

Abstract
Background
Different therapeutic strategies are available for the treatment of people with relapsing‐remitting multiple sclerosis (RRMS), including immunomodulators, immunosuppressants and biologics. Although there is consensus that these therapies reduce the frequency of relapses, their relative benefit in delaying new relapses or disability worsening remains unclear due to the limited number of direct comparison trials.
Objectives
To compare the benefit and acceptability of interferon beta‐1b, interferon beta‐1a (Avonex, Rebif), glatiramer acetate, natalizumab, mitoxantrone, fingolimod, teriflunomide, dimethyl fumarate, alemtuzumab, pegylated interferon beta‐1a, daclizumab, laquinimod, azathioprine and immunoglobulins for the treatment of people with RRMS and to provide a ranking of these treatments according to their benefit and acceptability, defined as the proportion of participants who withdrew due to any adverse event.
Search methods
We searched the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group Trials Register, which contains trials from CENTRAL (2014, Issue 9), MEDLINE (1966 to 2014), EMBASE (1974 to 2014), CINAHL (1981 to 2014), LILACS (1982 to 2014), clinicaltrials.gov and the WHO trials registry, and US Food and Drug Administration (FDA) reports. We ran the most recent search in September 2014.
Selection criteria
Randomised controlled trials (RCTs) that studied one or more of the 15 treatments as monotherapy, compared to placebo or to another active agent, for use in adults with RRMS.
Data collection and analysis
Two authors independently identified studies from the search results and performed data extraction. We performed data synthesis by pairwise meta‐analysis and network meta‐analysis. We assessed the quality of the body of evidence for outcomes within the network meta‐analysis according to GRADE, as very low, low, moderate or high.
Main results
We included 39 studies in this review, in which 25,113 participants were randomised. The majority of the included trials were short‐term studies, with a median duration of 24 months. Twenty‐four (60%) were placebo‐controlled and 15 (40%) were head‐to‐head studies.
Network meta‐analysis showed that, in terms of a protective effect against the recurrence of relapses in RRMS during the first 24 months of treatment, alemtuzumab, mitoxantrone, natalizumab, and fingolimod outperformed other drugs. The most effective drug was alemtuzumab (risk ratio (RR) versus placebo 0.46, 95% confidence interval (CI) 0.38 to 0.55; surface under the cumulative ranking curve (SUCRA) 96%; moderate quality evidence), followed by mitoxantrone (RR 0.47, 95% CI 0.27 to 0.81; SUCRA 92%; very low quality evidence), natalizumab (RR 0.56, 95% CI 0.47 to 0.66; SUCRA 88%; high quality evidence), and fingolimod (RR 0.72, 95% CI 0.64 to 0.81; SUCRA 71%; moderate quality evidence).
Disability worsening was based on a surrogate marker, defined as irreversible worsening confirmed at three‐month follow‐up, measured during the first 24 months in the majority of included studies. Both direct and indirect comparisons revealed that the most effective treatments were mitoxantrone (RR versus placebo 0.20, 95% CI 0.05 to 0.84; SUCRA 96%; low quality evidence), alemtuzumab (RR 0.35, 95% CI 0.26 to 0.48; SUCRA 94%; low quality evidence), and natalizumab (RR 0.64, 95% CI 0.49 to 0.85; SUCRA 74%; moderate quality evidence).
Almost all of the agents included in this review were associated with a higher proportion of participants who withdrew due to any adverse event compared to placebo. Based on the network meta‐analysis methodology, the corresponding RR estimates versus placebo over the first 24 months of follow‐up were: mitoxantrone 9.92 (95% CI 0.54 to 168.84), fingolimod 1.69 (95% CI 1.32 to 2.17), natalizumab 1.53 (95% CI 0.93 to 2.53), and alemtuzumab 0.72 (95% CI 0.32 to 1.61).
Information on serious adverse events (SAEs) was scanty, characterised by heterogeneous results and based on a very low number of events observed during the short‐term duration of the trials included in this review.
Authors' conclusions
Conservative interpretation of these results is warranted, since most of the included treatments have been evaluated in few trials. The GRADE approach recommends providing implications for practice based on moderate to high quality evidence. Our review shows that alemtuzumab, natalizumab, and fingolimod are the best choices for preventing clinical relapses in people with RRMS, but this evidence is limited to the first 24 months of follow‐up. For the prevention of disability worsening in the short term (24 months), only natalizumab shows a beneficial effect on the basis of moderate quality evidence (all of the other estimates were based on low to very low quality evidence). Currently, therefore, insufficient evidence is available to evaluate treatments for the prevention of irreversible disability worsening.
There are two additional major concerns that have to be considered. First, the benefit of all of these treatments beyond two years is uncertain and this is a relevant issue for a disease with a duration of 30 to 40 years. Second, short‐term trials provide scanty and poorly reported safety data and do not provide useful evidence in order to obtain a reliable risk profile of treatments. In order to provide long‐term information on the safety of the treatments included in this review, it will be necessary also to evaluate non‐randomised studies and post‐marketing reports released from the regulatory agencies. Finally, more than 70% of the studies included in this review were sponsored by pharmaceutical companies and this may have influenced the results.
There are three needs that the research agenda should address. First, randomised trials of direct comparisons between active agents would be useful, avoiding further placebo‐controlled studies. Second, follow‐up of the original trial cohorts should be mandatory. Third, more studies are needed to assess the medium and long‐term benefit and safety of immunotherapies and the comparative safety of different agents.
Plain language summary
Immunomodulators and immunosuppressants for relapsing‐remitting multiple sclerosis: a network meta‐analysis
Background
Different therapeutic strategies are available for the treatment of people with relapsing‐remitting multiple sclerosis (RRMS), including immunomodulators, immunosuppressants, and biologics. Although there is consensus that these therapies may reduce the frequency of relapses, their relative benefit (effectiveness compared to each other) in delaying new relapses or disability worsening remains unclear due to the limited number of direct comparison studies (i.e. studies comparing two or more active agents with each other).
Objectives
We aimed to assess and rank the benefit from and the extent of adverse events associated with 15 drugs, i.e. interferon beta‐1b, interferon beta‐1a (Avonex, Rebif), glatiramer acetate, natalizumab, mitoxantrone, fingolimod, teriflunomide, dimethyl fumarate, alemtuzumab, pegylated interferon beta‐1a, daclizumab, laquinimod, azathioprine, and immunoglobulins.
Study characteristics
We included 39 studies up to September 2014 in this review, comprising a total of 25,113 participants suffering from RRMS. The majority of the included studies were short‐term, with a median duration of 24 months.
Key results and quality of the evidence
For preventing relapses, alemtuzumab, natalizumab, and fingolimod are more effective than the other drugs, based on moderate to high quality evidence.
For preventing irreversible disability worsening, insufficient evidence is currently available.
Almost all of the agents included in this review were associated with a higher proportion of participants who withdrew due to any adverse event compared to placebo.
It is worth noting the following:
‐ The benefit of all of these treatments beyond two years is uncertain and this is a very relevant issue for people with a lifelong disease such as multiple sclerosis, who will possibly need long‐term treatments.
‐ Safety data from these short‐term studies are scanty, poorly reported and cannot provide enough evidence for us to obtain a reliable risk profile of the treatments included in this review.
‐ Most of the included studies were sponsored by pharmaceutical companies and this is a known potential source of bias.
Summary of findings
Summary of findings for the main comparison. Summary of findings for the main comparisons of treatment effects against placebo.
|
Patient or population: patients with relapsing‐remitting multiple sclerosis (RRMS) Settings: secondary healthcare centres Intervention: any immunomodulators or immunosuppressants used for RRMS Comparison: placebo | |||||||
| Intervention | Illustrative comparative risks* | Relative effect (95% CI) | SUCRA | No of participants (studies)# | Confidence in the evidence (GRADE) | Reasons for downgrading our confidence in the evidence° | |
| Assumed risk with placebo | Corresponding risk with intervention (95% CI) | ||||||
| CHANCE OF EXPERIENCING ONE OR MORE RELAPSES OVER 12 MONTHS | |||||||
| Alemtuzumab | Low |
RR 0.40 (0.31 to 0.51) |
97% | — | Moderate | Downgraded one level due to risk of bias ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains | |
| 41 per 100 | 16 per 100 (13 to 21) | ||||||
| High | |||||||
| 89 per 100 | 36 per 100 (28 to 45) | ||||||
| Mitoxantrone | Low |
RR 0.40 (0.20 to 0.76) |
93% | 51 (1 study) | Low | Downgraded two levels due to risk of bias ‐ the singular study contributing to this estimate at high risk of bias in blinding of outcome assessor domain | |
| 41 per 100 | 16 per 100 (8 to 31) | ||||||
| High | |||||||
| 89 per 100 | 36 per 100 (18 to 68) | ||||||
| Natalizumab | Low |
RR 0.56 (0.43 to 0.73) |
85% | 942 (1 study) | High | — | |
| 41 per 100 | 23 per 100 (18 to 30) | ||||||
| High | |||||||
| 89 per 100 | 50 per 100 (38 to 65) | ||||||
| Fingolimod | Low |
RR 0.63 (0.53 to 0.74) |
80% | 2355 (2 studies) | Low | Downgraded one level due to risk of bias and one level due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; I2 = 82% (P value = 0.02) | |
| 41 per 100 | 26 per 100 (22 to 30) | ||||||
| High | |||||||
| 89 per 100 | 56 per 100 (47 to 66) | ||||||
| Dimethyl fumarate | Low |
RR 0.78 (0.65 to 0.93) |
55% | 2307 (2 studies) | Moderate | Downgraded one level due to inconsistency ‐ wide predictive interval | |
| 41 per 100 | 32 per 100 (27 to 38) | ||||||
| High | |||||||
| 89 per 100 | 69 per 100 (58 to 83) | ||||||
| Immunoglobulins | Low |
RR 0.78 (0.61 to 1.00) |
53% | 219 (3 studies) | Very low | Downgraded one level due to risk of bias, two levels due to inconsistency, and one level due to imprecision ‐ the majority of studies at unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; I2 = 83% (P value = 0.003) and differences between pairwise and common τ2 (0.18 versus 0.01); wide CIs | |
| 41 per 100 | 32 per 100 (25 to 41) | ||||||
| High | |||||||
| 89 per 100 | 69 per 100 (54 to 89) | ||||||
| Glatiramer acetate | Low |
RR 0.80 (0.68 to 0.93) |
52% | 2416 (4 studies) | Moderate | Downgraded one level due to inconsistency ‐ wide predictive interval | |
| 41 per 100 | 33 per 100 (28 to 38) | ||||||
| High | |||||||
| 89 per 100 | 71 per 100 (61 to 83) | ||||||
| Daclizumab | Low |
RR 0.79 (0.61 to 1.02) |
52% | 621 (1 study) | Moderate | Downgraded one level due to imprecision ‐ wide CIs | |
| 41 per 100 | 32 per 100 (25 to 42) | ||||||
| High | |||||||
| 89 per 100 | 70 per 100 (54 to 91) | ||||||
| Teriflunomide | Low |
RR 0.84 (0.72 to 0.99) |
42% | 2257 (2 studies) | Low | Downgraded one level due to risk of bias and one level due to inconsistency ‐ the majority of studies at unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; wide predictive interval | |
| 41 per 100 | 34 per 100 (30 to 41) | ||||||
| High | |||||||
| 89 per 100 | 75 per 100 (64 to 88) | ||||||
| Azathioprine | Low |
RR 0.87 (0.58 to 1.31) |
39% | 59 (1 study) | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and two levels due to imprecision ‐ the singular study contributing to this estimate at unclear risk of bias in allocation concealment domain; indirectness of population (one monocentric study); wide CIs | |
| 41 per 100 | 36 per 100 (24 to 54) | ||||||
| High | |||||||
| 89 per 100 | 77 per 100 (52 to 100) | ||||||
| Interferon beta‐1a (Rebif) | Low |
RR 0.87 (0.76 to 1.01) |
36% | 853 (2 studies) | Low | Downgraded one level due to risk of bias and one level due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; I2 = 88% (P value = 0.004) | |
| 41 per 100 | 36 per 100 (31 to 41) | ||||||
| High | |||||||
| 89 per 100 | 77 per 100 (68 to 90) | ||||||
| Pegylated interferon beta‐1a | Low |
RR 0.89 (0.70 to 1.13) |
33% | 1512 (1 study) | Low | Downgraded one level due to risk of bias and one level due to imprecision ‐ the singular study contributing to this estimate at unclear risk of bias in blinding of outcome assessor domain; wide CIs | |
| 41 per 100 | 36 per 100 (29 to 46) | ||||||
| High | |||||||
| 89 per 100 | 79 per 100 (62 to 100) | ||||||
| Interferon beta‐1b (Betaseron) | Low |
RR 0.98 (0.54 to 1.75) |
27% | — | Very low | Downgraded one level due to risk of bias and two levels due to imprecision ‐ the majority of studies at unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; wide CIs | |
| 41 per 100 | 40 per 100 (22 to 72) | ||||||
| High | |||||||
| 89 per 100 | 87 per 100 (48 to 100) | ||||||
| Interferon beta‐1a (Avonex) | Low |
RR 0.93 (0.78 to 1.10) |
25% | 301 (1 study) | Moderate | Downgraded one level due to risk of bias ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains | |
| 41 per 100 | 38 per 100 (32 to 45) | ||||||
| High | |||||||
| 89 per 100 | 83 per 100 (69 to 98) | ||||||
| Interferons beta (Avonex, Rebif or Betaseron) | Low |
RR 1.05 (0.61 to 1.79) |
20% | — | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and two levels due to imprecision ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; indirectness of population (one monocentric study contributing 50% to this estimate); wide CIs | |
| 41 per 100 | 43 per 100 (25 to 73) | ||||||
| High | |||||||
| 89 per 100 | 93 per 100 (54 to 100) | ||||||
| CHANCE OF EXPERIENCING ONE OR MORE RELAPSES OVER 24 MONTHS | |||||||
| Alemtuzumab | Low |
RR 0.46 (0.38 to 0.55) |
96% | — | Moderate | Downgraded one level due to risk of bias ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains | |
| 57 per 100 | 26 per 100 (22 to 31) | ||||||
| High | |||||||
| 85 per 100 | 39 per 100 (32 to 47) | ||||||
| Mitoxantrone | Low |
RR 0.47 (0.27 to 0.81) |
92% | 51 (1 study) | Very low | Downgraded two levels due to risk of bias and one level due to inconsistency ‐ the singular study contributing to this estimate at high risk of bias in blinding of outcome assessor domain; wide predictive interval | |
| 57 per 100 | 27 per 100 (15 to 46) | ||||||
| High | |||||||
| 85 per 100 | 40 per 100 (23 to 69) | ||||||
| Natalizumab | Low |
RR 0.56 (0.47 to 0.66) |
88% | 942 (1 study) | High | — | |
| 57 per 100 | 32 per 100 (27 to 38) | ||||||
| High | |||||||
| 85 per 100 | 48 per 100 (40 to 56) | ||||||
| Fingolimod | Low |
RR 0.72 (0.64 to 0.81) |
71% | 2355 (2 studies) | Moderate | Downgraded one level due to risk of bias ‐ studies at unclear risk of bias in allocation concealment domain | |
| 57 per 100 | 41 per 100 (36 to 46) | ||||||
| High | |||||||
| 85 per 100 | 61 per 100 (54 to 69) | ||||||
| Immunoglobulins | Low |
RR 0.74 (0.60 to 0.91) |
66% | 190 (2 studies) | Moderate | Downgraded one level due to inconsistency ‐ wide predictive interval | |
| 57 per 100 | 42 per 100 (34 to 52) | ||||||
| High | |||||||
| 85 per 100 | 63 per 100 (51 to 77) | ||||||
| Azathioprine | Low |
RR 0.77 (0.55 to 1.07) |
57% | 59 (1 study) | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and one level due to imprecision ‐ the singular study contributing to this estimate at unclear risk of bias in allocation concealment domain; indirectness of population (one monocentric study); wide CIs | |
| 57 per 100 | 44 per 100 (31 to 61) | ||||||
| High | |||||||
| 85 per 100 | 65 per 100 (47 to 91) | ||||||
| Glatiramer acetate | Low |
RR 0.83 (0.75 to 0.91) |
48% | 1024 (3 studies) | Moderate | Downgraded one level due to inconsistency ‐ wide predictive interval | |
| 57 per 100 | 47 per 100 (43 to 52) | ||||||
| High | |||||||
| 85 per 100 | 71 per 100 (64 to 77) | ||||||
| Interferon beta‐1b (Betaseron) | Low |
RR 0.85 (0.77 to 0.94) |
42% | 372 (1 study) | Very low | Downgraded one level due to risk of bias and two levels due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; wide predictive interval and inconsistent loops of evidence | |
| 57 per 100 | 48 per 100 (44 to 54) | ||||||
| High | |||||||
| 85 per 100 | 72 per 100 (65 to 80) | ||||||
| Interferon beta‐1a (Rebif) | Low |
RR 0.86 (0.77 to 0.95) |
39% | 560 (1 study) | Low | Downgraded one level due to risk of bias and one level due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; wide predictive interval | |
| 57 per 100 | 49 per 100 (44 to 54) | ||||||
| High | |||||||
| 85 per 100 | 73 per 100 (65 to 81) | ||||||
| Interferons beta (Avonex, Rebif or Betaseron) | Low |
RR 0.89 (0.56 to 1.42) |
33% | — | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and one level due to imprecision ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; indirectness of population (one monocentric study contributing for 50% to this estimate); wide CIs | |
| 57 per 100 | 51 per 100 (32 to 81) | ||||||
| High | |||||||
| 85 per 100 | 76 per 100 (48 to 100) | ||||||
| Teriflunomide | Low |
RR 0.88 (0.75 to 1.03) |
32% | 1088 (1 study) | Very low | Downgraded two levels due to risk of bias and one level due to imprecision ‐ the singular study contributing to this estimate at high risk of bias in blinding of outcome assessor domain; wide CIs | |
| 57 per 100 | 50 per 100 (43 to 59) | ||||||
| High | |||||||
| 85 per 100 | 75 per 100 (64 to 88) | ||||||
| Laquinimod | Low |
RR 0.88 (0.79 to 0.99) |
31% | 1990 (2 studies) | Very low | Downgraded one level due to risk of bias and two levels due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; I2 = 66% (P value = 0.09), wide predictive interval and inconsistent loops of evidence | |
| 57 per 100 | 50 per 100 (45 to 56) | ||||||
| High | |||||||
| 85 per 100 | 75 per 100 (67 to 84) | ||||||
| Dimethyl fumarate | Low |
RR 0.89 (0.81 to 0.98) |
30% | 2307 (2 studies) | Moderate | Downgraded one level due to inconsistency ‐ wide predictive interval | |
| 57 per 100 | 51 per 100 (46 to 56) | ||||||
| High | |||||||
| 85 per 100 | 76 per 100 (69 to 83) | ||||||
| Interferon beta‐1a (Avonex) | Low |
RR 0.91 (0.82 to 1.02) |
22% | 1198 (2 studies) | Low | Downgraded one level due to risk of bias and one level due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; inconsistent loops of evidence | |
| 57 per 100 | 52 per 100 (47 to 58) | ||||||
| High | |||||||
| 85 per 100 | 77 per 100 (70 to 87) | ||||||
| CHANCE OF DISABILITY GETTING WORSE OVER 24 MONTHS | |||||||
| Mitoxantrone | Low |
RR 0.20 (0.05 to 0.84) |
96% | 51 (1 study) | Low | Downgraded one level due to indirectness and one level due to inconsistency ‐ surrogate outcome unclear; wide predictive interval | |
| 25 per 100 | 5 per 100 (1 to 21) | ||||||
| High | |||||||
| 52 per 100 | 10 per 100 (3 to 44) | ||||||
| Alemtuzumab | Low |
RR 0.35 (0.26 to 0.48) |
94% | — | Low | Downgraded one level due to risk of bias and one level due to indirectness ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; surrogate outcome in the majority of studies contributing to this estimate | |
| 25 per 100 | 9 per 100 (6 to 12) | ||||||
| High | |||||||
| 52 per 100 | 18 per 100 (14 to 25) | ||||||
| Natalizumab | Low |
RR 0.64 (0.49 to 0.85) |
74% | 942 (1 study) | Moderate | Downgraded one level due to indirectness ‐ surrogate outcome | |
| 25 per 100 | 16 per 100 (12 to 21) | ||||||
| High | |||||||
| 52 per 100 | 33 per 100 (25 to 44) | ||||||
| Azathioprine | Low |
RR 0.64 (0.30 to 1.37) |
64% | 59 (1 study) | Very low | Downgraded one level due to risk of bias, two levels due to indirectness, and two levels due to imprecision ‐ the singular study contributing to this estimate at unclear risk of bias in allocation concealment domain; indirectness of population (one monocentric study) and surrogate outcome unclear; wide CIs | |
| 25 per 100 | 16 per 100 (8 to 34) | ||||||
| High | |||||||
| 52 per 100 | 33 per 100 (16 to 71) | ||||||
| Glatiramer acetate | Low |
RR 0.77 (0.64 to 0.92) |
58% | 1024 (3 studies) | Very low | Downgraded one level due to indirectness and two levels due to inconsistency ‐ surrogate outcome in the majority of studies contributing to this estimate; wide predictive interval and inconsistent loops of evidence | |
| 25 per 100 | 19 per 100 (16 to 23) | ||||||
| High | |||||||
| 52 per 100 | 40 per 100 (33 to 48) | ||||||
| Immunoglobulins | Low |
RR 0.70 (0.39 to 1.27) |
56% | 190 (2 studies) | Very low | Downgraded one level due to indirectness, one level due to inconsistency, and two levels due to imprecision ‐ surrogate outcome in the majority of studies contributing to this estimate; wide predictive interval; wide CIs | |
| 25 per 100 | 18 per 100 (10 to 32) | ||||||
| High | |||||||
| 52 per 100 | 36 per 100 (20 to 66) | ||||||
| Interferon beta‐1b (Betaseron) | Low |
RR 0.79 (0.65 to 0.97) |
51% | 372 (1 study) | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and two levels due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; surrogate outcome in the majority of studies contributing to this estimate; wide predictive interval and inconsistent loops of evidence | |
| 25 per 100 | 20 per 100 (16 to 24) | ||||||
| High | |||||||
| 52 per 100 | 41 per 100 (34 to 50) | ||||||
| Dimethyl fumarate | Low |
RR 0.80 (0.67 to 0.94) |
50% | 2307 (2 studies) | Low | Downgraded one level due to indirectness and one level due to inconsistency ‐ surrogate outcome in the majority of studies contributing to this estimate; wide predictive interval | |
| 25 per 100 | 20 per 100 (17 to 23) | ||||||
| High | |||||||
| 52 per 100 | 42 per 100 (35 to 49) | ||||||
| Interferons beta (Avonex, Rebif or Betaseron) | Low |
RR 0.83 (0.34 to 2.07) |
40% | — | Very low | Downgraded one level due to indirectness, one level due to inconsistency, and two levels due to imprecision ‐ indirectness of population and surrogate outcome unclear (one study contributing for 50% to this estimate); wide predictive interval; wide CIs | |
| 25 per 100 | 21 per 100 (9 to 52) | ||||||
| High | |||||||
| 52 per 100 | 43 per 100 (18 to 100) | ||||||
| Interferon beta‐1a (Rebif) | Low |
RR 0.86 (0.69 to 1.06) |
36% | 560 (1 study) | Very low | Downgraded one level due to risk of bias, one level due to indirectness, one level due to inconsistency, and one level due to imprecision ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; surrogate outcome in the majority of studies contributing to this estimate; inconsistent loops of evidence; wide CIs | |
| 25 per 100 | 22 per 100 (17 to 26) | ||||||
| High | |||||||
| 52 per 100 | 45 per 100 (36 to 55) | ||||||
| Fingolimod | Low |
RR 0.86 (0.73 to 1.03) |
34% | 2355 (2 studies) | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and one level due to imprecision ‐ studies at unclear risk of bias in allocation concealment domain; surrogate outcome; wide CIs | |
| 25 per 100 | 22 per 100 (18 to 26) | ||||||
| High | |||||||
| 52 per 100 | 45 per 100 (38 to 54) | ||||||
| Laquinimod | Low |
RR 0.87 (0.72 to 1.04) |
34% | 1990 (2 studies) | Low | Downgraded one level due to indirectness and one level due to imprecision ‐ surrogate outcome in the majority of studies contributing to this estimate; wide CIs | |
| 25 per 100 | 22 per 100 (18 to 26) | ||||||
| High | |||||||
| 52 per 100 | 45 per 100 (37 to 54) | ||||||
| Teriflunomide | Low |
RR 0.87 (0.69 to 1.10) |
34% | 1088 (1 study) | Low | Downgraded one level due to indirectness and one level due to imprecision ‐ surrogate outcome; wide CIs | |
| 25 per 100 | 22 per 100 (17 to 28) | ||||||
| High | |||||||
| 52 per 100 | 45 per 100 (36 to 57) | ||||||
| Interferon beta‐1a (Avonex) | Low |
RR 0.93 (0.77 to 1.13) |
21% | 1198 (2 studies) | Very low | Downgraded one level due to risk of bias, one level due to indirectness, and two levels due to inconsistency ‐ the majority of studies at high or unclear risk of bias in allocation concealment and/or blinding of outcome assessor domains; surrogate outcome in the majority of studies contributing to this estimate; I2 = 57% (P value = 0.13), and inconsistent loops of evidence | |
| 25 per 100 | 23 per 100 (19 to 28) | ||||||
| High | |||||||
| 52 per 100 | 48 per 100 (40 to 59) | ||||||
| *The corresponding risk with intervention (and its 95% confidence interval) is based on the assumed risk with placebo and the relative effect of the intervention (and its 95% CI). Two values were chosen for the assumed risk with placebo, i.e. the second highest and second lowest placebo group risks in the included studies, defined as low and high assumed risk. #No of Participants (studies) is not available when the nature of the evidence is indirect. °We did not downgrade for reasons of reporting bias as insufficient studies contributed to network treatment estimates to draw meaningful conclusions. CI: confidence interval; RR: risk ratio; SUCRA: surface under the cumulative ranking curve | |||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||||
Background
Description of the condition
Multiple sclerosis (MS) is an inflammatory disease of the brain and spinal cord resulting from interaction between unidentified environmental factors and susceptibility genes. Several pathological processes occur in MS, involving the immune system, T‐cell‐mediated and B‐cell‐mediated mechanisms, demyelination, remyelination, microglial activation, and chronic neurodegeneration (Bennett 2009; Compston 2008). The sequential involvement of these processes influences the clinical course, which is characterised by attacks of neurological dysfunction with recovery, attacks leaving persistent deficits, and progression that causes permanent physical and cognitive disability. MS is among the most common causes of neurological disability in young people, with an annual incidence ranging from 2 to 10 cases per 100,000 persons per year and a north‐south gradient, with lower incidence closer to the equator. Its clinical manifestations typically occur between 20 and 40 years of age, with symptoms and signs involving different regions of the central nervous system: optic nerve, brainstem, cerebellum, cerebral hemispheres, and spinal cord.
MS has a chronic course that evolves over 30 to 40 years. The clinical phenotypes include relapsing‐remitting MS (RRMS), secondary‐progressive MS (SPMS), primary‐progressive MS (PPMS), and progressive‐relapsing MS (PRMS) (Lublin 1996). The development of progression after a relapsing‐remitting course is responsible for permanent long‐term disability; it supervenes in about 80% of RRMS people by 20 to 25 years from disease onset (Kremenchutzky 2006). Times to need assistance to walk, be confined to bed, or have died were 14, 24, and 45 median years from disease onset and 3, 12, and 30 median years from onset of secondary progression, respectively (Scalfari 2014).
Male sex, older age at onset, and high early relapse frequency (more than three attacks during the first three years) predict higher risk of unremitting disability worsening (Scalfari 2014). In people with RRMS, the onset of secondary progression is the determinant of long‐term prognosis, and its prevention is the key therapeutic goal.
According to the older Poser criteria (Poser 1983), MS can be clinically diagnosed by demonstrating two separate clinical attacks (dissemination in time) involving at least two different areas of the central nervous system (dissemination in space). The 2001 McDonald criteria and their 2005 and 2010 revisions incorporate magnetic resonance imaging (MRI) criteria for dissemination in space and time, allowing a MS diagnosis at the time of first symptoms (McDonald 2001; Polman 2005; Polman 2011). Dissemination in space is demonstrated by greater than or equal to one MRI lesion in at least two MS typical central nervous system regions (periventricular, juxtacortical, infratentorial, spinal cord). Dissemination in time is demonstrated by: (i) simultaneous asymptomatic contrast‐enhancing and non‐enhancing MRI lesions at any time; or (ii) a new lesion and/or contrast‐enhancing lesions(s) on follow‐up MRI, irrespective of its timing. The diagnostic criteria include exclusion of other possible diagnoses.
A declining trend in on‐study relapse rate (one of the most commonly used primary outcomes in MS trials) of placebo participants in trials has been observed (Inusah 2010; Nicholas 2012; Steinvorth 2013; Stellmann 2012). This decline is thought to result from decreasing pretrial relapse rates and a shorter time period over which pretrial relapse rates were calculated in recent trials (Steinvorth 2013; Stellmann 2012). Pre‐study relapse rate was found to be the best predictor for on‐study relapse rate. Other participant characteristics have changed in newer trials. Participants were older and had a longer disease duration, whereas their baseline Expanded Disability Status Scale (EDSS) scores were similar to those reported in the older trials. In newer trials the introduction of the new McDonald diagnostic criteria led to inclusion of participants who had had earlier diagnosis and were later in their disease course, which was less severe compared to people in older studies (Steinvorth 2013). These changes may explain the decrease in pretrial relapse rate and the associated decrease in on‐trial relapse rate. Unwelcome consequences of the expected decreased relapse rate were that the sample size of newer trials has been inflated and follow‐up periods shortened.
Another difference between older and newer studies is that the latter may have included participants who had made prior use of immunomodulators or immunosuppressants.
Description of the intervention
Several treatments are available for people with RRMS. For this review we considered all immunomodulators and immunosuppressants that, since 1966 up to September 2014, have been studied in people with RRMS in randomised clinical trials (RCTs) with more than six months' follow‐up.
Interferon beta‐1b (EMEA 2002; FDA 1993), interferon beta‐1a (Rebif) (EMEA 1998; FDA 2002), interferon beta‐1a (Avonex) (EMEA 1997; FDA 2003), and glatiramer acetate (FDA 1996) were the first agents approved by national regulatory agencies. Interferon beta‐1b, interferon beta‐1a (Rebif), and glatiramer acetate are administered by subcutaneous injection, interferon beta‐1a (Avonex) by intramuscular injection. The main adverse effects of interferons beta are local injection site reactions and flu‐like symptoms with hyperthermia.
Natalizumab was initially approved by the US Food and Drug Administration (FDA) in November 2004 (FDA 2004), but was withdrawn by the manufacturer in February 2005, after three participants in the drug's clinical trials developed progressive multifocal leukoencephalopathy (PML), a rare and serious viral infection of the brain. Two of the participants died. Following a re‐examination of the participants in the previous clinical trials, the FDA allowed a clinical trial of natalizumab to proceed in February 2006. No additional cases of PML were reported and marketing of the drug for severe RRMS resumed (EMA 2006; FDA 2006; Yousry 2006). Natalizumab is administered by intravenous infusion, as a dose of 300 mg every four weeks.
Mitoxantrone was approved in 2000 under the indication "for reducing neurological disability and/or the frequency of clinical relapses in people with worsening RRMS, SPMS or PRMS" (FDA 2000). Safety issues of concern for people treated with mitoxantrone are cardiotoxicity and acute leukaemia.
Fingolimod was the first oral treatment approved for people with RRMS to reduce the frequency of relapses and delay the accumulation of physical disability (EMA 2011; FDA 2010). Even at the recommended low dose of 0.5 mg once daily, the FDA and European Medicines Agency (EMA) warned about decrease in heart rate following initiation of fingolimod treatment, recommending that all patients be monitored for at least six hours for signs and symptoms of bradycardia, considering that in some patients the nadir of heart frequency can be observed up to 24 hours after the first dose.
Teriflunomide was the second oral agent approved for people with RRMS (EMA 2013a; FDA 2012). It is taken orally as a 7 mg or 14 mg tablet once daily. Warnings issued with this drug were hepatotoxicity and risk of teratogenicity.
Dimethyl fumarate has been approved as a first‐line oral treatment for people with RRMS (EMA 2014a; FDA 2013). The recommended dose is 240 mg twice a day. The most commonly reported adverse events leading to discontinuation in clinical trials were flushing and gastrointestinal events.
Alemtuzumab has been approved for treatment of people with RRMS who have had an inadequate response to two or more drugs indicated for the treatment of MS (EMA 2013b; FDA 2014a). The drug is administered by intravenous infusion, as a dose of 12 mg/day for five consecutive days (60 mg total dose) followed by 12 mg/day for three consecutive days (36 mg total dose) administered 12 months after the initial treatment course. Particular warnings and precautions have to be taken into account for the treatment with alemtuzumab, since serious and sometimes fatal autoimmune conditions, life‐threatening infusion reactions, and increased risk of malignancies were observed in people treated with alemtuzumab.
Peg‐interferon beta‐1a, which has been designed to maintain the effects of interferon beta in the body for a longer period of time, was approved by the FDA and EMA for people with RRMS (EMA 2014b; FDA 2014b). It is administered by subcutaneous injection at a dose of 125 µg every 14 days. The most common adverse reactions are injection site erythema, influenza‐like illness, pyrexia, headache, myalgia, chills, injection site pain, asthenia, injection site pruritus, and arthralgia.
Daclizumab is currently being investigated in clinical trials for RRMS, but it has not yet been approved for MS by regulatory agencies. It is administered by subcutaneous or intravenous injections. Risks of serious infections and autoimmune diseases are increased with daclizumab.
Ocrelizumab is in development for the treatment of RRMS, with two active phase clinical trials ongoing. It is administered by intravenous injections.
Laquinimod is an immunomodulator that is currently under evaluation for the treatment of RRMS. It is taken orally as a 0.6 mg tablet once daily. The EMA recommended refusal of the marketing authorisation for laquinimod as a treatment for RRMS due to concerns about potentially increased risks of cancer and teratogenicity in humans, especially given that the drug's mechanism of action is unclear (EMA 2014c). Further studies of laquinimod as a monotherapy and an add‐on therapy in people with RRMS are ongoing.
Azathioprine has been used for the treatment of MS in many countries on the basis of placebo‐controlled RCTs published more than two decades ago. However, since the approval of interferons beta, azathioprine is no longer recommended as first‐line therapy (Goodin 2002). It is taken orally as a 2 mg/kg or 3 mg/kg tablet daily. It was reported that chronic immunosuppression with azathioprine increases the risk of malignancy in humans (FDA 2014c).
Intravenous immunoglobulins may have a role for people with severe and frequent relapses for whom other treatments are contraindicated (Association of British Neurologists 2005). Severe adverse events, including thrombosis of the jugular vein and allergic reaction leading to treatment discontinuation, were noted in 4% of 84 treatment courses with a total 341 infusions under routine clinical conditions (Elovaara 2008).
How the intervention might work
Immunosuppressive or immunomodulatory effects are common to all treatments included in the review.
The mechanism of action of interferons beta in MS is incompletely understood. Interferons beta are naturally occurring cytokines possessing antiviral activity and a wide range of anti‐inflammatory properties. Recombinant forms of interferons beta are believed to directly increase expression and concentration of anti‐inflammatory agents, while down‐regulating the expression of pro‐inflammatory cytokines (Kieseier 2011).
Glatiramer acetate has an immunomodulatory action by inducing tolerance or anergy of myelin‐reactive lymphocytes (Schmied 2003). It is furthermore believed to promote neuroprotective repair processes (Aharoni 2014).
Natalizumab is a monoclonal antibody against the alfa4 integrin on the surface of lymphocytes. This integrin is essential in the process by which lymphocytes gain access to the brain by allowing the cells to penetrate the blood brain barrier. Natalizumab blocks the action of the alfa4 integrin so that lymphocytes are unable to enter the brain and attack myelin protein (Yednock 1992).
Mitoxantrone is a cytotoxic drug that intercalates with DNA and inhibits both DNA and RNA synthesis, thus reducing the number of lymphocytes (Fox 2004).
Fingolimod acts as a functional antagonist of sphingosine‐1‐phosphate(S1P) receptor on lymphocytes, resulting in a reduced egress of lymphocytes from the lymph nodes. In particular, auto‐aggressive T‐cells are prevented from recirculating to the central nervous system (Mandala 2002).
Teriflunomide is an inhibitor of dihydroorotate dehydrogenase (DHODH), a mitochondrial enzyme involved in new pyrimidine synthesis for DNA replication. Consequently, the drug reduces T‐ and B‐lymphocytes activation, proliferation, and function in response to autoantigens. The exact mechanism of action in MS is not fully understood. The drug is thought to reduce the number of activated lymphocytes, which would cause inflammation and damage myelin in the central nervous system (Claussen 2012).
Dimethyl fumarate is a derivative of fumaric acid. It acts primarily by triggering the activation of a nuclear factor (Nrf2) transcriptional pathway, the primary cellular defence against the cytotoxic effects of oxidative stress. It promotes anti‐inflammatory activity and can inhibit expression of pro‐inflammatory cytokines and adhesion molecules (Wilms 2010).
Alemtuzumab is a monoclonal antibody against the CD52 antigen expressed on lymphocytes and monocytes. Its effects in MS are thought to be mediated by an extended lymphocyte depletion and change in the composition of lymphocytes that accompanies lymphocyte reconstitution (Hill‐Cawthorne 2012).
Pegylated interferon beta‐1a has a polyethylene glycol group attached to the α‐amino group of the N terminus of interferon beta‐1a (Avonex). Pegylation of interferon beta‐1a may improve its pharmacokinetic and pharmacodynamic properties, allowing for reduced dosing frequency while maintaining the clinical effectiveness and safety of the intramuscular interferon beta‐1a (Hu 2012).
Daclizumab is a monoclonal antibody against the CD25 antigen (interleukin 2 receptor) expressed on immune cells. The mechanisms by which the drug exerts effects in MS are not clear. Daclizumab leads to expansion of regulatory CD56 natural killer T lymphocytes, which may be an important mechanism of action in MS. Furthermore, daclizumab modulates the function of dendritic cells, resulting in decreased T‐cell activation (Wuest 2011).
Ocrelizumab is a monoclonal antibody against the CD20 antigen expressed on B‐lymphocytes. The antibody depletes circulating B‐lymphocytes predominately through antibody‐mediated cytotoxicity (Oh 2013).
Exactly how laquinimod works is unknown, but it is believed to have an immunomodulatory effect on the peripheral and central nervous systems. Data from animal studies indicate that laquinimod has a primary effect on innate immunity. The drug modulates the function of various myeloid antigen‐presenting cell populations, which then down regulate pro‐inflammatory T‐cell responses. Furthermore, data indicate that laquinimod acts directly on resident cells within the central nervous system to reduce demyelination and axonal damage (Varrin‐Doyer 2014).
Azathioprine is a classical cytotoxic immunosuppressive drug that acts as a prodrug for mercaptopurine, inhibiting an enzyme that is required for DNA synthesis. Thus it most strongly affects proliferating cells, such as the T‐cells and B‐cells of the immune system (Tiede 2003).
The mechanism of action of intravenous immunoglobulins in MS remains unclear, although remyelination of demyelinated axons may occur through the mediation of the effects of cytokines (Stangel 1999).
Why it is important to do this review
Although there is consensus that immunotherapies reduce the frequency of relapses in MS, their relative benefit in delaying new relapses or disability worsening remains unclear. This uncertainty is due to the limited number of direct comparison trials, which provide the most rigorous and valid research evidence on the relative benefit and safety of different, competing treatments. A summary of the results, including both direct and indirect comparisons, may help to clarify the stated uncertainty (Caldwell 2005; Glenny 2005).
Objectives
To compare the benefit and acceptability of interferon beta‐1b, interferon beta‐1a (Avonex, Rebif), glatiramer acetate, natalizumab, mitoxantrone, fingolimod, teriflunomide, dimethyl fumarate, alemtuzumab, pegylated interferon beta‐1a, daclizumab, laquinimod, azathioprine and immunoglobulins for the treatment of people with RRMS and to provide a ranking of these treatments according to their benefit and acceptability, defined as the proportion of participants who withdrew due to any adverse event.
Methods
Criteria for considering studies for this review
Types of studies
We included all RCTs that studied one or more of the agents for use in RRMS and compared them to placebo or to another active agent. We also included trials for which it was unclear whether the method of randomisation provided adequate allocation concealment or open‐label studies, but we took the quality of these studies into account. We excluded RCTs with follow‐up of less than or equal to six months because these trials measured too short‐term outcomes that are not clinically relevant to patients with MS. We excluded non‐randomised studies.
Types of participants
We included participants 18 years of age or older with a diagnosis of RRMS according to Poser (Poser 1983) or McDonald (McDonald 2001; Polman 2005; Polman 2011) diagnostic criteria. We included all participants regardless of sex, degree of disability, and disease duration.
Types of interventions
We included all immunomodulators or immunosuppressants (even if they were not licensed in any country). We excluded: (i) combination treatments; (ii) trials in which a drug regimen was compared with a different regimen of the same drug without another active agent or placebo as a control arm; (iii) all non‐pharmacological treatments; and (iv) interventions with over‐the‐counter drugs.
We included RCTs that evaluated one or more of the following pharmacological interventions as monotherapy, compared to placebo or to another active agent:
interferon beta‐1b
interferon beta‐1a (Avonex, Rebif)
glatiramer acetate
natalizumab
mitoxantrone
fingolimod
teriflunomide
dimethyl fumarate
alemtuzumab
pegylated interferon beta‐1a
daclizumab
ocrelizumab
laquinimod
azathioprine
immunoglobulins
We included regimens as defined in primary studies irrespective of their dose.
We assumed that any patient who met the inclusion criteria was, in principle, equally likely to have been randomised to any of the eligible interventions.
Types of outcome measures
Primary outcomes
We estimated the relative effects of the competing interventions according to the following primary outcomes:
Benefit
Relapses: proportion of participants who experienced new relapses over 12, 24, or 36 months after randomisation or at the end of the study. A relapse is defined as newly developed or recently worsened symptoms of neurologic dysfunction that last for at least 24 hours, occurring in the absence of fever or other acute diseases and separated in time from any previous episode by more than 30 days (McDonald 2001; Polman 2005). A more stringent 48‐hour criterion has been used in some RCTs. A relapse can resolve either partially or completely.
Disability worsening: proportion of participants who experienced disability worsening over 24 or 36 months after randomisation or at the end of the study. Worsening is defined as at least a 1‐point Expanded Disability Status Scale (EDSS) increase or a 0.5‐point increase if the baseline EDSS was greater than or equal to 5.5, confirmed during two subsequent neurological examinations separated by at least a six‐month interval free of attacks (Kurtzke 1983). Disability worsening confirmed after only three months of follow‐up is considered a surrogate marker for unremitting disability. The EDSS is a common measure of MS disability (where 0 is normal, 3 mild disability, 6 care requirement, 7 wheelchair use, and 10 is death from MS) and is used to measure disability worsening in clinical trials for MS.
Acceptability
We used treatment discontinuation due to adverse events to assess acceptability and we measured it by the number of participants who withdrew due to any adverse event over 12, 24, or 36 months after randomisation or at the end of the study out of the total number of participants randomly assigned to each treatment arm.
Secondary outcomes
The total number of serious adverse events (SAEs). If not enough studies reported the total number of SAEs and person‐years, we planned to use the number of participants with at least one SAE as defined in the study.
Search methods for identification of studies
We searched for all possible comparisons formed by the interventions of interest. We applied no language restrictions to the search.
Electronic searches
The Trials Search Co‐ordinator searched the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group Trials Register (30 September 2014) which, among other sources, contains trials from:
Cochrane Central Register of Controlled Trials (CENTRAL 2014, Issue 9);
MEDLINE (PubMed) (1966 to 30 September 2014);
EMBASE (EMBASE.com) (1974 to 30 September 2014);
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (EBSCOhost) (1981 to 30 September 2014);
Latin American and Caribbean Health Science Information Database (LILACS) (Bireme) (1982 to 30 September 2014);
-
Clinical trial registries:
clinicaltrials.gov;
World Health Organization (WHO) International Clinical Trials Registry Portal (apps.who.int/trialsearch/).
Information on the Trials Register of the Review Group and details of the search strategies used to identify trials can be found in the 'Specialised Register' section within the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group module.
The keywords used to search for trials for this review are listed in Appendix 1.
Searching other resources
We extended the search to other resources, including:
contact with principal authors of the included trials for additional information;
searching FDA reports on all of the treatments included in this review (www.fda.gov).
Data collection and analysis
Selection of studies
We used the search strategy described above to obtain titles and abstracts of studies that were relevant to the review. Two review authors independently screened the titles and abstracts and discarded studies that were not applicable; however, we initially retained studies and reviews that might have included relevant data or information on trials. Two review authors independently assessed the retrieved abstracts and, when necessary, the full text of these studies to determine which studies satisfied the inclusion criteria. We compared multiple reports of the same study and used the most comprehensive report. We linked together multiple publications as companion reports, but excluded true duplicates. We resolved discrepancies in judgement by discussion with a third author.
Data extraction and management
Two authors (IP, IT) independently extracted data using a predefined data extraction form in an Excel spreadsheet. We resolved disagreements by discussion with a third author (GF).
Outcome data
We extracted from each included study the number of participants who:
had relapses or disability worsening at 12, 24, and 36 months;
withdrew due to any adverse event at 12, 24, and 36 months;
dropped out at each time point;
had at least one SAE.
We extracted the authors' definition of relapses and disability worsening. We extracted arm‐level data when possible. When arm‐level data were not available we extracted effect sizes.
When outcomes were not reported at the predefined time points, we extracted data as close as possible to that time point. When numbers of dropouts were not reported or unclear in the primary studies, we consulted reports from the FDA or asked the trial author to supply data.
Data on potential effect modifiers
We extracted from each included study data on the following potential effect modifiers:
population: diagnostic criteria (Poser or McDonald criteria), baseline mean age, prior immunomodulator or immunosuppressant treatments (yes, no), definition of relapse, pre‐trial relapse rate and number of years over which the pretrial relapse rate was calculated;
intervention: dose, frequency, or duration of treatment;
risk of bias: allocation concealment, blinding of outcome assessors, incomplete outcome data;
funding source.
Other data
We extracted from each included study data on the following additional information:
study: first author or acronym, number of centres, year of publication, years that the study was conducted (recruitment and follow‐up), publication (full‐text publication, abstract publication, unpublished data);
study design: inclusion criteria, number of randomised participants, duration of follow‐up (12, 24, or 36 months), sequence generation, blinding of participants, selective outcome reporting, early termination of trial.
Assessment of risk of bias in included studies
We assessed the risk of bias of each included study using The Cochrane Collaboration criteria (Higgins 2011). These include: random sequence generation, allocation concealment, blinding of participants, blinding of outcome assessor, incomplete outcome data, and selective outcome reporting. Other potential risks of bias included the role of the sponsor. We explicitly judged the risk of bias of each study on each criterion and classified it as at 'low', 'high', or 'unclear' risk of bias. We judged incomplete outcome data at low risk of bias when numbers and causes of dropouts were balanced (i.e. in the absence of a significant difference) between arms and appeared to be unrelated to the studied outcomes. Furthermore, we stated for each included study and for each outcome the accuracy of reporting dropouts, i.e. identifying studies that provided (or did not provide) complete and clear reporting of dropout data. We assessed selective outcome reporting bias by comparing outcomes reported in the study protocol along with published outcome results. When a study protocol was not available, we assigned low risk of bias if the study results included the two primary outcomes relevant to the review, i.e. relapse and disability worsening.
To summarise the quality of the evidence we considered allocation concealment, blinding of outcome assessor, and incomplete outcome data in order to classify each study as at: low risk of bias when we judged all of the three criteria as at low risk of bias; high risk of bias when we judged at least one criterion as at high risk of bias; unclear risk of bias when we judged all of the three criteria as at unclear risk of bias; and moderate risk of bias in the remaining cases.
We assessed characteristics associated with the monitoring and reporting of adverse events considering specific factors that may have a large influence on adverse event data. We evaluated methods of monitoring and detecting adverse events in each primary study: Did the researchers actively monitor for adverse events, or did they simply provide spontaneous reporting of adverse events that arose? Did the authors define adverse events according to an accepted international classification and report the number of SAEs? We reported this information in an additional table called 'Assessment of Adverse Events Monitoring'.
Three authors (IP, IT, GF) assessed the risk of bias of each study independently andresolved any disagreement by discussion to reach consensus.
Measures of treatment effect
Relative treatment effects
We estimated, through pairwise meta‐analysis, the treatment effects of the competing interventions using risk ratio (RR) with 95% confidence intervals (95% CI) for each outcome at each time point. We presented results from network meta‐analysis as summary relative effect sizes (RR) for each possible pair of treatments.
Relative treatment ranking
We estimated the ranking probabilities for all treatments of being at each possible rank for each intervention. We then obtained a treatment hierarchy using the surface under the cumulative ranking curve (SUCRA) and mean ranks. SUCRA can also be expressed as the percentage of benefit/acceptability of a treatment that would be ranked first without uncertainty (Salanti 2011).
Unit of analysis issues
Cluster and cross‐over trials have not been carried out to evaluate immunomodulator and immunosuppressant treatments for MS.
We performed separate analyses for participants who had relapses at 12, 24, and 36 months and disability worsening at 24 and 36 months.
Studies with multiple treatment groups
For multi‐arm trials, the intervention groups of relevance were all those that could be included in a pairwise comparison of intervention groups which, if investigated alone, would have met the criteria for including studies in the review. For example, if we identified a study comparing 'interferon beta versus natalizumab versus interferon beta plus natalizumab', only one comparison ('interferon beta versus natalizumab') addresses the review objective, and no comparison involving combination therapy does. Thus, the 'interferon beta plus natalizumab' therapy group was not relevant to the review. However, if the study had compared 'interferon beta‐1b versus interferon beta‐1a (Rebif) versus interferon beta‐1a (Avonex)', all three pairwise comparisons of interventions are relevant to the review. In this case we treated the multi‐arm studies as multiple independent two‐arm studies in pairwise meta‐analysis; we accounted for the correlation between the effect sizes from multi‐arm studies in network meta‐analysis. We converted multi‐arm trials involving the same agent at different doses compared to a control treatment into a single arm by merging of doses and summing the number of events and the sample size.
Dealing with missing data
In order to assess the effect of missing outcome data, we analysed data according to a likely scenario, i.e. we assumed that treated and control group participants who contributed to missing outcome data both had an unfavourable outcome (relapse or disability worsening).
Assessment of heterogeneity
Assessment of clinical heterogeneity within treatment comparisons
To evaluate the presence of heterogeneity deriving from different characteristics of study participants, we assessed differences in age, disease duration, and baseline EDSS scores across the trials using information reported in the table 'Characteristics of included studies'.
Assessment of transitivity across treatment comparisons
We expected that the transitivity assumption held, assuming that all pairwise comparisons did not differ with respect to the distribution of effect modifiers. We evaluated the assumption of transitivity by comparing potential effect modifiers, which are reported in the 'Data extraction and management' section, across the different pairwise comparisons.
Assessment of reporting biases
Considering that it is not mandatory to publish results of clinical trials, it is difficult to have an estimate of the number of unpublished trials in MS. We evaluated the possibility of reporting bias by means of contour‐enhanced funnel plots (Peters 2008). Contour‐enhanced funnel plots show areas of statistical significance, and they can help in distinguishing reporting bias from other possible reasons for asymmetry. In a network of interventions, each study estimates the relative effect of different interventions, so asymmetry in the funnel plot cannot be judged. To account for this, we used an adaptation of the funnel plot by subtracting from each study‐specific effect size the mean of meta‐analysis of the study‐specific comparison and plotted it against the study's standard error (Chaimani 2012; Chaimani 2013). We employed the comparison‐adjusted funnel plot for all placebo‐controlled trials. Note that any asymmetry in the plot indicates the presence of small study effects and not necessarily reporting bias.
Data synthesis
Methods for direct treatment comparisons
We performed conventional pairwise meta‐analyses for each primary outcome using a random‐effects model for each treatment comparison with at least two studies (DerSimonian 1986).
Methods for indirect and mixed comparisons
We performed network meta‐analysis for primary outcomes (relapses, disability worsening, and acceptability), using a random‐effects model within a frequentist setting assuming equal heterogeneity across all comparisons, and we accounted for correlations induced by multi‐arm studies (Miladinovic 2014; Salanti 2012). The models enabled us to estimate the probability for each intervention to be at each possible rank for each outcome, given the relative effect sizes as estimated in network meta‐analysis. We summarised the probabilities of a treatment being at each possible rank using SUCRAs. By using the cluster analysis technique, we grouped the treatments according to the SUCRA values for both benefit and acceptability outcomes and presented them in a plot. We performed network meta‐analysis in Stata 13 using the 'mvmeta' command and self programmed Stata routines available at http://www.mtm.uoi.gr (Chaimani 2013; White 2011; White 2012).
Assessment of statistical heterogeneity
Assumptions when estimating heterogeneity
As we expected to have few studies (around two to four) in each direct comparison, in standard pairwise meta‐analysis we assumed a common heterogeneity variance for all direct comparisons. In network meta‐analysis we assumed a common estimate for the heterogeneity variance across the different comparisons.
Measures and tests for heterogeneity
We statistically assessed the presence of heterogeneity for all direct pairwise comparisons using the common τ2 and I2 statistic.
The assessment of statistical heterogeneity in the entire network was based on the magnitude of the heterogeneity variance parameter (τ2) estimated from the network meta‐analysis models (Jackson 2014).
Assessment of statistical inconsistency
Consistency in a network of treatments refers to the agreement between direct and indirect estimates. Joint analysis of treatments can be misleading if the network is substantially inconsistent. Inconsistency can be present if the trials in the network have different protocols and their inclusion/exclusion criteria are not comparable or may result as an uneven distribution of the effect modifiers across groups of trials that compare different treatments.
Local approaches for evaluating inconsistency
To evaluate the presence of inconsistency locally we used the loop‐specific approach. This method evaluates the consistency assumption in each closed loop of the network separately as the difference between direct and indirect estimates for a specific comparison in the loop (inconsistency factor) (Veroniki 2013). The magnitude of the inconsistency factors and their 95% CIs can then be used to infer the presence of inconsistency in each loop. We assumed a common heterogeneity estimate within each loop. We presented the results of this approach graphically in a forest plot using the 'ifplot' command in Stata (Chaimani 2013).
Global approaches for evaluating inconsistency
We used the 'design‐by‐treatment' model to evaluate the assumption of consistency in the entire network (Higgins 2012). This method accounts for different sources of inconsistency that can occur when studies with different designs (two‐arm trials versus three‐arm trials) give different results, as well as disagreement between direct and indirect evidence. Using this approach we inferred the presence of inconsistency from any source in the entire network based on a Chi2 test. We performed the design‐by‐treatment model in Stata using the 'mvmeta' command. Inconsistency and heterogeneity are interwoven; to distinguish between these two sources of variability we employed the I2 for inconsistency, which measures the percentage of variability that cannot be attributed to random error or heterogeneity (Jackson 2014).
Subgroup analyses
We performed subgroup analyses for benefit at 12, 24, and 36 months' follow‐up by using the following effect modifiers as possible sources of inconsistency or heterogeneity, or both:
diagnostic criteria (Poser or McDonald criteria);
previous treatment with immunomodulators or immunosuppressants (no or yes), i.e. first‐ or second‐line treatments;
definition of relapse (24‐hour definition or 48‐hour definition);
pre‐trial relapse rate and number of years over which the pre‐trial relapse rate was calculated (relapse rate of one or greater than one during the year before randomisation, one or greater than one during the two years before randomisation, two or greater than two during the two/three years before randomisation).
Sensitivity analysis
We performed the following sensitivity analyses:
including only trials with low risk of bias;
excluding studies that did not provide complete and clear reporting of dropout data (see 'Assessment of risk of bias in included studies' section);
excluding trials with a total sample size of fewer than 50 randomised participants to detect potential small study effects.
'Summary of findings' table
We presented the main results of the review in a 'Summary of findings' (SoF) table, according to recommendations described in Chapter 11 of the Cochrane Handbook for Systematic Reviews of Interventions (version 5.1.0) (Schünemann 2011). We provided estimates from the network meta‐analysis based on the methodology developed from the GRADE Working Group (GRADE Working Group 2004). For more details, see Salanti 2014. We included an overall grading of the evidence for three patient‐important outcomes:
proportion of people who experienced new relapses over 12 months;
proportion of people who experienced new relapses over 24 months;
proportion of people who experienced disability worsening over 24 months.
For each outcome, we chose two values for the assumed risk with placebo, i.e. the second highest and second lowest placebo group risks in the included studies.
We graded the quality of evidence for each outcome considering study limitations, indirectness, inconsistency, imprecision of effect estimates, and risk of reporting bias. Since we chose a likely scenario, accounting for incomplete outcome data, for the overall analyses, the grading of the evidence related to the study limitations was based on allocation concealment and blinding of outcome assessor only, and not on incomplete outcome data. According to the software GRADEpro 2008, we assigned four levels of quality of evidence: high, moderate, low, and very low.
Results
Description of studies
Results of the search
Figure 1 shows the results of the electronic search. We identified 415 articles through the search strategy (CENTRAL 10, MEDLINE 131, EMBASE 254, CINAHL 2, clinical trials registries 18). We excluded 356 articles on the basis of abstracts that we considered not pertinent.
1.

Study flow diagram.
We provisionally selected a total of 56 articles and three ongoing trials as potentially fulfilling the inclusion criteria. After full‐text review, we included 39 studies and three ongoing trials, and excluded 17 studies.
Included studies
We included 39 studies involving 25,113 participants and published between 1987 and 2014 in this review (Achiron 1998; ADVANCE 2014; AFFIRM 2006; ALLEGRO 2012; BECOME 2009; BEYOND 2009; Bornstein 1987; BRAVO 2014; CAMMS223 2008; CARE‐MS I 2012; CARE‐MS II 2012; CombiRx 2013; Comi 2001; CONFIRM 2012; DEFINE 2012; Etemadifar 2007; EVIDENCE 2007; Fazekas 1997; Fazekas 2008; FREEDOMS 2010; FREEDOMS II 2014; GALA 2013; Goodkin 1991; IFNB MS Group 1993; INCOMIN 2002; Johnson 1995; Koch‐Henriksen 2006; Lewanska 2002; MAIN TRIAL; Millefiorini 1997; MSCRG 1996; OWIMS 1999; PRISMS 1998; REGARD 2008; SELECT 2013; TEMSO 2011; TENERE 2014; TOWER 2014; TRASFORMS 2010). The table 'Characteristics of included studies' provides details of included studies. Median follow‐up was 24 months (12‐month follow‐up from 12 studies, 24‐month follow‐up from 25 studies, and 36‐month follow‐up from two studies). Twenty‐four (60%) were placebo‐controlled and 15 (40%) were head‐to‐head studies.
We identified three ongoing trials (DECIDE; NCT01247324; NCT01412333). We will include these studies in a future update of this review. 'Characteristics of ongoing studies' provides details on the characteristics of these studies.
Excluded studies
After full‐text review we excluded 17 studies (see 'Characteristics of excluded studies'): seven studies for insufficient duration (CHOICE 2010; Kappos 2006; Kappos 2008; Kappos 2011; Knobler 1993; Saida 2012; Sorensen 2014), five studies evaluating combination therapies (ACT 2009; Freedman 2012; Havrdova 2009; Khoury 2010; SENTINEL 2006), two studies evaluating treatments that are not included in this review (Ashtari 2011; ATAMS 2014), two studies that were non‐randomised (Calabrese 2012; Etemadifar 2006), and one dose‐finding study without a control group (FORTE 2011).
Risk of bias in included studies
The risks of bias of the included studies are summarised in Figure 2 and Figure 3. Considering our predefined criteria (allocation concealment, blinding of outcome assessor, and incomplete outcome data) to assess the overall risk of bias of a study, we judged three out of 39 (8%) trials at low risk of bias (AFFIRM 2006; Fazekas 1997; PRISMS 1998), we judged 16 (41%) at moderate risk of bias (Achiron 1998; BECOME 2009; BEYOND 2009; BRAVO 2014; Comi 2001; Etemadifar 2007; EVIDENCE 2007; Fazekas 2008; GALA 2013; Goodkin 1991; IFNB MS Group 1993; Johnson 1995; Lewanska 2002; MSCRG 1996; REGARD 2008; SELECT 2013), and we judged 20 (51%) at high risk of bias (ADVANCE 2014; ALLEGRO 2012; Bornstein 1987; CAMMS223 2008; CARE‐MS I 2012; CARE‐MS II 2012; CombiRx 2013; CONFIRM 2012; DEFINE 2012; FREEDOMS 2010; FREEDOMS II 2014; INCOMIN 2002; Koch‐Henriksen 2006; MAIN TRIAL; Millefiorini 1997; OWIMS 1999; TEMSO 2011; TENERE 2014; TOWER 2014; TRASFORMS 2010).
2.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Five trials (13%) did not provide enough information to assess sequence generation (unclear risk), and 34 (87%) reported adequate methods (low risk).
Of 39 included studies, 21 (54%) reported adequate methods of allocation concealment (low risk), 17 (44%) did not provide sufficient information to enable a risk of bias judgment (unclear risk), and one trial used an unconcealed procedure (high risk) (Bornstein 1987).
Blinding
Twelve studies (31%) reported that participants and investigators were blinded (low risk), 15 studies (38%) reported that they were not blinded (high risk), and the remaining 12 studies (31%) did not provide sufficient information to enable assessment (unclear risk). We suspected that most participants and treating physicians had become aware of the treatment they were receiving during the course of the trial because most of the agents included in this review have well‐documented side effects, for example injection site reactions and influenza‐like symptoms after interferon beta injection.
Nineteen studies (49%) were at low risk of detection bias (i.e. they reported that outcome assessors were blinded), seven studies (18%) were at high risk, and the remaining 13 studies (33%) did not provide sufficient information to enable assessment (unclear risk).
Incomplete outcome data
We judged 20 of 39 (51%) included studies to meet the criteria for low risk of incomplete outcome data (balanced numbers across intervention groups with similar reasons for loss to follow‐up), 14 studies (36%) were at high risk, and the remaining five studies (13%) did not provide sufficient information to assess risk of incomplete outcome data (unclear risk). The percentage of people who were lost‐to follow‐up among the 39 studies varied from 0% to 43%, with an average of 13.5% (standard deviation 9.1%), and a median of 11.9%.
Selective reporting
All the studies reported all pre‐specified primary benefit outcomes, with the exception of three trials (CONFIRM 2012; DEFINE 2012; TEMSO 2011), in which disability worsening confirmed at six months was not reported in the published report, but was reported in the FDA reports, and thus we considered them at high risk of reporting bias.
Other potential sources of bias
Other bias
We judged 33 studies (85%) at high risk of other bias; this includes the role of the sponsor in authorship of the study report or in data management or analysis (27/39), and incomplete or unclear reporting of data on outcomes and/or study discontinuation (27/39), which make it impossible to understand how the corresponding analyses were performed (e.g. annualised relapse rate estimation).
Method of adverse event monitoring
(See Table 2). In 28 trials (72%), adverse events were actively monitored and we judged the risk of bias to be low. Eight trials (21%) reported insufficient information about the method of adverse event monitoring so that it was uncertain whether or not adverse events were monitored appropriately. We judged the risk of bias to be unclear in these studies. Spontaneous reporting of adverse events as they occurred was reported in three studies and thus we judged them at high risk of bias (Bornstein 1987; EVIDENCE 2007; Goodkin 1991).
1. Assessment of adverse events monitoring.
| Study | Risk of bias | Did the researchers actively monitor for adverse events (AEs) or did they simply provide spontaneous reporting of AEs that arose? | Risk of bias | Did the authors define serious AEs (SAEs) according to an accepted international classification and report the number of SAEs? |
| Achiron 1998 | Unclear | Not reported | High | SAEs not reported |
| ADVANCE 2014 | Unclear | Not reported | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| AFFIRM 2006 | Low | "Treating neurologists were responsible for all aspects of patient care, including the management of adverse events". Participants"visited the clinic every 12 weeks for ... blood chemical and hematologic analyses, evaluation of adverse events..." (Page 901) | Unclear | Insufficient information on SAEs definition |
| ALLEGRO 2012 | Low | "Safety assessments were performed at screening, at baseline, and every 3 months until month 24" (Page 1002) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| BECOME 2009 | Low | "After the initial interim analysis failed to raise any safety concerns with the use of monthly triple dose gadolinium, all patients still in the study were offered the option of obtaining additional monthly MRI scans for a second year of treatment" (Page 1977) | High | SAEs not reported |
| BEYOND 2009 | Low | "Clinic visits were scheduled every 3 months to assess ... safety, and tolerability. The occurrence of new neurological symptoms and adverse events was assessed by telephone, 6 weeks after each visit" (Page 891) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Bornstein 1987 | High | "Self‐evaluation reported to a clinical assistant" (Page 409) | High | SAEs not reported |
| BRAVO 2014 | Low | "Patients were evaluated at 12 scheduled visits: months ‐1 (screening), 0 (baseline), 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24. Safety assessments (laboratory measures, vital signs) were performed at all visits, and electrocardiograms (ECGs) were performed at months ‐1, 0, 1, 2, 3, 6, 12, 18, and 24/early termination" (Page 775) | Unclear | Insufficient information on SAEs definition |
| CAMMS223 2008 | Low | "Safety was assessed quarterly by the treating neurologist, who was aware of study‐group assignment" (Page 1787), "Thyroid function and levels of antithyrotropinreceptor antibodies and lymphocyte subpopulations were measured quarterly at a central laboratory", and "All adverse events with an onset up to 36 months are reported. In addition, all serious adverse events and autoimmune‐associated disorders occurring before March 1, 2008, are listed" (Page 1788) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| CARE‐MS I 2012 | Low | "To assess safety, we undertook monthly questionnaire follow‐up of patients, and did complete blood counts, serum creatinine, urinalysis, and microscopy monthly (every three months in patients in the interferon beta 1a group), and thyroid function tests every 3 months", "Circulating lymphocyte subsets were assessed every 3 months in all patients and 1 month after alemtuzumab administration. We screened for antialemtuzumab antibodies with a bridging ELISA before and at 1 month, 3 months, and 12 months after each dosing", and "We measured interferon beta 1a‐neutralising antibodies at baseline and at 24 months with a cytopathic effect inhibition assay" (Page 1821) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). |
| CARE‐MS II 2012 | Low | "To assess safety, we undertook monthly questionnaire follow‐up of patients, and did complete blood counts, serum creatinine, and urinalysis with microscopy monthly (every 3 months in patients in the interferon beta 1a group), and thyroid function tests every 3 months", "We assessed circulating lymphocyte subsets every 3 months in all patients and 1 month after every course of alemtuzumab. We screened for anti‐alemtuzumab antibodies with ELISA before and at 1 month, 3 months and 12 months after each dosing", and "We measured interferon beta 1a‐neutralising antibodies at baseline and at 24 months with a cytopathic effect inhibition assay" (Page 1832) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| CombiRx 2013 | Low | "Safety was assessed by recording all adverse events, serious and nonserious" (Page 329) | Unclear | No information on SAE definition |
| Comi 2001 | Unclear | "The treating physician monitored safety..." (Page 291) | Unclear | Insufficient information on SAEs definition |
| CONFIRM 2012 | Low | "Throughout the course of the study, every effort was made to remain alert to possible adverse events (AEs)" and "Any AE or SAE experienced by the subject was recorded on the CRF, regardless of the severity of the event or its relationship to study treatment" (Pages 66‐7 of Protocol) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| DEFINE 2012 | Low | "Study visits were scheduled every 4 weeks for safety assessments, including the monitoring of laboratory values" (Page 1100) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Etemadifar 2007 | Low | "Adverse events, vital signs and blood tests were monitored monthly" (Page 1724) | High | SAEs not reported |
| EVIDENCE 2007 | High | "Adverse events were determined by spontaneous reporting and monthly laboratory testing during the comparative phase" (Page 2031) | Unclear | Insufficient information on SAEs definition |
| Fazekas 1997 | Low | Participants "asked about safety monthly..." (Page 590) | High | SAEs not reported |
| Fazekas 2008 | Unclear | Not reported | Unclear | Insufficient information on SAEs definition |
| FREEDOMS 2010 | Low | "An independent data and safety monitoring board evaluated the safety" and "Study visits, including safety assessments, were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomization" (Page 389) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| FREEDOMS II 2014 | Low | "...safety assessments, were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomization" (Appendix, Page 2) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| GALA 2013 | Low | "Safety assessments included adverse events (AEs), standard clinical laboratory tests, vital signs, and electrocardiographic (ECG) measurements" (Page 707) | Unclear | No information on SAE definition |
| Goodkin 1991 | High | "Side effect were reported to the treating neurologist every 6 months" (Page 21) | High | SAEs not reported |
| IFNB MS Group 1993 | Low | "Treating neurologist reviewed side effects, laboratory findings for toxicity ..." (Page 656) | High | SAEs not reported |
| INCOMIN 2002 | Low | "Safety assessments included adverse events, vital signs, physical examination, and concomitant medications. Patients underwent haematology and biochemical tests, including liver‐function tests, every 2 weeks for the first 8 weeks, and then every 3 months" (Page 1455) | High | SAEs not reported |
| Johnson 1995 | Low | "The evaluating physician monitored safety every 3 month..." (Page 1270) | Unclear | Insufficient information on SAEs definition |
| Koch‐Henriksen 2006 | Low | "Patients were interviewed about side effects and had routine blood tests including hematology and liver function tests every 3 months and thyroid tests and neutralizing antibodies every 6 months" (Page 1057) | High | SAEs not reported |
| Lewanska 2002 | Unclear | "Laboratory safety examinations were made at the beginning and at the end of the study period" (Page 566) | Unclear | Insufficient information on SAEs definition |
| MAIN TRIAL | Low | "At scheduled (quarterly) and unscheduled (i.e., at the onset of new symptoms or complications) follow‐up visits the treating neurologist recorded symptoms, blood test results, clinical AEs and their management" | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| Millefiorini 1997 | Low | "The safety of the treatment was assessed on the basis of adverse events volunteered by the patient either spontaneously or on questioning and monitoring of the main laboratory parameters" (Page 155) | Unclear | Insufficient information on SAEs definition |
| MSCRG 1996 | Low | "Study visits were scheduled at baseline and every 6 months. Treating physicians reviewed toxicity test results, examined patients, and made all medical decision" (Page 286) | Unclear | Insufficient information on SAEs definition |
| OWIMS 1999 | Unclear | "The treating physician recorded and treated AEs..." (Page 680) | Unclear | Insufficient information on SAEs definition |
| PRISMS 1998 | Unclear | "A “treating” neurologist was responsible for overall medical management of the patient, including treatment of any side‐effects" (Page 1499) | Unclear | Insufficient information on SAEs definition |
| REGARD 2008 | Unclear | "Adverse events (including pregnancy), withdrawals owing to adverse events, serious adverse events, and laboratory results were obtained for safety comparisons" (Page 905) | Unclear | Insufficient information on SAEs definition |
| SELECT 2013 | Low | "Safety parameters were assessed at all visits" (Page 2168) | Unclear | No information on SAE definition |
| TEMSO 2011 | Low | "A treating neurologist at each site was responsible for recording and managing adverse events and monitoring safety assessments" and "Safety was evaluated on the basis of adverse events reported by study participants or investigators. Laboratory tests were performed at the time of screening, at baseline, every 2 weeks for the first 24 weeks, and then every 6 weeks until study completion. Physical and neurologic examinations were performed at week 12 and then every 24 weeks. An abdominal ultrasonographic examination to asses for pancreatic abnormalities was performed before the study and then every 24 weeks, because of previous infrequent reports of pancreatitis associated with leflunomide use" (Pages 1294‐5) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| TENERE 2014 | Low | "Safety and tolerability were assessed using AE reporting, vital signs and laboratory assessments. Adverse event reports were collected at randomisation, Weeks 2, 6, 12, 18, 24, 36 and every 12 weeks thereafter. Vital signs were documented at screening, randomisation and every 12 weeks thereafter; clinical laboratory results were assessed throughout the study. Adverse events and vital signs were also recorded during unscheduled relapse visits" (Page 707) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) [information provided on request by Genzyme] |
| TOWER 2014 | Low | "Safety was assessed through adverse event reporting (upon occurrence), clinical laboratory tests (every 2 weeks until week 24, then every 6 weeks while still on treatment), vital signs (at weeks 2 and 6, then every 6 weeks until week 24, then every 12 weeks while still on treatment), abdominal ultrasonography (at week 24, then every 24 weeks), and electrocardiography (at baseline and end of treatment)" (Page 248) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
| TRASFORMS 2010 | Low | "An independent data and safety monitoring board evaluated overall safety in the fingolimod phase 3 program" and "Safety assessments were conducted during screening, at baseline, and at months 1, 2, 3, 6, 9, and 12" (Page 404) | Low | Categorisation of SAEs conformed to ICH guidelines (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) |
Serious adverse event (SAE) definition and reporting
In nine trials (23%) SAEs were not reported and we judged the risk of bias to be high. In 15 trials (38%) SAEs were reported but insufficient information on their definition was given and we judged the risk of bias to be unclear. Fifteen studies (38%) provided a definition of SAEs and we judged the risk of bias to be low.
Effects of interventions
See: Table 1
Table 1 provides overall estimates of treatment effects compared with placebo and the quality of the available evidence for the three benefit outcomes (chance of experiencing one or more relapses over 12 months, chance of experiencing one or more relapses over 24 months, chance of disability getting worse over 24 months), obtained through a network meta‐analysis. Figure 4 shows the networks of evidence for the benefit and acceptability of immunomodulators and immunosuppressants included in the review. Each line links the treatments that have been directly compared in studies. The thickness of the line is proportional to the number of participants included in the comparison and the width of each circle is proportional to the number of studies included in the comparison. Figure 5 and Figure 6 show, respectively, the estimates of benefit and acceptability of each treatment against placebo within the networks. Analysis 3.1 provides the summary of treatment safety compared with placebo within pairwise comparisons. Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11 show the network meta‐analysis estimates of primary benefit and acceptability outcomes for each comparison.
4.

Network plots of treatment comparisons for benefit and acceptability outcomes.
5.

Network meta‐analysis (NMA) estimates of treatment benefit against placebo: relapses over 12 and 24 months, and disability worsening over 24 months.
CI: confidence interval; RR: risk ratio.
6.

Network meta‐analysis (NMA) estimates of treatment acceptability against placebo: treatment discontinuation due to AEs over 12 and 24 months.
AEs: adverse events; CI: confidence interval; RR: risk ratio.
3.1. Analysis.

Comparison 3 Treatment safety against placebo within pairwise comparisons, Outcome 1 Serious adverse events.
7.

Network meta‐analysis (NMA) estimates of treatment benefit (lower triangle) and acceptability (upper triangle) over 12 months for each comparison: relapses and treatment discontinuation due to adverse events (AEs) over 12 months.
Drugs are reported in order of primary benefit ranking. Comparisons should be read from left to right. The estimate (risk ratio, RR) is located at the intersection of the column‐defining treatment and the row‐defining treatment. A RR value below 1 favours the column‐defining treatment for lower triangle, and the row‐defining treatment for upper triangle. To obtain RRs for comparisons in the opposing direction, reciprocals should be taken. Significant results are bolded and underscored.
Alemtuz: alemtuzumab; Avonex: interferon beta‐1a (Avonex); Aza: azathioprine; Betaseron: interferon beta‐1b (Betaseron); Dacliz: daclizumab; Dimethyl: dimethyl fumarate; Fingolim: fingolimod; Glatir: glatiramer acetate; IFNß: interferons beta; Immunogl: immunoglobulins; Mitoxan: mitoxantrone; Nataliz: natalizumab; PegIFNß: pegylated interferon beta‐1a; Rebif: interferon beta‐1a (Rebif); Terifl: teriflunomide.
8.

Network meta‐analysis (NMA) estimates of treatment benefit (lower triangle) and acceptability (upper triangle) over 24 months for each comparison: relapses and treatment discontinuation due to adverse events (AEs) over 24 months. Drugs are reported in order of primary benefit ranking. Comparisons should be read from left to right. The estimate (risk ratio, RR) is located at the intersection of the column‐defining treatment and the row‐defining treatment. A RR value below 1 favours the column‐defining treatment for lower triangle, and the row‐defining treatment for upper triangle. To obtain RRs for comparisons in the opposing direction, reciprocals should be taken. Significant results are bolded and underscored.
Alemtuz: alemtuzumab; Avonex: interferon beta‐1a (Avonex); Aza: azathioprine; Betaseron: interferon beta‐1b (Betaseron); Dimethyl: dimethyl fumarate; Fingolim: fingolimod; Glatir: glatiramer acetate; IFNß: interferons beta; Immunogl: immunoglobulins; Laquin: laquinimod; Mitoxan: mitoxantrone; Nataliz: natalizumab; Rebif: interferon beta‐1a (Rebif); Terifl: teriflunomide.
9.

Clustered ranking plot based on cluster analysis of surface under the cumulative ranking curve (SUCRA) values for benefit (relapses) and acceptability (treatment discontinuation due to AEs) over 24 months. Each colour represents a group of treatments that belong to the same cluster. Treatments lying in the upper right corner are more effective and acceptable than the other treatments.
10.

Network meta‐analysis (NMA) estimates of treatment benefit (lower triangle) and acceptability (upper triangle) over 24 months for each comparison: disability worsening and treatment discontinuation due to adverse events (AEs) over 24 months. Drugs are reported in order of primary benefit ranking. Comparisons should be read from left to right. The estimate is located at the intersection of the column‐defining treatment and the row‐defining treatment. A RR value below 1 favours the column‐defining treatment for lower triangle, and the row‐defining treatment for upper triangle. To obtain RRs for comparisons in the opposing direction, reciprocals should be taken. Significant results are bolded and underscored.
Alemtuz: alemtuzumab; Avonex: interferon beta‐1a (Avonex); Aza: azathioprine; Betaseron: interferon beta‐1b (Betaseron); Dimethyl: dimethyl fumarate; Fingolim: fingolimod; Glatir: glatiramer acetate; IFNß: interferons beta; Immunogl: immunoglobulins; Laquin: laquinimod; Mitoxan: mitoxantrone; Nataliz: natalizumab; Rebif: interferon beta‐1a (Rebif); Terifl: teriflunomide.
11.

Clustered ranking plot based on cluster analysis of surface under the cumulative ranking curve (SUCRA) values for benefit (disability worsening) and acceptability (treatment discontinuation due to AEs) over 24 months. Each colour represents a group of treatments that belong to the same cluster. Treatments lying in the upper right corner are more effective and acceptable than the other treatments.
1. Primary outcomes
1.1 Benefit
Relapses over 12 and 24 months and disability worsening over 24 months
Pairwise meta‐analysis (direct comparisons)
Treatment estimates for pairwise meta‐analyses are reported in Analysis 1.1, Analysis 1.2, and Analysis 1.3.
1.1. Analysis.
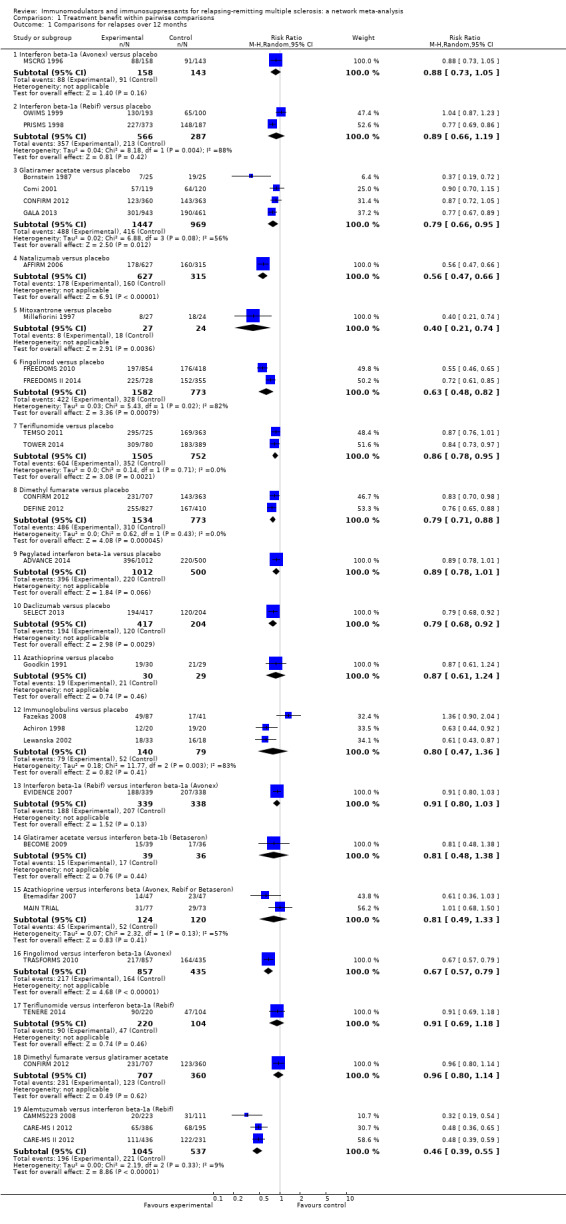
Comparison 1 Treatment benefit within pairwise comparisons, Outcome 1 Comparisons for relapses over 12 months.
1.2. Analysis.

Comparison 1 Treatment benefit within pairwise comparisons, Outcome 2 Comparisons for relapses over 24 months.
1.3. Analysis.

Comparison 1 Treatment benefit within pairwise comparisons, Outcome 3 Comparisons for disability worsening over 24 months.
Network meta‐analysis estimates (combination of direct and indirect comparisons) of treatment effects against placebo
We did not find any evidence that important variables varied across comparisons or altered the effectiveness of the treatments. Accordingly, none of the corresponding analyses provided evidence that any potential effect modifiers were possible sources of inconsistency or heterogeneity. However, few studies per comparison were available and the results from sensitivity and subgroup analyses were very uncertain, so no firm conclusion can be drawn about the presence or absence of transitivity and heterogeneity.
See: Table 1, Figure 4, Figure 5, Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11.
a) Relapses over 12 months were provided in 29 studies involving 17,897 participants with relapsing‐remitting multiple sclerosis (RRMS) (71.3% of the participants in this review) (Achiron 1998; ADVANCE 2014; AFFIRM 2006; BECOME 2009; Bornstein 1987; CAMMS223 2008; CARE‐MS I 2012; CARE‐MS II 2012; Comi 2001; CONFIRM 2012; DEFINE 2012; Etemadifar 2007; EVIDENCE 2007; Fazekas 2008; FREEDOMS 2010; FREEDOMS II 2014; GALA 2013; Goodkin 1991; Lewanska 2002; MAIN TRIAL; Millefiorini 1997; MSCRG 1996; OWIMS 1999; PRISMS 1998; SELECT 2013; TEMSO 2011; TENERE 2014; TOWER 2014; TRASFORMS 2010). Nineteen studies of 12 treatments involving 12,100 participants were placebo‐controlled trials, nine studies of 12 treatments involving 4367 participants were head‐to‐head trials directly comparing active treatments, and one study involving 1430 participants had both a placebo and two active treatment arms. Five of 15 treatments (33%) were compared to placebo only. The majority of direct comparisons between active treatments were not assessed in any trial (Figure 4). Alemtuzumab was the best drug (risk ratio (RR) versus placebo 0.40, 95% confidence interval (CI) 0.31 to 0.51; SUCRA = 97%; moderate quality evidence), followed by mitoxantrone (RR versus placebo 0.40, 95% CI 0.20 to 0.76; SUCRA = 93%; low quality evidence), natalizumab (RR versus placebo 0.56, 95% CI 0.43 to 0.73; SUCRA = 85%; high quality evidence), and fingolimod (RR versus placebo 0.63, 95% CI 0.53 to 0.74; SUCRA = 80%; low quality evidence). The heterogeneity τ2 for this network overall was 0.01, which we considered low heterogeneity.
b) Relapses over 24 months were provided in 26 studies and 16,800 participants with RRMS (67% of those included in this review) (Achiron 1998; AFFIRM 2006; ALLEGRO 2012; BECOME 2009; BEYOND 2009; Bornstein 1987; BRAVO 2014; CAMMS223 2008; CARE‐MS I 2012; CARE‐MS II 2012; CONFIRM 2012; DEFINE 2012; Fazekas 1997; FREEDOMS 2010; FREEDOMS II 2014; Goodkin 1991; IFNB MS Group 1993; INCOMIN 2002; Johnson 1995; Koch‐Henriksen 2006; MAIN TRIAL; Millefiorini 1997; MSCRG 1996; PRISMS 1998; REGARD 2008; TEMSO 2011). Fifteen studies of 12 treatments involving 8562 participants were placebo‐controlled trials, nine studies of seven treatments involving 5477 participants were head‐to‐head trials directly comparing active treatments, and two studies involving 2761 participants had both a placebo and two active treatment arms each. Five of 14 treatments (36%) were compared to placebo only. The majority of direct comparisons between active treatments were not assessed in any trial (Figure 4). As for the relapse over 12 months outcome, alemtuzumab was the best drug (RR versus placebo 0.46, 95% CI 0.38 to 0.55; SUCRA = 96%; moderate quality evidence), followed by mitoxantrone (RR versus placebo 0.47, 95% CI 0.27 to 0.81; SUCRA = 92%; very low quality evidence), natalizumab (RR versus placebo 0.56, 95% CI 0.47 to 0.66; SUCRA = 88%; high quality evidence), and fingolimod (RR versus placebo 0.72, 95% CI 0.64 to 0.81; SUCRA = 71%; moderate quality evidence). The heterogeneity τ2 for this network overall was 0.0036, which we considered low heterogeneity.
c) Disability worsening over 24 months was available from 26 studies and 16,800 participants with RRMS (67% of those included in this review) (Achiron 1998; AFFIRM 2006; ALLEGRO 2012; BECOME 2009; BEYOND 2009; Bornstein 1987; BRAVO 2014; CAMMS223 2008; CARE‐MS I 2012; CARE‐MS II 2012; CONFIRM 2012; DEFINE 2012; Fazekas 1997; FREEDOMS 2010; FREEDOMS II 2014; Goodkin 1991; IFNB MS Group 1993; INCOMIN 2002; Johnson 1995; Koch‐Henriksen 2006; MAIN TRIAL; Millefiorini 1997; MSCRG 1996; PRISMS 1998; REGARD 2008; TEMSO 2011). The network geometry for disability worsening over 24 months was as for relapses over 24 months (Figure 4). Mitoxantrone was the best drug (RR versus placebo 0.20, 95% CI 0.05 to 0.84; SUCRA = 96%; low quality evidence), followed by alemtuzumab (RR versus placebo 0.35, 95% CI 0.26 to 0.48; SUCRA = 94%; low quality evidence), and natalizumab (RR versus placebo 0.64, 95% CI 0.49 to 0.85; SUCRA = 74%; moderate quality evidence). The heterogeneity τ2 for this network overall was 0.0081, which we considered low heterogeneity.
Relapses and disability worsening over 36 months
Relapses and disability worsening over 36 months were available from two studies only: one on glatiramer acetate versus interferon beta‐1a (Avonex), with a RR of 0.71 (95% CI 0.57 to 0.88) for relapses, and a RR of 0.91 (95% CI 0.75 to 1.10) for disability worsening (CombiRx 2013); one on alemtuzumab versus interferon beta‐1a (Rebif), with a RR of 0.48 (95% CI 0.33 to 0.68) for relapses, and a RR of 0.42 (95% CI 0.30 to 0.57) for disability worsening (CAMMS223 2008). We judged both studies at high risk of bias (Figure 3).
1.2 Acceptability
Treatment discontinuation due to adverse events over 12 and 24 months
Pairwise meta‐analysis (direct comparisons)
Treatment estimates for pairwise meta‐analyses are reported in Analysis 2.1 and Analysis 2.2.
2.1. Analysis.

Comparison 2 Treatment acceptability within pairwise comparisons, Outcome 1 Comparisons for treatment discontinuation due to AEs over 12 months.
2.2. Analysis.

Comparison 2 Treatment acceptability within pairwise comparisons, Outcome 2 Comparisons for treatment discontinuation due to AEs over 24 months.
Network meta‐analysis estimates (combination of direct and indirect comparisons) of treatment effects against placebo
See: Figure 4, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11.
Acceptability over 12 months was reported in 13 studies on 10 treatments involving 8105 participants: nine studies of seven treatments involving 5718 participants were placebo‐controlled trials, and four studies of six treatments involving 2387 participants were head‐to‐head trials directly comparing active treatments. Four of 10 treatments (40%) were compared to placebo only. The majority of direct comparisons between active treatments were not assessed in any trial (Figure 4). The network geometry for acceptability over 24 months was as for relapses and disability worsening over 24 months (Figure 4). The network meta‐analysis showed that over 12 months, compared to placebo, several treatments had a significantly higher proportion of participants who withdrew due to any adverse event, such as teriflunomide (RR versus placebo 2.24, 95% CI 1.50 to 3.34), peg‐interferon beta (RR versus placebo 2.80, 95% CI 1.39 to 5.64), interferon beta‐1a (Avonex) (RR versus placebo 4.36, 95% CI 1.98 to 9.60), interferon beta‐1a (Rebif) (RR versus placebo 4.83, 95% CI 2.59 to 9.00), and fingolimod (RR versus placebo 8.26, 95% CI 3.25 to 20.97). Over 24 months, the network meta‐analysis showed that, compared to placebo, only fingolimod had a significantly higher proportion of participants who withdrew due to any adverse event (RR versus placebo 1.69, 95% CI 1.32 to 2.17). The heterogeneity τ2 for these networks overall was < 0.0001, which we considered low heterogeneity.
1.3 Relationship between benefit and acceptability outcomes for each comparison (network meta‐analysis estimates)
See: Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11.
a) Relapses and acceptability over 12 months (Figure 7). Compared to placebo and all other active agents, excluding mitoxantrone and natalizumab, alemtuzumab showed a significantly lower proportion of participants who experienced new relapses over 12 months, but no data were available on the acceptability of alemtuzumab over 12 months. Compared to placebo and several other active agents, mitoxantrone, natalizumab, and fingolimod showed a significantly lower proportion of participants who experienced new relapses over 12 months. However, data on the acceptability of these treatments over 12 months were available for fingolimod only, showing a significantly higher proportion of participants who withdrew due to any adverse event over 12 months with fingolimod compared to glatiramer acetate, teriflunomide, interferon beta‐1a (Avonex), and placebo.
b) Relapses and acceptability over 24 months (Figure 8 and Figure 9). Compared to placebo and all other active agents, excluding mitoxantrone and natalizumab, alemtuzumab showed a significantly lower proportion of participants who experienced new relapses over 24 months, and did not show a significantly higher proportion of participants who withdrew due to any adverse event over 24 months. Compared to placebo and several other active agents, mitoxantrone, natalizumab, and fingolimod showed a significantly lower proportion of participants who experienced new relapses over 24 months, and did not show a significantly higher proportion of participants who withdrew due to any adverse event over 24 months, with the exception of fingolimod versus placebo (RR 1.69, 95% CI 1.32 to 2.17). Similar results are shown in the plot representing the groups of treatments obtained from the cluster analysis according to the SUCRA values for both benefit and acceptability.
c) Disability worsening and acceptability over 24 months (Figure 10 and Figure 11). Compared to placebo and all other active agents, excluding mitoxantrone, alemtuzumab showed a significantly lower proportion of participants who experienced disability worsening over 24 months, and did not show a significantly higher proportion of participants who withdrew due to any adverse event over 24 months. Compared to placebo and a few other active agents, mitoxantrone showed a significantly lower proportion of participants who experienced disability worsening over 24 months, and did not show a significantly higher proportion of participants who withdrew due to any adverse event over 24 months. Similar results are shown in the plot representing the groups of treatments obtained from the cluster analysis according to the SUCRA values for both benefit and acceptability.
2. Secondary outcomes
2.1 Safety
Serious adverse events (SAEs)
Pairwise meta‐analysis (direct comparisons)
Compared to the placebo group there was not a significant difference in the proportion of participants with serious adverse events. Nevertheless, information on serious adverse events was scanty, based on a very low number of events, poorly reported and characterised by heterogeneous results (Analysis 3.1).
3. Assessment of heterogeneity and inconsistency within the network analyses
We performed an assessment of heterogeneity and inconsistency within the network analyses for relapses over 12 and 24 months, for disability worsening over 24 months and for acceptability at 12 and 24 months.
We observed evidence of local statistical inconsistency, estimated as a difference between direct and indirect treatment estimates in networks, for two loops for relapses over 24 months and for three for disability worsening over 24 months, and none for relapses over 12 months and acceptability at 12 and 24 months (Figure 12 and Figure 13). However, due to the presence of imprecise direct and network estimates, the absence of statistically significant inconsistency is not evidence against the presence of inconsistency. The values for common heterogeneity (τ2) for the network for each outcome seem to show no evidence of heterogeneity. When evaluating the inconsistency in the networks as a whole, there is no indication of global inconsistency within any network (global test for inconsistency: P value = 0.99 for relapses over 12 months; P value = 0.97 for relapses over 24 months; P value = 0.08 for disability worsening over 24 months), but we expected the power to be low with few studies per comparison and few closed loops. Hence, we decided to downgrade the quality of the evidence for inconsistency in most comparisons.
12.
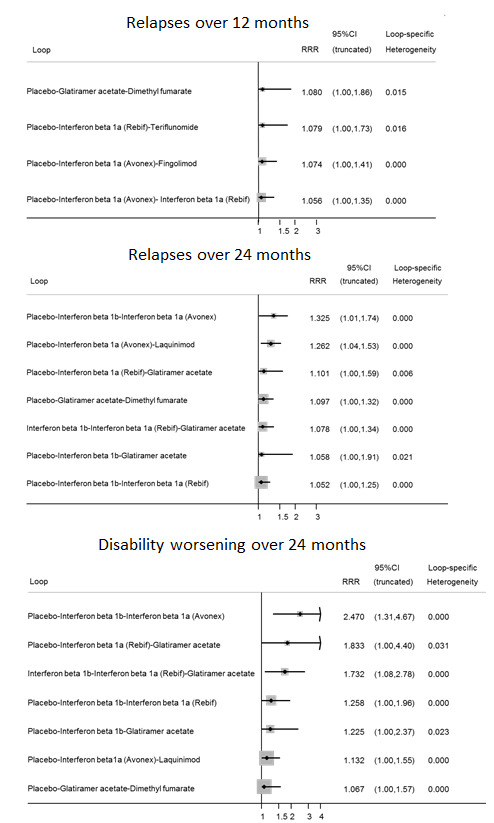
Inconsistency plots for relapses over 12 and 24 months and disability worsening over 24 months assuming loop‐specific heterogeneity estimates. RRR is calculated as the risk ratio for direct evidence over the risk ratio for indirect evidence in the loop and it is reported together with its 95% confidence interval (CI). RRR values close to one indicate the absence of evidence for disagreement between direct and indirect evidence.
13.

Inconsistency plots for acceptability over 12 and 24 months assuming loop‐specific heterogeneity estimates. RRR is calculated as the risk ratio for direct evidence over the risk ratio for indirect evidence in the loop and it is reported together with its 95% confidence interval (CI). RRR values close to one indicate the absence of evidence for disagreement between direct and indirect evidence.
4. Subgroup and sensitivity analyses
None of the pre‐defined subgroup analyses (i.e. by diagnostic criteria, previous treatments, definition of relapse and pre‐trial relapse rate) and sensitivity analyses (i.e. including only trials at low risk of bias, excluding studies that did not provide complete and clear reporting of dropout data, and excluding trials with a total sample size of fewer than 50 randomised participants) that we performed provided any significantly different results, compared to the overall analyses (see Table 3 and Table 4 for the corresponding network meta‐analysis estimates for relapse outcome over 24 months for the three best drugs, based on moderate to high quality evidence, i.e. alemtuzumab, natalizumab, and fingolimod). However, few studies per comparison were available and limitations in study reporting cannot exclude differences between subgroups.
2. Subgroup analyses: network meta‐analysis estimates for relapse outcome over 24 months for the three best drugs based on moderate to high quality evidence.
| Intervention | Subgroup analysis by | |||||||
| Diagnostic criteria RR (95% CI) |
Previous treatments RR (95% CI) |
Definition of relapse RR (95% CI) |
Pre‐trial relapse rate RR (95% CI) |
|||||
| Poser criteria | McDonald criteria | No | Yes | 24‐hour definition | 48‐hour definition |
≥ 1 during the year before randomisation |
≥ 2 during the 2/3 years before randomisation |
|
| Alemtuzumab | — | 0.48 (0.33 to 0.68) | 0.46 (0.28 to 0.76) | 0.47 (0.27 to 0.79) | — | 0.46 (0.27 to 0.78) | 0.63 (0.48 to 0.81) | 0.28 (0.16 to 0.49) |
| Natalizumab | — | 0.56 (0.45 to 0.69) | — | 0.70 (0.56 to 0.88) | 0.63 (0.52 to 0.77) | — | 0.68 (0.54 to 0.85) | — |
| Fingolimod | — | 0.72 (0.63 to 0.83) | — | 0.72 (0.65 to 0.80) | 0.81 (0.67 to 0.97) | — | — | 0.72 (0.60 to 0.87) |
CI: confidence interval; NMA: network meta‐analysis; RR: risk ratio.
3. Sensitivity analyses: NMA estimates for relapse outcome over 24 months for the three best drugs based on moderate to high quality evidence.
| Intervention | Sensitivity analysis | ||
| Including only trials of low risk of bias RR (95% CI) |
Excluding studies that did not provide complete and clear reporting of dropout data RR (95% CI) |
Excluding trials with a total sample size of fewer than 50 randomised participants RR (95% CI) |
|
| Alemtuzumab | — | 0.47 (0.35 to 0.63) | 0.46 (0.39 to 0.56) |
| Natalizumab | 0.66 (0.54 to 0.81) | — | 0.56 (0.47 to 0.66) |
| Fingolimod | — | 0.72 (0.65 to 0.80) | 0.72 (0.64 to 0.81) |
CI: confidence interval; NMA: network meta‐analysis; RR: risk ratio.
5. Reporting bias
We did not produce a contour‐enhanced funnel plot for each pairwise comparison due to the low number of studies. We employed a comparison‐adjusted funnel plot for all placebo‐controlled trials for relapses over 12 and 24 months and disability worsening over 24 months. Small study effects (not necessarily due to reporting bias) appeared to be present for relapses over 12 and 24 months, but not for disability worsening over 24 months (data not shown).
6. Grading of the evidence
We graded the evidence for relapses over 12 and 24 months and for disability worsening over 24 months for each network estimate of an immunomodulator or immunosuppressant used for RRMS versus placebo (Table 1) according to the approach proposed by Salanti 2014. We considered the following domains: study limitations, indirectness, inconsistency, imprecision of effect estimates, and risk of reporting bias.
We assessed the study limitations for each network estimate by first evaluating the risk of bias for each direct estimate and then by integrating these judgements with the contribution of each direct estimate to the network estimates (Figure 14). The percentage contribution of each direct estimate to the network estimates for any considered outcome are reported in Figure 15, Figure 16 and Figure 17. We also took these percentages into account for the evaluation of the other domains, and determined the confidence in an overall treatment ranking from each network meta‐analysis.
14.

Study limitations distribution for each network estimate for pairwise comparisons versus placebo on relapses over 12 and 24 months and disability worsening over 24 months outcomes. Calculations are based on the contributions of direct evidence to the network estimates and the overall risks of bias considering our predefined criteria (allocation concealment, blinding of outcome assessor, and incomplete outcome data) within studies contributing to the direct evidence. The colours represent risk (green, low; yellow, moderate; red, high). The direct comparisons against placebo are described in the vertical axis.
15.

Contribution matrix: percentage contribution of each direct estimate to the NMA estimates versus placebo for relapses over 12 months outcome. Rows correspond to NMA risk ratios of each treatment versus placebo (separated for mixed and indirect evidence) and the columns correspond to direct meta‐analysis risk ratios. The last row shows the number of included direct comparisons. The names of the treatment comparisons are shown in the first column. For example, for relapses over 12 months, information for the network estimate of interferon beta 1a (Avonex) versus placebo is derived from both direct and indirect evidence (generating a mixed estimate). Of this mixed network estimate, trials directly comparing interferon beta 1a (Avonex) versus placebo contribute 31.2% of the information to the network estimate of effect and trials directly comparing interferon beta 1a (Rebif) versus interferon beta 1a (Avonex) contribute 18.8% of the network estimated effect, etc. The contribution matrix shows how much each direct comparison in the network contributes to each network (mixed or indirect) estimate and to the entire network.
16.

Contribution matrix: percentage contribution of each direct estimate to the NMA estimates versus placebo for relapses over 24 months outcome.
17.

Contribution matrix: percentage contribution of each direct estimate to the NMA estimates versus placebo for disability worsening over 24 months outcome.
We assessed indirectness by evaluating both indirectness of populations, interventions, and outcomes as in standard GRADE, and transitivity by comparing the distribution of known effect modifiers across comparisons that contribute evidence to estimation of the network meta‐analysis treatment effect, and across networks that contribute evidence to estimation of the overall treatment ranking from each network meta‐analysis.
We assessed inconsistency by judging the extent of heterogeneity (i.e. considering the comparison‐specific heterogeneity variance with other relevant metrics such as the I2 statistic, the network meta‐analysis estimate of variance and a prediction interval), and by evaluating the extent to which the comparison under evaluation was involved in inconsistent loops of evidence, and the overall inconsistency in the networks using global tests of inconsistency.
Evaluation of imprecision was focused on width of the CIs of the network meta‐analysis treatment effect estimates, and visually examining ranking probabilities (i.e. rankograms) for overlap to assess the precision of treatment rankings.
Due to the low number of studies for each comparison, we assessed reporting bias through a comparison‐adjusted funnel plot for the treatment ranking estimates only.
For relapses at 24 months we judged the confidence in the treatment estimate to be high for natalizumab only, moderate for alemtuzumab, fingolimod, dimethyl fumarate, glatiramer acetate, daclizumab, and interferon beta‐1a (Avonex), and low or very low for all the other treatment estimates. For disability worsening at 24 months the confidence in almost all of the treatment estimates varied from low to very low, except for natalizumab, which we evaluated as moderate quality.
In the ranking of the treatments we judged our confidence as low for relapses over 12 and 24 months due to study limitations and reporting bias, and low for disability worsening at 24 months due to indirectness and imprecision.
Discussion
Summary of main results
This review of the effects of treatments for relapsing‐remitting multiple sclerosis (RRMS) included 39 studies involving 25,113 randomised adult participants. The majority of studies were short‐term trials, with the median randomised controlled trial (RCT) duration being 24 months. Only two studies reported a 36‐month follow‐up, therefore the effects of these treatments beyond two years remain uncertain.
1. Recurrence of relapses
Alemtuzumab, mitoxantrone, natalizumab, and fingolimod outperformed other drugs, being statistically significantly more effective than placebo and the majority of other drugs, with risk ratios (RRs) versus placebo ranging between 0.40 and 0.72 at both 12 and 24 months. Direct comparisons contributed to these network estimates: 40% to alemtuzumab (three trials versus interferon beta‐1a (Rebif) involving 1582 participants), and 100% to fingolimod (two trials versus placebo involving 2355 participants), to natalizumab (one study versus placebo involving 942 participants), and to mitoxantrone (one study versus placebo involving 51 participants). Furthermore, based on the GRADE approach for network meta‐analysis, we assessed our confidence in the evidence for their beneficial effects as high for natalizumab, moderate for alemtuzumab, moderate to low for fingolimod, and low to very low for mitoxantrone.
2. Disability worsening
The measurement of this outcome was based on a surrogate marker in the majority of the included studies, since they used disability worsening confirmed at only three months' follow‐up, thus reflecting an effect on relapse‐related disability. Both direct and indirect comparisons revealed that almost none of the treatments included in this review were effective in preventing disability worsening over two years, with the exception of mitoxantrone, alemtuzumab, and natalizumab. Nevertheless, our confidence in the beneficial effects was moderate for natalizumab and low for mitoxantrone and alemtuzumab.
3. Acceptability and safety
Almost all of the agents included in this review were associated with a higher proportion of participants who withdrew due to any adverse event compared to placebo at 12 and 24 months. All the treatments for which information on serious adverse events was available were associated with a non‐significantly higher proportion of people with at least one SAE compared with placebo during a median two‐year follow‐up period. Lack of statistical significance in our analyses was likely to have been caused by a low number of events and short‐term trials. Moreover, information on serious adverse events was scanty, poorly reported and characterised by heterogeneous results.
Overall completeness and applicability of evidence
Many of the trials included in this review provided evidence on the proportion of participants who experienced new relapses, disability worsening, and adverse events over 12 or 24 months' follow‐up, but only two studies reported data on these outcomes over 36 months. This is an unwelcome finding considering that multiple sclerosis (MS) is a disease of 30 to 40 years' duration. Moreover, scanty and poorly reported safety data did not provide complete evidence, leading to uncertainty about the risk profile of the treatments included in the review.
Evidence on 15 treatments included in this review was derived from 39 trials (searched for up to September 2014), which is a small number of studies relative to the number of treatments. Furthermore, the evidence was derived primarily from 16 trials (16,162 patients; 64.4% of those included in this review) on new drugs mostly compared with placebo (60% of the trials included in this review). There is therefore uncertainty that the results of the review fit into the context of current practice since about 50% of patients are treated with at least one of these treatments (Carroll 2014).
The reasons why there are few randomised studies for RRMS, and these are mostly placebo‐controlled, are probably due to: i) approval of treatments for RRMS by many national regulatory agencies based on results from as little as one placebo‐controlled trial; ii) the consequent lack of interest of pharmaceutical companies in conducting additional expensive studies; iii) the unlikely advantage of pharmaceutical companies in conducting head‐to‐head trials directly comparing active treatments.
Our review was not intended to be a comprehensive review of all effects of treatments for RRMS. We focused on three main clinical outcomes (relapses, disability worsening, and acceptability of treatment) that we considered meaningful to patients and clinicians. Patient‐reported outcomes, such as behavioural functions or quality of life, were not included. They are certainly important outcomes for participants but are reported rarely in clinical trials, often without adequate monitoring and availability of appropriate published results. The different scales used and different assessment time points do not allow comparisons to be made. Moreover, these measures may be susceptible to bias in trials in which many, if not most, treated participants have become aware of the treatment they are receiving owing to the well‐documented side effects of the treatments included in our review.
Although magnetic resonance imaging (MRI) measures are widely used in trials of MS, we decided to include only clinical outcomes in this review.
Quality of the evidence
We frequently downgraded the quality of the evidence for relapses at 12 and 24 months from high quality due to study limitations and then either due to inconsistency or imprecision, resulting in moderate or low quality evidence for most of the comparisons. We only judged three out of 39 included trials (8%) to be at low risk of bias, when criteria for allocation concealment, blinding of outcome assessors, and complete outcome data were met. The frequency of downgrading the quality of the evidence regarding benefit for preventing relapses at 12 and 24 months was 40% and 57% of treatment estimates respectively for inconsistency, and 40% and 21% respectively for imprecision.
Reasons for downgrading the quality of the evidence regarding benefit for preventing disability worsening were similar to those reported for relapses. Moreover, the majority of the included trials required only three months' follow‐up to confirm sustained disability worsening. Although we had to accept the definition given in the original papers, we considered the three months criterion to be at high risk of bias because this definition meant that participants who recovered slowly from relapses were regarded as having unremitting disability worsening (Ebers 2008). Thus, for disability worsening at 24 months the confidence in almost all the treatment estimates varied from low to very low, except for natalizumab, which we evaluated as moderate quality.
Potential biases in the review process
1. Transitivity assumption
We assumed that any patient who met the inclusion criteria was, in principle, equally likely to have been randomised to any of the eligible interventions. However, as we discussed in the Background section, several participant characteristics have changed in newer trials, and thus a transitivity hypothesis may have not been a reasonable assumption to make due to differences in patient or trial characteristics. Thus, we evaluated the assumption of transitivity by assessing differences in patient characteristics such as age, disease duration, and baseline Expanded Disability Status Scale (EDSS) scores across the trials, and by comparing the pre‐defined potential effect modifiers across the different comparisons in the networks. We did not find any evidence that important variables varied across comparisons or altered the effectiveness of the treatments; although some confounders may be hidden and unmeasured, it might be reasonable to analyse the network as a whole. Thus, we assumed that the transitivity held and a network meta‐analytical approach was reasonable. However, few studies per comparison were available and limitations in study reporting cannot exclude the possibility of intransitivity.
2. Heterogeneity and inconsistency
We did not find any strong evidence of the presence of heterogeneity either in direct pairwise comparisons or in the entire networks. Similarly, the loop‐specific approach and the 'design‐by‐treatment' model did not provide any clear indication of the presence of inconsistency either locally or in the entire networks. Thus, we believe that the consistency assumption is reasonable for this type of data. However, the power of these tests and approaches to detect inconsistency is low, particularly for networks with a small number of included studies per comparison. Accordingly, we decided to downgrade the evidence for inconsistency on many occasions.
3. Subgroup and sensitivity analyses
None of the analyses performed on any of the hypothesised effect modifiers, such as different diagnostic criteria, prevalence in the included trials of participants who had received first‐ or second‐line treatments, and definitions of relapse and pre‐trial relapse rates, provided any significantly different results compared to the overall analyses. This unexpected result was probably due to the fact that, although there are differences in the characteristics of participants included in older and newer studies, the relative effects of treatments are not affected by any of the effect modifiers we hypothesised.
4. Reporting bias
The possible presence of reporting bias, partially supported by the contour‐enhanced funnel plot for relapses over 12 and 24 months, could not be totally excluded.
Agreements and disagreements with other studies or reviews
In this review, which included RCTs of 15 pharmacological treatments for patients with RRMS, we found that patients receiving natalizumab or alemtuzumab had significantly lower risk of experiencing new relapses over 12 or 24 months compared to placebo, based on high or moderate quality evidence respectively. Currently there is insufficient evidence concerning the superiority of any included treatment in preventing irreversible disability worsening over 24 months compared to placebo; such evidence is of low quality for all the included treatments, with the exception moderate qualityevidence for natalizumab.
In our previous Cochrane review of RCTs, including only trials of the nine drugs firstly approved for RRMS, we evaluated the quality of evidence for benefit derived from pairwise comparisons because the GRADE approach was available only for traditional meta‐analysis (Filippini 2013). In that review we found high quality evidence that natalizumab and interferon beta‐1a (Rebif) were superior to all other treatments for preventing new relapses in RRMS over 12 and 24 months compared to placebo. Moderate quality evidence also supported a protective effect of natalizumab and interferon beta‐1a (Rebif) against disability worsening in RRMS over 24 months compared to placebo. In this new review we were able to assess the quality of the evidence using an adaptation of the standard GRADE approach to the results from network meta‐analysis, which is now available (Salanti 2014). By using this method we could confirm the superiority of natalizumab for preventing new relapses over 12 and 24 months (high quality evidence) and for disability worsening over 24 months (moderate quality evidence). We cannot confirm the previous results for interferon beta‐1a (Rebif), which we now judge to be low or very low quality evidence. Accordingly, for relapses over 12 months interferon beta‐1a (Rebif) has been compared in trials with placebo, interferon beta‐1a (Avonex), teriflunomide, and alemtuzumab. We have judged the evidence for interferon beta‐1a (Rebif) versus placebo as low quality due to limitations of the studies included in the loops where interferon beta‐1a (Rebif) was involved. Heterogeneity was also present within the loops. We found a similar scenario for relapses and disability worsening over 24 months.
One network meta‐analysis examined 48 RCTs (20,455 participants), published before 12 November 2012, of interferons beta, glatiramer acetate, natalizumab, fingolimod, teriflunomide, and mitoxantrone for patients with RRMS, secondary‐progressive MS (SPMS) with relapses and progressive‐relapsing MS (PRMS) or combinations of the previous types of MS (Hadjigeorgiou 2013). The direct analysis showed that fingolimod was more beneficial than interferon beta‐1a (Avonex) (odds ratio (OR) 2.02, 95% confidence interval (CI) 1.46 to 2.79) for preventing new relapses, and interferon beta‐1b was more beneficial than interferon beta‐1a (Avonex) (OR 2.77, 95% CI 1.33 to 5.88) for preventing disability worsening. The indirect analysis indicated that natalizumab may have better relative benefit compared with the other treatments for preventing new relapses. The authors reported that no data were available for any comparisons regarding adverse events with those treatments. Most of our findings cannot be compared to Hadjigeorgiou's network meta‐analysis since this focused on different types of participants and did not include all of the treatments that are now available for RRMS, which we have included in our review. Moreover, the authors did not assess the quality of the evidence for the results arising from their network meta‐analysis.
The network meta‐analysis published by Zintzaras 2012 is a previous version of the article published by Hadjigeorgiou 2013. In Zintzaras 2012, treatments without marketing authorisation have been also included, resulting in 109 studies comparing different therapies commonly used for MS, but also many agents that are not currently in clinical use, such as bovine myelin, or that were rejected by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) because they were found to cause toxicity, such as cladribine. The meta‐analysis considered 145 arms as different treatments (that is one for each dose of each treatment) compared to interferon beta‐1b (250 µg) (that is the chosen reference treatment for analysis) and provided about 90 estimates based on eight direct comparisons with interferon beta‐1b. Thus, the remaining estimates were obtained through the use of indirect analysis. The authors reported that their results needed to be interpreted with caution because the network was dominated by indirect comparisons, but they claimed that combination therapies could be more promising than monotherapies. Important facts invalidate this conclusion in our opinion. First, this claim came only from indirect comparisons. Second, combined therapies were not superior to a single compared treatment or resulted in a worst outcome. For example, methylprednisolone in combination with interferon beta‐1a did not improve disability worsening any more than interferon beta‐1a alone (Ravnborg 2010), or atorvastatin combined with interferon beta‐1a resulted in increased MRI and clinical disease activity (Birnbaum 2008). Third, some of the primary studies included in the indirect analysis were small phase two trials (Birnbaum 2008; Goodman 2009; Weiner 1993), or used no validated clinical outcomes to assess treatment effects (Khoury 2010). Fourth, combination therapies increased the frequency of serious adverse events.
Authors' conclusions
Implications for practice.
Some conservative interpretation of these results is warranted, since most of the included treatments have been evaluated in few trials. Nevertheless, we used a comprehensive, transparent, and pragmatic system for rating the quality of the evidence (i.e. the GRADE approach), so the results of this review may provide guidance to clinicians and patients. According to the GRADE approach, implications for practice should be based on moderate to high quality evidence since any estimate of effect based on low to very low quality evidence is very uncertain and further research is likely to change the estimate. The results of this review show that for preventing clinical relapses in the short term (24 months), alemtuzumab, natalizumab, and fingolimod are superior to several other treatments, on the basis of moderate to high quality evidence. For preventing disability worsening in the short term (24 months) natalizumab is superior to placebo on the basis of moderate quality evidence only.
In addition to the available evidence for benefit provided above, there are two major concerns that have to be considered. First, the benefit of all of these treatments beyond two years is uncertain and this is a relevant issue for a disease with a duration of 30 to 40 years. Second, short‐term trials provide scanty and poorly reported safety data and do not provide useful evidence to obtain a reliable risk profile of treatments. In order to provide information on the long‐term safety of the treatments included in this review, it will be necessary also to evaluate non‐randomised studies.
Finally, more than 70% of the studies included in this review were sponsored by pharmaceutical companies and this may have influenced the results.
Implications for research.
There are three needs that the research agenda should address. First, randomised trials of direct comparisons between active agents would be useful, avoiding further placebo‐controlled studies that do not now comply with the principle of clinical equipoise for relapsing‐remitting multiple sclerosis (RRMS). Second, follow‐up of the original trial cohorts should be mandatory. Third, more studies are needed to evaluate the medium and long‐term benefit and safety of immunotherapies and the comparative safety of the different agents. As the number of drugs, including biologics, that are available for the treatment of RRMS increases, more options will become available to participants and clinicians. In the absence of comparative trials, national and international registries and other types of large non‐randomised studies might be relevant sources for providing complementary data regarding the long‐term benefit and safety of immunotherapies for RRMS.
Acknowledgements
We thank Andrea Fittipaldo for developing the search strategy methods for identification of studies, Ilaria Pacchetti for contributing to data collection, and Silvana Simi, Hwanhee Hong, and Chris Cameron for their useful comments during the preparation of the Cochrane review. We also express our gratitude for the assistance and guidance of Toby Lasserson and Nuala Livingstone for reviewing the review.
Appendices
Appendix 1. Keywords for searching the Cochrane Multiple Sclerosis Group Register
{interferon\*} OR {interferon beta} OR {beta‐1 interferon} OR {beta 1 interferon} OR {interferon beta‐1\*} OR {rebif} OR {avonex} OR {Betaseron} OR {beta‐seron} OR {betaferon} OR {beta‐IFN‐1\*} OR {interferon beta‐1\*} OR {Interferon‐beta\*} OR {interferon beta\*} OR {recombinant interferon beta‐1\*} OR {beta‐1a interferon} OR {beta 1a interferon} OR {interferon beta‐1a} OR {beta 1b interferon} OR {interferon beta1b } OR {IFNb‐1b} OR {IFNbeta‐1b} OR {interferon beta‐1b} OR {novantrone} OR {novantron} OR {onkotrone} OR {pralifan} OR {mitozantrone} OR {mitoxantrone} OR {copolymer‐1} OR {cop‐1} OR {copaxone} OR {glatiramer acetate} OR {cpx} OR {cop1} OR {copolymer} OR {glatiramer} OR {immunomodulation\*} OR {immunomodulator\*} OR {immunosuppression} OR {antegren} OR {natalizumab} OR {tysabri} OR {monoclonal antibody*} OR {Antibodies, Monoclonal} OR {fingolimod} OR {FTY720} OR {FTY 720} OR {fingolimod hydrochloride} OR {FTY‐720} OR {2‐amino‐2‐(2‐(4‐octylphenyl)ethyl)‐1,3‐propanediol hydrochloride} OR {Gilenya} OR {sphingosine‐fosphate‐receptor antagonist} OR {HMR1726} OR {A77 1726} OR {Leflunomide} OR {Arava} OR {teriflunomide} OR {TFN} OR {teriflunomide‐D4} OR {A771726} OR {Dihydroorotate dehydrogenase (DHODH) inhibitors} OR {(Z)‐2‐Cyano‐3‐hydroxy‐N‐[4‐(trifluoromethyl)phenyl]‐2‐butenamide} OR {dimethylfumarate} OR {Fumaderm} OR {FAG 201} OR {FAG201} OR {FAG‐201} OR {BG 00012} OR {BG00012} OR {BG‐00012} OR {BG 12 compound} OR {BG12 compound} OR {BG‐12 compound} OR {BG‐12} OR {tecfidera} OR {Nrf2 activator} OR {oral fumarate} OR {fumaric acid eaters} OR {alemtuzumab} OR {Campath 1G} OR {Campath‐1G} OR {Campath‐1‐G} OR {Campath 1M} OR {Campath‐1M} OR {MabCampath} OR {Schering brand of alemtuzumab} OR {Campath} OR {Berlex brand of alemtuzumab} OR {Campath 1H} OR {monoclonal antibody Campath‐1H} OR {Campath‐1H} OR {monoclonal antibody*} OR {Antibodies, Monoclonal} OR {lemtrada} OR {daclizumab} OR {antigen} OR {zenapax} OR {dacliximab} OR {monoclonal antibody} OR {monoclonal antibodies} OR {antigens} OR {Laquinimod} OR {azathioprine} OR {azathioprine} OR {immuran} OR {imuran} OR {imurel} OR {immunoglobulin\*} OR {intravenous immunoglobulin\*} OR {iV immunoglobulin\*} {intravenous} OR {Intravenous IG} OR {Intravenous Antibodies} {ivig} OR {igiv} OR {adrenal cortex hormones} OR {steroid\*} OR {methylprednisolone} OR {prednisolone} OR {dexamethasone} OR {corticosteroid\*} OR {acth} OR {prednisone} OR {Adrenocorticotropic Hormone} OR {polyethylene glycol‐interferon‐beta‐1a} OR {PEG IFN‐beta‐1a} OR {Pegylated interferon beta‐1a} OR {Ocrelizumab} OR {placebo\*}
AND
{relapsing remitting} OR {relapsing‐remitting } OR {remitting‐relapsing} OR {remitting relapsing} OR {relapses} OR {relapsing} OR {relapse} OR {RR‐MS}
Data and analyses
Comparison 1. Treatment benefit within pairwise comparisons.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Comparisons for relapses over 12 months | 29 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Interferon beta‐1a (Avonex) versus placebo | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.73, 1.05] |
| 1.2 Interferon beta‐1a (Rebif) versus placebo | 2 | 853 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.66, 1.19] |
| 1.3 Glatiramer acetate versus placebo | 4 | 2416 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.66, 0.95] |
| 1.4 Natalizumab versus placebo | 1 | 942 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.47, 0.66] |
| 1.5 Mitoxantrone versus placebo | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.21, 0.74] |
| 1.6 Fingolimod versus placebo | 2 | 2355 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.48, 0.82] |
| 1.7 Teriflunomide versus placebo | 2 | 2257 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.78, 0.95] |
| 1.8 Dimethyl fumarate versus placebo | 2 | 2307 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.71, 0.88] |
| 1.9 Pegylated interferon beta‐1a versus placebo | 1 | 1512 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.78, 1.01] |
| 1.10 Daclizumab versus placebo | 1 | 621 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| 1.11 Azathioprine versus placebo | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.61, 1.24] |
| 1.12 Immunoglobulins versus placebo | 3 | 219 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.47, 1.36] |
| 1.13 Interferon beta‐1a (Rebif) versus interferon beta‐1a (Avonex) | 1 | 677 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.80, 1.03] |
| 1.14 Glatiramer acetate versus interferon beta‐1b (Betaseron) | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.48, 1.38] |
| 1.15 Azathioprine versus interferons beta (Avonex, Rebif or Betaseron) | 2 | 244 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.49, 1.33] |
| 1.16 Fingolimod versus interferon beta‐1a (Avonex) | 1 | 1292 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.57, 0.79] |
| 1.17 Teriflunomide versus interferon beta‐1a (Rebif) | 1 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.69, 1.18] |
| 1.18 Dimethyl fumarate versus glatiramer acetate | 1 | 1067 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.80, 1.14] |
| 1.19 Alemtuzumab versus interferon beta‐1a (Rebif) | 3 | 1582 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.39, 0.55] |
| 2 Comparisons for relapses over 24 months | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Interferon beta‐1b (Betaseron) versus placebo | 1 | 372 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.81, 0.99] |
| 2.2 Interferon beta‐1a (Avonex) versus placebo | 2 | 1198 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.76, 1.04] |
| 2.3 Interferon beta‐1a (Rebif) versus placebo | 1 | 560 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.77, 0.92] |
| 2.4 Glatiramer acetate versus placebo | 3 | 1024 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.75, 0.98] |
| 2.5 Natalizumab versus placebo | 1 | 942 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.49, 0.64] |
| 2.6 Mitoxantrone versus placebo | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.27, 0.80] |
| 2.7 Fingolimod versus placebo | 2 | 2355 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.67, 0.78] |
| 2.8 Teriflunomide versus placebo | 1 | 1088 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.79, 0.98] |
| 2.9 Dimethyl fumarate versus placebo | 2 | 2307 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.81, 0.97] |
| 2.10 Laquinimod versus placebo | 2 | 1990 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.75, 0.99] |
| 2.11 Azathioprine versus placebo | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.56, 1.05] |
| 2.12 Immunoglobulins versus placebo | 2 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.61, 0.90] |
| 2.13 Interferon beta‐1a (Avonex) versus interferon beta‐1b (Betaseron) | 1 | 188 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [1.06, 1.71] |
| 2.14 Interferon beta‐1a (Rebif) versus interferon beta‐1b (Betaseron) | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.89, 1.11] |
| 2.15 Glatiramer acetate versus interferon beta‐1b (Betaseron) | 2 | 2319 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.47, 1.38] |
| 2.16 Glatiramer acetate versus interferon beta‐1a (Rebif) | 1 | 764 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.78, 1.09] |
| 2.17 Dimethyl fumarate versus glatiramer acetate | 1 | 1067 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.98, 1.21] |
| 2.18 Alemtuzumab versus interferon beta‐1a (Rebif) | 3 | 1582 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.39, 0.65] |
| 2.19 Laquinimod versus interferon beta‐1a (Avonex) | 1 | 881 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.97, 1.32] |
| 2.20 Azathioprine versus interferons beta (Avonex, Rebif or Betaseron) | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.16] |
| 3 Comparisons for disability worsening over 24 months | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Interferon beta‐1b (Betaseron) versus placebo | 1 | 372 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.72, 1.32] |
| 3.2 Interferon beta‐1a (Avonex) versus placebo | 2 | 1198 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.70, 1.09] |
| 3.3 Interferon beta‐1a (Rebif) versus placebo | 1 | 560 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.61, 0.96] |
| 3.4 Glatiramer acetate versus placebo | 3 | 1024 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.61, 1.09] |
| 3.5 Natalizumab versus placebo | 1 | 942 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.52, 0.80] |
| 3.6 Mitoxantrone versus placebo | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.05, 0.83] |
| 3.7 Fingolimod versus placebo | 2 | 2355 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.76, 0.99] |
| 3.8 Teriflunomide versus placebo | 1 | 1088 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.75, 1.01] |
| 3.9 Dimethyl fumarate versus placebo | 2 | 2307 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.72, 0.90] |
| 3.10 Laquinimod versus placebo | 2 | 1990 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.73, 0.95] |
| 3.11 Azathioprine versus placebo | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.31, 1.34] |
| 3.12 Immunoglobulins versus placebo | 2 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.39, 1.24] |
| 3.13 Interferon beta‐1a (Avonex) versus interferon beta‐1b (Betaseron) | 1 | 188 | Risk Ratio (M‐H, Random, 95% CI) | 2.23 [1.29, 3.83] |
| 3.14 Interferon beta‐1a (Rebif) versus interferon beta‐1b (Betaseron) | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.78, 1.25] |
| 3.15 Glatiramer acetate versus interferon beta‐1b (Betaseron) | 2 | 2319 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.86, 1.13] |
| 3.16 Glatiramer acetate versus interferon beta‐1a (Rebif) | 1 | 764 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.39, 0.85] |
| 3.17 Dimethyl fumarate versus glatiramer acetate | 1 | 1067 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.83, 1.22] |
| 3.18 Alemtuzumab versus interferon beta‐1a (Rebif) | 3 | 1582 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.32, 0.54] |
| 3.19 Laquinimod versus interferon beta‐1a (Avonex) | 1 | 881 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.85, 1.33] |
| 3.20 Azathioprine versus interferons beta (Avonex, Rebif or Betaseron) | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.49, 1.23] |
Comparison 2. Treatment acceptability within pairwise comparisons.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Comparisons for treatment discontinuation due to AEs over 12 months | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Interferon beta‐1a (Avonex) 30 µg versus placebo | 1 | 301 | Risk Ratio (M‐H, Random, 95% CI) | 3.17 [0.67, 15.00] |
| 1.2 Interferon beta‐1a (Rebif) 22 µg versus placebo | 1 | 195 | Risk Ratio (M‐H, Random, 95% CI) | 3.16 [0.13, 76.54] |
| 1.3 Interferon beta‐1a (Rebif) 44 µg versus placebo | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 11.22 [0.63, 200.27] |
| 1.4 Glatiramer acetate 20 mg daily versus placebo | 1 | 239 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.26, 8.89] |
| 1.5 Glatiramer 40 mg three times per week versus placebo | 1 | 1404 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [0.99, 5.65] |
| 1.6 Teriflunomide 7 mg versus placebo | 1 | 797 | Risk Ratio (M‐H, Random, 95% CI) | 2.06 [1.31, 3.24] |
| 1.7 Teriflunomide 14 mg versus placebo | 1 | 761 | Risk Ratio (M‐H, Random, 95% CI) | 2.51 [1.61, 3.91] |
| 1.8 Pegylated interferon beta‐1a every 4 weeks versus placebo | 1 | 1000 | Risk Ratio (M‐H, Random, 95% CI) | 2.78 [1.31, 5.89] |
| 1.9 Pegylated interferon beta‐1a every 2 weeks versus placebo | 1 | 1012 | Risk Ratio (M‐H, Random, 95% CI) | 2.82 [1.34, 5.96] |
| 1.10 Daclizumab 150 mg versus placebo | 1 | 412 | Risk Ratio (M‐H, Random, 95% CI) | 3.43 [0.72, 16.33] |
| 1.11 Daclizumab 300 mg versus placebo | 1 | 413 | Risk Ratio (M‐H, Random, 95% CI) | 4.39 [0.96, 20.08] |
| 1.12 Immunoglobulins 0.2 g versus placebo | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 2.14 [0.23, 19.96] |
| 1.13 Immunoglobulins 0.4 g versus placebo | 1 | 34 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.15, 76.93] |
| 1.14 Interferon beta‐1a (Rebif) 44 µg versus interferon beta‐1a (Avonex) 30 µg | 1 | 677 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.56, 1.97] |
| 1.15 Fingolimod 0.5 mg versus interferon beta‐1a (Avonex) 30 µg | 1 | 866 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.81, 2.54] |
| 1.16 Fingolimod 1.25 mg versus interferon beta‐1a (Avonex) 30 µg | 1 | 861 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.40, 3.98] |
| 1.17 Teriflunomide 7 mg versus interferon beta‐1a (Rebif) 44 µg | 1 | 213 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.19, 0.81] |
| 1.18 Teriflunomide 14 mg versus interferon beta‐1a (Rebif) 44 µg | 1 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.27, 0.98] |
| 1.19 Azathioprine versus interferons beta (Avonex, Rebif or Betaseron) | 1 | 94 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.21, 4.70] |
| 2 Comparisons for treatment discontinuation due to AEs over 24 months | 23 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Interferon beta‐1b (Betaseron) 50 µg versus placebo | 1 | 248 | Risk Ratio (M‐H, Random, 95% CI) | 4.92 [0.58, 41.51] |
| 2.2 Interferon beta‐1b (Betaseron) 250 µg versus placebo | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 9.92 [1.29, 76.32] |
| 2.3 Interferon beta‐1a (Avonex) 30 µg versus placebo | 1 | 897 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.81, 2.54] |
| 2.4 Interferon beta‐1a (Rebif) 22 µg versus placebo | 1 | 376 | Risk Ratio (M‐H, Random, 95% CI) | 2.31 [0.61, 8.79] |
| 2.5 Interferon beta‐1a (Rebif) 44 µg versus placebo | 1 | 371 | Risk Ratio (M‐H, Random, 95% CI) | 3.05 [0.84, 11.08] |
| 2.6 Glatiramer acetate 20 mg daily versus placebo | 3 | 1024 | Risk Ratio (M‐H, Random, 95% CI) | 1.74 [0.49, 6.13] |
| 2.7 Natalizumab versus placebo | 1 | 942 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.93, 2.53] |
| 2.8 Mitoxantrone versus placebo | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 9.82 [0.57, 168.84] |
| 2.9 Fingolimod 0.5 mg versus placebo | 2 | 1556 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.89, 2.25] |
| 2.10 Fingolimod 1.25 mg versus placebo | 2 | 1572 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [1.48, 2.52] |
| 2.11 Teriflunomide 7 mg versus placebo | 1 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.77, 1.96] |
| 2.12 Teriflunomide 14 mg versus placebo | 1 | 722 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.86, 2.15] |
| 2.13 Dimethyl fumarate 480 mg versus placebo | 2 | 1546 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.91, 1.51] |
| 2.14 Dimethyl fumarate 720 mg versus placebo | 2 | 1534 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.93, 1.54] |
| 2.15 Laquinimod versus placebo | 2 | 1990 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.96, 2.00] |
| 2.16 Azathioprine versus placebo | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 5.8 [0.74, 45.26] |
| 2.17 Immunoglobulins 0.15 to 0.20 g versus placebo | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.32, 28.19] |
| 2.18 Interferon beta‐1a (Avonex) 30 µg versus interferon beta‐1b (Betaseron) 250 µg | 1 | 188 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.02, 1.75] |
| 2.19 Glatiramer acetate 20 mg daily versus interferon beta‐1b (Betaseron) 250 μg | 2 | 1420 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.56, 2.53] |
| 2.20 Glatiramer acetate 20 mg daily versus interferon beta‐1b (Betaseron) 500 μg | 1 | 1347 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.37, 1.68] |
| 2.21 Glatiramer acetate 20 mg daily versus interferon beta‐1a (Rebif) 44 µg | 1 | 764 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.47, 1.52] |
| 2.22 Dimethyl fumarate 480 mg versus glatiramer acetate 20 mg daily | 1 | 722 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.80, 1.84] |
| 2.23 Dimethyl fumarate 720 mg versus glatiramer acetate 20 mg daily | 1 | 705 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.80, 1.85] |
| 2.24 Alemtuzumab 12 mg versus interferon beta‐1a (Rebif) 44 µg | 3 | 1472 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.22, 0.68] |
| 2.25 Alemtuzumab 24 mg versus interferon beta‐1a (Rebif) 44 µg | 2 | 625 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.10, 1.09] |
| 2.26 Laquinimod versus interferon beta‐1a (Avonex) 30 µg | 1 | 881 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.49, 1.45] |
| 2.27 Azathioprine versus interferons beta (Avonex, Rebif or Betaseron) | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 2.21 [0.90, 5.45] |
Comparison 3. Treatment safety against placebo within pairwise comparisons.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Serious adverse events | 16 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Interferons beta (Avonex, Rebif or Betaseron) versus placebo | 3 | 870 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.67, 2.37] |
| 1.2 Glatiramer acetate versus placebo | 2 | 490 | Risk Ratio (M‐H, Random, 95% CI) | 1.87 [0.73, 4.74] |
| 1.3 Natalizumab versus placebo | 1 | 939 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.81, 1.73] |
| 1.4 Fingolimod versus placebo | 2 | 1572 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.72, 1.30] |
| 1.5 Teriflunomide versus placebo | 1 | 718 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.87, 1.83] |
| 1.6 Dimethyl fumarate versus placebo | 2 | 1531 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.76, 1.55] |
| 1.7 Pegylated interferon beta‐1a versus placebo | 1 | 1012 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.57, 1.68] |
| 1.8 Daclizumab versus placebo | 1 | 413 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.77, 3.10] |
| 1.9 Laquinimod versus placebo | 2 | 1988 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.67, 1.41] |
| 1.10 Immunoglobulins versus placebo | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.11, 3.70] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Achiron 1998.
| Methods | RCT | |
| Participants | Age: 19 to 60 years; definite RRMS; mean disease duration 4 years; mean EDSS 3.0; prior use of DMT not reported | |
| Interventions | Loading dose of immunoglobulins 0.4 g/kg body weight intravenously daily for 5 consecutive days followed by additional booster doses of immunoglobulins 0.4 g/kg body weight intravenously daily every 2 months for 24 months (n = 20) Placebo consisting of 0.9% saline administered with the same schedule as the active treatment (n = 20) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Miles Inc. Cutter Biological, Bayer and Promedico | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were assigned to receive immunoglobulin or placebo by a block‐stratified randomisation procedure, designed to ensure groups balanced for YER, age, and disease duration" (Page 399) |
| Allocation concealment (selection bias) | Unclear risk | "Randomization was performed at the pharmacy, and the bottles of immunoglobulin or placebo were wrapped in sealed opaque bags and brought to the patients' rooms. The entire IV set was covered by an opaque plastic bag to ensure that any possible fluid turbidity or frothing would not be evident to the investigators or patients" (Page 399) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All patients and evaluators were blinded to treatment" (Page 399) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A relapse was confirmed only when the patient's symptoms were accompanied by objective changes on neurologic examination by the treating neurologist who was blind to the patient's treatment", and "Upon entry, and monthly thereafter, every patient underwent a neurologic examination by two examining neurologists, and an independent EDSS score was recorded by each" (Page 399) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 5.0% were lost to follow‐up (5.0% in immunoglobulins and 5.0% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Triton Biosciences and the role of the study sponsor was unclear ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of patients who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse and disability worsening confirmed at 3 months outcomes were reported incompletely, and disability worsening confirmed at 6 months was not assessed |
ADVANCE 2014.
| Methods | RCT | |
| Participants | Age: 18 to 65 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.5; prior use of any MS medication at any time prior to the start of study: 17% | |
| Interventions | Peg‐interferon beta‐1a 125 μg subcutaneously once every 2 weeks for 12 months (n = 512) Peg‐interferon beta‐1a 125 μg subcutaneously once every 4 weeks for 12 months (n = 500) Placebo subcutaneously once every 2 weeks for 12 months (n = 500) |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned (1:1:1) to receive subcutaneous injections with pre‐filled syringes of placebo, peginterferon beta‐1a at a dose of 125 μg once every 2 weeks, or peginterferon beta‐1a 125 μg once every 4 weeks, stratified by site" (Page 658) |
| Allocation concealment (selection bias) | Low risk | "Randomisation was done by a centralised interactive voice response and web system. Placebo was a matched diluent, given with a matched pre‐filled syringe. Patients received either study drug or placebo every 2 weeks to maintain masking; those assigned to receive study drug every 4 weeks received alternate injections of placebo and peginterferon beta‐1a every 2 weeks" (Page 658) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All study management and site personnel, investigators, and patients were masked to treatment assignment" (Page 658) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Each site had separate examining and treating neurologists, thereby maintaining rater masking for all treatment groups" and "relapse was confirmed by the independent neurological evaluation committee" (Page 658) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 11.9% were lost‐to follow‐up (14.5% in peg‐interferon beta‐1a 125 μg every 2 weeks, 12.4% in peg‐interferon beta‐1a 125 μg every 4 weeks, and 8.8% in placebo), with some indication of the differences in reasons: adverse events of 4.8% in peg‐interferon beta‐1a 125 μg every 2 weeks, 4.7% in peg‐interferon beta‐1a 125 μg every 4 weeks, and 1.0% in placebo |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Biogen Idec, "Biogen Idec collected, analysed, and contributed to the interpretation of the data" (Page 659), and 5 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of patients who discontinued the study and in time to discontinuation, which they did not report |
AFFIRM 2006.
| Methods | RCT | |
| Participants | Age: 18 to 50 years; definite RRMS; median disease duration 5 years (range, 0 to 34 years); mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | Natalizumab 300 mg by intravenous infusion once every 4 weeks for up to 116 weeks (n = 627) Placebo (unspecified) (n = 315) | |
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Biogen Idec, Inc. and Elan Pharmaceutica | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned to treatment that was stratified according to study site in blocks of three (two active, one placebo) with the use of a computer‐generated block randomization schedule" (Page 900) |
| Allocation concealment (selection bias) | Low risk | "A multidigit identification number, implemented by an interactive voice‐response system was used" (Page 900) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All study personnel, patients, sponsor personnel involved in the conduct of the study, and the investigator advisory committee were unaware of treatment assignments throughout the study", and "Treating neurologists were responsible for all aspects of patient care, including the management of adverse events and the treatment of relapsing disease" (Pages 900‐1) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Examining neurologists performed objective evaluation with use of the EDSS and neurologic examination during all study visits; they were not in contact with patients in any other capacity, so as to reduce the possibility of being unblinded by side effects or laboratory assessments", "Patients visited the clinic every 12 weeks for scoring on the EDSS", and "If a relapse was suspected, the patient was referred to the examining neurologist, who evaluated the patient within five days after the event" (Page 901) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 9.1% were lost‐to follow‐up (8.3% in natalizumab and 10.8% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Biogen Idec and Elan Pharmaceuticals, "Data were analyzed by Biogen Idec and Elan Pharmaceuticals" (Page 909) and 4 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse and disability worsening confirmed at 3 months outcomes were reported incompletely |
ALLEGRO 2012.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 9 years; mean EDSS 2.6; prior use of DMT at any time prior to the start of study: 39.0% (38.2% in laquinimod and 39.7% in placebo) | |
| Interventions | Laquinimod 0.6 mg oral capsule once daily for 24 months (n = 550) Placebo oral capsule once daily for 24 months (n = 556) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomization list, stratified according to study center, was computer‐generated" (Page 1002) |
| Allocation concealment (selection bias) | Low risk | "The subject was allocated a screening number by the investigator using an Interactive Voice Response System (IVRS)" (Page 44 of Protocol) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients and investigators were unaware of the study assignments" (Page 1002) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Neurologic assessments and general medical evaluations were conducted by two neurologists in order to minimize the possibility of unblinding: an examining neurologist assessed neurologic condition, and the treating neurologist determined whether a patient had a relapse", and "the treating neurologist was unaware of the study‐group assignment" (Page 1002) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 21.9% were lost‐to follow‐up (20.5% in laquinimod and 23.2% in placebo), with some indication of the differences in reasons: adverse event(s) in 7.6% in laquinimod and 5.0% in placebo |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ "The sponsor designed and monitored the study" and "The data were collected and analyzed by the sponsor" (Page 1001) ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse and disability worsening confirmed at 6 months outcomes were reported incompletely, and no additional data were provided on request |
BECOME 2009.
| Methods | RCT | |
| Participants | Age: 18 to 55; definite RRMS or CIS; median time since MS onset 1 year; mean EDSS 2.0; all participants (except 1) were previously untreated patients | |
| Interventions | Interferon beta‐1b (Betaseron) 250 μg subcutaneously every other day for 24 months (n = 36) Glatiramer acetate 20 mg subcutaneous daily for 24 months (n = 39) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Bayer Schering Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was stratified by clinical site (Newark or Teaneck) and the presence of enhancement on screening MRI" (Page 1977) |
| Allocation concealment (selection bias) | Unclear risk | Nothing was said about allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients could not be blinded because of the characteristic injection reactions to IFN‐1b or GA" (Page 1977) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Subjective relapses that were confirmed by a blinded examining neurologist using worsening scores on either the Scripps Neurological Rating Scale (SNRS) or the Expanded Disability Status Scale (EDSS) were considered objective relapses" (Page 1977). However, it is not clear how and when the examining neurologist evaluated subjective relapses and EDSS scores |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 14.7% were lost‐to follow‐up (19.4% in interferon beta‐1b and 10.3% in glatiramer acetate), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ "The BECOME study was supported by Bayer Schering Pharma, distributors of IFN‐1b, but was investigator‐initiated and remains the intellectual property of New Jersey Medical School/University of Medicine & Dentistry of New Jersey. The sponsor of the study was allowed to comment on data interpretation and had the opportunity to review and comment on the final manuscript prior to submission. The sponsor was not allowed to participate in any of the following phases of the study: conduct of the study, data collection, data management, data analysis, and preparation of the manuscript" (Page 1981) ‐ Relapse outcome was reported incompletely, and no additional data were provided on request |
BEYOND 2009.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 5 years; mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | Interferon beta‐1b (Betaseron) 250 µg subcutaneous every other day for 24 months (n = 897) Interferon beta‐1b (Betaseron) 500 µg subcutaneous every other day for 24 months (n = 899) Glatiramer acetate 20 mg subcutaneous daily for 24 months (n = 448) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: Bayer HealthCare Pharmaceuticals | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Use of SAS‐based block randomisation with regional stratification" (Page 890) |
| Allocation concealment (selection bias) | Unclear risk | "Patients were randomly assigned in a 2:2:1 ratio ... by the central randomisation group..." (Page 890) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Physicians and patients were double‐blind to comparisons between the two doses of IFNß‐1b... Ibuprofen or acetaminophen were given at the same time as random assignment to IFNß‐1b, at least during the first 3 months, to reduce flu‐like symptoms. The treating physicians and the patients were therefore aware of treatment assignments" (Page 891) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The masked evaluating physicians did all neurological assessments and ascertained functional system and EDSS scores...The evaluating physicians were not involved in the care of patients and had no access to patient files or previous assessments", and "Patients covered their injection sites during neurological examination and did not discuss any adverse events with the evaluating physician" (Pages 891‐2) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 16.0% were lost‐to follow‐up (19.2% in interferon beta‐1b 500 µg, 12.6% in interferon beta‐1b 250 µg, and 16.5% in glatiramer acetate), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | Relapse and disability worsening outcomes were reported incompletely |
Bornstein 1987.
| Methods | RCT | |
| Participants | Age: 20 to 35 years; definite RRMS; mean disease duration 6 years; mean EDSS 3.1; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneous daily for 24 months (n = 25) Placebo bacteriostatic saline subcutaneous daily for 24 months (n = 25) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: grants from the NINCDS and the NIH, Bethesda, Md | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The random assignment of the first patient of a pair determined the assignment of both" (Page 409) |
| Allocation concealment (selection bias) | High risk | An open allocation schedule was used: "Treatment assignments were made known to the clinical assistant responsible for the production, labelling and distribution of medication" (Page 409) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "The patient's self evaluation of ... side effects were reported to the clinical assistant, who was not blinded to the treatment" (Page 409) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Patients visited the clinic every three months for two years. At each visit, a neurologist unaware of the patient's treatment group completed a neurologic examination and status evaluation" and "Patients were also seen at the time of suspected exacerbations ... the neurologist verified exacerbations on the basis of study criteria" (Page 409) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 4.0% were lost‐to follow‐up (0% in glatiramer acetate and 8.0% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | Low risk | The study appears to be free of other sources of bias |
BRAVO 2014.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; median disease duration 5 years; median EDSS 2.5; prior use of DMT at any time prior to the start of study: 7.4% (6.9% in laquinimod, 9.4% in interferon beta‐1a and 6.0% in placebo) | |
| Interventions | Laquinimod 0.6 mg oral capsule once daily for 24 months (n = 434) Interferon beta‐1a (Avonex) 30 µg intramuscular once a week for 24 months (n = 447) Placebo oral capsule once daily for 24 months (n = 450) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The computer‐generated randomization scheme prepared by the Teva Global Biostatistics Unit" (Page 775) |
| Allocation concealment (selection bias) | Unclear risk | "1:1:1 treatment assignment ratio stratified by study center, to laquinimod 0.6 mg capsule once‐daily, matching oral placebo, or IFNß‐1a IM 30 µg once‐weekly injection" (Page 775) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients and treating neurologists were blinded to oral treatment assignment (laquinimod or placebo), but not to IFNb‐1a IM assignment", and "All patients, including those receiving oral treatment, wore clothing and/or a robe that ensured coverage of all potential IM injection sites during examination and were instructed not to discuss adverse events (AEs), routes of administration, or treatment assignments with the examining neurologist" (Page 775) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The examining neurologist was blinded to all treatments", and "The examining neurologist performed an EDSS assessment for relapse confirmation within 7 days of symptom onset" (Page 775) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 18.1% were lost‐to follow‐up (18.7% in laquinimod, 15.4% in interferon beta‐1a, and 20.2% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ "N. Sasson of Teva Pharmaceutical Industries provided statistical support for the manuscript" (Page 773), and 2 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
CAMMS223 2008.
| Methods | RCT | |
| Participants | Age: 18 to 50; definite RRMS; median time since first relapse 1 year; mean EDSS 1.9; all participants were previously untreated patients | |
| Interventions | Alemtuzumab 24 mg per day intravenously on 5 consecutive days during the first month and on 3 consecutive days at months 12 and 24 (n = 110) Alemtuzumab 12 mg per day intravenously on 5 consecutive days during the first month and on 3 consecutive days at months 12 and 24 (n = 113) Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 36 months (n = 111) All participants received 1 g of intravenous methylprednisolone for 3 days at baseline and at months 12 and 24 |
|
| Outcomes | Relapse at 12, 24, and 36 months. Disability worsening at 24 and 36 months | |
| Notes | Funding: Genzyme (a Sanofi company) and Bayer Schering Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Eligible patients were randomly assigned in a 1:1:1 ratio to receive alemtuzumab (at a dose of either 12 mg per day or 24 mg per day) or interferon beta‐1a with the use of the Pocock and Simon minimization algorithm to balance the study groups with regard to age (<30 years or ≥30 years), sex, and baseline EDSS score (<2.0 or ≥2.0)" (Page 1787) |
| Allocation concealment (selection bias) | Low risk | "Patients were allocated via an interactive voice response system (IVRS)" (Information provided on request by Genzyme) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients wore clothing that covered injection sites", and "Safety was assessed quarterly by the treating neurologist, who was aware of study‐group assignment" (Page 1787) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "EDSS scores were determined quarterly in a blinded fashion by a neurologist who also adjudicated possible relapses. Patients wore clothing that covered injection sites" (Page 1787). It is not clear how potential relapses were assessed |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 25.1% were lost to follow‐up (16.4% in alemtuzumab 24 mg, 18.6% in alemtuzumab 12 mg, and 40.5% in interferon beta‐1a), with some indication of the differences in reasons: adverse events of 0.01% in alemtuzumab 24 mg, 1.8% in alemtuzumab 12 mg, and 11.7% in interferon beta‐1a; and lack of benefit of 1.8% in alemtuzumab 24 mg, 1.8% in alemtuzumab 12 mg, and 14.4% in interferon beta‐1a |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes. Missing data not reported in the published paper were provided on request by Genzyme |
| Other bias | High risk | "Genzyme employees analyzed the data" (Page 1789), and 5 co‐authors of the published paper were affiliated to the pharmaceutical company |
CARE‐MS I 2012.
| Methods | RCT | |
| Participants | Age: 18 to 50 years; definite RRMS; mean disease duration 2 years; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | Alemtuzumab 12 mg per day intravenously on 5 consecutive days at month 0 and 3 consecutive days at month 12 (n = 386) Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 24 months (n = 195) Participants in both groups received 1 g per day of intravenous methylprednisolone on 3 consecutive days at baseline and at month 12. After a protocol amendment in January 2009, alemtuzumab patients received oral aciclovir 200 mg twice daily during alemtuzumab infusion and for 28 days thereafter as prophylaxis against herpes infection |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "We randomly allocated patients in a 2:1 ratio" and "Randomisation was stratified by site" (Page 1820) |
| Allocation concealment (selection bias) | Low risk | "We randomly allocated patients using an interactive voice response system" (Page 1820) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Because both study drugs have adverse effects that precluded masking of patients and treating clinicians to treatment assignment, and because subcutaneous interferon beta 1a was available only in proprietary prefilled syringes that could not effectively be duplicated for placebo..." (Page 1820) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "We secured clinical data integrity by stringent clinical and MRI rater masking, and adjudication of relapses by a committee comprising six independent and masked neurologists. In the absence of a masked rater, unmasked raters could submit EDSS assessments" (Page 1820). Moreover, it is not clear how and when the committee evaluated potential relapses |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 7.1% were lost to follow‐up (4.9% in alemtuzumab 12 mg and 11.3% in interferon beta‐1a), with some indication of the differences in reasons: adverse events of 2.6% in alemtuzumab and 0% in interferon beta‐1a |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | "The study sponsor (Genzyme) was involved in the design and undertaking of the trial, data analysis and interpretation, writing of the manuscript, and the decision to submit the manuscript for publication. Bayer Schering Pharma participated in the design and oversight of the trial", "The sponsor did the statistical analyses" (Page 1822), and 4 co‐authors of the published paper were affiliated to the pharmaceutical company |
CARE‐MS II 2012.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 5 years; mean EDSS 2.7; all patients were previously treated: "at least one relapse while on interferon beta or glatiramer after at least 6 months of treatment" | |
| Interventions | Alemtuzumab 24 mg per day intravenously on 5 consecutive days at month 0 and 3 consecutive days at month 12 (n = 170; data presented for safety assessment only) Alemtuzumab 12 mg per day intravenously on 5 consecutive days at month 0 and 3 consecutive days at month 12 (n = 436) Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 24 months (n = 231) Participants in both groups received 1 g per day of intravenous methylprednisolone on 3 consecutive days at baseline and at month 12. After a protocol amendment in December 2008, alemtuzumab patients received oral aciclovir 200 mg twice daily during alemtuzumab infusion and for 28 days thereafter as prophylaxis against herpes infection |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "2:1 randomisation allocation stratified by site" (Pages 1830‐1) |
| Allocation concealment (selection bias) | Low risk | "We randomly allocated patients with an interactive voice response system" (Page 1830) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Because both study drugs had adverse effects that precluded double‐blinding, and interferon beta 1a proprietary syringes could not effectively be duplicated for placebo..." (Page 1831) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Clinical data integrity was secured by stringent rater‐masking and independent adjudication of relapses. Raters, who were masked to treatment‐group assignment, did the EDSS assessments every 3 months and when a relapse was suspected" and "In the absence of a masked rater, unmasked raters could submit EDSS assessments" (Page 1831). Moreover, it is not clear how and when the raters evaluated potential relapses |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 11.4% were lost to follow‐up (4.6% in alemtuzumab 12 mg and 24.2% in interferon beta‐1a), with some indication of the differences in reasons: lack of benefit of 0% in alemtuzumab 12 mg and 2.6% in interferon beta‐1a |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ "Genzyme (Sanofi) was involved in the design and undertaking of the trial, data analysis and interpretation, writing of the manuscript, and the decision to submit the manuscript for publication" (Page 1833), and 4 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ Sample size reported in the article was not that estimated in the protocol but calculated after an amendment in December 2008 |
CombiRx 2013.
| Methods | RCT | |
| Participants | Age: 18 to 60 years; definite RRMS; mean disease duration 1 year; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | Interferon beta‐1a (Avonex) 30 µg intramuscular once a week with matched placebo preparation for 36 months (n = 250) Glatiramer acetate 20 mg subcutaneous daily with matched placebo preparation for 36 months (n = 259) |
|
| Outcomes | Relapse at 36 months. Disability worsening at 36 months | |
| Notes | Funding: National Institutes of Health (NIH) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Participants were randomized via a computerized data entry system using a permuted block design within sites with block sizes of 6 and 12" (Page 328) |
| Allocation concealment (selection bias) | Low risk | "Participants were randomized via a computerized data entry system that masked treatment arm allocation" (Page 328) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Participants were randomized via a computerized data entry system that masked drug dispensing to participants and all site personnel for the entire duration of the trial period" (Page 328) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Treating clinician and an examining clinician were both blinded to treatment assignment", "confirmed progression was assessed by the blinded EDSS examiner and confirmed centrally", and "The designation of the type of relapse was determined centrally according to data entered onto a relapse assessment form and the change in EDSS" (Page 328‐329). The blinding of central commission was not reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 18.1% were lost to follow‐up (22.4% in interferon beta‐1a and 13.9% in glatiramer acetate; P value for proportion terminating early = 0.029), with some indication of the differences in reasons: adverse event(s) of 7.2% in interferon beta‐1a and 4.6% in glatiramer acetate |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of patients who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse and disability worsening confirmed at 6 months outcomes were reported incompletely, and no additional data were provided on request |
Comi 2001.
| Methods | RCT | |
| Participants | Age 18 to 50 years; definite RRMS; mean disease duration 8 years; mean EDSS 2.4; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneous daily for 9 months (n = 119) Placebo (not described) (n = 120) |
|
| Outcomes | Relapse at 9 months | |
| Notes | Funding: Teva Pharmaceutical | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomization list, stratified by centers, was computer‐generated by the TEVA Statistical Data Management Department. Equal allocation of the two treatment groups was used" (Page 291) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "A treating neurologist was responsible for the overall medical management of the patient including safety monitoring ... All personnel were unaware of treatment allocation... both the treating neurologist and the patient were informed on the importance of not discussing safety issue with the examining neurologist" (Page 291) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "An examining neurologist was responsible for all scheduled neurological examinations and exacerbation follow‐up" (Page 291) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 5.9% were lost to follow‐up (5.9% in glatiramer acetate and 5.8% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Teva Pharmaceutical, and "Drs Stark, Gurevich, Kadosh, Zak, Pinchassi, and Ladkani are employees of Teva Pharmaceutical, Ltd., involved in trial design and execution, study management, database management, and statistical analysis" (Page 296) ‐ Relapse outcome was reported incompletely |
CONFIRM 2012.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration (time since diagnosis) 5 years; mean EDSS 2.6; prior use of any MS medication at any time prior to the start of study: 40% to 41% across study groups | |
| Interventions | Dimethyl fumarate 240 mg oral capsule 3 times daily for 24 months (n = 345) Dimethyl fumarate 240 mg oral capsule 2 times daily for 24 months (n = 362) Glatiramer acetate 20 mg subcutaneous daily for 24 months (n = 350) Placebo oral capsule 3 times daily for 24 months (n = 363) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned in a 1:1:1:1 ratio to receive oral placebo, BG‐12 at a dose of 240 mg two times daily, BG‐12 at a dose of 240 mg three times daily, or subcutaneous daily injections of 20 mg of glatiramer acetate for 96 weeks" (Page 1088); and "The randomization was stratified by site" (Page 33 of Protocol) |
| Allocation concealment (selection bias) | Low risk | "Randomization took place across all study sites using a centralized Interactive Voice Response System (IVRS)" (Page 33 of Protocol) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients receiving glatiramer acetate were aware of their treatment assignment. All study management and site personnel, investigators, and patients were unaware of assignment to the BG‐12 and placebo groups", and "To ensure that the assignments to the BG‐12 and placebo groups would not be revealed, patients in those groups were instructed not to take the study medication within 4 hours before each study visit, since a flushing reaction is known to be more common with BG‐12" (Page 1088). Since flushing is a known side effect of dimethyl fumarate, patients were possibly not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "An independent neurologic evaluation committee, whose members were unaware of the study‐group assignments, provided confirmation of relapses of multiple sclerosis" and "examining neurologists and members of the independent neurologic evaluation committee were unaware of all study‐group assignments" (Page 1088) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 20.3% were lost to follow‐up (20.8% in dimethyl fumarate 240 mg 3 times daily, 20.7% in dimethyl fumarate 240 mg 2 times daily, 16.1% in glatiramer acetate, and 23.4% in placebo), with some indication of the differences in reasons: adverse events of 8.1% in dimethyl fumarate 240 mg 3 times daily, 6.1% in dimethyl fumarate 240 mg 2 times daily, 3.6% in glatiramer acetate, and 3.3% in placebo |
| Selective reporting (reporting bias) | High risk | The published report included all pre‐specified primary benefit outcomes. However, disability confirmed at 6 months was not reported in the published report, it was reported by the FDA in terms of survival probabilities |
| Other bias | High risk | ‐ The study was sponsored by Biogen Idec, "data were analyzed by the sponsor" (Page 1088), and 6 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
DEFINE 2012.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration (time since diagnosis) 6 years; mean EDSS 2.4; prior use of DMT at any time prior to the start of study: 40.7% (40.4% in dimethyl fumarate 240 mg 3 times daily, 39.5% in dimethyl fumarate 240 mg 2 times daily, and 42.2% in placebo) | |
| Interventions | Dimethyl fumarate 240 mg oral capsule 3 times daily for 24 months (n = 416) Dimethyl fumarate 240 mg oral capsule 2 times daily for 24 months (n = 411) Placebo oral capsule 3 times daily for 24 months (n = 410) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Biogen Idec | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned, in a 1:1:1 ratio, to receive BG‐12 at a dose of 240 mg twice daily, BG‐12 at a dose of 240 mg three times daily, or placebo. Randomization was performed centrally and was stratified according to site" (Page 1100) |
| Allocation concealment (selection bias) | Low risk | "Randomization was performed centrally" (Page 1100), and "Randomization took place across all study sites using a centralized Interactive Voice Response System (IVRS)" (Page 33 of Protocol) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double‐blind", and "To ensure that the study‐group assignments would not be revealed, patients were instructed to take the assigned study drug at least 4 hours before study visits, in case patients in the BG‐12 groups had a side effect of flushing" (Page 1100). Since flushing is a known side effect of dimethyl fumarate, patients were possibly not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "To maintain concealment of the study‐group assignments, each study center used separate examining and treating neurologists (all of whom remained unaware of the assignments throughout the trial). The examining neurologists conducted neurologic assessments, including assessment of the EDSS score, whereas the treating neurologists were responsible for all aspects of patient care, including the treatment of relapses and other disease symptoms" and "relapses were evaluated by an independent neurologic evaluation committee, whose members reviewed a standardized set of blinded clinical records (which did not include MRI data) from the treating and examining neurologists" (Page 1100) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 23.0% were lost to follow‐up (23.1% in dimethyl fumarate 240 mg 3 times daily, 23.4% in dimethyl fumarate 240 mg 2 times daily, and 22.7% in placebo), with some indication of the differences in reasons: adverse events of 8.7% in dimethyl fumarate 240 mg 3 times daily, 9.8% in dimethyl fumarate 240 mg 2 times daily, and 5.4% in placebo |
| Selective reporting (reporting bias) | High risk | The published report included all pre‐specified primary benefit outcomes. However, disability confirmed at 6 months was not reported in the published report, it was reported by the FDA in terms of survival probabilities |
| Other bias | High risk | The study was sponsored by Biogen Idec, "data were analyzed by the sponsor" (Page 1099), and 4 co‐authors of the published paper were affiliated to the pharmaceutical company |
Etemadifar 2007.
| Methods | RCT | |
| Participants | Age: 13 to 50 years; definite RRMS; mean disease duration not reported ("short duration"); mean EDSS 1.5; all participants were previously untreated patients | |
| Interventions | Azathioprine 3 mg/kg body weight oral daily for 12 months (n = 47) Interferons beta (Betaseron, Avonex, or Rebif) for 12 months (n = 47: 15 Betaseron 250 μg subcutaneously every other day, 19 Avonex 30 µg intramuscular once a week, 13 Rebif 44 µg subcutaneous 3 times a week) |
|
| Outcomes | Relapse at 12 months | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomized according to a preexisting list produced by a computer program that differed from a random number generator only in that it assigned equal numbers of patients into each treatment group" (Page 1724) |
| Allocation concealment (selection bias) | Unclear risk | "The first treatment group received IFNβ products regimen. The second group received AZA" (Page 1724) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "The trial was single blinded in that patients were aware but physicians who assessed the outcome were unaware of treatment type that the patient was receiving" (Page 1724) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The trial was single blinded in that patients were aware but physicians who assessed the outcome were unaware of treatment type that the patient was receiving", and "Two neurologists (ME and VS) who do not know which patients had received which treatment clinically evaluated all patients" (Page 1724‐5) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 6.4% were lost to follow‐up (6.4% in azathioprine and 6.4% in interferon beta), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | Low risk | The study appears to be free of other sources of bias |
EVIDENCE 2007.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 12 months (n = 339) Interferon beta‐1a (Avonex) 30 µg intramuscular once a week for 12 months (n = 338) |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Serono Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated scheme with block size of 6 followed by block size of 4" (Page 2033) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients and a treating physician who was not involved in end point assessment were aware of treatment assignments" (Page 2033) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Evaluating physicians who were blinded to the patients' treatment and symptoms performed all clinical exams" (Page 2033) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 10.6% were lost to follow‐up (11.8% in Rebif and 9.5% in Avonex), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Serono Inc., "The sponsor designed and implemented the study and managed the data" (Page 2047) ‐ Relapse outcome was reported incompletely |
Fazekas 1997.
| Methods | RCT | |
| Participants | Age: 15 to 64 years; definite RRMS; mean disease duration 7 years; mean EDSS 3.3; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.15 to 0.20 g/kg body weight intravenously monthly for 24 months (n = 75) Placebo intravenously monthly for 24 months (n = 75) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: Sero‐Merieux (Vienna, Austria) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Centralised computer‐generated randomisation schedule with stratification by centre, age, sex, and deterioration rate" (Page 590) |
| Allocation concealment (selection bias) | Low risk | "Randomly and centrally allocated" and "Infusions of IVIg and placebo were identical in appearance and were stored in plastic bags for concealment during administration" (Page 590) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "At each monthly visit a neurologist who was aware of treatment allocation (treating physician) administered the study medication and asked the patient about any side‐effects" (Page 590) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Patients were assessed on the first day of treatment, every 6 months, and at the end of the 2‐year study by a different neurologist (assessing physician) who was unaware of treatment allocation", and "All patients were told to contact their centre as soon as there was any change in their condition. In such cases, the assessing physician examined the patient to confirm a possible relapse and to assess the severity of the disability" (Page 590) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 1.3% were lost to follow‐up (0% in immunoglobulins and 2.7% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | Unclear risk | ‐ The study was sponsored by Triton Biosciences and the role of the study sponsor was unclear ‐ Definition of sustained disability worsening was not clearly reported |
Fazekas 2008.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 3 years; mean EDSS 2.0; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.2 g/kg body weight intravenously monthly for 12 months (n = 45) Immunoglobulins 0.4 g/kg body weight intravenously monthly for 12 months (n = 42) Placebo intravenously monthly for 12 months (n = 41) | |
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Bayer HealthCare AG | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The random code number was computer generated by the Statistics and Data System Department of Bayer" (Page 266) |
| Allocation concealment (selection bias) | Unclear risk | "Randomisation performed by an unblinded pharmacist who assigned code numbers from sealed envelopes in a sequential manner" (Page 266) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Considerable effort was made to achieve optimal blinding, including the provision that all patients received a total volume of 4 mL/kg body weight per infusion, which was adjusted by the addition of dextrose 5%" (Page 266) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Endpoints "assessed by an evaluating physician who was otherwise not involved in patient care" (Page 266) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 12.5% were lost to follow‐up (9.5% in immunoglobulins 0.4 g/kg, 17.8% in immunoglobulins 0.2 g/kg, and 9.8% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Bayer HealthCare AG and the role of the study sponsor was unclear. 3 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ Relapse outcome was reported incompletely |
FREEDOMS 2010.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 8 years; mean EDSS 2.4; prior use of DMT at any time prior to the start of study: 40.9% (39.6% in fingolimod 1.25 mg, 42.6% in fingolimod 0.5 mg, and 40.4% in placebo) | |
| Interventions | Fingolimod 1.25 mg oral capsule once daily for 24 months (n = 429) Fingolimod 0.5 mg oral capsule once daily for 24 months (n = 425) Placebo oral capsule once daily for 24 months (n = 418) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned, in a 1:1:1 ratio, to receive oral fingolimod capsules in a dose of 0.5 mg or 1.25 mg or matching placebo ... Randomization was performed ... with the use of stratification according to site, with a block size of six within each site" (Page 388) |
| Allocation concealment (selection bias) | Unclear risk | "Randomization was performed centrally, with the use of a validated system " (Page 388) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double blind" (Page 388) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "To ensure that all assessments remained unbiased regarding the study‐group assignments (i.e., unaffected by awareness of them), an independent, specially trained and certified examining neurologist determined all the EDSS scores" (Page 388). "Relapses were verified by the examining neurologist within 7 days after the onset of symptoms" (Page 389) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 18.8% were lost to follow‐up (22.6% in fingolimod 1.25 mg, 13.2% in fingolimod 0.5 mg, and 20.6% in placebo), with some indication of the differences in reasons: unsatisfactory therapeutic effect 3.0% in fingolimod 1.25 mg, 1.4% in fingolimod 0.5 mg, and 6.0% in placebo; and abnormal laboratory values(s) 4.7% in fingolimod 1.25 mg, 2.1% in fingolimod 0.5 mg, and 0.2% in placebo |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Novartis Pharma, "data were analyzed by the sponsor" (Page 388), and 4 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
FREEDOMS II 2014.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 11 years; mean EDSS 2.4; prior use of DMT at any time prior to the start of study: 74.8% (77.6% in fingolimod 1.25 mg, 73.7% in fingolimod 0.5 mg, and 73.0% in placebo) | |
| Interventions | Fingolimod 1.25 mg oral capsule once daily for 24 months (n = 370) Fingolimod 0.5 mg oral capsule once daily for 24 months (n = 358) Placebo oral capsule once daily for 24 months (n = 355) "After review of data from the FREEDOMS and TRANSFORMS phase 3 studies, completed on Nov 12, 2009, after consultation with and at the recommendation of the data and safety monitoring board, we decided to stop the 1·25 mg dose. Patients on the high dose were subsequently switched to the 0·5 mg dose in a blinded manner" (Page 546) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "We randomly allocated patients (1:1:1; stratified by study centre) to receive oral fingolimod capsules in a dose of 0.5 mg or 1.25 mg or matching placebo, once daily for 24 months. The randomisation sequence was generated with an automated system under the supervision of the Novartis Drug Supply Management team" (Page 546) |
| Allocation concealment (selection bias) | Unclear risk | "To mask treatment allocation, both fingolimod and placebo were dispensed in hard gelatin capsules of identical colour and size and packed in identical bottles" (Page 546) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients, investigators, site personnel, independent evaluating physician, first dose administrator and all Novartis personnel were blinded to the study medication assignments from the time of randomisation until the database lock and data analysis for the double‐blind Treatment Phase was completed" (Appendix, Page 2) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The efficacy assessments (ie, confirmation of relapses, scheduled EDSS, ...) were done by an independent, specially trained, and certified assessor not otherwise involved in the treatment of patients)" (Page 546), "Patients were instructed not to discuss adverse events with the independent evaluating physician", "Another physician not otherwise involved in the care of the study patient monitored patients for 6 or more hours after administration of the first dose of the study drug to maintain blind for the known heart rate decrease with fingolimod upon first dose administration", "Clinical assessments were performed at screening and at randomization (baseline), and study visits were scheduled at 2 weeks and 1, 2, 3, 6, 9, 12, 15, 18, 21, and 24 months after randomization", and "In the case of MS relapse EDSS assessment was required at every unscheduled visit to confirm relapse" (Appendix, Page 2) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 28.2% were lost to follow‐up (32.2% in fingolimod 1.25 mg, 24.0% in fingolimod 0.5 mg, and 28.2% in placebo), with some indication of the differences in reasons: unsatisfactory therapeutic effect 2.7% in fingolimod 1.25 mg, 1.7% in fingolimod 0.5 mg, and 4.8% in placebo; and adverse events or abnormal laboratory values(s) 12.7% in fingolimod 1.25 mg, 10.1% in fingolimod 0.5 mg, and 5.1% in placebo |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Novartis Pharma, "The study sponsor participated in the design of the study, conduct of the study, data collection, data management, data analysis and interpretation, and preparation, review, and approval of the paper" (Page 550), and 4 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
GALA 2013.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 8 years; mean EDSS 2.8; prior use of DMT at any time prior to the start of study: 13.6% (13.6% in glatiramer acetate and 13.7% in placebo) | |
| Interventions | Glatiramer acetate 40 mg subcutaneous 3 times a week for 12 months (n = 943) Placebo subcutaneous 3 times a week for 12 months (n = 461) |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Teva Pharmaceutical Industries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Eligible patients were assigned to treatment groups in a 2:1 ratio (GA 40mg tiw or placebo) according to the randomization scheme produced. The randomization scheme used constrained blocks stratified by center" (Page 706) |
| Allocation concealment (selection bias) | Unclear risk | "Study drugs were packaged and labeled in a way that maintained the masked nature of the study; the appearance, shape, color, and smell were identical" (Page 706) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The investigators, the sponsor, and any personnel involved in patients’ assessments, monitoring, analysis, and data management were blinded to treatment assignment" (Page 706) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Patients’ general medical assessments were performed separately from the neurological assessments by 2 neurologists or physicians. The examining neurologist/physician was responsible for all neurological assessments" and "All follow‐up neurological examinations were performed by the blinded examining neurologist" (Page 706‐7) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 8.2% were lost to follow‐up (8.9% in glatiramer acetate and 6.7% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ "This study was funded by Teva Pharmaceutical Industries, Petah Tikva, Israel. All members of the clinical advisory board, the country principal investigators, the Data Monitoring Committee (DMC), and the MRI Reading Center were reimbursed for their specific services on a contractual basis by Teva Pharmaceutical Industries" (Page 711) ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report. ‐ Relapse outcome was reported incompletely, and no additional data were provided on request |
Goodkin 1991.
| Methods | RCT | |
| Participants | Age: 18 to 65 years; definite RRMS; mean disease duration 6 years; mean EDSS 3.5; prior use of DMT not reported | |
| Interventions | Azathioprine 3.0 mg/kg body weight oral daily for 24 months (n = 30) Placebo oral daily for 24 months (n = 29) | |
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Wellcome Company | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomised by the statistician using random number tables" (Page 21) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Patients and personnel were blinded, "group PLC received indistinguishable placebo", and "whenever the treating physician made a dose change for an AZA patient, a similar dose change was simultaneously made for a matched placebo patient to preserve the blind" (Pages 20‐1) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Each patient had the same masked examining neurologist and unmasked treating neurologist for the duration of the study. Standardized neurologic examinations were recorded at study entry and at 6 month intervals by the examining neurologist unless the patient reported subjective worsening, in which case an examination was performed as soon as was practical" (Page 21) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 11.9% were lost to follow‐up (10.0% in azathioprine and 13.8% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | Unclear risk | Definition of sustained disability worsening not clearly reported |
IFNB MS Group 1993.
| Methods | RCT | |
| Participants | Age: 18 to 50 years; definite RRMS; mean disease duration (time since diagnosis) 4 years; mean EDSS 2.9; prior use of DMT not reported | |
| Interventions | Interferon beta‐1b (Betaseron) 250 µg subcutaneous every other day for 24 months (n = 124) Interferon beta‐1b (Betaseron) 50 µg subcutaneous every other day for 24 months (n = 125) Placebo subcutaneous every other day for 24 months (n = 123) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: Triton Biosciences, Inc., Alameda, CA and Berlex Laboratories Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Each placebo vial contained only similar quantity of albumin and dextrose", "All personnel were blinded to treatment categories", and "One treating neurologist who knew about side effects, reviewed laboratory findings for toxicity, and was responsible for overall care" (Page 656) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "One neurologist who was not aware of drug side effects to do the periodic examinations" (Page 656). However, it is not clear how and when potential relapses and EDSS were assessed |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 9.1% were lost to follow‐up (7.3% in interferon beta‐1b 250 µg, 11.2% in interferon beta‐1b 50 µg, and 8.9% in placebo). Nothing was said about the reasons for study discontinuation |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Triton Biosciences and the role of the study sponsor was unclear ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report ‐ Disability worsening confirmed at 3 months outcome was reported incompletely, and disability worsening confirmed at 6 months was not assessed |
INCOMIN 2002.
| Methods | RCT | |
| Participants | Age: 18 to 50 years; definite RRMS; mean disease duration (time since diagnosis) 6 years; mean EDSS 2.0; all participants were previously untreated patients | |
| Interventions | Interferon beta‐1b (Betaseron) 250 µg subcutaneous every other day for 24 months (n = 96) Interferon beta‐1a (Avonex) 30 µg intramuscular once a week for 24 months (n = 92) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: The Italian Ministry of Health and the Italian MS Society | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation followed computer‐generated random sequences of digits that were different for each centre and for each sex, to achieve centre and sex stratification" (Page 1454) |
| Allocation concealment (selection bias) | Low risk | "The codes were randomly assigned to treatments by an independent team of statisticians unaware of the patient’s clinical characteristics" (Page 1454) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "All clinical outcomes were assessed in an open‐label manner" (Page 1454) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "All clinical outcomes were assessed in an open‐label manner" (Page 1454) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 3.2% were lost to follow‐up (2.1% in interferon beta‐1b and 4.3% in interferon beta‐1a). Nothing was said about the reasons for study discontinuation |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | Unclear risk | Relapse and disability worsening outcomes were reported incompletely |
Johnson 1995.
| Methods | RCT | |
| Participants | Age: 18 to 45 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.6; prior use of DMT not reported | |
| Interventions | Glatiramer acetate 20 mg subcutaneous daily for 24 months (n = 125) Placebo (not described) (n = 126) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: Teva Pharmaceutical | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "A centralized randomization scheme was used" (Page 1270) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Treating neurologists were blinded" (Page 1270) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Examining neurologists were blinded" (Page 1270) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 14.3% were lost to follow‐up (15.2% in glatiramer acetate and 13.5% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse and disability worsening confirmed at 3 months outcomes were reported incompletely, and disability worsening confirmed at 6 months was not assessed |
Koch‐Henriksen 2006.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 8 years; mean EDSS 2.9; prior use of DMT not reported | |
| Interventions | Interferon beta‐1b (Betaseron) 250 µg subcutaneous every other day for 24 months (n = 158) Interferon beta‐1a (Rebif) 22 µg subcutaneous once a week for 24 months (n = 143) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomization algorithm was adjusted to reduce deviations from a 50/50 result in each center" (Page 1057) |
| Allocation concealment (selection bias) | Low risk | "A central computerized randomization schedule assigned patients to treatment" (Page 1057) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Blinding was abandoned because it could not be maintained owing to the different administration schemes of the two study drugs" (Page 1057) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Open‐label trial" (Page 1057) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 25.6% were lost to follow‐up (27.8% in interferon beta‐1b and 23.1% in interferon beta‐1a), with some indication of the differences in reasons: "The main cause of withdrawal in the IFN‐1b 250 g arm was side effects (24/158, 15.2%), and treatment failure was the most frequent cause in the IFN‐1a arm (15/143, 10.5%)" (Page 1057) |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ It is unclear if the study was sponsored ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse and disability worsening confirmed at 3 months outcomes were reported incompletely, and disability worsening confirmed at 6 months was not assessed ‐ Rebif at very low dose that is not used in clinical practice |
Lewanska 2002.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 9 years; mean EDSS 3.0; prior use of DMT not reported | |
| Interventions | Immunoglobulins 0.2 g/kg body weight intravenously monthly for 12 months (n = 17) Immunoglobulins 0.4 g/kg body weight intravenously monthly for 12 months (n = 16) Placebo intravenously monthly for 12 months (n = 18) | |
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Supported by the KBN (State Research Committee) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The generation of allocation sequence was based on random‐number table" (Page 566) |
| Allocation concealment (selection bias) | Unclear risk | "Infusions of intravenous immunoglobulins and placebo were stored in identical opaque plastic bags for concealment during administration" (Page 566) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Evaluating physician was unaware of the actual treatment allocation. Before entry to the study, and monthly thereafter during the study and 3 months after the end of the study, each patient was examined blindly by the same neurologist who was unaware of treatment allocation. Monitoring and recording of relapses, concomitant treatment, side‐effects or other medical events were documented throughout the study" (Page 566) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 3.9% were lost to follow‐up (6.3% in immunoglobulins 0.4 g/kg, 0% in immunoglobulins 0.2 g/kg, and 5.6% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report. ‐ Relapse outcome was reported incompletely |
MAIN TRIAL.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 6 years; mean EDSS 1.9; prior use of DMT at any time prior to the start of study: 6.0% (6.5% in azathioprine and 5.5% in interferon beta) | |
| Interventions | Azathioprine 3 mg/kg body weight oral daily for 24 months (n = 77) Interferons beta (Betaseron, Avonex, or Rebif) for 24 months (n = 73: 5 Betaseron 250 μg subcutaneously every other day, 26 Avonex 30 µg intramuscular once a week, 35 Rebif 22 µg subcutaneous 3 times a week, 7 Rebif 44 µg subcutaneous 3 times a week) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: AIFA (Italian medicines agency) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were selected for AZA or IFNs using a randomization list (1:1 ratio), in blocks of four and stratified by disability score (EDSS≤3.5 or>3.5)" |
| Allocation concealment (selection bias) | Low risk | "Patients were selected for AZA or IFNs using a computer generated central randomization list" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Single‐masked" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Patients were assessed by an un‐masked treating and a masked examining neurologist at their centers", and "The masked examining neurologist was responsible for the neurological examination and EDSS scoring at scheduled (every six months) and unscheduled visits, requested by the treating neurologist to confirm relapses". Relapse assessment was not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 15.3% were lost to follow‐up (19.5% in azathioprine and 11.0% in interferon beta), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Millefiorini 1997.
| Methods | RCT | |
| Participants | Age: 18 to 45 years; definite RRMS; mean disease duration 5 years; mean EDSS 3.6; prior use of DMT not reported | |
| Interventions | Mitoxantrone 8 mg/m² of body surface intravenously monthly for 12 months (total dosage of 96 mg/m² of body surface over 12 months) (n = 27) Placebo intravenously monthly for 12 months (n = 24) | |
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomized to MTX or placebo using a scheme stratified on age, sex and EDSS which resulted in eight different age/sex/EDSS strata. According to the study protocol, within each stratum the allocation of patients to treatment or placebo was balanced by using a block design of size eight" (Page 154) |
| Allocation concealment (selection bias) | Low risk | "Central allocation and the intravenous bag and tubing were black to ensure no differences between the treatment groups" (Page 154) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Treating physicians were not blinded. Unclear blinding of patients |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Monitoring and recording of exacerbations, concomitant therapy or other medical events were documented throughout the study by a treating physician selected in each centre before the beginning of the study. The treating physician was not blinded to study treatment", and "In order to maintain blindness, the interaction of the EDSS physicians with the patient was strictly restricted to the neurological examination. The neurologist was not allowed to talk with the patient about adverse events, or any other issue which could potentially disclose the patient’s treatment" (Page 154) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None were lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ It is unclear if this study was sponsored ‐ Definition of sustained disability worsening was not clearly reported |
MSCRG 1996.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.4; all participants were previously untreated patients | |
| Interventions | Interferon beta‐1a (Avonex) 30 µg intramuscular once a week for 24 months (n = 158) Placebo intramuscular once a week for 24 months (n = 143) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Biogen, Inc, Cambridge, MA | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomisation performed at statistical centre of Buffalo General Hospital, one of the participating centres (biased coin assignment used for sequence generation)" (Page 286) |
| Allocation concealment (selection bias) | Unclear risk | "schedule sent to each clinical centre, included patients were sequentially assigned the next ID number from the schedule"(Page 286) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Personnel and participants were blinded to treatment status" (Page 286) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Evaluating physicians were blinded to treatment status" (Page 286) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 42.9% were lost to follow‐up (46.2% in interferon beta‐1a and 39.2% in placebo). The study stopped early for benefit without a formal‐stopping rule |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | The study was sponsored by Biogen and "Personnel of the study sponsor (Biogen) were involved in the conduct and data analysis" (Page 293) |
OWIMS 1999.
| Methods | RCT | |
| Participants | Age: 18 to 50 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.6; prior use of DMT not reported | |
| Interventions | Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 12 months (n = 98) Interferon beta‐1a (Rebif) 22 µg subcutaneous 3 times a week for 12 months (n = 95) Placebo subcutaneous 3 times a week for 12 months (n = 100) |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Ares‐Serono International SA, Geneva, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation performed at Corporate Biometrics Department of Ares‐Serono (computer‐generated list)" (Page 680) |
| Allocation concealment (selection bias) | Low risk | "The randomization code for each patient was delivered to the investigator in sealed envelopes" (Page 680) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "If desired, patients could remain on blinded study medication for another 24 weeks", "Both active treatment and placebo were administered as ready‐to‐use solutions in a volume of 0.5 mL", and "To preserve blinding, patients were instructed to cover injection sites and to refrain from discussing any symptoms that might be in any way related to treatment when visiting the evaluating physician" (Page 681) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The evaluating physician was responsible for neurologic assessments, both at scheduled visits and during exacerbations. Throughout the study, the evaluating physician remained unaware of adverse event profiles and any changes in safety assessments" (Page 681) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 8.2% were lost to follow‐up (13.3% in interferon beta‐1a 44 µg, 8.4% in interferon beta‐1a 22 µg, and 3.0% in placebo), with some indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Ares‐Serono International SA, Geneva, Switzerland ‐ Relapse outcome was reported incompletely |
PRISMS 1998.
| Methods | RCT | |
| Participants | Age: 18 to 50 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.5; prior use of DMT: "Only 3% of patients had received previous immunosuppressive therapy" | |
| Interventions | Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 24 months (n = 184) Interferon beta‐1a (Rebif) 22 µg subcutaneous 3 times a week for 24 months (n = 189) Placebo subcutaneous 3 times a week for 24 months (n = 187) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Ares‐Serono International SA, Geneva, Switzerland | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation at Corporate Biometrics Department of Ares‐Serono (computer‐generated list, stratified by centre, equal allocation of the treatment groups by a block size of 6)" (Page 1499) |
| Allocation concealment (selection bias) | Low risk | "The study drug was packed accordingly to the randomisation list and delivered to the centres so that treatment allocation remained concealed" (Page 1499) |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "All personnel involved in the study were unaware of treatment allocation", and "All injection sites were covered up at neurological examinations to ensure that masking was not compromised because of local reactions" (Page 1499) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "All personnel involved in the study were unaware of treatment allocation", "Patients were assessed by two physicians. A “treating” neurologist was responsible for overall medical management of the patient, including treatment of any side‐effects, and an “assessing” neurologist was responsible for neurological assessments and follow‐up of relapses", and "All patients had a neurological assessment every 3 months. Additional assessments were done during relapses" (Page 1499) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 4.8% were lost to follow‐up (2.7% in interferon beta‐1a 44 µg, 6.3% in interferon beta‐1a 22 µg, and 5.3% in placebo), without indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Ares‐Serono International SA, Geneva, Switzerland ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report ‐ Relapse outcome was reported incompletely |
REGARD 2008.
| Methods | RCT | |
| Participants | Age: 18 to 60 years; definite RRMS; mean disease duration 6 years; mean EDSS 2.3; prior use of DMT not reported | |
| Interventions | Interferon beta‐1a (Rebif) 44 µg subcutaneous 3 times a week for 24 months (n = 386) Glatiramer acetate 20 mg subcutaneous daily for 24 months (n = 378) |
|
| Outcomes | Relapse at 24 months. Disability worsening at 24 months | |
| Notes | Funding: EMD Serono and Pfizer | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated randomisation list stratified by centre" (Page 904) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Neither the patients nor the treating physicians were blinded to treatment" (Page 904) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The physicians who assessed patients ...were blinded to treatment and communicated with the patients only as needed to complete the EDSS, Kurtzke functional scale (KFS), and relapse assessments. Patients were asked not to discuss their treatment with the assessing physician and they covered their injection sites" (Page 904) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 3.3% were lost to follow‐up (5.2% in interferon beta‐1a and 1.3% in glatiramer acetate). Nothing was said about the reasons for study discontinuation |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ "The study protocol was drafted and developed by the study sponsors, EMD Serono and Pfizer, in conjunction with the investigator steering committee. Data management and analysis were done by the study sponsors" (Page 907), and 2 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ Disability worsening confirmed at 6 months outcome was reported incompletely |
SELECT 2013.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; median disease duration (since diagnosis) 3 years; mean EDSS 2.7; prior use of DMT at any time prior to the start of study: 23.7% (22.5% in daclizumab 300 mg, 25.5% in daclizumab 150 mg, and 24.0% in placebo) | |
| Interventions | Daclizumab 300 mg subcutaneously once every 4 weeks for 12 months (n = 209) Daclizumab 150 mg subcutaneously once every 4 weeks for 12 months (n = 208) Placebo subcutaneously once every 4 weeks for 12 months (n = 204) |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Biogen Idec and AbbVie Biotherapeutics Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned in a 1:1:1 ratio" (Page 2168) |
| Allocation concealment (selection bias) | Low risk | "Patients were randomly assigned via a centralised interactive voice response system" (Page 2168) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All personnel and patients were masked to treatment assignment" (Page 2168) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Three members of an independent neurology assessment committee, consisting of multiple sclerosis neurologists who were masked to group assignment, adjudicated whether the protocol definition of relapse was satisfied" (Page 2168) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 7.1% were lost to follow‐up (5.7% in daclizumab 300 mg, 7.7% in daclizumab 150 mg, and 7.8% in placebo). Nothing was said about the reasons for study discontinuation |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Biogen Idec and AbbVie Biotherapeutics Inc, "The sponsor of the study provided assistance in manuscript preparation. The study was designed by the sponsor; the sponsor held and analysed data" (Page 2169), and 5 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
TEMSO 2011.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 9 years; mean EDSS 2.7; prior use of DMT in the previous 2 years: 27.0% (28.4% in teriflunomide 14 mg, 27.9% in teriflunomide 7 mg, and 24.8% in placebo) | |
| Interventions | Teriflunomide 14 mg oral capsule once daily for 25 months (n = 359) Teriflunomide 7 mg oral capsule once daily for 25 months (n = 366) Placebo oral capsule once daily for 25 months (n = 363) |
|
| Outcomes | Relapse at 12 and 24 months. Disability worsening at 24 months | |
| Notes | Funding: Sanofi‐Aventis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Eligible patients were randomly assigned (in a 1:1:1 ratio) to receive a once‐daily oral dose of placebo, 7 mg of teriflunomide, or 14 mg of teriflunomide for 108 weeks. Randomization was stratified according to the baseline EDSS score (≤3.5 or >3.5) and according to trial site, with a block size of 6." (Page 1294) |
| Allocation concealment (selection bias) | Low risk | "The treatment allocation was determined according to the randomization code provided by an interactive voice response system (IVRS)" (Page 74 of Medical Review of FDA) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double blind" (Page 1294), and at Page 40 of the Protocol they described blinding, packaging and labeling ("Each medication kit was labeled with a two‐part tear‐off label..."). "Unblinding of 40 patients in TEMSO study, and the reasons provided do not appear to justify the need of unblinding" (Page 230 of Statistical Review of FDA) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "A treating neurologist at each site was responsible for evaluating patient eligibility, supervising the administration of study medication, recording and managing adverse events, assessing relapses, and monitoring safety assessments. An independent, specially trained and certified examining neurologist determined all the EDSS scores and performed all assessments of functional systems. Both treating and examining neurologists were unaware of treatment assignments; only the treating neurologist was aware of any side effects that could potentially be related to active therapy" (Pages 1294‐5), "Each episode of relapse was to be confirmed by the treating neurologist (unblinded), based on the objective assessments by an independent examining neurologist (blinded)" (Page 207 of Statistical Review of FDA) and "Patients were required to visit the study site within 7 days after the onset of a suspected relapse, for assessments by the examining neurologist" (Page 1295). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Overall, 20.1% were lost to follow‐up (21.2% in teriflunomide 14 mg, 19.1% in teriflunomide 7 mg, and 20.1% in placebo). Nothing was said about the reasons for study discontinuation. "Some patients discontinued study at the time of blind broken, although it is not clear whether or not the discontinuation was due to unblinding" (Page 208 of Statistical Review of FDA) |
| Selective reporting (reporting bias) | High risk | The published report included all pre‐specified primary benefit outcomes. However, disability confirmed at 6 months was not reported in the published report, it was reported by the FDA in terms of survival probabilities |
| Other bias | High risk | ‐ The study was sponsored by Sanofi‐Aventis, "data were analyzed by the sponsor" (Page 1294), and 3 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
TENERE 2014.
| Methods | RCT | |
| Participants | Age: 18 years and older; definite RRMS; mean disease duration 7 years; mean EDSS 2.1; prior use of DMT in the previous 2 years: 18.8% (11.7% in teriflunomide 14 mg, 21.1% in teriflunomide 7 mg, and 24.0% in interferon beta‐1a) | |
| Interventions | Teriflunomide 14 mg oral capsule once daily for at least 12 months (n = 111) Teriflunomide 7 mg oral capsule once daily for at least 12 months (n = 109) Interferon beta‐1a (Rebif) 44 µg ("when the 44 μg dose was not tolerated, the dose was reduced to 22 μg") subcutaneous 3 times a week for at least 12 months (n = 104) The study was completed 48 weeks after the last patient was randomised, resulting in a variable duration of follow‐up |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomised 1:1:1 to teriflunomide 7 mg or 14 mg or IFNβ‐1a, and stratified by country (Americas, Eastern Europe, Western Europe and Africa) and baseline EDSS score (≤3.5 or >3.5)" (Page 706) |
| Allocation concealment (selection bias) | Low risk | "A phone interactive voice response system was used to randomize patients" (information provided on request by Genzyme) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients were randomised 1:1:1 to teriflunomide 7 mg or 14 mg (double‐blind) or IFNβ‐1a (open‐label)" (Page 706) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "The treating neurologist was responsible for relapse assessments, while an examining neurologist scored the EDSS. The examining neurologist remained blinded to treatment and associated AEs", and "Each relapse was confirmed by the treating neurologist based on the objective assessment of the examining neurologist" (Page 706) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, at 1 year 17.9% were lost to follow‐up (17.1% in teriflunomide 14 mg, 10.1% in teriflunomide 7 mg, and 26.9% in interferon beta‐1a) (data provided on request by Genzyme), with some indication of the differences in reasons |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes. Missing data not reported in the published paper were provided on request by Genzyme |
| Other bias | High risk | "This study was funded by Genzyme, a Sanofi company. Editorial support was provided by Meg Church, Fishawack Communications, Ltd, also funded by Genzyme, a Sanofi company" (Page 716), and 3 co‐authors of the published paper were affiliated to the pharmaceutical company |
TOWER 2014.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 8 years; mean EDSS 2.7; prior use of DMT in the previous 2 years: 32.8% (33.9% in teriflunomide 14 mg, 30.1% in teriflunomide 7 mg, and 34.7% in placebo) | |
| Interventions | Teriflunomide 14 mg oral capsule once daily for at least 12 months (n = 372) Teriflunomide 7 mg oral capsule once daily for at least 12 months (n = 408) Placebo oral capsule once daily for at least 12 months (n = 389) The study was completed 48 weeks after the last patient was randomised, resulting in a variable duration of follow‐up |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Genzyme (a Sanofi company) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was done using a permuted‐block randomisation schedule with stratification according to study site and baseline EDSS score (≤3.5 or >3.5)" (Page 248) |
| Allocation concealment (selection bias) | Low risk | "Randomisation was done centrally, via an interactive voice recognition system that generated an allocation sequence" and "investigators used the allocation sequence to randomly assign eligible patients in a 1:1:1 ratio to receive once‐daily oral placebo, teriflunomide 7 mg, or teriflunomide 14 mg (identical in taste and appearance)" (Page 248) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients and individuals administering the interventions were masked to treatment assignment" (Page 248) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Those assessing the outcomes were masked to treatment assignment" and "A treating neurologist was responsible for recording of adverse events, and assessment of relapses. An examining neurologist assigned EDSS scores at screening, randomisation, and every 12 weeks until the last treatment visit, and on any unscheduled visits for assessment of suspected relapse or disability worsening" (Page 248) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Overall, 29.8% were lost to follow‐up (30.6% in teriflunomide 14 mg, 29.2% in teriflunomide 7 mg, and 29.6% in placebo), with some indication of the differences in reasons: adverse events of 15.6% in teriflunomide 14 mg, 13.2% in teriflunomide 7 mg, and 6.7% in placebo; and lack of benefit of 5.4% in teriflunomide 14 mg, 7.4% in teriflunomide 7 mg, and 9.5% in placebo |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes |
| Other bias | High risk | ‐ The study was sponsored by Genzyme, "data were analyzed by the sponsor" (Page 250), and 4 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
TRASFORMS 2010.
| Methods | RCT | |
| Participants | Age: 18 to 55 years; definite RRMS; mean disease duration 7 years; mean EDSS 2.2; prior use of DMT at any time prior to the start of study: 56.7% (58.5% in fingolimod 1.25 mg, 55.2% in fingolimod 0.5 mg, and 56.3% in interferon beta‐1a) | |
| Interventions | Fingolimod 1.25 mg oral capsule once daily for 12 months (n = 426) Fingolimod 0.5 mg oral capsule once daily for 12 months (n = 431) Interferon beta‐1a (Avonex) 30 µg intramuscular once a week for 12 months (n = 435) |
|
| Outcomes | Relapse at 12 months | |
| Notes | Funding: Novartis Pharma | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed in blocks of six within each site and was stratified according to site" (Page 403) |
| Allocation concealment (selection bias) | Low risk | "Randomization was performed centrally" and "Study‐group assignments were performed with the use of an interactive voice‐response system" (Page 403) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Capsules, syringes and packaging materials for active and placebo treatments were indistinguishable", "During the trial, patients, study personnel, steering‐committee members, and the study statistician were unaware of study‐group assignments and leukocyte counts", and "An independent physician monitored patients after the first dose of the oral study drug was administered and was instructed not to discuss heart‐rate changes with patients or study personnel" (Page 404) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "At each site, a treating neurologist supervised medical management", "Patients were instructed to not to discuss adverse events with clinical evaluators", and "Potential relapses triggered an unscheduled visit and were confirmed by the treating neurologist on the basis of blinded examination by the examining neurologist" (Pages 403‐4) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall, 10.8% were lost to follow‐up (13.4% in fingolimod 1.25 mg, 7.7% in fingolimod 0.5 mg, and 11.3% in interferon beta‐1a), with few indications of the differences in reasons: unsatisfactory therapeutic effect of 0.7% in fingolimod 1.25 mg, 0.7% in fingolimod 0.5 mg, and 1.6% in interferon beta‐1a; adverse event(s) of 6.1% in fingolimod 1.25 mg, 2.1% in fingolimod 0.5 mg, and 2.1% in interferon beta‐1a; and abnormal laboratory values(s) of 0.9% in fingolimod 1.25 mg, 1.4% in fingolimod 0.5 mg, and 0.2% in interferon beta‐1a |
| Selective reporting (reporting bias) | Low risk | The published report included all pre‐specified primary benefit outcomes. Missing data not reported in the published paper were provided on request by Novartis Pharma |
| Other bias | High risk | ‐ The study was sponsored by Novartis Pharma, "data were analyzed by the sponsor" (Page 403), and 5 co‐authors of the published paper were affiliated to the pharmaceutical company ‐ The primary benefit outcome measure for relapse (ARR) was strongly affected by differences among treatment groups both in the number of participants who discontinued the study and in time to discontinuation, which they did not report |
ARR: annualised relapse rate CIS: clinically isolated syndrome DMT: disease modifying therapy EDSS: Expanded Disability Status Scale FDA: (US) Food and Drug Administration MS: multiple sclerosis RCT: randomised controlled trial RRMS: relapsing‐remitting multiple sclerosis
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ACT 2009 | Study evaluating combination therapy (interferon beta‐1a combined with methotrexate, methylprednisolone, or both) |
| Ashtari 2011 | Study on interferon beta‐1a versus methotrexate; methotrexate is not relevant to the review |
| ATAMS 2014 | Study on atacicept versus placebo; atacicept is not relevant to the review |
| Calabrese 2012 | Non‐randomised study |
| CHOICE 2010 | Follow‐up of 6 months |
| Etemadifar 2006 | Non‐randomised study |
| FORTE 2011 | Study evaluating 2 doses of glatiramer acetate (40 mg compared to 20 mg) without a control group |
| Freedman 2012 | Study evaluating combination therapy (interferon beta‐1a alone and combined with teriflunomide), with a follow‐up of 6 months |
| Havrdova 2009 | Study evaluating combination therapy (interferon beta‐1a alone and combined with low‐dose azathioprine alone or low‐dose azathioprine and low‐dose corticosteroids) |
| Kappos 2006 | Follow‐up of 6 months The patients were possibly included in the FREEDOMS study |
| Kappos 2008 | Follow‐up of 6 months |
| Kappos 2011 | Follow‐up of 6 months |
| Khoury 2010 | Study evaluating combination therapy (glatiramer acetate alone and combined with albuterol) |
| Knobler 1993 | Follow‐up of 6 months |
| Saida 2012 | Follow‐up of 6 months |
| SENTINEL 2006 | Study evaluating combination therapy (natalizumab combined with interferon beta‐1a versus interferon beta‐1a alone) |
| Sorensen 2014 | Follow‐up of 6 months |
Characteristics of ongoing studies [ordered by study ID]
DECIDE.
| Trial name or title | Multicenter, double‐blind, randomized, parallel‐group, monotherapy, active‐control study to determine the efficacy and safety of daclizumab high yield process (DAC HYP) versus Avonex® (interferon β 1a) in patients with relapsing‐remitting multiple sclerosis |
| Methods | RCT |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Daclizumab 150 mg subcutaneously once every 4 weeks for 24 to 36 months Interferon beta‐1a (Avonex) 30 µg intramuscular once a week for 24 to 36 months |
| Outcomes | Primary outcome measures:
Secondary outcome measures (time frame: 2 years):
|
| Starting date | May 2010 |
| Contact information | Biogen Idec |
| Notes | Sponsor: Biogen Idec ClinicalTrials.gov Identifier: NCT01064401 |
NCT01247324.
| Trial name or title | A randomized, double‐blind, double‐dummy, parallel‐group study to evaluate the efficacy and safety of Ocrelizumab in comparison to Interferon Beta‐1a (Rebif®) in patients with relapsing multiple sclerosis |
| Methods | RCT |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Ocrelizumab 600 mg intravenously every 24 weeks for 24 months Interferon beta‐1a (Rebif) 8.8 µg (weeks 1 + 2)/22 µg (weeks 3 + 4)/44 µg (week 5 and following) subcutaneous 3 times a week for 24 months |
| Outcomes | Primary outcome measures:
Secondary outcome measures (time frame: 2 years):
|
| Starting date | August 2011 |
| Contact information | Hoffmann‐La Roche |
| Notes | Sponsor: Hoffmann‐La Roche ClinicalTrials.gov Identifier: NCT01247324 |
NCT01412333.
| Trial name or title | A randomized, double‐blind, double‐dummy, parallel‐group study to evaluate the efficacy and safety of ocrelizumab in comparison to interferon beta‐1a (Rebif®) in patients with relapsing multiple sclerosis |
| Methods | RCT |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Ocrelizumab 600 mg intravenously every 24 weeks for 24 months Interferon beta‐1a (Rebif) 8.8 µg (weeks 1 + 2)/22 µg (weeks 3 + 4)/44 µg (week 5 and following) subcutaneous 3 times a week for 24 months |
| Outcomes | Primary outcome measures:
Secondary outcome measures (time frame: 2 years):
|
| Starting date | September 2011 |
| Contact information | Hoffmann‐La Roche |
| Notes | Sponsor: Hoffmann‐La Roche ClinicalTrials.gov Identifier: NCT01412333 |
EDSS: Expanded Disability Status Scale MRI: magnetic resonance imaging MS: multiple sclerosis RCT: randomised controlled trial
Differences between protocol and review
We excluded the route of administration of treatments (oral, subcutaneous, intravenous) from the effect modifiers that were possible sources of inconsistency or heterogeneity, since it was not clinically expected.
Contributions of authors
Concept: GF, GS
Title registration: GF
Protocol draft: GF, IT, CDG, GS
Protocol editing: GF, IT, CDG, GS, RD
Title and abstract review: GF, IT
Data abstraction: IT, IP
Data entry: IT, IP
Data analysis: CDG, IT
Drafting the review: GF, IT
Editing and revising the review: GF, IT, CDG, GS, RD
Sources of support
Internal sources
Fondazione Istituto Neurologico Carlo Besta ‐ Milan, Italy.
External sources
Ministero della Salute, Italy.
Declarations of interest
GF: none
GS: none
CDG: none
IT: none
RD: none
New
References
References to studies included in this review
Achiron 1998 {published data only}
- Achiron A, Gabbay U, Gilad R, Hassin B, Barak Y, Gornish M, et al. Intravenous immunoglobulin treatment in multiple sclerosis. Effect on relapses. Neurology 1998;50(2):398‐402. [DOI] [PubMed] [Google Scholar]
ADVANCE 2014 {published data only}
- Calabresi PA, Kieseier BC, Arnold DL, Balcer LJ, Boyko A, Pelletier J, et al. Pegylated interferon beta‐1a for relapsing‐remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double‐blind study. Lancet Neurology 2014;13:657‐65. [DOI] [PubMed] [Google Scholar]
AFFIRM 2006 {published data only}
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. New England Journal of Medicine 2006;354(9):899‐910. [DOI] [PubMed] [Google Scholar]
ALLEGRO 2012 {published data only}
- Comi G, Jeffery D, Kappos L, Montalban X, Boyko A, Rocca MA, et al. Placebo‐controlled trial of oral laquinimod for multiple sclerosis. New England Journal of Medicine 2012;366(11):1000‐9. [DOI] [PubMed] [Google Scholar]
BECOME 2009 {published data only}
- Cadavid D, Wolansky LJ, Skurnick J, Lincoln J, Cheriyan J, Szczepanowski K, et al. Efficacy of treatment of MS with IFNbeta‐1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology 2009;72(23):1976‐83. [DOI] [PubMed] [Google Scholar]
BEYOND 2009 {published data only}
- O'Connor P, Filippi M, Arnason B, Comi G, Cook S, Goodin D, et al. 250 microg or 500 microg interferon beta‐1b versus 20 mg glatiramer acetate in relapsing‐remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurology 2009;8(10):889‐97. [DOI] [PubMed] [Google Scholar]
Bornstein 1987 {published data only}
- Bornstein MB, Miller A, Slagle S, Weitzman M, Crystal, Drexler E, et al. A pilot trial of Cop 1 in exacerbating‐remitting multiple sclerosis. New England Journal of Medicine 1987;317(7):408‐14. [DOI] [PubMed] [Google Scholar]
BRAVO 2014 {published data only}
- Vollmer TL, Sorensen PS, Selmaj K, Zipp F, Havrdova E, Cohen JA, et al. A randomized placebo‐controlled phase III trial of oral laquinimod for multiple sclerosis. Journal of Neurology 2014;261(4):773‐83. [DOI] [PubMed] [Google Scholar]
CAMMS223 2008 {published data only}
- CAMMS223 Trial Investigators, Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, et al. Alemtuzumab vs. interferon beta‐1a in early multiple sclerosis. New England Journal of Medicine 2008;359(17):1786‐801. [DOI] [PubMed] [Google Scholar]
CARE‐MS I 2012 {published data only}
- Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first‐line treatment for patients with relapsing‐remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012;380(9856):1819‐28. [DOI] [PubMed] [Google Scholar]
CARE‐MS II 2012 {published data only}
- Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease‐modifying therapy: a randomised controlled phase 3 trial. Lancet 2012;380(9856):1829‐39. [DOI] [PubMed] [Google Scholar]
CombiRx 2013 {published data only}
- Lublin FD, Cofield SS, Cutter GR, Conwit R, Narayana PA, Nelson F, et al. Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Annals of Neurology 2013;73(3):327‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Comi 2001 {published data only}
- Comi G, Filippi M, Wolinsky JS. European/Canadian multicenter, double‐blind, randomized, placebo‐controlled study of the effects of glatiramer acetate on magnetic resonance imaging‐‐measured disease activity and burden in patients with relapsing multiple sclerosis. European/Canadian Glatiramer Acetate Study Group. Annals of Neurology 2001;49(3):290‐7. [PUBMED: 11261502] [PubMed] [Google Scholar]
CONFIRM 2012 {published data only}
- Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo‐controlled phase 3 study of oral BG‐12 or glatiramer in multiple sclerosis. New England Journal of Medicine 2012;367(12):1087‐97. [DOI] [PubMed] [Google Scholar]
DEFINE 2012 {published data only}
- Gold R, Kappos L, Arnold DL, Bar‐Or A, Giovannoni G, Selmaj K, et al. Placebo‐controlled phase 3 study of oral BG‐12 for relapsing multiple sclerosis. New England Journal of Medicine 2012;367(12):1098‐107. [DOI] [PubMed] [Google Scholar]
Etemadifar 2007 {published data only}
- Etemadifar M, Janghorbani M, Shaygannejad V. Comparison of interferon beta products and azathioprine in the treatment of relapsing‐remitting multiple sclerosis. Journal of Neurology 2007;254(12):1723‐8. [DOI] [PubMed] [Google Scholar]
EVIDENCE 2007 {published data only}
- Schwid S, Panitch H. Full results of the Evidence of Interferon Dose‐Response‐European North American Comparative Efficacy (EVIDENCE) study: a multicenter, randomized, assessor‐blinded comparison of low‐dose weekly versus high‐dose, high‐frequency interferon beta‐1a for relapsing multiple sclerosis. Clinical Therapeutics 2007;29(9):2031‐48. [DOI] [PubMed] [Google Scholar]
Fazekas 1997 {published data only}
- Fazekas F, Deisenhammer F, Strasser‐Fuchs S, Nahler G, Mamoli B. Randomised placebo‐controlled trial of monthly intravenous immunoglobulin therapy in relapsing‐remitting multiple sclerosis. Lancet 1997;349(9052):589‐93. [DOI] [PubMed] [Google Scholar]
Fazekas 2008 {published data only}
- Fazekas F, Lublin F, Li D, Freedman M, Hartung H, Rieckmann P, et al. Intravenous immunoglobulin in relapsing‐remitting multiple sclerosis: a dose‐finding trial. Neurology 2008;71(4):265‐71. [DOI] [PubMed] [Google Scholar]
FREEDOMS 2010 {published data only}
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):387‐401. [DOI] [PubMed] [Google Scholar]
FREEDOMS II 2014 {published data only}
- Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing‐remitting multiple sclerosis (FREEDOMS II): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Neurology 2014;13(6):545‐56. [DOI] [PubMed] [Google Scholar]
GALA 2013 {published data only}
- Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R, GALA Study Group. Three times weekly glatiramer acetate in relapsing‐remitting multiple sclerosis. Annals of Neurology 2013;73(6):705‐13. [DOI] [PubMed] [Google Scholar]
Goodkin 1991 {published data only}
- Goodkin D, Bailly R, Teetzen M, Hertsgaard D, Beatty W. The efficacy of azathioprine in relapsing‐remitting multiple sclerosis. Neurology 1991;41:20‐5. [DOI] [PubMed] [Google Scholar]
IFNB MS Group 1993 {published data only}
- IFNB MSG. Interferon beta‐1b is effective in relapsing‐remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double‐blind, placebo‐controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology 1993;43(4):655‐61. [DOI] [PubMed] [Google Scholar]
INCOMIN 2002 {published data only}
- Durelli L, Verdun E, Barbero P, Bergui M, Versino E, Ghezzi A, et al. Every‐other‐day interferon beta‐1b versus once‐weekly interferon beta‐1a for multiple sclerosis: results of a 2‐year prospective randomised multicentre study (INCOMIN). Lancet 2002;359:1453‐60. [DOI] [PubMed] [Google Scholar]
Johnson 1995 {published data only}
- Johnson K, Brooks B, Cohen J, Ford C, Goldstein J, Lisak R, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing‐remitting multiple sclerosis: results of a phase III multicenter, double‐blind placebo‐controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995;45(7):1268‐76. [DOI] [PubMed] [Google Scholar]
Koch‐Henriksen 2006 {published data only}
- Koch‐Henriksen N, Sørensen P, Christensen T, Frederiksen J, Ravnborg M, Jensen K, et al. A randomised study of two interferon‐beta treatments in relapsing‐remitting multiple sclerosis. Neurology 2006;66(7):1056‐60. [DOI] [PubMed] [Google Scholar]
Lewanska 2002 {published data only}
- Lewanska M, Siger‐Zajdel M, Selmaj K. No difference in efficacy of two different doses of intravenous immunoglobulins in MS: clinical and MRI assessment. European Journal of Neurology 2002;9(6):565‐72. [DOI] [PubMed] [Google Scholar]
MAIN TRIAL {published data only}
- Massacesi L, Tramacere I, Amoroso S, Battaglia MA, Benedetti MD, Filippini G, et al. Azathioprine versus beta interferons for relapsing‐remitting multiple sclerosis: a multicentre randomized non‐inferiority trial. PLoS One 2014;9(11):e113371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Millefiorini 1997 {published data only}
- Millefiorini E, Gasperini C, Pozzilli C, D'Andrea F, Bastianello S, Trojano M, et al. Randomized placebo‐controlled trial of mitoxantrone in relapsing‐remitting multiple sclerosis: 24‐month clinical and MRI outcome. Journal of Neurology 1997;244(3):153‐9. [DOI] [PubMed] [Google Scholar]
MSCRG 1996 {published data only}
- Jacobs L, Cookfair D, Rudick R, Herndon R, Richert J, Salazar A, et al. Intramuscular interferon beta‐1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Annals of Neurology 1996;39:285‐94. [DOI] [PubMed] [Google Scholar]
OWIMS 1999 {published data only}
- OWIMS. Evidence of interferon beta‐1a dose response in relapsing‐remitting MS: the OWIMS Study. The Once Weekly Interferon for MS Study Group. Neurology 1999;53(4):679‐86. [DOI] [PubMed] [Google Scholar]
PRISMS 1998 {published data only}
- PRISMS. Randomised double‐blind placebo‐controlled study of interferon beta‐1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta‐1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet 1998;352(9139):1498‐504. [PubMed] [Google Scholar]
REGARD 2008 {published data only}
- Mikol D, Barkhof F, Chang P, Coyle P, Jeffery D, Schwid S, et al. Comparison of subcutaneous interferon beta‐1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open‐label trial. Lancet Neurology 2008;7(10):903‐14. [DOI] [PubMed] [Google Scholar]
SELECT 2013 {published data only}
- Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW, et al. Daclizumab high‐yield process in relapsing‐remitting multiple sclerosis (SELECT): a randomised, double‐blind, placebo‐controlled trial. Lancet 2013;381(9884):2167‐75. [DOI] [PubMed] [Google Scholar]
TEMSO 2011 {published data only}
- O'Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. New England Journal of Medicine 2011;365(14):1293‐303. [DOI] [PubMed] [Google Scholar]
TENERE 2014 {published data only}
- Vermersch P, Czlonkowska A, Grimaldi LM, Confavreux C, Comi G, Kappos L, et al. Teriflunomide versus subcutaneous interferon beta‐1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Multiple Sclerosis (Houndmills, Basingstoke, England) 2014;20(6):705‐16. [DOI] [PubMed] [Google Scholar]
TOWER 2014 {published data only}
- Confavreux C, O'Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurology 2014;13(3):247‐56. [DOI] [PubMed] [Google Scholar]
TRASFORMS 2010 {published data only}
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. New England Journal of Medicine 2010;362(5):402‐15. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
ACT 2009 {published data only}
- Cohen JA, Imrey PB, Calabresi PA, Edwards KR, Eickenhorst T, Felton WL 3rd, et al. Results of the Avonex Combination Trial (ACT) in relapsing‐remitting MS. Neurology 2009;72(6):535‐41. [DOI] [PubMed] [Google Scholar]
Ashtari 2011 {published data only}
- Ashtari F, Savoj MR. Effects of low dose methotrexate on relapsing‐remitting multiple sclerosis in comparison to Interferon β‐1α: a randomized controlled trial. Journal of Research in Medical Sciences 2011;16(4):457‐62. [PMC free article] [PubMed] [Google Scholar]
ATAMS 2014 {published data only}
- Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurology 2014;13(4):353‐63. [DOI] [PubMed] [Google Scholar]
Calabrese 2012 {published data only}
- Calabrese M, Bernardi V, Atzori M, Mattisi I, Favaretto A, Rinaldi F, et al. Effect of disease‐modifying drugs on cortical lesions and atrophy in relapsing‐remitting multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2012;18(4):418‐24. [DOI] [PubMed] [Google Scholar]
CHOICE 2010 {published data only}
- Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double‐blind, placebo‐controlled, add‐on trial with interferon beta. Lancet Neurology 2010;9(4):381‐90. [DOI] [PubMed] [Google Scholar]
Etemadifar 2006 {published data only}
- Etemadifar M, Janghorbani M, Shaygannejad V. Comparison of Betaferon, Avonex, and Rebif in treatment of relapsing‐remitting multiple sclerosis. Acta Neurologica Scandinavica 2006;113(5):283‐7. [DOI] [PubMed] [Google Scholar]
FORTE 2011 {published data only}
- Comi G, Cohen JA, Arnold DL, Wynn D, Filippi M, FORTE Study Group. Phase III dose‐comparison study of glatiramer acetate for multiple sclerosis. Annals of Neurology 2011;69(1):75‐82. [DOI] [PubMed] [Google Scholar]
Freedman 2012 {published data only}
- Freedman MS, Wolinsky JS, Wamil B, Confavreux C, Comi G, Kappos L, et al. Teriflunomide added to interferon‐β in relapsing multiple sclerosis: a randomized phase II trial. Neurology 2012;78(23):1877‐85. [DOI] [PubMed] [Google Scholar]
Havrdova 2009 {published data only}
- Havrdova E, Zivadinov R, Krasensky J, Dwyer MG, Novakova I, Dolezal O, et al. Randomized study of interferon beta‐1a, low‐dose azathioprine, and low‐dose corticosteroids in multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2009;15(8):965‐76. [DOI] [PubMed] [Google Scholar]
Kappos 2006 {published data only}
- Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. New Egyptian Journal of Medicine 2006;355(11):1124‐40. [DOI] [PubMed] [Google Scholar]
Kappos 2008 {published data only}
- Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, et al. Efficacy and safety of oral fumarate in patients with relapsing‐remitting multiple sclerosis: a multicentre, randomised, double‐blind, placebo‐controlled phase IIb study. Lancet 2008;372(9648):1463‐72. [DOI] [PubMed] [Google Scholar]
Kappos 2011 {published data only}
- Kappos L, Li D, Calabresi PA, O'Connor P, Bar‐Or A, Barkhof F, et al. Ocrelizumab in relapsing‐remitting multiple sclerosis: a phase 2, randomised, placebo‐controlled, multicentre trial. Lancet 2011;378(9805):1779‐87. [DOI] [PubMed] [Google Scholar]
Khoury 2010 {published data only}
- Khoury SJ, Healy BC, Kivisäkk P, Viglietta V, Egorova S, Guttmann CR, et al. A randomized controlled double‐masked trial of albuterol add‐on therapy in patients with multiple sclerosis. Archives of Neurology 2010;67(9):1055‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Knobler 1993 {published data only}
- Knobler RL, Greenstein JI, Johnson KP, Lublin FD, Panitch HS, Conway K, et al. Systemic recombinant human interferon‐beta treatment of relapsing‐remitting multiple sclerosis: pilot study analysis and six‐year follow‐up. Journal of Interferon Research 1993;13(5):333‐40. [DOI] [PubMed] [Google Scholar]
Saida 2012 {published data only}
- Saida T, Kikuchi S, Itoyama Y, Hao Q, Kurosawa T, Nagato K, et al. A randomized, controlled trial of fingolimod (FTY720) in Japanese patients with multiple sclerosis. Multiple Sclerosis (Houndmills, Basingstoke, England) 2012;18(9):1269‐77. [DOI] [PubMed] [Google Scholar]
SENTINEL 2006 {published data only}
- Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab plus interferon beta‐1a for relapsing multiple sclerosis. New England Journal of Medicine 2006;354(9):911‐23. [DOI] [PubMed] [Google Scholar]
Sorensen 2014 {published data only}
- Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing‐remitting multiple sclerosis: a phase 2 study. Neurology 2014;82(7):573‐81. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
DECIDE {published data only}
- Multicenter, double‐blind, randomized, parallel‐group, monotherapy, active‐control study to determine the efficacy and safety of daclizumab high yield process (DAC HYP) versus Avonex® (interferon β 1a) in patients with relapsing‐remitting multiple sclerosis. https://clinicaltrials.gov/ct2/show/study/NCT01064401 (accessed 30 September 2014).
NCT01247324 {published data only}
- A randomized, double‐blind, double‐dummy, parallel‐group study to evaluate the efficacy and safety of Ocrelizumab in comparison to interferon beta‐1a (Rebif®) in patients with relapsing multiple sclerosis. https://clinicaltrials.gov/ct2/show/study/NCT01247324 (accessed 30 September 2014).
NCT01412333 {published data only}
- A randomized, double‐blind, double‐dummy, parallel‐group study to evaluate the efficacy and safety of Ocrelizumab in comparison to Interferon Beta‐1a (Rebif®) in patients with relapsing multiple sclerosis. https://clinicaltrials.gov/ct2/show/study/NCT01412333 (accessed 30 September 2014).
Additional references
Aharoni 2014
- Aharoni R. Immunomodulation neuroprotection and remyelination. The fundamental therapeutic effects of glatiramer acetate: a critical review. Journal of Autoimmunity 2014;54:81‐92. [DOI: 10.1016/j.jaut.2014.05.005] [DOI] [PubMed] [Google Scholar]
Association of British Neurologists 2005
- Association of British Neurologists 2005. Guidelines for the use of intravenous immunoglobulin in neurological diseases. www.theabn.org/documents/IVIg‐Guidelines (accessed October 2013).
Bennett 2009
- Bennett J, Stüve O. Update on inflammation, neurodegeneration, and immunoregulation in multiple sclerosis: therapeutic implications. Clinical Neuropharmacology 2009;32(3):121‐32. [DOI] [PubMed] [Google Scholar]
Birnbaum 2008
- Birnbaum G, Cree B, Altafullah I, Zinser M, Reder A. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology 2008;71(18):1390–5. [DOI] [PubMed] [Google Scholar]
Caldwell 2005
- Caldwell D, Ades A, Higgins J. Simultaneous comparison of multiple treatments: combining direct and indirect evidence as well. BMJ 2005;331(7521):897‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Carroll 2014
- Carroll CA, Fairman KA, Lage MJ. Updated cost‐of‐care estimates for commercially insured patients with multiple sclerosis: retrospective observational analysis of medical and pharmacy claims data. BMC Health Services Research 2014;14:286. [http://www.biomedcentral.com/1472‐6963/14/286] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chaimani 2012
- Chaimani A, Salanti G. Using network meta‐analysis to evaluate the existence of small‐study effects in a network of interventions. Research Synthesis Methods 2012;3(2):161‐76. [DOI] [PubMed] [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta‐analysis in Stata. PLoS One 2013;8(10):e76654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Claussen 2012
- Claussen MC, Korn T. Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clinical Immunology 2012;142:49‐56. [DOI] [PubMed] [Google Scholar]
Compston 2008
- Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502‐17. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7:177‐88. [DOI] [PubMed] [Google Scholar]
Ebers 2008
- Ebers G, Heigenhauser L, Daumer M, Lederer C, Noseworthy J. Disability as an outcome in MS clinical trials. Neurology 2008;71:624–31. [DOI] [PubMed] [Google Scholar]
Elovaara 2008
- Elovaara I, Apostolski S, Doorn P, Gilhus N, Hietaharju A, Honkaniemi J, et al. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. European Journal of Neurology 2008;15(9):893‐908. [DOI] [PubMed] [Google Scholar]
EMA 2006
- European Agency for the Evaluation of Medicinal Products. Committee for proprietary medicinal products European public assessment report: Tysabri. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000603/wapp/Post‐authorisation/human_wapp_000166.jsp&mid=WC0b01ac058001d128 (accessed June 2014).
EMA 2011
- European Medicines Agency. Committee for proprietary medicinal products European public assessment report: Gilenya. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002202/human_med_001433.jsp&mid=WC0b01ac058001d124 (accessed June 2014).
EMA 2013a
- European Medicines Agency. Committee for proprietary medicinal products European public assessment report: Aubagio. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002514/human_med_001645.jsp&mid=WC0b01ac058001d124 (accessed June 2014).
EMA 2013b
- European Medicines Agency. Committee for proprietary medicinal products European public assessment report: Lemtrada. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/003718/WC500150521.pdf (accessed June 2014).
EMA 2014a
- European Medicines Agency. Committee for proprietary medicinal products European public assessment report: Tecfidera. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002601/WC500162069.pdf (accessed June 2014).
EMA 2014b
- European Medicines Agency. Committee for proprietary medicinal products European public assessment report: Plegridy. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Summary_for_the_public/human/002827/WC500170305.pdf (accessed June 2015).
EMA 2014c
- European Medicines Agency. Refusal of the marketing authorisation for Nerventra (laquinimod). http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_‐_Initial_authorisation/human/002546/WC500160120.pdf (accessed June 2014).
EMEA 1997
- European Agency for the Evaluation of Medicinal Products. Committee for proprietary medicinal products European public assessment report: Avonex. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Summary_for_the_public/human/000102/WC500029423.pdf (accessed June 2015).
EMEA 1998
- European Agency for the Evaluation of Medicinal Products. Committee for proprietary medicinal products European public assessment report: Rebif. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Assessment_Report_‐_Variation/human/000136/WC500122183.pdf (accessed June 2014).
EMEA 2002
- European Agency for the Evaluation of Medicinal Products. Committee for proprietary medicinal products European public assessment report: Betaferon. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000081/human_med_000673.jsp&mid=WC0b01ac058001d124 (accessed June 2014).
FDA 1993
- U.S. Food, Drug Administration. Betaseron interferon beta‐1b subcutaneous. Drug Approval Package ‐ Licensing Action 1993. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/103471s5063s5067s5079s5088s5120s5124s5136s5138lbl.pdf (accessed June 2014).
FDA 1996
- U.S. Food, Drug Administration. Glatiramer acetate (Capoxane) Product Approval Information ‐ Licensing Action 1996. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#apphist (accessed June 2014).
FDA 2000
- U.S. Food, Drug Administration. Mitoxantrone (Novantrone) Product Approval Information ‐ Licensing Action 2000. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Overview&DrugName=NOVANTRONE&CFID=17505855&CFTOKEN=fca8add70180e2b1‐F1C49694‐F504‐892A‐3B263F68EAFF59DC (accessed June 2014).
FDA 2002
- U.S. Food, Drug Administration. Interferon beta‐1a (Rebif) Product Approval Information ‐ Licensing Action 2002. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm080737.htm (accessed June 2014).
FDA 2003
- U.S. Food, Drug Administration. Interferon beta‐1a (Avonex) Product Approval Information ‐ Licensing Action 2003. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/103628s5021TOC.cfm (accessed June 2014).
FDA 2004
- U.S. Food, Drug Administration. Tysabri (Natalizumab) Product Approval Information 2004. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/125104s000_Natalizumab.cfm (accessed June 2014).
FDA 2006
- U.S. Food, Drug Administration. FDA Approves Resumed Marketing of Tysabri Under a Special Distribution Program. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108662.htm (accessed June 2014).
FDA 2010
- U.S. Food, Drug Administration. Gilenya (Fingolimod) Product Approval Information 2010. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=022527&DrugName=GILENYA&ActiveIngred=FINGOLIMOD&SponsorApplicant=NOVARTIS&ProductMktStatus=1&goto=Search.DrugDetails (accessed June 2014).
FDA 2012
- U.S. Food, Drug Administration. Aubagio (Teriflunomide) Product Approval Information 2012. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202992&DrugName=AUBAGIO&ActiveIngred=TERIFLUNOMIDE&SponsorApplicant=SANOFI%20AVENTIS%20US&ProductMktStatus=1&goto=Search.DrugDetails (accessed June 2014).
FDA 2013
- U.S. Food, Drug Administration. Tecfidera (Dimethyl fumarate) Product Approval Information 2013. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=204063&DrugName=TECFIDERA&ActiveIngred=DIMETHYL%20FUMARATE&SponsorApplicant=BIOGEN%20IDEC%20INC&ProductMktStatus=1&goto=Search.DrugDetails (accessed June 2014).
FDA 2014a
- U.S. Food, Drug Administration. Alemtuzumab (Lemtrada) Product Approval Information. Licensing Action 2014. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2014/103948Orig1s5139ltr.pdf (accessed Sept 2014).
FDA 2014b
- U.S. Food, Drug Administration. Peginterferon beta‐1a (Plegridy) Product Approval Information. Licensing Action 2014. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/biologics/2004/125083ltr.pdf (accessed September 2014).
FDA 2014c
- U.S. Food, Drug Administration. Imuran Product Information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/016324s037,017391s016lbl.pdf (accessed June 2015).
Filippini 2013
- Filippini G, Giovane C, Vacchi L, D’Amico R, Pietrantonj C, Beecher D, et al. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta‐analysis. Cochrane Database of Systematic Reviews 2013, Issue 6. [DOI: 10.1002/14651858.CD008933] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fox 2004
- Fox E. Mechanism of action of mitoxantrone. Neurology 2004;12:15‐8. [DOI] [PubMed] [Google Scholar]
Glenny 2005
- Glenny A, Altman D, Song F, Sakarovitch C, Deeks J, D'Amico R, et al. Indirect comparisons of competing interventions. Health Technology Assessment 2005;9:1‐34. [DOI] [PubMed] [Google Scholar]
Goodin 2002
- Goodin DS, Frohman EM, Garmany GP Jr, Halper J, Likosky WH, Lublin FD, et al. Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology 2002;58(2):169‐78. [DOI] [PubMed] [Google Scholar]
Goodman 2009
- Goodman A, Rossman H, Bar‐Or A, Miller A, Miller D, Schmierer K, et al. GLANCE: results of a phase 2, randomized, double‐blind, placebo‐controlled study. Neurology 2009;72(9):806–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE Working Group 2004
- GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328:1490‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro 2008 [Computer program]
- Brozek J, Oxman A, Schünemann H. GRADEpro. Version 3.2 for Windows. Brozek J, Oxman A, Schünemann H, 2008.
Hadjigeorgiou 2013
- Hadjigeorgiou GM, Doxani C, Miligkos M, Ziakas P, Bakalos G, Papadimitriou D, et al. A network meta‐analysis of randomized controlled trials for comparing the effectiveness and safety profile of treatments with marketing authorization for relapsing multiple sclerosis. Journal of Clinical Pharmacy and Therapeutics 2013;38(6):433‐9. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2012
- Higgins JPT, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta‐analysis: concepts and models for multi‐arm studies. Research Synthesis Methods 2012;3:98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hill‐Cawthorne 2012
- Hill‐Cawthorne GA, Button T, Tuohy O, Jones JL, May K, Somerfield J, et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. Journal of Neurology, Neurosurgery, and Psychiatry 2012;83:298‐304. [DOI] [PubMed] [Google Scholar]
Hu 2012
- Hu X, Miller L, Richman S, Hitchman S, Glick G, Liu S, et al. A novel PEGylated interferon beta‐1a for multiple sclerosis: safety, pharmacology, and biology. Journal of Clinical Pharmacology 2012;52:798–808. [DOI] [PubMed] [Google Scholar]
Inusah 2010
- Inusah S, Sormani MP, Cofield SS, Aban IB, Musani SK, Srinivasasainagendra V, et al. Assessing changes in relapse rates in multiple sclerosis. Multiple Sclerosis 2010;16:1414–21. [DOI] [PubMed] [Google Scholar]
Jackson 2014
- Jackson D, Barrett JK, Rice S, White IR, Higgins JP. A design‐by‐treatment interaction model for network meta‐analysis with random inconsistency effects. Statistics in Medicine 2014;33(21):3639‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kieseier 2011
- Kieseier BC. The mechanism of action of interferon‐β in relapsing multiple sclerosis. CNS Drugs 2011;25:491‐502. [DOI] [PubMed] [Google Scholar]
Kremenchutzky 2006
- Kremenchutzky M, Rice G, Baskerville J, Wingerchuk D, Ebers G. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain 2006;129:584‐94. [DOI] [PubMed] [Google Scholar]
Kurtzke 1983
- Kurtzke J. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444‐52. [DOI] [PubMed] [Google Scholar]
Lublin 1996
- Lublin F, Reingold S. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996;46:907‐11. [DOI] [PubMed] [Google Scholar]
Mandala 2002
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, et al. Alteration of lymphocyte trafficking by sphingosine‐1‐phosphate receptor agonists. Science 2002;296:346‐9. [DOI] [PubMed] [Google Scholar]
McDonald 2001
- McDonald W, Compston A, Edan G, Goodkin D, Hartung H, Lublin F. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Annals of Neurology 2001;50:121‐7. [DOI] [PubMed] [Google Scholar]
Miladinovic 2014
- Miladinovic B, Hozo I, Chaimani A, Djulbegovic B. Indirect treatment comparison. Stata Journal 2014;14(1):76‐86. [Google Scholar]
Nicholas 2012
- Nicholas R, Straube S, Schmidli H, Pfeiffer S, Friede T. Time‐patterns of annualized relapse rates in randomized placebo‐controlled clinical trials in relapsing multiple sclerosis: a systematic review and meta‐analysis. Multiple Sclerosis 2012;18:1290‐6. [DOI] [PubMed] [Google Scholar]
Oh 2013
- Oh J, Calabresi PA. Emerging injectable therapies for multiple sclerosis. Lancet Neurology 2013;12:1115–26. [DOI] [PubMed] [Google Scholar]
Peters 2008
- Peters J, Sutton A, Jones D, Abrams K, Rushton L. Contour‐enhanced meta‐analysis funnel plots help distinguish publication bias from other causes of asymmetry. Journal of Clinical Epidemiology 2008;61(10):991‐6. [DOI] [PubMed] [Google Scholar]
Polman 2005
- Polman C, Reingold S, Edan G, Filippi M, Hartung H, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the 'McDonald Criteria'. Annals of Neurology 2005;58:840‐6. [DOI] [PubMed] [Google Scholar]
Polman 2011
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Annals of Neurology 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Poser 1983
- Poser C, Paty D, Scheinberg L, McDonald W, Davis F, Ebers G, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Annals of Neurology 1983;13:227‐31. [DOI] [PubMed] [Google Scholar]
Ravnborg 2010
- Ravnborg M, Sørensen P, Andersson M, Celius E, Jongen P, Elovaara I, et al. Methylprednisolone in combination with interferon beta‐1a for relapsing‐remitting multiple sclerosis (MECOMBIN study): a multicentre, double blind, randomised, placebo‐controlled, parallel‐group trial. Lancet Neurology 2010;9:672‐80. [DOI] [PubMed] [Google Scholar]
Salanti 2011
- Salanti G, Ades A, Ioannidis J. Graphical methods and numerical summaries for presenting results from multiple‐treatment meta‐analysis: an overview and tutorial. Journal of Clinical Epidemiology 2011;64:163‐71. [DOI] [PubMed] [Google Scholar]
Salanti 2012
- Salanti G. Indirect and mixed‐treatment comparison, network, or multiple‐treatments meta‐analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Research Synthesis Methods 2012;3(2):80‐97. [DOI] [PubMed] [Google Scholar]
Salanti 2014
- Salanti G, Giovane C, Chaimani A, Caldwell DM, Higgins JP. Evaluating the quality of evidence from a network meta‐analysis. PLoS One 2014;9:e99682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scalfari 2014
- Scalfari A, Neuhaus A, Daumer M, Muraro PA, Ebers GC. Onset of secondary progressive phase and long‐term evolution of multiple sclerosis. Journal of Neurology, Neurosurgery & Psychiatry 2014;85:67‐75. [DOI] [PubMed] [Google Scholar]
Schmied 2003
- Schmied M, Duda PW, Krieger JI, Trollmo C, Hafler DA. In vitro evidence that subcutaneous administration of glatiramer acetate induces hyporesponsive T cells in patients with multiple sclerosis. Clinical Immunology 2003;106:163‐74. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann H, Oxman A, Higgins J, Vist G, Glasziou P, Guyatt G. Chapter 11: Presenting results and ‘Summary of findings’ tables. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Stangel 1999
- Stangel M, Toyka K, Gold R. Mechanisms of high‐dose intravenous immunoglobulins in demyelinating diseases. Archives of Neurology 1999;56(6):661‐3. [DOI] [PubMed] [Google Scholar]
Steinvorth 2013
- Steinvorth SM, Röver C, Schneider S, Nicholas R, Straube S, Friede T. Explaining temporal trends in annualised relapse rates in placebo groups of randomised controlled trials in relapsing multiple sclerosis: systematic review and meta‐regression. Multiple Sclerosis 2013;19:1580‐6. [DOI] [PubMed] [Google Scholar]
Stellmann 2012
- Stellmann JP, Neuhaus A, Herich L, Schippling S, Roeckel M, Daumer M, et al. Placebo cohorts in phase‐3 MS treatment trials, predictors for on‐trial disease activity 1990‐2010 based on a meta‐analysis and individual case data. PLoS One 2012;7(11):e50347. [DOI: 10.1371/journal.pone.0050347] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tiede 2003
- Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, et al. CD28‐dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. Journal of Clinical Investigation 2003;111:1133‐45. [PUBMED: 12697733] [DOI] [PMC free article] [PubMed] [Google Scholar]
Varrin‐Doyer 2014
- Varrin‐Doyer M, Zamvil SS, Schulze‐Topphoff U. Laquinimod, an up‐and‐coming immunomodulatory agent for treatment of multiple sclerosis. Experimental Neurology 2014;262:66‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Veroniki 2013
- Veroniki AA, Vasiliadis HS, Higgins JP, Salanti G. Evaluation of inconsistency in networks of interventions. Internal Journal Epidemiology 2013;42(1):332‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weiner 1993
- Weiner H, Mackin G, Matsui M, Orav E, Khoury S, Dawson D, et al. Double‐blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science 1993;259(5099):1321–4. [DOI] [PubMed] [Google Scholar]
White 2011
- White IR. Multivariate random‐effects meta‐regression: updates to mvmeta. Stata Journal 2011;11:255‐70. [Google Scholar]
White 2012
- White IR, Barrett JK, Jackson D, Higgins JPT. Consistency and inconsistency in network meta‐analysis: model estimation using multivariate meta‐regression. Research Synthesis Methods 2012;3(2):111‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilms 2010
- Wilms H, Sievers J, Rickert U, Rostami‐Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL‐1beta, TNF‐alpha and IL‐6 in an in‐vitro model of brain inflammation. Journal of Neuroinflammation 2010;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wuest 2011
- Wuest SC, Edwan JH, Martin JF, Han S, Perry JS, Cartagena CM, et al. A role for interleukin‐2 trans‐presentation in dendritic cell‐mediated T cell activation in humans, as revealed by daclizumab therapy. Nature Medicine 2011;17:604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yednock 1992
- Yednock TA, Cannon C, Fritz LC, Sanchez‐Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992;356:63‐6. [DOI] [PubMed] [Google Scholar]
Yousry 2006
- Yousry T, Major E, Ryschkewitsch C, Fahle G, Fischer S, Hou J, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. New England Journal of Medicine 2006;354(9):924‐33. [PUBMED: 16510746] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zintzaras 2012
- Zintzaras E, Doxani C, Mprotsis T, Schmid CH, Hadjigeorgiou G. Network analysis of randomized controlled trials in multiple sclerosis. Clinical Therapeutics 2012;34(4):857–69. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Tramacere 2014
- Tramacere I, Giovane C, Salanti G, D'Amico R, Pacchetti I, Filippini G. Immunomodulators and immunosuppressants for relapsing‐remitting multiple sclerosis: a network meta‐analysis. Cochrane Database of Systematic Reviews 2014, Issue 11. [DOI: 10.1002/14651858.CD011381] [DOI] [PMC free article] [PubMed] [Google Scholar]