

Abstract
Shiga-like toxins (Stxs), produced by pathogenic Escherichia coli, are a major virulence factor involved in severe diseases in human and animals. These toxins are ribosome-inactivating proteins, and treatment for diseases caused by them is not available. Therefore, there is an urgent need for agents capable of effectively targeting this lethal toxin. In this study, we identified baicalin, a flavonoid compound used in Chinese traditional medicine, as a compound against Shiga-like toxin 2 (Stx2). We found that baicalin significantly improves renal function and reduces Stx2-induced lethality in mice. Further experiments revealed that baicalin induces the formation of oligomers by the toxin by direct binding. We also identified the residues important for such interactions and analyzed their roles in binding baicalin by biophysical and biochemical analyses. Our results establish baicalin as a candidate compound for the development of therapeutics against diseases caused by Stxs.
INTRODUCTION
Shiga toxin-producing Escherichia coli (STEC), particularly E. coli O157:H7, can cause several diseases, ranging from bloody diarrhea to life-threatening hemolytic uremic syndrome (HUS), which is the leading cause of acute renal failure in children and the elderly (1). STEC infection often is caused by ingestion of contaminated food and drink or by direct contact with infected animals (2). The mortality of STEC infection can reach 50% or more in susceptible populations (3, 4). The emergence of antibiotic-resistant E. coli strains has made the treatment of STEC infection more difficult. Moreover, therapies with antimicrobial agents for this infection have been shown to carry a higher risk of patients' developing HUS (5, 6). As a result, STEC infections have become a worldwide challenge. Shiga-like toxins (Stxs) are considered the most important virulence factor of STEC. These toxins can be divided into two types: Stx1 and Stx2 (7). Although Stx1 and Stx2 possess similar mechanisms of action, Stx2 has been proved to be more toxic than Stx1 (4, 8). In addition, several studies have demonstrated that Stx2 is able to induce bloody diarrhea and hemolytic-uremic syndrome (HUS) in animal models (9, 10).
Stxs are highly toxic class II ribosome-inhibiting proteins (RIPs) (11), which consist of the enzymatic A chain, which has N-glycosidase activity, and the B chain, which is essential for the toxin's entry into the cell (12). The A subunit cleaves an adenine base at position 4,324 of the 28S rRNA, leading to the inhibition of protein synthesis of target cells (4).
Death caused by Stx poisoning normally occurs 3 to 5 days after exposure in a mouse model of HUS, but efficacious therapeutics are not available for postexposure treatments (13). Similarly, there is no therapy for treating STEC infections that has been approved by any country. Therefore, there is an urgent need for the development of countermeasures against this severe threat. A number of antibodies capable of neutralizing the toxicity of Stxs have been developed (14). Small molecules with similar inhibitory effects have also been described (15). The availability of high-resolution structure of Stxs has greatly facilitated the development of inhibitory molecules by rational compound design (16). For example, agents capable of interfering with the RNA N-glycosidase activity of both Shiga-like toxin and ricin have been identified via such methods (17). Moreover, inhibitors that block the retrograde trafficking between endosome and the trans-Golgi network (TGN) have been shown to be effective in protecting mice from killing by ricin and Stxs (18, 19). Despite this progress, there is an urgent need for compounds that are able to treat Stx poisoning after individuals have been exposed to the toxins.
Here, we described the identification of baicalin (BAI), a flavonoid compound isolated from Scutellaria baicalensis Georgi, as an agent capable of inhibiting poisoning by Stx2. Similar to its effects on ricin (20), we found that BAI inhibits the activity of Stx2 by inducing the formation of oligomers by the toxin. Our results established BAI as a potentially useful compound in treating STEC infections.
MATERIALS AND METHODS
Chemicals and reagents.
Baicalin (BAI; purity, 98.5%) (Fig. 1A) was obtained from Sigma-Aldrich (St. Louis, MO, USA), and the stock solution at a concentration of 40.96 mg/ml was prepared in dimethyl sulfoxide (DMSO) (Sigma-Aldrich). For in vivo studies, BAI was dissolved in sterile phosphate-buffered saline (PBS) at a concentration of 100 mg/ml.
FIG 1.

Inhibitory effect of baicalin against rStx2. (A) Chemical structure of baicalin. (B) Inhibition of cell death by baicalin. rStx2 was added to HeLa cells treated with the indicated concentrations of baicalin for 72 h. Cytotoxicity was evaluated by measuring extracellular LDH 72 h after addition of the toxins. (C) rStx2 premixed with baicalin can protect HeLa cells. rStx2 mixed with the indicated concentrations of baicalin was added to HeLa cells, and the LDH release was measured after incubating for 72 h. (D) Effects of baicalin on the inhibition of protein synthesis by rStx2. Baicalin was added to in vitro protein synthesis with rStx2, and the expression of luciferase was measured. All data are means and standard errors from three independent experiments. *, P < 0.05; **, P < 0.01.
Expression and purification of rStx2. (i) Construction of plasmid encoding Stx2.
The recombinant plasmid was constructed as described previously (21). Briefly, the E. coli stx2 gene, coding for the mature Stx2 protein, was amplified from the genome of E. coli O157:H7 strain ATCC 43895 with the primer pair 5′-CCGGAATTCATGAAGTGTATATTATTTAAATG-3′ (forward) and 5′-ACGCGTCGACTTAGTGGTGGTGGTGGTGGTGGTCAT TATTAAACTGCACTTC-3′ (reverse). An EcoRI site and a SalI site (underlined) were added to the 5′ and 3′ ends of the primers and used for subcloning of the gene into the expression vector pET32a (Merck, Germany). The resulting plasmid, pET32a-stx2, was transformed into strain BL21(DE3) for protein production.
(ii) Procedure of protein expression and purification.
Cells of strain BL21(DE3)(pET32a-stx2) grown in LB to saturation were diluted 1:100 into 1,000 ml of LB broth with 0.08% glucose. Protein expression was induced with 1 mM IPTG when the optical density at 600 nm (OD600) of the culture reached 0.6 to 0.8 for 10 h at 30°C. Cells harvested by centrifugation at 3,800 × g for 30 min at 4°C were suspended in lysis buffer (1× PBS, 1 mM dithiothreitol [DTT], and 1 mM phenylmethylsulfonyl fluoride [PMSF]) and broken by sonication. The lysates were cleared by centrifugation at 16,000 × g for 40 min at 4°C. The supernatant was loaded onto a self-packaged nickel-nitrilotriacetic acid (Ni-NTA) affinity column (2 ml Ni-NTA Hi-Bind resin; Qiagen). Unbound proteins were removed by washing with 10 column volumes of washing buffer (lysis buffer containing 60 mM imidazole). The protein was eluted with 200 mM imidazole in lysis buffer. The protein was further purified with a Superdex 75 16/60 column (GE Healthcare) on an AKTA system. The purity of all proteins was greater than 95%, as assessed by SDS-PAGE and Coomassie bright blue staining.
Cell-free translation assay.
The TNT coupled reticulocyte lysate systems and luciferase assay system (Promega, Madison, WI) were employed to investigate the ability of BAI to block protein synthesis inhibition imposed by rStx2 (22). One nanogram of rStx2 was used in all reactions, and the concentration of BAI ranged from 9 μM to 72 μM. The reaction was allowed to proceed for 90 min at 30°C in a water bath and was terminated by placing the plates on ice. Luciferase activity was measured per the manufacturer's instructions.
Cell-based assay for protection against rStx2.
We examined the effects of BAI on cell toxicity of Stx2 with HeLa cells as described previously (23). HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 3 mM glutamine, antibiotics (100 U/ml penicillin and 100 U/ml streptomycin), and 10% heat-inactivated fetal bovine serum, at 37°C in 5% CO2 in a humidified incubator. Cells for experiments were plated on a 96-well plate at a density of 1.5 × 104 cells per well in DMEM and incubated for 16 h at 37°C in 5% CO2.
The cells were incubated with 50 pg/ml rStx2 (capable of killing about 80% cells) (see Table S1 in the supplemental material) in the presence of BAI at different concentrations for 72 h at 37°C. Samples without BAI or toxin were established as controls. Cytotoxicity was evaluated by measuring lactate dehydrogenase (LDH) release using the cytotoxicity detection kit (LDH) (Roche, Basel, Switzerland) according to the manufacturer's directions. Plates were read on a microplate reader (Tecan, Austria) at 490 nm.
Ethics statement.
All animal studies were performed according to the Regulations for the Administration of Affairs Concerning Experiments Animals (1988.11). The experimental protocols were approved and supervised by the Institutional Animal Care and Use Committee (IACUC) of Jilin University.
Mice injection protocols.
Assays for lethality, clinical chemistry, histopathology, and cytokine secretion were performed to detect the ability of BAI to protect mice from rStx2. Eight-week-old BALB/c mice weighing 16 to 18 g were obtained from the Experimental Animal Center of Jilin University (Changchun, China). Mice were injected intraperitoneally with 100 μl of either sterile PBS or various amounts of rStx2. For mortality studies, each mouse was injected with a single dose of rStx2 at 100 ng (about twice the dose required for killing 50% of the animals [LD50]) (see Table S2 in the supplemental material); for other studies, mice were administered a single dose of rStx2 (50 ng per mouse). For mortality studies, the infected mice were treated with 100 mg BAI/kg of body weight subcutaneously 6 h after injection and then at 6-h intervals for a total of 6 days. Mice were weighed at 24-h intervals for 8 days after receiving all the treatments.
Hematology and clinical chemistry analysis.
For blood studies, mice were euthanized and blood was collected 72 h after toxin injection. For blood urea nitrogen (BUN) and serum creatinine (Cr) level analysis, blood samples were coagulated for 10 min at 37°C and then were centrifuged at 2,900 × g for 10 min at 4°C to obtain serum. BUN and Cr levels were determined with a Hitachi 7600 biochemical analyzer. For hemoglobin level determinations, 100 μl serum was used to obtain an absorbance from 300 nm to 600 nm with a spectrophotometer.
Histopathology analysis.
Mice receiving the different treatments were anesthetized and sacrificed by cervical dislocation 72 h after rStx2 injection. Kidneys were removed and fixed in 1% formalin. Fixed kidneys were then stained with hematoxylin and eosin (H&E) and were visualized by light microscopy.
Cytokine detection in kidneys.
Kidneys were dissected from mice treated with the different regimens 72 h after toxin injection. Kidney tissues were homogenized in PBS buffer. After centrifugation, the supernatants were collected and stored at −20°C until analysis. Cytokine levels were measured using an enzyme-linked immunosorbent assay (ELISA) by specific mouse ELISA kits (BioLegend, CA).
Cell-free translation assays for mutant proteins.
Cell-free translation assays were performed as described above. Briefly, 1 ng rStx2 was mixed with BAI at a concentration of 36 μM, and the mixture was then added to the reaction system according to the manufacturer's directions. After incubation for 90 min at 30°C in a water bath, the reaction was terminated by placing the plates on ice and analyzed by the luciferase assay system.
Statistical analysis.
The experimental data were assessed using independent Student's t test with SPSS 14.0 statistical software (SPSS Inc., Chicago, IL), and a P value less than 0.05 was considered statistically significant.
RESULTS
BAI protects against rStx2-induced cytotoxicity.
BAI is able to neutralize the toxicity of ricin (20). Because Stxs are also ribosome-inactivating enzymes, we first evaluated whether BAI is able to protect cultured cells caused by Stx2. To this end, we incubated various amounts of BAI with HeLa cells treated with 50 pg/ml rStx2 and evaluated the integrity of cell membranes by measuring LDH release. In samples from mice that did not receive BAI, 82.96% LDH release was detected (Fig. 1B). Significant protection was detected when BAI was used at 4.5 μM, and maximal protection was achieved with 36 μM under our experimental conditions (Fig. 1B).
Similar protection was achieved when BAI was mixed with rStx2 30 min prior to addition to the cells under the same experimental conditions. Again, 4.5 μM BAI was able to significantly protect HeLa cells, and maximal protection was achieved when the concentration used was 36 μM (Fig. 1C). These results suggest that BAI is able to block the activity of the toxin, and this inhibition likely occurs by direct interactions.
Stxs induce cell death by inhibiting protein synthesis. We thus employed the cell-free translation assay to evaluate the effects BAI on the inhibition of ribosomal activity. Under our experimental conditions, 36 μM BAI significant rescued protein synthesis in reactions received 1 ng rStx2 (Fig. 1D). Taken together, these results establish that BAI is able to neutralize the cytotoxicity of rStx2 in vitro.
BAI significantly reduces rStx2-induced lethality in mice.
The strong protective effects of BAI against rStx2-induced cytotoxicity in in vitro systems prompted us to explore its potential usefulness in therapeutics. To this end, we employed the mouse model for hemolytic-uremic syndrome (HUS) (9). Under our experimental conditions, a single dose (∼2 LD50s, 100 ng per mouse) of rStx2 caused 100% animal deaths within 6 days (Fig. 2A). Mice receiving rStx2 alone showed tremors and ataxia 2 days postinjection, and death began to occur on the third day (Fig. 2A). In mice receiving BAI subcutaneously at 6 h postexposure and subsequently at 6-hour intervals for a total of 23 doses, all at 100 mg/kg, the death rate at day 6 was about 30% (Fig. 2A), indicating that this compound can provide approximately 70% protection.
FIG 2.

Baicalin protects mice against lethality induced by rStx2. (A) Groups (n = 10) of mice injected with rStx2 were treated with PBS solution or with BAI 6 h after toxin injection, and survival of mice was monitored for 6 days. The death of mice was followed up to 6 days after rStx2 injection, with no additional mortalities shown in the figure. The curves for BAI-treated mice are statistically significantly different from those for rStx2-injected mice, as evaluated by the log rank test (P = 0.0002 for 100 mg/kg of BAI against rStx2). (B) Effects of BAI on body weight caused by sublethal doses of rStx2. Groups (n = 10) of mice receiving Stx2 or a control solution were administered BAI or PBS 6 h after toxin injection. The body weight was monitored at 24-h intervals for 8 days. Similar results were obtained from more than three independent experiments. *, P < 0.05 for rStx versus rStx plus BAI and PBS at day 6; **, P < 0.01 for rStx2 versus PBS at day 7 and for rStx2 versus rStx plus BAI and PBS at day 8.
We further evaluated the ability of BAI to protect against the weight loss caused by sublethal doses of rStx2 (9). BAI alone, even at high doses, does not detectably affect mouse body weight over 8 days (24). In our experiments, mice injected with PBS or a single dose of rStx2 (50 ng per mouse) were monitored for body weight at 24-h intervals. In the treated group, 100 mg/kg BAI was administered 6 h after toxin injection and subsequently at 6-h intervals for 8 days. Untreated mice in the group injected with the toxin exhibited decreased food and water intake, body weight loss became apparent on day 3, and weights reached the lowest point at day 6 (Fig. 2B). In agreement with the lethality protection results, BAI treatment delayed the occurrence of body weight loss for 24 h; further, the severity of the loss was significantly lower throughout the experiment (Fig. 2B). As expected, consistent with their normal health status, the body weights of control mice receiving PBS displayed detectable increases throughout the experiment (Fig. 2B).
Treatment with BAI improves renal function and blood pathology induced by rStx2.
Severe renal damage is another symptom associated with Stxs toxicity (5); we thus performed a pathology study to evaluate whether BAI treatment can alleviate the manifestation of renal injury. Mice injected with 50 ng rStx2 were administered BAI at 100 mg/kg or with a PBS buffer 6 h after receiving the toxin and subsequently at 6-h intervals as described above. Kidneys from mice in the different treatment groups were sectioned for pathological examination 72 h after toxin injection. In mice not receiving BAI treatment, significant damage was observed in tubules, characterized by swelling and casts in the lumen; the glomerulus also appeared to be hyperemic and smaller (Fig. 3A). Such renal injury was not observed in kidneys from mice treated with BAI after toxin challenge, which was similar to those from control animals not receiving the toxin (Fig. 3A).
FIG 3.

Treatment with baicalin alleviated the pathologies induced by rStx2. (A) Baicalin reduced tissue damage in kidney. Sections of kidneys from representative rStx2-treated mice receiving BAI or control PBS solution are shown. Note the swelling and casts in the lumen and the smaller glomerulus in the tissue of untreated mice (left). In contrast, the morphology of tissues from mice receiving Stx and treated with BAI (middle) was similar to that of tissues from control mice (right). Each group contained 10 sections, and 5 fields were observed in each section. (B and C) Effects of baicalin on blood urea nitrogen (BUN) (B) and creatinine (Cr) (C) induced by rStx2. The blood of mice in the relevant groups was evaluated for BUN and Cr at 72 h after treatment. Each circle represents the BUN or Cr level from one mouse (n = 5). (D) Baicalin treatment reduced the level of hemoglobin in the serum. Sera of relevant mice were evaluated for hemoglobin spectrophotometrically. Similar results were obtained in multiple independent experiments. (E) Baicalin reduced the production of several cytokines induced by rStx2. Kidney tissues from relevant mice were measured for cytokines by ELISA. Note the significant reduction of cytokine production after baicalin treatment. **, P < 0.01.
To further determine kidney function after BAI treatment, we examined levels of blood urea nitrogen (BUN) and creatinine (Cr), the two important indicators of kidney function, 72 h after injection (9). The BUN level in mice receiving rStx2 was 6.56 ± 0.32 mmol/liter. BAI treatment significantly reduced the BUN level (4.56 ± 0.28 mmol/liter). In fact, this level is comparable to the 4.53 ± 0.43-mmol/liter concentration observed in control mice that were not injected with the toxin (Fig. 3B). Similar results were observed when Cr was measured (Fig. 3C). Significantly higher levels of hemoglobin were detected at 72 h in the serum of untreated mice that had been injected with the toxin; similarly, treatment with BAI led to reduction of hemolysis (Fig. 3D).
We also examined the levels of several relevant cytokines to determine whether BAI reduces inflammation accompanied by kidney damage. As expected, rStx2 caused elevated levels of interleukin 1β (IL-1β), IL-4, IL-6, tumor necrosis factor alpha (TNF-α), and gamma interferon (IFN-γ) (Fig. 3E). Consistent with its effect in protecting kidney damage caused by Stx2, the levels of these cytokines significantly decreased in mice treated with BAI at 72 h after injection (Fig. 3E). Taken together, these results indicate that BAI is able to effectively reduce the damage to the kidney caused by rStx2.
BAI inhibits rStx2 activity by directly interacting with the toxin to induce protein oligomerization.
Our results from both in vitro and in vivo experiments suggest that BAI exerts its protective activity by targeting rStx2 directly. Our previous study revealed that BAI inhibits the activity of ricin, another RIP, by inducing the A subunit of ricin (RTA) to form enzymatically less active oligomers (20). Given the fact that the activities of RTA and Stx2a are highly similar and that these two toxins share high-level resemblance in their structures (Fig. 4), we attempted to identify residues on Stx2 potentially involved in binding baicalin. Comparative structural analysis allowed us to identify R179, Q180, S183, E184, and V218 as candidate residues involved in the binding of Stx2 to BAI (Fig. 5). We thus replaced each of these sites with an alanine residue and studied the relevant phenotypes associated with the mutant proteins. In HeLa cells, only the Q180A mutant lost considerable toxicity (Fig. 6A), and it was still sensitive to BAI. The S183A mutant maintained toxicity but was sensitive to BAI (Fig. 6B). In contrast, the R179A, E184A, and V218A mutants retained toxicity similar to that of wild-type rStx2 (Fig. 6B). Importantly, all these mutations abolished or significantly reduced the sensitivity of rStx2 to BAI (Fig. 6B), suggesting the involvement of these residues in engaging BAI. Taken together, these results suggest that BAI interacts with Stx2a directly, probably by inducing the formation of oligomers with markedly reduced activity.
FIG 4.
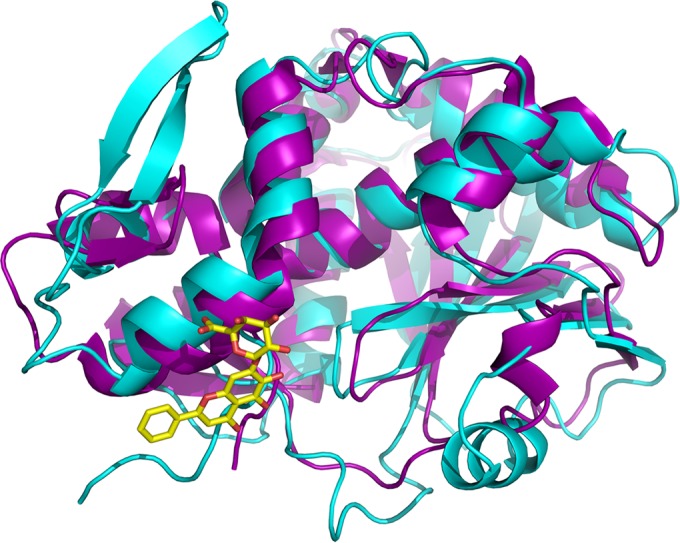
Overlay of structures of ricin subunit A (cyan) and Shiga-like toxin 2 subunit A1 (purple). The crystal structures of RTA and Stx2A1 are displayed in ribbon representation, with coils for α-helices and arrows for β strands. BAI is superimposed in stick mode, with carbon and oxygen atoms in yellow and red, respectively.
FIG 5.

Structure-based sequence alignment of the A subunit of ricin and the A1 subunit of Shiga-like toxin 2. Identical residues are highlighted in red, and similar residues are shown in red font. Secondary structures are shown for both proteins above and below their respective sequences, with coils representing α/η-helices and arrows representing β-strands.
FIG 6.

Validation of the involvement of rStx2 residues in its interaction with baicalin. (A) Purified rStx2 mutants were measured for their toxicity to HeLa cells by determining LDH release upon treatment of the cells with recombinant holotoxin. (B) Ability of the mutants to inhibit protein synthesis in a cell-free system. The effects of baicalin were evaluated by including this compound in parallel reactions. The results are expressed as the percentage of protein synthesis in control reactions without toxin or baicalin. All data are means and standard errors from three independent experiments. *, P < 0.05; ** P < 0.01.
DISCUSSION
Pathogenic E. coli is one of the most common pathogens that often is contracted from contaminated food and water as well as animal and human effluents (25). Shiga-like toxin (Stx)-producing E. coli (STEC), particularly enterohemorrhagic E. coli (EHEC) strains such as O157:H7, are a leading cause of outbreaks and sporadic cases of bloody diarrhea and hemolytic-uremic syndrome (HUS) (26). These infections pose grave challenges to public health. In the United States, 265,000 STEC infections occur each year, of which about 36% are caused by EHEC O157:H7. Although antibiotics can be used when oligoanuric HUS is established, the wide spread of resistance has threated the effectiveness of such treatments (6, 27). Furthermore, in some cases, the problem is further is compounded by the fact that conventional treatments such as antibiotics aggravate symptoms (5, 28). Thus, there is an urgent need for agents complementary to antibiotics that are clinically effective in treating diseases caused by these pathogens. With better understanding of how bacteria cause diseases, some new approaches based on our understanding of virulence mechanisms have been established in recent years (29). Among these, agents that target virulence factors hold great promise.
Shiga-like toxins (Stxs) are the key virulence factor associated with severe diseases caused by STEC infections, and Shiga-like toxin 2 (Stx2) is the primary virulence factor for HUS (30). Stx2 thus has been recommended as a potent target for drug development (15). Recently, several excellent reports described the identification of novel small-molecule inhibitors and the development of specific antibodies against Stx2. Significant achievements have been made in therapies against STEC infections using specific neutralizing monoclonal antibodies, and several antibodies have been tested in phase I clinical trials (14, 25, 31). Small-molecule inhibitors presumably have some advantages over neutralizing antibodies, such as having a lower cost, being relatively easier to store and transport among different locations, and being less likely to be contaminated. To date, only two small-molecule inhibitors for Stx2 toxicity had been described. These compounds, called Retro-1 and Retro-2, have been shown to protect mice from ricin- and Stx1-induced lethality (18). However, to achieve the protection, the animals need to receive these compounds at least 1 h prior to toxin exposure (18). Clearly, these inhibitors are not appropriate for therapeutic purposes, where the treatment of infection or toxin exposure is the focus. Furthermore, these two compounds function by targeting the retrograde trafficking of the host cells (18), the potential side effects of which are unknown.
Our identification of baicalin (BAI) as an inhibitor for the activity of Stx2 and the observation that this compound at a low concentration is able to significantly protect mice from toxin-induced mortality after toxin exposure represent a significant step forward in this research direction. The successful use of the structure information of the ricin-BAI complex to identify the key residues on Stx2 important for its interactions with BAI suggests that BAI targets Stx2 with a similar mechanism, namely, induction of protein oligomerization to occlude its active site (20). These results also suggest that chemical modification of BAI to obtain molecules with higher affinity for the toxins is feasible and clearly is a direction worth pursuing in future research. Moreover, BAI is reported as a safe agent in mice; the dosage can reach 15 g/kg of body weight when administered intragastrically and 2.74 g/kg of body weight when injected intraperitoneally (24, 32).
Given the similarity in the mechanism of action between ricin and Stxs, the activity of BAI against Stx2 is not completely unexpected. Because toxin released from dead bacteria killed by antibiotics can potentially compound the disease symptoms, agents that target the toxin directly will be very beneficial. It will be interesting to determine how a combination of antibiotics and BAI functions in the treatment of HUS and other infectious diseases caused by Stx toxin-producing pathogens. Because BAI directly targets the toxin released by the bacteria killed by the antibiotics, such a treatment regimen may be more efficacious than BAI administered alone.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Basic Research Program of China (grant 2013CB127205 to X.D.), the National Nature Science Foundation of China (grant 31130053 to X.D.), and the National 863 program (grant 2012AA020303).
We declare that no competing interests exist.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01416-15.
REFERENCES
- 1.Griffin PM, Tauxe RV. 1991. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol Rev 13:60–98. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien AD, LaVeck GD, Thompson MR, Formal SB. 1982. Production of Shigella dysenteriae type 1-like cytotoxin by Escherichia coli. J Infect Dis 146:763–769. doi: 10.1093/infdis/146.6.763. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien AD, Holmes RK. 1987. Shiga and Shiga-like toxins. Microbiol Rev 51:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O'Brien AD, Samuel JE. 1993. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect Immun 61:3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao YL, Cen XB, Ito M, Yokoyama K, Takagi K, Kitaichi K, Nadai M, Ohta M, Hasegawa T. 2002. Shiga-like toxin II derived from Escherichia coli O157:H7 modifies renal handling of levofloxacin in rats. Antimicrob Agents Chemother 46:1522–1528. doi: 10.1128/AAC.46.5.1522-1528.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis TK, McKee R, Schnadower D, Tarr PI. 2013. Treatment of Shiga toxin-producing Escherichia coli infections. Infect Dis Clin North Am 27:577–597. doi: 10.1016/j.idc.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 7.Melton-Celsa A, Mohawk K, Teel L, O'Brien A. 2012. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr Top Microbiol Immunol 357:67–103. doi: 10.1007/82_2011_176. [DOI] [PubMed] [Google Scholar]
- 8.Louise CB, Kaye SA, Boyd B, Lingwood CA, Obrig TG. 1995. Shiga toxin-associated hemolytic uremic syndrome: effect of sodium butyrate on sensitivity of human umbilical vein endothelial cells to Shiga toxin. Infect Immun 63:2766–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sauter KA, Melton-Celsa AR, Larkin K, Troxell ML, O'Brien AD, Magun BE. 2008. Mouse model of hemolytic-uremic syndrome caused by endotoxin-free Shiga toxin 2 (Stx2) and protection from lethal outcome by anti-Stx2 antibody. Infect Immun 76:4469–4478. doi: 10.1128/IAI.00592-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stearns-Kurosawa DJ, Collins V, Freeman S, Tesh VL, Kurosawa S. 2010. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect Immun 78:2497–2504. doi: 10.1128/IAI.01435-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsnes S, Reisbig R, Eiklid K. 1981. Subunit structure of Shigella cytotoxin. J Biol Chem 256:8732–8738. [PubMed] [Google Scholar]
- 12.Yamasaki C, Natori Y, Zeng XT, Ohmura M, Yamasaki S, Takeda Y. 1999. Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 (Stx1) and Stx2 but not by non-toxic mutant Stx1 which lacks N-glycosidase activity. FEBS Lett 442:231–234. doi: 10.1016/S0014-5793(98)01667-6. [DOI] [PubMed] [Google Scholar]
- 13.Silberstein C, Lucero MS, Zotta E, Copeland DP, Lingyun L, Repetto HA, Ibarra C. 2011. A glucosylceramide synthase inhibitor protects rats against the cytotoxic effects of shiga toxin 2. Pediatr Res 69:390–394. doi: 10.1203/PDR.0b013e318211dd57. [DOI] [PubMed] [Google Scholar]
- 14.Wahome PG, Robertus JD, Mantis NJ. 2012. Small-molecule inhibitors of ricin and Shiga toxins. Curr Top Microbiol Immunol 357:179–207. doi: 10.1007/82_2011_177. [DOI] [PubMed] [Google Scholar]
- 15.Pang YP, Park JG, Wang S, Vummenthala A, Mishra RK, McLaughlin JE, Di R, Kahn JN, Tumer NE, Janosi L, Davis J, Millard CB. 2011. Small-molecule inhibitor leads of ribosome-inactivating proteins developed using the doorstop approach. PLoS One 6:e17883. doi: 10.1371/journal.pone.0017883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MN. 2004. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem 279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 17.Miller DJ, Ravikumar K, Shen H, Suh JK, Kerwin SM, Robertus JD. 2002. Structure-based design and characterization of novel platforms for ricin and shiga toxin inhibition. J Med Chem 45:90–98. doi: 10.1021/jm010186s. [DOI] [PubMed] [Google Scholar]
- 18.Stechmann B, Bai SK, Gobbo E, Lopez R, Merer G, Pinchard S, Panigai L, Tenza D, Raposo G, Beaumelle B, Sauvaire D, Gillet D, Johannes L, Barbier J. 2010. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141:231–242. doi: 10.1016/j.cell.2010.01.043. [DOI] [PubMed] [Google Scholar]
- 19.Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, Weibezahn J, Horlbeck MA, Chen S, Mann M, Hyman AA, Leproust EM, McManus MT, Weissman JS. 2013. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 152:909–922. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong J, Zhang Y, Chen Y, Niu X, Li R, Yang C, Wang Q, Li X, Deng X. 2015. Baicalin inhibits the lethality of ricin in mice by inducing protein oligomerization. J Biol Chem 290:12899–12907. doi: 10.1074/jbc.M114.632828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tu W, Cai K, Gao X, Xiao L, Chen R, Shi J, Liu H, Hou X, Wang Q, Wang H. 2009. Improved production of holotoxin Stx2 with biological activities by using a single-promoter vector and an auto-induction expression system. Protein Expr Purif 67:169–174. doi: 10.1016/j.pep.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Endo Y, Tsurugi K. 1987. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem 262:8128–8130. [PubMed] [Google Scholar]
- 23.Challa S, Tzipori S, Sheoran A. 2014. Selective evolution of ligands by exponential enrichment to identify RNA aptamers against Shiga toxins. J Nucleic Acids 2014:214929. doi: 10.1155/2014/214929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Cheng Q, Zhang Y. 2006. Acute toxicity test of baicalin capsule in mice. J Med Res 35:17–19. [Google Scholar]
- 25.Tzipori S, Sheoran A, Akiyoshi D, Donohue-Rolfe A, Trachtman H. 2004. Antibody therapy in the management of shiga toxin-induced hemolytic uremic syndrome. Clin Microbiol Rev 17:926–941. doi: 10.1128/CMR.17.4.926-941.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matussek A, Lauber J, Bergau A, Hansen W, Rohde M, Dittmar KE, Gunzer M, Mengel M, Gatzlaff P, Hartmann M, Buer J, Gunzer F. 2003. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 102:1323–1332. doi: 10.1182/blood-2002-10-3301. [DOI] [PubMed] [Google Scholar]
- 27.Beceiro A, Tomas M, Bou G. 2013. Antimicrobial resistance and virulence: a successful or deleterious association in the bacterial world? Clin Microbiol Rev 26:185–230. doi: 10.1128/CMR.00059-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. 2000. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med 342:1930–1936. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clatworthy AE, Pierson E, Hung DT. 2007. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 30.Slutsker L, Ries AA, Maloney K, Wells JG, Greene KD, Griffin PM. 1998. A nationwide case-control study of Escherichia coli O157:H7 infection in the United States. J Infect Dis 177:962–966. doi: 10.1086/515258. [DOI] [PubMed] [Google Scholar]
- 31.Bitzan M, Poole R, Mehran M, Sicard E, Brockus C, Thuning-Roberson C, Riviere M. 2009. Safety and pharmacokinetics of chimeric anti-Shiga toxin 1 and anti-Shiga toxin 2 monoclonal antibodies in healthy volunteers. Antimicrob Agents Chemother 53:3081–3087. doi: 10.1128/AAC.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Cheng Q, Shen P, Yan W, Li Q, Wang F, He Q. 2007. Empirical study on safety and acute toxicity of baicalin injection. J Med Res 36:15–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.