients with SUDEP in SCN2A encephalopathy are low, but our observations suggest an increased risk of SUDEP.
There are now 46 patients reported with SCN2A encephalopathy (12 here, 39 reported; 5 patients reported twice).4–20 Our analysis suggests that patients fall into 3 phenotypic groups: a severe neonatal-infantile phenotype, an intermediate neonatal-infantile phenotype, and a childhood phenotype (figure 4). The numbers in the latter groups are small; further cases are required to confirm distinct subgroups or, alternatively, a spectrum of severity. Clustering of different nonepilepsy clinical features across groups, such as the type of movement disorder, may favor distinct subgroups. Two remaining groups of SCN2A phenotypes exist: the self-limited autosomal dominant syndrome of BFNIS (with excellent outcome and seizure resolution) and moderate to severe intellectual disability without epilepsy2,3,21–23,32,33; autism may be present.21–23 The expanded phenotypic spectrum of SCN2A mutations parallels that seen with KCNQ2 and SCN1A and may be broader than currently appreciated.34–36
Figure 4. Clinical features in SCN2A epilepsy phenotypic groups.

Gray scale denotes severity: light color = mild; darker color = severe. Ep = epilepsy; ID = intellectual disability; MD = movement disorder.
As yet, no clear genotype-phenotype correlation explains the phenotypic heterogeneity. Although mutations identified in patients with BFNIS and SCN2A encephalopathy show different inheritance patterns and Grantham scores,37 and no BFNIS-associated mutations have been found in patients with SCN2A encephalopathy, mutations are found throughout the gene (figure 1) and affect both transmembrane domains and linker regions in both phenotypes. No genotypic differences were seen between the postulated severe, intermediate, and childhood phenotypes of SCN2A encephalopathy. Indeed, patients 3 and 10, who share the same mutation, show phenotypic variability suggesting that modifier genes have a role. Research is required to correlate the molecular dysfunction of specific mutations with the phenotypic groups.
The differential diagnosis of SCN2A encephalopathy includes many genetic EEs, including other sodium channelopathies. While Dravet syndrome is differentiated by age at onset and seizure types, rare cases of EIMFS due to SCN1A mutations are described.26,36,38 SCN8A encephalopathy can present with early-onset EE and responds to sodium channel blockers27,30,39 in contradistinction to SCN1A diseases.38,40 However, the median age at onset of 5 months for SCN8A is later,4–17,19,27 which may reflect the respective patterns of expression of the subunits. SCN8A gradually replaces SCN2A as the major subunit in excitatory neurons during maturation.1
SCN2A is an important cause of EE with an emerging distinctive picture, particularly for the severe phenotype. The observation of improved seizure control with phenytoin requires more detailed study and ideally prospective trials, which may lead to improved treatment of this disorder.
ACKNOWLEDGMENT
The authors are grateful to the patients and their families for participating in the research.
GLOSSARY
- BFNIS
benign familial neonatal infantile seizures
- EE
epileptic encephalopathy
- EIMFS
epilepsy of infancy with migrating focal seizures
- MIP
molecular inversion probe
- SUDEP
sudden unexpected death in epilepsy
- WES
whole-exome sequencing
AUTHOR CONTRIBUTIONS
K.B.H. participated in the conception and design of the study, provided phenotyping data, analyzed the data, and wrote and edited the manuscript. J.M.M. provided phenotyping data. G.L.C. and D.T. provided molecular data. M.T.M., V.R.-C., R.W., D.C., J.L.F., S.C., and H.E.O. provided phenotyping data. S.M. analyzed imaging data. A.P. and H.C.M. provided molecular data. A.S.H. and I.E.S. participated in the conception and design of the study, provided phenotyping data, analyzed the data, and wrote and edited the manuscript. All authors edited the manuscript.
STUDY FUNDING
K.B.H. is supported by the Gustav Nossal National Health and Medical Research Council of Australia (NHMRC) Postgraduate Scholarship and the Clifford PhD Scholarship. A.P. is supported by the National Institute of Neurological Disorders and Stroke (NINDS). H.C.M. is a recipient of the Burroughs Wellcome Fund Career Award for Medical Scientists and is supported by funding from the NIH/NINDS. I.E.S. is supported by an NHMRC Program Grant and an NHMRC Practitioner Fellowship.
DISCLOSURE
K. Howell, J. McMahon, G. Carvill, D. Tambunan, M. Mackay, V. Rodriguez-Casero, R. Webster, D. Clark, J. Freeman, S. Calvert, H. Olson, S. Mandelstam, A. Poduri, H. Mefford, and A. Harvey report no disclosures relevant to the manuscript. I. Scheffer has served on scientific advisory boards for UCB and Janssen-Cilag EMEA; serves on the editorial boards of the Annals of Neurology, Neurology®, and Epileptic Disorders; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has received speaker honoraria from GlaxoSmithKline, Athena Diagnostics, UCB, Biocodex, and Janssen-Cilag EMEA; has received funding for travel from Athena Diagnostics, UCB, Biocodex, GlaxoSmithKline, and Janssen-Cilag EMEA; and receives/has received research support from the National Health and Medical Research Council of Australia, NIH, Australian Research Council, Health Research Council of New Zealand, CURE, American Epilepsy Society, US Department of Defense Autism Spectrum Disorder Research Program, the Jack Brockhoff Foundation, the Shepherd Foundation, Perpetual Charitable Trustees, and The University of Melbourne. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia 2012;53:1849–1859. [DOI] [PubMed] [Google Scholar]
- 2.Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002;360:851–852. [DOI] [PubMed] [Google Scholar]
- 3.Berkovic SF, Heron SE, Giordano L, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 2004;55:550–557. [DOI] [PubMed] [Google Scholar]
- 4.Kamiya K, Kaneda M, Sugawara T, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci 2004;24:2690–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi X, Yasumoto S, Nakagawa E, et al. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev 2009;31:758–762. [DOI] [PubMed] [Google Scholar]
- 6.Ogiwara I, Ito K, Sawaishi Y, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology 2009;73:1046–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi K, Ohzono H, Shinohara M, et al. Acute encephalopathy with a novel point mutation in the SCN2A gene. Epilepsy Res 2012;102:109–112. [DOI] [PubMed] [Google Scholar]
- 8.Touma M, Joshi M, Connolly MC, et al. Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings. Epilepsia 2013;54:e81–e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sundaram SK, Chugani HT, Tiwari VN, Huq AH. SCN2A mutation is associated with infantile spasms and bitemporal glucose hypometabolism. Pediatr Neurol 2013;49:46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kodera H, Kato M, Nord AS, et al. Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia 2013;54:1262–1269. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura K, Kato M, Osaka H, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 2013;81:992–998. [DOI] [PubMed] [Google Scholar]
- 13.Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet 2012;49:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhamija R, Wirrell E, Falcao G, et al. Novel de novo SCN2A mutation in a child with migrating focal seizures of infancy. Pediatr Neurol 2013;49:486–488. [DOI] [PubMed] [Google Scholar]
- 15.Martin HC, Kim GE, Pagnamenta AT, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet 2014;23:3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baasch AL, Hüning I, Gilissen C, et al. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia 2014;55:e25–e29. [DOI] [PubMed] [Google Scholar]
- 17.Matalon D, Goldberg E, Medne L, Marsh ED. Confirming an expanded spectrum of SCN2A mutations: a case series. Epileptic Disord 2014;16:13–18. [DOI] [PubMed] [Google Scholar]
- 18.Liao Y, Anttonen AK, Liukkonen E, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 2010;75:1454–1458. [DOI] [PubMed] [Google Scholar]
- 19.Liao Y, Deprez L, Maljevic S, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010;133:1403–1414. [DOI] [PubMed] [Google Scholar]
- 20.de Ligt J, Willemsen MH, van Bon BWM, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929. [DOI] [PubMed] [Google Scholar]
- 21.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674–1682. [DOI] [PubMed] [Google Scholar]
- 22.Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012;485:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavassoli T, Kolevzon A, Wang AT, et al. De novo SCN2A splice site mutation in a boy with autism spectrum disorder. BMC Med Genet 2014;15:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu TW, Chahrour MH, Coulter ME, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron 2013;77:259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012;44:1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McTague A, Appleton R, Avula S, et al. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain 2013;136:1578–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsen J, Carvill GL, Gardella E, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015;84:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim YO, Korff CM, Villaluz MMG, et al. Head stereotypies in STXBP1 encephalopathy. Dev Med Child Neurol 2013;55:769–772. [DOI] [PubMed] [Google Scholar]
- 29.Roger J, Bureau M, Dravet C, Genton P. Epileptic Syndromes in Infancy, Childhood, and Adolescence. Montrouge, France: John Libbey Eurotext; 2005. [Google Scholar]
- 30.Veeramah KR, O'Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012;90:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakauchi M, Oguni H, Kato I, et al. Retrospective multiinstitutional study of the prevalence of early death in Dravet syndrome. Epilepsia 2011;52:1144–1149. [DOI] [PubMed] [Google Scholar]
- 32.Herlenius E, Heron SE, Grinton BE, et al. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia 2007;48:1138–1142. [DOI] [PubMed] [Google Scholar]
- 33.Striano P, Bordo L, Lispi ML, et al. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia 2006;47:218–220. [DOI] [PubMed] [Google Scholar]
- 34.Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 1998;18:25–29. [DOI] [PubMed] [Google Scholar]
- 35.Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25. [DOI] [PubMed] [Google Scholar]
- 36.Mulley JC, Scheffer IE, Petrou S, et al. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–542. [DOI] [PubMed] [Google Scholar]
- 37.Brunklaus A, Ellis R, Reavey E, et al. Genotype phenotype associations across the voltage-gated sodium channel family. J Med Genet 2014;51:650–658. [DOI] [PubMed] [Google Scholar]
- 38.Brunklaus A, Ellis R, Reavey E, et al. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012;135:2329–2336. [DOI] [PubMed] [Google Scholar]
- 39.Ohba C, Kato M, Takahashi S, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 2014;55:994–1000. [DOI] [PubMed] [Google Scholar]
- 40.Guerrini R, Dravet C, Genton P, et al. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998;39:508–512. [DOI] [PubMed] [Google Scholar]