

Abstract
The rostral ventromedial medulla (RVM) exerts both inhibitory and excitatory controls over nociceptive neurons in the spinal cord and medullary dorsal horn. Selective ablation of mu-opioid receptor (MOR)-expressing neurons in the RVM using saporin conjugated to the MOR agonist dermorphin–saporin (derm-sap) attenuates stress and injury–induced behavioral hypersensitivity, yet the effect of RVM derm-sap on the functional integrity of the descending inhibitory system and the properties of RVM neurons remain unknown. Three classes of RVM neurons (on-cells, off-cells, and neutral cells) have been described with distinct responses to noxious stimuli and MOR agonists. Using single unit recording in lightly anesthetized rats, RVM neurons were characterized after microinjections of derm-sap or saporin. Derm-sap treatment resulted in a reduction in on-cells and off-cells when compared to saporin controls (P < 0.05). The number of neutral cells remained unchanged. After derm-sap treatment, RVM microinjections of the glutamate receptor agonist homocysteic acid increased tail-flick latencies, whereas the MOR agonist DAMGO had no effect. Furthermore, electrical stimulation of the periaqueductal gray produced analgesia in both derm-sap and saporin controls with similar thresholds. Microinjection of kynurenic acid, a glutamate receptor antagonist, into the RVM disrupted periaqueductal gray stimulation–produced analgesia in both saporin-treated and derm-sap– treated rats. These results indicate that MOR-expressing neurons in the RVM are not required for analgesia produced by either direct or indirect activation of neurons in the RVM.
Keywords: Nucleus raphe magnus, Periaqueductal gray, Opioid, Analgesia
1. Introduction
Pain-modulating neurons in the rostral ventromedial medulla (RVM), which includes the nucleus raphe magnus and surrounding paragigantocellularis pars alpha, inhibit and facilitate nociceptive transmission through direct projections to the medullary and spinal cord dorsal horn.22 Three classes of RVM neurons have been described with distinct responses to noxious stimuli and mu-opioid receptor (MOR) agonists.9,10 On-cells are characterized by an increase in activity before a nocifensive reflex and are inhibited by MOR agonists such as morphine and DAMGO.9,10,18,19 By contrast, off-cells cease firing just before a tail flick and are activated by MOR agonists.9,10,19 The activity of a third class of RVM neurons, neutral cells, remains unaffected by both noxious stimulation and MOR agonists.18
The inhibition of on-cells by MOR agonists seems to be due to direct, postsynaptic actions, whereas the excitation of off-cells by MOR agonists may be the result of presynaptic inhibition of GABAergic terminals.14,15,18,19,23 The microinjection of the neurotoxin dermorphin–saporin (derm-sap) has been used to selectively ablate MOR-expressing neurons in the RVM, demonstrated by in situ hybridization for MOR mRNA and immunohistochemistry for MOR protein.3,7,34,36 Additional studies have determined that MOR-expressing neurons in the RVM, presumably the on-cells, contribute to behavioral hyperalgesia produced by nerve injury, pancreatitis, intracolonic mustard oil, and chronic stress.3,34–36,39,40 Although it has been hypothesized that ablation of MOR-expressing neurons in the RVM selectively reduces the number of on-cells, this idea has not been directly tested.
The RVM represents the final output to a well-defined nociceptive modulatory circuit that includes a pathway from the amygdala to the ventrolateral periaqueductal gray (vlPAG) and projections from the vlPAG to the RVM.22,23,28 Activation of the vlPAG produces antinociception through an excitatory glutamatergic connection in the RVM.1,4,5,37 The ability of vlPAG or RVM stimulation to produce antinociception after the ablation of MOR-expressing neurons has not been explored. In this study, we examined the properties of neurons surviving after a single RVM injection of derm-sap or saporin (sap) control and determined the ability of these neurons to inhibit a nociceptive reflex when activated by either direct or vlPAG stimulation.
2. Methods
2.1. Animals
All experiments were performed on male Sprague-Dawley rats (280–350 g; Bantin and Kingman, Hayward, CA). Animals were group housed in a climate controlled environment with a 12 hours light–dark cycle and provided free access to food and water. Studies were approved by the Institutional Committees on Animal Research at the University of New England and the University of California, San Francisco, and animals were treated according to the policies and recommendations of the National Institutes of Health guidelines for the handling and use of laboratory animals.
2.2. Rostral ventromedial medulla cannula placement
Rats received microinjections of derm-sap or sap through cannulas positioned directly above the RVM. In some experiments, guide cannulas were implanted 5 to 7 days before injections to allow for repeated injections into the same location, whereas in other experiments, the guide cannula was not implanted. In these cases, the cannulas and guides were removed 5 minutes after completion of the derm-sap or sap injections into the RVM, the hole in the skull was filled with bone wax, the incision sutured, and the animals were allowed to recover for 17 to 21 days.
Guide cannulas were inserted under ketamine (100 mg/kg, intraperitoneally [i.p.]) and xylazine (10 mg/kg, i.p.) anesthesia, with additional boosters of ketamine administered as needed. The cannulas (26 ga, 1.2 mm separation) straddled midline and were positioned 1 mm above the RVM according to the atlas of Paxinos and Watson,33 2.0 mm caudal to the interaural line and 7.8 mm below the surface of cerebellum. Implanted cannulas were fixed to the skull using dental cement, and a stainless steel stylet was inserted into each cannula to keep it free of debris during the recovery period.
2.3. Dermorphin–saporin microinjections
Microinjections of derm-sap or sap (1.2 pmol in 400 nL on each side; Advanced Targeting Systems, San Diego, CA) were performed under isoflurane anesthesia.34 After removing the stylets, injector cannulas were inserted into the guide cannulas that protruded 1 mm beyond the tip of the guide. Injectors were backfilled with drug and connected to a Hamilton syringe through PE10 tubing. A small bubble in the tubing was used to track the progress of the microinjections. Injections, controlled by a syringe pump, began 5 minutes after insertion of the injector cannula and were performed over a 4-minute period. The injectors were replaced with the stylet 5 minutes after completion of the injection.
2.4. DAMGO and homocysteic acid microinjections
In lightly anesthetized animals, tail-flick latencies were assessed after DAMGO and homocysteic acid injections into the RVM 17 days after derm-sap or sap treatment. Isoflurane anesthesia was titrated (0.75%-1.0%) so that stable tail-flick latencies could be obtained using radiant heat applied to the blackened ventral surface of the tail 2 to 6 cm from the distal end. From a holding temperature of 35°C, the temperature rose to a plateau of 53°C and remained stable with a cutoff of 12 seconds to prevent tissue damage. Injectors backfilled with drug were inserted into the guide cannulas, and tail flicks were elicited every 2 minutes using heat applied to 3 alternating locations on the tail. After recording 6 stable baseline latencies, the MOR agonist DAMGO (350 nL on each side, 0.5 mg/mL) or the glutamate receptor agonist homocysteic acid (200 nL on each side, 100 mM) was microinjected into the RVM. Tail-flick latencies were assessed for 20 to 30 minutes after drug injections.
2.5. Stimulation of the ventrolateral periaqueductal gray
The ability of vlPAG stimulation to produce antinociception was determined 17 to 20 days after RVM microinjections of derm-sap or sap. Before vlPAG stimulation, rats were anesthetized with sodium pentobarbital (60–70 mg/kg, i.p.) and a catheter inserted into the external jugular vein for administration of anesthetics. After placement in a stereotaxic holder, anesthesia was maintained with a constant, continuous infusion of sodium methohexital (30–50 mg·kg−1·h−1, intravenously). A concentric bipolar stimulating electrode was stereotaxically guided into the vlPAG (coordinates), and two 26 ga guide cannulas were positioned above the RVM.
After stable baseline tail-flick latencies were established, stimulation-induced analgesia was examined using the methods of Aimone et al. (1987). Stimulation (0.1 millisecond, 100 Hz, 0–200 μA) began 5 seconds before onset of heat stimulus and continued until the tail flick or cutoff (8 seconds). The minimum current required to inhibit the tail-flick reflex was determined by increasing the current in 10 μA increments. If the tail flick was inhibited by vlPAG stimulation, a control tail flick was elicited before retesting with vlPAG stimulation. A total of 3 consecutive trials in which vlPAG stimulation produced inhibition of tail flick were necessary to establish the threshold for analgesia. Once the electrical threshold required for tail-flick inhibition was established, electrical thresholds were again determined 15 minutes after injection of the glutamate receptor antagonist kynurenic acid (10 mM, 800 nL) into the RVM. In additional experiments, the effect of microinjecting the presynaptic neurotransmitter blocker cobalt chloride (CoCl2, 100 mM, 800 nL) into the RVM was also determined in derm-sap–treated and saporin-treated rats.
2.6. Electrophysiological recordings
Extracellular recordings of RVM neurons commenced 17 to 21 days after microinjecting derm-sap or sap into the RVM. For recordings, rats were injected with sodium pentobarbital (60–70 mg/kg, i.p.) and a catheter inserted into the external jugular vein for administration of anesthetics (sodium methohexital, 30–50 mg·kg−1·h−1, intravenously). After placing the rat into a stereotaxic holder, a hole was drilled in the interparietal bone for insertion of an electrode into the medulla. The ventral surface of tail was blackened and body temperature was maintained at 37°C with a heating pad. Before recording, anesthesia level was adjusted so that tail flicks could be elicited using a feedback controlled projector lamp with a consistent latency (3.5–5.0 seconds) without any signs of discomfort. Electrophysiological recordings were initiated at least 60 minutes after completion of the surgery using tungsten electrodes (3 Mohm; FHC, Bowdoinham, ME) as previously described.29 To prevent selection bias, the experimenter was blind to the treatment of the animal.
Similar areas throughout the RVM were sampled by performing a total of 10 electrode penetrations in each animal at identical coordinates. Four tracks were made every 0.4 mm along midline beginning at −1.2 mm and ending at −2.4 mm caudal to the interaural line. Three additional tracks were made bilaterally, 0.4mm off midline, beginning at −1.4 mm and ending at −2.2 mm caudal to the interaural line. Extracellular single unit activity was recorded 7.5 to 9.0 mm below the surface of the cerebellum. Each discriminated unit with a greater than 3:1 signal-to-noise ratio was characterized using noxious heat stimulation of the tail, with data acquired and analyzed off-line using LabView (National Instruments, Austin, TX).
To search for neurons, periodic light brushing and pressure of the hind paw and also heat evoked tail withdrawals were used to activate neurons. Ample time was allowed between stimuli to allow ongoing activity of neurons suppressed by stimulation to return. For each isolated neuron, at least 2 tail flicks were elicited using radiant heat applied 2 to 6 cm from the distal end of the tail with a 3- to 5-minute interval. Each unit encountered was characterized as an on, off, or neutral cell according to its pattern of neuronal activity related to the tail flick.9 On-cells were identified by an onset of neuronal activity that occurred just before the tail flick (on-cell burst), whereas off-cells ceased firing before the tail flick (off-cell pause). Neutral cells did not demonstrate any change in activity associated with the tail flick. An electrolytic lesion was performed at the bottom of the final electrode track to mark the site for histological verification.
Baseline activity for all neurons was calculated as the average frequency over a 10-second period just before heat onset. Tail flick–related activity for on-cells was calculated as the average frequency of activity over a 10-second period after the heat onset. In addition, the peak frequency of activity was recorded during this same period. The duration of the off-cell pause in discharge was determined as the time between the last action potential before the tail flick and the first action potential to occur after the tail flick. Results were averaged across trials for each cell.
2.7. Data analysis
Comparisons between 2 treatment groups were performed using the Student t test. Multiple group comparisons were performed using 2-way analysis of variance (ANOVA) with repeated measures after it was determined that the data set conformed to a normal distribution with equal variances, followed by a Tukey–Kramer post hoc test. In cases where normality and equal variance tests failed, a Friedman repeated-measures 1-way ANOVA on ranks was performed. Post hoc multiple comparisons were made by comparing postdrug injection vs the baseline measurement using the Dunn’s method. A χ2 analysis was performed to compare the frequency distribution of each cell type in the different treatment groups. Data are presented as mean ± SEM, and differences between groups were considered statistically significant with P values of <0.05.
2.8. Histological verification
After each experiment, rats were deeply anesthetized with pentobarbital and perfused with 0.1 M phosphate-buffered saline followed by 10% buffered formalin. Brainstems were removed and postfixed in 10% formalin followed by 30% sucrose solution. Frozen brainstem sections were sliced at 50 μm and stained with cresyl violet to locate cannulae placements and electrolytic lesions.
3. Results
3.1. DAMGO and homocysteic acid microinjections into the RVM
Initial experiments examined the ability of derm-sap treatment to affect antinociception produced by the microinjection of the MOR agonist DAMGO or the glutamate receptor agonist homocysteic acid into the RVM. Microinjections were performed 14 days after the RVM injection of derm-sap or saporin. If derm-sap effectively ablated MOR-expressing neurons, then the microinjection of derm-sap into the RVM would be expected to attenuate antinociception produced by a MOR agonist. Tail-flick latencies in animals pretreated with derm-sap remained unchanged after DAMGO injections (Fig. 1A; P > 0.05; Friedman 1-way ANOVA on ranks; n = 7). In contrast, microinjection of DAMGO in saporin control animals increased tail-flick latencies beginning 15 minutes after injection (Fig. 1A; P < 0.001; Friedman 1-way ANOVA on ranks; Dunn’s post hoc; n = 8).
Figure 1.

Tail-flick latencies after rostral ventromedial medulla (RVM) microinjections of DAMGO and homocysteic acid (HCA) in animals pretreated with RVM microinjections of dermorphin–saporin or saporin. (A) The microinjection of DAMGO had no effect in derm-sap–treated animals, yet increased tail-flick latencies in saporin-treated animals. (B) Microinjection of HCA produced an increase in tail-flick latencies in both treatment groups. a, P <0.05 vs baseline (BSL) in saporin-treated animals; b, P < 0.05 vs BSL in derm-sap–treated animals.
The microinjection of derm-sap into the RVM should leave intact neurons that do not express MOR. To determine whether the activation of these surviving neurons is capable of producing antinociception, homocysteic acid was injected into the RVM 14 days after derm-sap or saporin treatment. The microinjections of homocysteic acid into the RVM produced comparable increases in tail-flick latencies in derm-sap (n = 7) and saporin (n = 8) treated animals (Fig. 1B; P < 0.05; Friedman 1-way ANOVA on ranks). Unlike the increases observed after DAMGO, tail-flick latencies typically did not reach cutoff after homocysteic acid injections. Post hoc analysis revealed significant increases in tail-flick latencies at 11 and 15 minutes after homocysteic acid injections in saporin-treated animals and at 3 and 17 minutes after injections in derm-sap–treated animals. From these results, it can be concluded that the neurons remaining in the RVM after derm-sap injections are functional in their ability to produce antinociception.
3.2. Periaqueductal gray stimulation–produced analgesia
Electrical stimulation of the vlPAG produces analgesia that is mediated by a relay through the RVM. The functional integrity of this circuit was examined 14 to 21 days after the injection of derm-sap (n = 7) or saporin (n = 7) into the RVM. All stimulation sites in derm-sap–treated and saporin-treated animals were located within the ventrolateral portion of PAG (Fig. 2A). Electrical stimulation thresholds required to produce analgesia were not significantly different, with an average threshold of 77.1±13.8 μA in derm-sap–treated animals and 72.9±6.4 μA in saporin-treated rats (Fig. 2B; P>0.05; 2-way ANOVA with repeated measures). In these same animals, the microinjection of the glutamate receptor antagonist kynurenic acid (10 mM, 800 nL) into the RVM disrupted stimulation-produced analgesia, significantly increasing electrical stimulation thresholds in both derm-sap (151.4 ± 17.0 μA) and saporin (174.3 ± 12.7 μA) treated rats (Fig. 2B; P < 0.001; 2-way ANOVA with repeated measures). After kynurenic acid injections, tail-flick latencies in 2/7 derm-sap–treated and 4/7 saporin-treated animals did not reach cutoff even at the maximum stimulus intensity of 200 μA.
Figure 2.
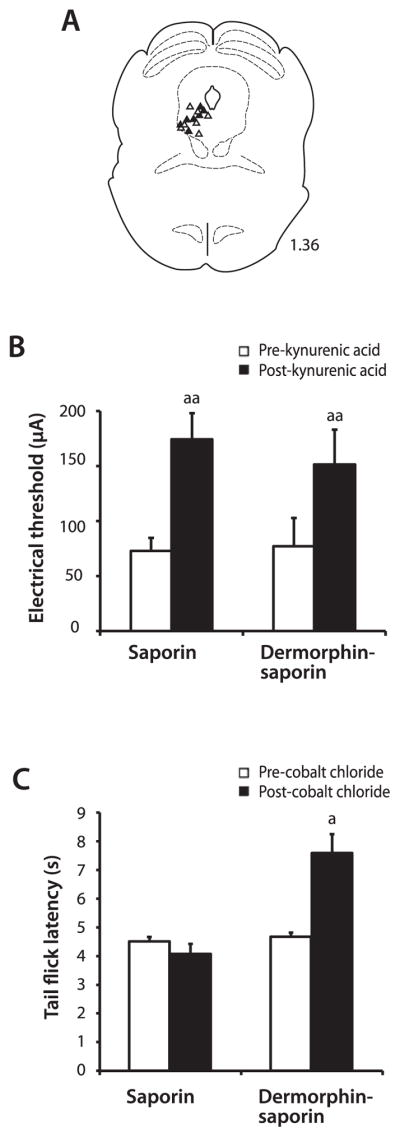
Electrical stimulation in the periaqueductal gray (PAG) produced analgesia in derm-sap–treated and saporin-treated animals. (A) Stimulation sites located in the PAG as verified histologically with electrolytic lesions. Lesions in saporin (closed triangles) and derm-sap (open triangles) treated rats were all located in the left ventrolateral PAG. Number to the right of the figure represents the distance of rostral from the interaural line in mm. (B) The microinjection of kynurenic acid into the rostral ventromedial medulla (RVM) produced comparable increases in PAG electrical stimulation–induced analgesia thresholds. (C) Tail-flick latencies preinjection and postinjection of cobalt chloride into the RVM in saporin and derm-sap–treated animals. Cobalt chloride increased tail-flick latencies only in derm-sap–treated animals. a, P < 0.05; aa, P < 0.01 vs pre–kynurenic acid or pre–cobalt chloride injections into the RVM.
In initial experiments, CoCl2 was used as an alternative strategy to kynurenic acid to block all synaptic transmissions in the RVM.31,38 The microinjection of CoCl2 into the RVM in derm-sap–treated animals caused a significant increase in tail-flick latencies in the absence of vlPAG stimulation (Fig.2C;P<0.05; 2-way ANOVA with repeated measures; n = 5). In contrast, CoCl2 had no effect on baseline tail-flick latencies in saporin-treated animals (Fig. 2C; P > 0.05; 2-way ANOVA with repeated measures; n = 6).
3.3. Electrophysiological properties of rostral ventromedial medulla neurons
The effect of derm-sap and saporin injections on the properties of RVM neurons was determined in lightly anesthetized rats. Recordings were performed in 8 derm-sap and 8 saporin control animals with a total of 10 electrode tracks performed in each animal (Fig. 3A). A total of 344 neurons were characterized in saporin-treated animals and 210 neurons in derm-sap–treated animals. The average number of neurons recorded per animal was significantly less after derm-sap when compared to saporin treatment (26.25 ± 2.7 vs 43 ± 3.3; P < 0.01; Student t test). Categorized by tail flick–related activity, a comparison of the average number of on-cells, off-cells, and neutral cells between treatment groups revealed significantly fewer on-cells and off-cells in derm-sap–treated animals, with no difference in the average number of neutral cells (Fig. 3B).
Figure 3.

Electrophysiological recordings in the rostral ventromedial medulla after saporin and derm-sap injections. (A) A total of 10 recording tracks were made in each animal. Each electrode penetration site is indicated by an “X” and the site of the microinjection cannulas is represented as circles. The number to the right indicates the distance of caudal from the interaural line. (B) The average number of on, off, and neutral cells recorded in each animal. a, P < 0.05 vs the saporin treatment group. (C) The frequency distribution of individual neutral cell ongoing activity in saporin-treated and derm-sap–treated animals.
The reduction in on-cells and off-cells was also apparent when each category of neuron was analyzed as a percentage of the total number of neurons recorded in each animal. An average of 85.3% ± 2.3% of the neurons recorded in derm-sap–treated animals was classified as neutral cells, a significantly greater proportion than the 64.3% ± 4.5% neutral cell population recorded in the saporin control group (P < 0.01; Student t test). Although the percentage of neutral cells in derm-sap animals was greater, the percentage of both on-cells and off-cells was lower when compared with saporin control animals (P<0.01; Student t test). A total of 10.8% ± 2.9% of the neurons characterized in derm-sap animals were on-cells, compared to 22.9% ± 4.7% of the neurons recorded in saporin-treated animals. The percentage of off-cells was only 3.9 ± 1.5 after derm-sap injections compared to 12.8 ± 3.0 after saporin.
The general properties of RVM neurons seemed similar in derm-sap–treated and saporin-treated animals. In neutral cells, the average ongoing activity and frequency distribution of this activity were not different between treatment groups (Fig. 3C, mean activity = 8.9 ± 0.7 spikes per second in saporin-treated animals, and 8.0 ± 0.6 spikes per second in derm-sap–treated animals; P > 0.05; Student t test). Likewise, on-cells from both treatment groups demonstrated comparable levels of baseline activity and heat-evoked activity (Fig. 4A; P > 0.05; Student t test). Derm-sap treatment also had no significant effect on off-cell baseline activity or in the duration of tail flick–related pause (Fig. 4B; P > 0.05; Student t test).
Figure 4.

The properties of on-cells and off-cells recorded in the rostral ventromedial medulla after saporin and derm-saporin treatment. (A) Average on-cell baseline and peak heat-evoked activity. (B) Average off-cell baseline activity and duration of the off-cell pause produced by heating the tail.
4. Discussion
Through projections to the spinal cord and medullary dorsal horn, neurons in the RVM can either increase or decrease nociceptive transmission.22 A loss of behavioral hypersensitivity in several animal models has been previously demonstrated after derm-sap injections into the RVM.3,34,35,39,40 Electrophysiological recordings of spinal cord dorsal horn nociceptive neurons after RVM derm-sap injections have revealed a decrease in mechanical and thermal stimulation-evoked activity.3,36 However, baseline nociceptive thresholds remain unchanged after derm-sap treatment, indicating a more prominent role for the RVM in state-dependent pain modulation.7,34 The effect of derm-sap on the function of RVM neurons involved in producing antinociception has received less attention. In this study, we found that RVM neurons and circuitry necessary for the production of analgesia remained largely intact and functional after microinjections of derm-sap.
Dermorphin–saporin causes the ablation of MOR-expressing neurons when microinjected into the RVM, as evidenced by a reduction in both MOR protein and mRNA.7,34,41 As a functional test for the effectiveness of derm-sap, we found that DAMGO analgesia was completely blocked after treatment with derm-sap but not saporin control. Although not surprising, similar results were reported after the RVM microinjection of cholecystokinin (CCK) conjugated to saporin.41 In the previous study, the injection of CCK-saporin attenuated RVM morphine-induced analgesia, indicating significant overlap in MOR-expressing and CCK2 receptor–expressing neurons.41
The absence of adverse side effects, with no alterations in respiration, blood pressure, and heart rate, has been reported after RVM derm-sap treatment.7 Furthermore, examination of glial cells after RVM injections of derm-sap, saporin, and phosphate-buffered saline found an increase in activation after derm-sap after 1-week, with similar levels of activation at later time points.7 Until this study, the potential for derm-sap to affect pain inhibition produced by the activation of RVM neurons had not been examined. Although derm-sap–abolished analgesia produced by the microinjection of DAMGO, analgesia produced by the excitation of RVM neurons with the glutamate receptor agonist homocysteic acid remained intact. These results indicate that the neurons remaining after derm-sap treatment are still functional in their ability to produce analgesia. It is unlikely that the analgesic effect of homocysteic acid was due to diffusion of the drug to regions outside that affected by the derm-sap because the injection was half the volume of the derm-sap injection.
In addition to directly activating RVM neurons with homocysteic acid, electrical stimulation of the vlPAG was performed to examine the functionality of the vlPAG-RVM circuit after derm-sap treatment. Previous studies demonstrated an increase in the release of glutamate within the RVM after vlPAG stimulation.5 Furthermore, antinociception produced by vlPAG stimulation or systemic morphine is attenuated by the microinjection of glutamate receptor antagonists into the RVM.1,4,17,21 The ability of vlPAG stimulation to produce analgesia in derm-sap–treated animals, and the attenuation of this analgesia by kynurenic acid injections into the RVM, indicates that the vlPAG-RVM analgesia circuit remains functional after the ablation of MOR-expressing neurons within the RVM.
Although analgesia produced by electrical stimulation of the PAG was unaffected, other forms of RVM-mediated analgesia could still be modified by derm-sap treatment, including stress-induced and/or endocannabinoid-induced analgesia. Stress-induced analgesia is attenuated by the microinjection of MOR antagonists into the RVM, as is analgesia produced by the microinjection of morphine into the vlPAG.11,26 Thus, there is evidence for the engagement of 2 distinct systems when the vlPAG activates RVM off-cells, one that entails the direct excitation of off-cells through the release of glutamate (Fig. 5) and another that involves the disinhibition of off-cells through the release of endogenous opioids. It might be expected that conditions that produce analgesia primarily through the disinhibition of off-cells would still be adversely affected by derm-sap treatment.
Figure 5.
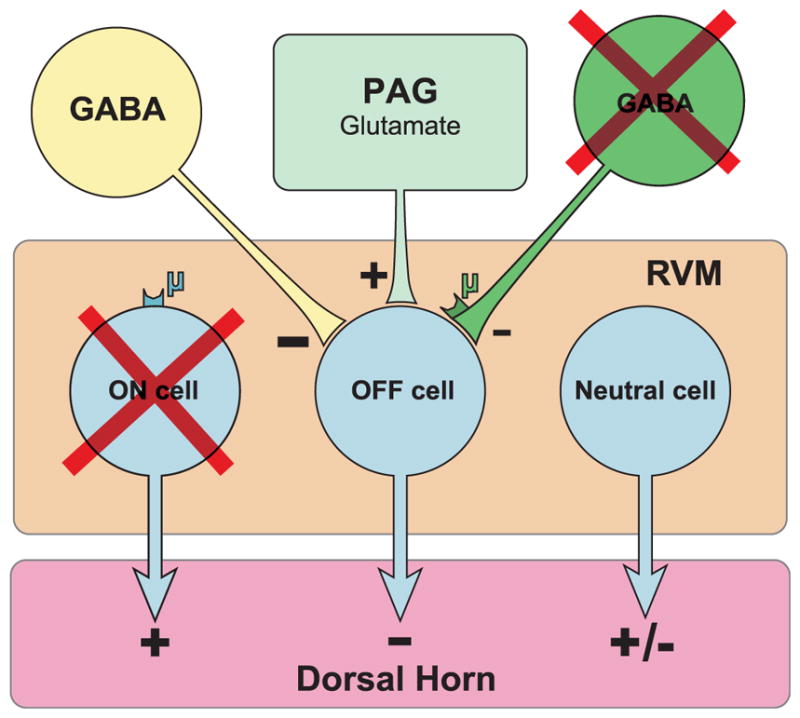
Model of the periaqueductal gray rostral ventromedial medulla (PAGRVM)- dorsal horn circuit after the injection of derm-sap into the RVM. Stimulation of the PAG produces analgesia through an excitatory glutamatergic synapse with off-cells in the RVM, a circuit that remains functional even after derm-sap injections. The ablation of μ-opioid receptor–expressing neurons includes pain-facilitating on-cells and possibly an additional population of inhibitory GABAergic neurons that participate in producing the off-cell pause. As a compensatory response, it is proposed that derm-sap treatment increases tonic GABAergic inhibition of off-cells.
The loss in pain facilitation produced by derm-sap has been attributed to the ablation of RVM on-cells. The on-cell is the only class of neuron in the RVM directly inhibited by MOR agonists, and activation of on-cells has been demonstrated to produce hyperalgesia.2,12,18,20,25,27,30 The reduction in the number of on-cells characterized in derm-sap animals is consistent with the loss in pain facilitation and supports the evidence that on-cells express MOR.18,32 Although significantly reduced, the on-cells still present after derm-sap treatment indicates that either the derm-sap treatment did not ablate all MOR-expressing neurons or that some on-cells do not express MOR. In addition to decreasing the number of on-cells, derm-sap treatment also reduced the number of off-cells. This plus the general reduction in the number of RVM neurons recorded after derm-sap raises the possibility that derm-sap lesions are not completely selective for the on-cell population.
The inability to characterize off-cells does not necessarily indicate that they were not present and functional. To be identified, off-cells require both ongoing activity and a nociceptive reflex-related pause in this activity.9,10 The absence of either ongoing activity or a tail flick–related pause would lead to fewer characterized off-cells. One possibility is that derm-sap caused the loss of inhibitory GABAergic neurons responsible for producing the off-cell pause.16,19,24 It is also possible that the derm-sap– induced damage in the MOR-expressing GABAergic terminals required for the off-cell pause. The source of the putative GABAergic input is unknown but may include RVM on-cells, although results from some studies have provided evidence against this notion.16 Alternatively, derm-sap injections could have caused either an increase in tonic inhibitory input or a decrease in excitatory input onto the off-cells, rendering them silent.
Cobalt chloride was used to block synaptic neurotransmission within the RVM, which would be expected to produce analgesia through an increase off-cell activity only if those neurons are under tonic GABAergic inhibition. Although CoCl2 had no effect in saporin control animals, RVM injections produced analgesia in derm-sap–treated animals. It is suggested that tonic inhibition of off-cells increases after derm-sap as a compensatory response to the loss of more phasic GABAergic inputs normally involved in producing the off-cell pause.15,24 The mechanism for this plasticity is unknown; however, the hypothesized increase in tonic GABAergic inhibition could be tested in future studies by examining the sensitivity of RVM neurons to a GABA receptor antagonist after derm-sap treatment.
Taking into consideration the results from this study, the following model of the vlPAG-RVM-dorsal horn circuit is proposed after derm-sap injections into the RVM (Fig. 5). The microinjection of derm-sap causes the predominant ablation of MOR-expressing on-cells located within the RVM and also GABAergic neurons or terminals responsible for the off-cell pause. These neurons not only may include RVM on-cells but also might be located outside the RVM with the MOR located on presynaptic terminals within the RVM. This would explain the decrease in on-cells and also the inability to identify off-cells after derm-sap treatment. Activation of the surviving neurons located within the RVM, either directly or through the activation of vlPAG glutamatergic neurons, is still able to produce analgesia indicating the continued presence of functional off-cells. Neutral cells are unlikely to account for the analgesia because morphine injections into the vlPAG do not activate neutral cells.8 Furthermore, activation of at least one subset of neutral cells, serotonergic neurons located within the RVM, produces hyperalgesia.6,13 Finally, the ability of RVM injections of CoCl2 to produce analgesia after derm-sap treatment is consistent with a compensatory increase in tonic GABAergic input onto the off-cells. The release from inhibition is likely sufficient to activate off-cells under these experimental conditions because the CoCl2 treatment would also block synaptic excitatory inputs.
In summary, using derm-sap to ablate MOR-expressing neurons in the RVM, we were able to directly demonstrate the loss of pain-facilitating on-cells. In addition, although fewer off-cells were identified, analgesia produced by the direct or indirect activation of RVM neurons indicates the continued presence of functional pain-inhibitory neurons after derm-sap treatment.
Acknowledgments
Funding was provided by the National Institute on Drug Abuse (R01DA014548 to I.D.M. and R01DA034975 to F.P.) and by an Institutional Development Award (IDeA) from the National Institute of General Medicine (P20GM103643 to I.D.M.).
Footnotes
Conflict of interest statement
The authors have no conflicts of interest to declare.
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- 1.Aimone L, Gebhart G. Stimulation-produced spinal inhibition from the midbrain in the rat is mediated by an excitatory asmino acid neurotransmitter in the medial medulla. J Neurosci. 1986;6:1803–13. doi: 10.1523/JNEUROSCI.06-06-01803.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bederson JB, Fields HL, Barbaro NM. Hyperalgesia during naloxone-precipitated withdrawal from morphine is associated with increased on-cell activity in the rostral ventromedial medulla. Somatosens Mot Res. 1990;7:185–203. doi: 10.3109/08990229009144706. [DOI] [PubMed] [Google Scholar]
- 3.Bee LA, Dickenson AH. Descending facilitation from the brainstem determines behavioural and neuronal hypersensitivity following nerve injury and efficacy of pregabalin. PAIN. 2008;140:209–23. doi: 10.1016/j.pain.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Behbehani MM, Fields HL. Evidence that an excitatory connection between the periaqueductal gray and nucleus raphe magnus mediates stimulation produced analgesia. Brain Res. 1979;170:85–93. doi: 10.1016/0006-8993(79)90942-9. [DOI] [PubMed] [Google Scholar]
- 5.Beitz A. Relationship of glutamate and aspartate to the periaqueductal gray-raphe magnus projection: analysis using immunocytochemistry and microdialysis. J Histochem Cytochem. 1990;38:1755–65. doi: 10.1177/38.12.1701457. [DOI] [PubMed] [Google Scholar]
- 6.Cai YQ, Wang W, Hou YY, Pan ZZ. Optogenetic activation of brainstem serotonergic neurons induces persistent pain sensitization. Mol Pain. 2014;10:70. doi: 10.1186/1744-8069-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao F, Chen SS, Yan XF, Xiao XP, Liu XJ, Yang SB, Xu AJ, Gao F, Yang H, Chen ZJ, Tian YK. Evaluation of side effects through selective ablation of the mu opioid receptor expressing descending nociceptive facilitatory neurons in the rostral ventromedial medulla with dermorphin-saporin. Neurotoxicology. 2009;30:1096–106. doi: 10.1016/j.neuro.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Cheng ZF, Fields HL, Heinricher MM. Morphine microinjected into the periaqueductal gray has differential effects on 3 classes of medullary neurons. Brain Res. 1986;375:57–65. doi: 10.1016/0006-8993(86)90958-3. [DOI] [PubMed] [Google Scholar]
- 9.Fields HL, Bry J, Hentall I, Zorman G. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J Neurosci. 1983;3:545–52. doi: 10.1523/JNEUROSCI.03-12-02545.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fields HL, Vanegas H, Hentall ID, Zorman G. Evidence that disinhibition of brain stem neurones contributes to morphine analgesia. Nature. 1983;306:684–6. doi: 10.1038/306684a0. [DOI] [PubMed] [Google Scholar]
- 11.Foo H, Helmstetter FJ. Hypoalgesia elicited by a conditioned stimulus is blocked by a mu, but not a delta or a kappa, opioid antagonist injected into the rostral ventromedial medulla. PAIN. 1999;83:427–31. doi: 10.1016/S0304-3959(99)00125-6. [DOI] [PubMed] [Google Scholar]
- 12.Foo H, Mason P. Discharge of raphe magnus ON and OFF cells is predictive of the motor facilitation evoked by repeated laser stimulation. J Neurosci. 2003;23:1933–40. doi: 10.1523/JNEUROSCI.23-05-01933.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao K, Mason P. Serotonergic Raphe magnus cells that respond to noxious tail heat are not ON or OFF cells. J Neurophysiol. 2000;84:1719–25. doi: 10.1152/jn.2000.84.4.1719. [DOI] [PubMed] [Google Scholar]
- 14.Heinricher MM, Drasner K. Lumbar intrathecal morphine alters activity of putative nociceptive modulatory neurons in rostral ventromedial medulla. Brain Res. 1991;549:338–41. doi: 10.1016/0006-8993(91)90478-e. [DOI] [PubMed] [Google Scholar]
- 15.Heinricher MM, Haws CM, Fields HL. Evidence for GABA-mediated control of putative nociceptive modulating neurons in the rostral ventromedial medulla: iontophoresis of bicuculline eliminates the off-cell pause. Somatosens Mot Res. 1991;8:215–25. doi: 10.3109/08990229109144745. [DOI] [PubMed] [Google Scholar]
- 16.Heinricher M, McGaraughty S. Analysis of excitatory amino acid transmission within the rostral ventromedial medulla: implications for circuitry. PAIN. 1998;75:247–55. doi: 10.1016/s0304-3959(97)00226-1. [DOI] [PubMed] [Google Scholar]
- 17.Heinricher MM, McGaraughty S, Farr DA. The role of excitatory amino acid transmission within the rostral ventromedial medulla in the antinociceptive actions of systemically administered morphine. PAIN. 1999;81:57–65. doi: 10.1016/s0304-3959(98)00271-1. [DOI] [PubMed] [Google Scholar]
- 18.Heinricher MM, Morgan MM, Fields HL. Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience. 1992;48:533–43. doi: 10.1016/0306-4522(92)90400-v. [DOI] [PubMed] [Google Scholar]
- 19.Heinricher MM, Morgan MM, Tortorici V, Fields HL. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience. 1994;63:279–88. doi: 10.1016/0306-4522(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 20.Heinricher MM, Neubert MJ. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. J Neurophysiol. 2004;92:1982–9. doi: 10.1152/jn.00411.2004. [DOI] [PubMed] [Google Scholar]
- 21.Heinricher MM, Schouten JC, Jobst EE. Activation of brainstem N-methyl-D-aspartate receptors is required for the analgesic actions of morphine given systemically. PAIN. 2001;92:129–38. doi: 10.1016/s0304-3959(00)00480-2. [DOI] [PubMed] [Google Scholar]
- 22.Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev. 2009;60:214–25. doi: 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmstetter FJ, Tershner SA, Poore LH, Bellgowan PS. Antinociception following opioid stimulation in the basolateral amygdala is expressed through the periaqueductal gray and rostral ventromedial medulla. Brain Res. 1998;779:104–18. doi: 10.1016/s0006-8993(97)01104-9. [DOI] [PubMed] [Google Scholar]
- 24.Heinricher MM, Tortorici V. Interference with GABA transmission in the rostral ventromedial medulla: disinhibition of off-cells as a central mechanism in nociceptive modulation. Neuroscience. 1994;63:533–46. doi: 10.1016/0306-4522(94)90548-7. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan H, Fields HL. Hyperalgesia during acute opioid abstinence: evidence for a nociceptive facilitating function of the rostral ventromedial medulla. J Neurosci. 1991;11:1433–9. doi: 10.1523/JNEUROSCI.11-05-01433.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiefel JM, Rossi GC, Bodnar RJ. Medullary mu and delta opioid receptors modulate mesencephalic morphine analgesia in rats. Brain Res. 1993;624:151–61. doi: 10.1016/0006-8993(93)90073-v. [DOI] [PubMed] [Google Scholar]
- 27.Kincaid W, Neubert MJ, Xu M, Kim CJ, Heinricher MM. Role for medullary pain facilitating neurons in secondary thermal hyperalgesia. J Neurophysiol. 2006;95:33–41. doi: 10.1152/jn.00449.2005. [DOI] [PubMed] [Google Scholar]
- 28.McGaraughty S, Farr DA, Heinricher MM. Lesions of the periaqueductal gray disrupt input to the rostral ventromedial medulla following microinjections of morphine into the medial or basolateral nuclei of the amygdala. Brain Res. 2004;1009:223–7. doi: 10.1016/j.brainres.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 29.Meng ID, Harasawa I. Chronic morphine exposure increases the proportion of on-cells in the rostral ventromedial medulla in rats. Life Sci. 2007;80:1915–20. doi: 10.1016/j.lfs.2007.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neubert MJ, Kincaid W, Heinricher MM. Nociceptive facilitating neurons in the rostral ventromedial medulla. PAIN. 2004;110:158–65. doi: 10.1016/j.pain.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Nuseir K, Heidenreich BA, Proudfit HK. The antinociception produced by microinjection of a cholinergic agonist in the ventromedial medulla is mediated by noradrenergic neurons in the A7 catecholamine cell group. Brain Res. 1999;822:1–7. doi: 10.1016/s0006-8993(98)01195-0. [DOI] [PubMed] [Google Scholar]
- 32.Pan Z, Williams J, Osborne P. Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. J Neurophysiol. 1990;427:519–32. doi: 10.1113/jphysiol.1990.sp018185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1986. [Google Scholar]
- 34.Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J Neurosci. 2001;21:5281–8. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynolds J, Bilsky EJ, Meng ID. Selective ablation of mu-opioid receptor expressing neurons in the rostral ventromedial medulla attenuates stress-induced mechanical hypersensitivity. Life Sci. 2011;89:313–19. doi: 10.1016/j.lfs.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 36.Sikandar S, Bannister K, Dickenson AH. Brainstem facilitations and descending serotonergic controls contribute to visceral nociception but not pregabalin analgesia in rats. Neurosci Lett. 2012;519:31–6. doi: 10.1016/j.neulet.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spinella M, Cooper M, Bodnar R. Excitatory amino acid antagonists in the rostral ventromedial medulla inhibit mesencephalic morphine analgesia in rats. PAIN. 1996;64:545–52. doi: 10.1016/0304-3959(95)00192-1. [DOI] [PubMed] [Google Scholar]
- 38.Trzebski A, Baradziej S. Role of the rostral ventrolateral medulla in the generation of synchronized sympathetic rhythmicities in the rat. J Auton Nerv Syst. 1992;41:129–39. doi: 10.1016/0165-1838(92)90135-4. [DOI] [PubMed] [Google Scholar]
- 39.Vera-Portocarrero LP, Xie JY, Kowal J, Ossipov MH, King T, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains visceral pain in rats with experimental pancreatitis. Gastroenterology. 2006;130:2155–64. doi: 10.1053/j.gastro.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 40.Vera-Portocarrero LP, Zhang ET, Ossipov MH, Xie JY, King T, Lai J, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains nerve injury-induced central sensitization. Neuroscience. 2006;140:1311–20. doi: 10.1016/j.neuroscience.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 41.Zhang W, Gardell S, Zhang D, Xie JY, Agnes RS, Badghisi H, Hruby VJ, Rance N, Ossipov MH, Vanderah TW, Porreca F, Lai J. Neuropathic pain is maintained by brainstem neurons co-expressing opioid and cholecystokinin receptors. Brain. 2009;132:778–87. doi: 10.1093/brain/awn330. [DOI] [PMC free article] [PubMed] [Google Scholar]