

Abstract
Extracellular nucleotides and nucleosides are important signaling molecules in the lung. Nucleotide and nucleoside concentrations in alveolar lining fluid are controlled by a complex network of surface ectonucleotidases. Previously, we demonstrated that influenza A/WSN/33 (H1N1) virus resulted in increased levels of the nucleotide ATP and the nucleoside adenosine in bronchoalveolar lavage fluid (BALF) of wild-type (WT) C57BL/6 mice. Influenza-induced acute lung injury (ALI) was highly attenuated in A1-adenosine receptor-knockout mice. Because AMP hydrolysis by the ecto-5′-nucleotidase (CD73) plays a central role in and is rate-limiting for generation of adenosine in the normal lung, we hypothesized that ALI would be attenuated in C57BL/6-congenic CD73-knockout (CD73-KO) mice. Infection-induced hypoxemia, bradycardia, viral replication, and bronchoconstriction were moderately increased in CD73-KO mice relative to WT controls. However, postinfection weight loss, pulmonary edema, and parenchymal dysfunction were not altered. Treatment of WT mice with the CD73 inhibitor 5'-(α,β-methylene) diphosphate (APCP) also had no effect on infection-induced pulmonary edema but modestly attenuated hypoxemia. BALF from CD73-KO and APCP-treated WT mice contained more IL-6 and CXCL-10/IFN-γ-induced protein 10, less CXCL-1/keratinocyte chemoattractant, and fewer neutrophils than BALF from untreated WT controls. BALF from APCP-treated WT mice also contained fewer alveolar macrophages and more transforming growth factor-β than BALF from untreated WT mice. These results indicate that CD73 is not necessary for development of ALI following influenza A virus infection and suggest that tissue-nonspecific alkaline phosphatase may be responsible for increased adenosine generation in the infected lung. However, they do suggest that CD73 has a previously unrecognized immunomodulatory role in influenza.
Keywords: influenza, adenosine, CD73, acute lung injury, mouse
each year, ∼20% of children and 5% of adults worldwide will develop symptomatic influenza infections (9). It has been estimated that ∼200,000 hospitalizations and >36,000 excess deaths per year result from seasonal influenza in the United States alone, at an annual cost to the economy of more than $80 billion (4, 17). In addition, reoccurring pandemics have resulted in devastating losses worldwide (32). Emergent highly pathogenic H5N1 and H7N9 avian influenza A virus strains pose a potentially escalating threat to the human population, particularly as these strains have very high mortality rates (36).
Nucleotides and nucleosides are important physiological regulators of lung function (21). Both are normally present at very low concentrations in the bronchoalveolar lining fluid (BALF), but we and others have reported that large amounts of ATP are released apically by respiratory epithelial cells in response to viral infections (2, 12, 23, 24). ATP and ADP in the BALF can be metabolized to adenosine by enzymes expressed on the apical surface of respiratory epithelial cells. The ecto-5′-nucleotidase (CD73) plays a central role in and is rate-limiting for this process (40). Expression of CD73 is high in lung (33) and is increased in influenza-infected human middle ear epithelial cells in vitro (34). Adenosine metabolism is important for regulation of pulmonary fluid dynamics (22) and lung inflammation (7). However, bioavailability of this nucleoside is limited by reuptake into cells, degradation by adenosine deaminase to inosine, or rephosphorylation by adenosine kinase (39).
Recently, we showed that infection of wild-type (WT) C57BL/6 mice with an acutely lethal dose of influenza A/WSN/33 (H1N1) resulted in a significant increase in BALF ATP and adenosine content (3, 38). We also showed that ligation of A1-subtype adenosine receptors (AdoRs) by adenosine was central to development of acute lung injury (ALI) in WT mice following infection (3). Given that CD73 is believed to be the rate-limiting step in conversion of ATP to adenosine in the normal lung, we hypothesized that the influenza viral infection-induced rise in BALF ATP content would result in increased CD73-mediated catabolism to adenosine by mass action. The aim of this study was therefore to build on our earlier findings by showing that CD73 activity is required for catabolism of ATP to adenosine and, therefore, is necessary for development of ALI in influenza-infected mice. We also wished to determine whether pharmacological inhibition of CD73 over the course of infection would reduce ALI severity. We were surprised to find that adenosine generation and ALI pathogenesis did not differ significantly between influenza A virus-infected WT controls and C57BL/6-congenic CD73-knockout (CD73-KO) mice. Similarly, treatment with the CD73 inhibitor adenosine 5'-(α,β-methylene) diphosphate (APCP) did not alter BALF adenosine and had limited beneficial effects on disease severity in influenza-infected WT mice. We attribute these findings to the functional redundancy for adenosine generation between CD73 and tissue-nonspecific alkaline phosphatase (TNAP) proteins, which were unaffected by cd73 gene deletion or APCP treatment. Interestingly, however, we did find some significant differences in the cellular and humoral innate immune response to infection between WT controls, CD73-KO mice, and APCP-treated WT mice. Taken together, our data indicate that CD73 is not required for adenosine generation and development of ALI in influenza A virus-infected mice but is necessary to maintain a normal immune response to infection.
MATERIALS AND METHODS
Mice.
C57BL/6AnNCr WT mice were purchased from the National Cancer Institute (Frederick, MD). C57BL/6-congenic CD73-KO mice were generated by Dr. Linda Thompson (University of Oklahoma, Norman, OK) (26) and kindly provided to us by Dr. Prosper Boyaka (The Ohio State University). These animals breed normally, have average-sized litters, and have no obvious phenotype (10). All procedures were approved by The Ohio State University Animal Care and Use Committee. For ethical reasons, survival studies were not performed.
Preparation of viral inoculum.
All studies used influenza A/WSN/33 (H1N1) virus, which is a mouse-adapted, pneumotropic influenza A strain (15). Virus was grown in embryonated chicken eggs according to a standard protocol. Virus preparations were checked for absence of contamination with Mycoplasma pulmonis by PCR (Charles River Research Animal Diagnostic Services, Wilmington, MA). Absence of endotoxin contamination was confirmed by a standard Limulus amebocyte assay (Lonza, Basel, Switzerland).
Mouse infection.
At 8–12 wk of age, WT and CD73-KO mice were inoculated intranasally with 10,000 plaque-forming units of H1N1 influenza A/WSN/33 in 50 μl of PBS with 0.1% BSA under ketamine-xylazine anesthesia, as described in our previous studies (38). Median time to death in WT mice infected with this inoculum is 7 days, with 100% mortality at day 8 (1, 2), and no viral replication is detectable in brain tissue from infected animals (3). Mice were individually marked and weighed every 2 days. Data for each experimental group were derived from a minimum of two independent infections.
Quantitative real-time PCR.
Mice were euthanized by intraperitoneal administration of ketamine (87 mg/kg)-xylazine (13 mg/kg). After bilateral thoracotomy, lungs were removed and homogenized. Total RNA was extracted using the RNeasy system (Qiagen, Alameda, CA). RNA was reverse-transcribed into cDNA using the high-capacity cDNA reverse-transcription kit (Applied Biosystems, Grand Island, NY). Quantitative real-time PCR amplification of cDNA was performed using the TaqMan gene expression system (Applied Biosystems). Expression of cd73 and tnap mRNA was determined by the cycle threshold (ΔΔCt) method and normalized to the endogenous control 18s rrna, which was not altered by infection (19).
CD73 enzymatic activity assay.
Whole lung homogenate ecto-5′-nucleotidase activity was measured by the AMP catabolism assay (37), in which free inorganic phosphate released by CD73-mediated degradation of AMP to adenosine is quantified colorimetrically using malachite green (8). To confirm specificity of the CD73 activity assay, it was repeated in the presence of a highly specific quinolinyl-benzenesulfonamide TNAP inhibitor (Calbiochem, Billerica, MA).
Lung wet-to-dry weight ratio.
Lung wet-to-dry weight ratio was measured as previously described (12). Briefly, mice were euthanized and exsanguinated, and their lungs were removed, weighed, dried in an oven at 55°C for 3 days, and weighed again. Wet-to-dry weight ratio was then calculated without correction for blood content.
Measurement of lung mechanics.
Mechanical properties of the mouse lung were assessed in diazepam (Valium)-ketamine-anesthetized mice using the forced-oscillation technique (20), as described in our previous studies (35). Anesthetized mice were mechanically ventilated on a flexiVent computer-controlled piston ventilator (SCIreq, Montreal, PC, Canada) with 8 ml/kg tidal volume at a frequency of 150 breaths/min against 2–3 cmH2O positive end-expiratory pressure. Pressure and flow data (reflective of airway and tissue dynamics) were used to calculate lung resistance, static lung compliance, and dynamic lung compliance at baseline with use of the single-compartment model (20). As in prior studies, maximal airway responsiveness to bronchoconstrictors was measured following exposure to increasing doses of nebulized methacholine (0.1–50 mg/ml) (13).
Measurement of BALF inflammatory mediators.
Bronchoalveolar lavage was performed as previously described (38). BALF protein content was determined by a standard bicinchoninic acid assay. BALF IFN-γ, IL-1β, IL-6, IL-10, IL-12, CXCL-1/keratinocyte chemoattractant (KC), and TNF-α were quantified by an ultrasensitive multiplex electrochemiluminescence assay (Meso Scale Discovery, Gaithersburg, MD). Murine CXCL-10/IFN-γ-induced protein 10, CCL-2/monocyte chemotactic protein-1, CCL-5/regulated on activation, normal T cell expressed and secreted (RANTES), and transforming growth factor (TGF)-β levels were measured using Quantikine ELISA kits (R & D Systems, Minneapolis, MN). All assays were performed in accordance with the manufacturer's instructions.
Measurement of BALF adenosine.
BALF adenosine was measured using a fluorometric assay (Biovision, San Francisco, CA) in accordance with the manufacturer's instructions.
APCP treatment regimen.
WT mice were gavaged every other day from 1 day before infection to 5 days postinfection (dpi) with 200 μl of saline containing the CD73 inhibitor APCP (20 mg/kg; Sigma-Aldrich, St. Louis, MO) (30). Treatment controls were gavaged at the same frequency with 200 μl of saline only.
Western blotting.
Western blotting and densitometry were performed as described in our previous studies (3). TNAP was detected using a mouse monoclonal IgG1 antibody (catalog no. sc73524, Santa Cruz Biotechnology, Dallas, TX).
Measurement of TNAP activity.
Alkaline phosphatase activity was measured using a colorimetric assay kit (Abcam, San Francisco, CA) in accordance with the manufacturer's instructions.
Other measurements.
Carotid arterial O2 saturation, heart rate, viral titers, and BALF leukocytes were measured as previously described (2).
Statistical analyses.
Descriptive statistics (mean and SE) were calculated using Instat software (GraphPad, San Diego, CA). Gaussian data distribution was verified by the method of Kolmogorov and Smirnov. An unpaired Student's t-test was used when two groups were compared. Statistical analyses of data sets containing more than two groups were made by ANOVA, with a post hoc Tukey-Kramer multiple-comparison posttest. All data are presented as means ± SE. P < 0.05 was considered statistically significant.
RESULTS
Influenza A/WSN/33 (H1N1) virus infection reduces cd73 gene expression, but not CD73 enzymatic activity, in WT mouse lung.
Infection of WT mice with influenza A/WSN/33 (10,000 plaque-forming units/mouse) significantly decreased whole lung homogenate cd73 gene expression (relative to uninfected levels) at 6 dpi (Fig. 1A). In contrast, infection had no effect on whole lung homogenate CD73 enzymatic activity (Fig. 1B). However, CD73 enzymatic activity appeared to be more variable at 6 dpi than other time points. Addition of a specific TNAP inhibitor had no significant effect, indicating that the assay is specific for CD73 activity (not shown).
Fig. 1.

Influenza A/WSN/33 (H1N1) virus infection reduces cd73 gene expression, but not CD73 enzymatic activity, in wild-type (WT) mouse lung. A and B: effect of intranasal infection of WT C57BL/6 mice with H1N1 influenza A/WSN/33 (10,000 plaque-forming units/mouse) on whole lung homogenate ecto-5′-nucleotidase (cd73) gene expression (fold change relative to uninfected mice, n = 3 per time point) and whole lung homogenate CD73 activity (nmol Pi·mg protein−1·min−1, n = 8 per time point). *P < 0.05 vs. uninfected WT mice. Values are means ± SE.
Cardiopulmonary dysfunction and viral replication are not significantly altered in influenza-infected CD73-KO mice.
Uninfected CD73-KO mice were phenotypically normal. The rate and extent of weight loss caused by infection with influenza A virus did not differ between WT and CD73-KO mice (Fig. 2A). Infection-induced hypoxemia (Fig. 2B) and bradycardia (Fig. 2C) tended to be more severe in CD73-KO mice, although this effect was only statistically significant at 4 dpi. Lung homogenate viral titers were higher in CD73-KO mice at 6 dpi (Fig. 2D), although this small increase in viral replication is unlikely to be of biological significance.
Fig. 2.

Cardiopulmonary dysfunction and viral replication are not significantly altered in influenza-infected C57BL/6-congenic CD73-knockout (CD73-KO) mice. A–C: effect of infection of CD73-KO mice with H1N1 influenza A/WSN/33 virus on body weight (BWT, %change from day 0, n > 15 per mouse strain), carotid arterial O2 saturation (SaO2, n > 15 per mouse strain), and heart rate (beats/min, n > 15 per mouse strain) at 0–6 days postinfection (dpi). D: effect on viral titers in lung homogenates at 6 dpi (n = 8–9 per mouse strain). #P < 0.001 vs. WT mice. Values are means ± SE.
CD73-KO mice are not protected from pulmonary edema following influenza infection.
There was no difference in lung water content (wet-to-dry weight ratio) between uninfected WT and CD73-KO mice (Fig. 3). Lung water content was significantly and comparably increased in both mouse strains at 6 dpi.
Fig. 3.
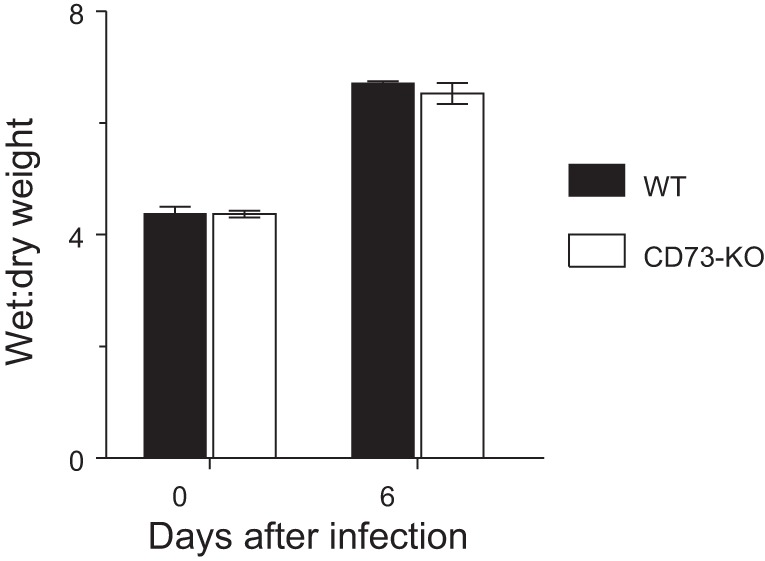
CD73-KO mice are not protected from pulmonary edema following influenza infection. Effects of H1N1 influenza A/WSN/33 infection for 6 days on lung water content (wet-to-dry weight ratio) in WT controls and CD73-KO mice (n = 11 per mouse strain). Values are means ± SE.
Detrimental effects of influenza infection on pulmonary compliance and resistance are not attenuated in CD73-KO mice.
Interstitial edema can result in a decrease in pulmonary compliance, which is a measurement of distensibility of the lung parenchyma. Static and dynamic lung compliance (Fig. 4, A and B) did not differ between uninfected WT and CD73-KO mice. Influenza A virus infection resulted in significant and comparable decreases in static and dynamic lung compliance at 6 dpi in both mouse strains.
Fig. 4.

Detrimental effects of influenza infection on pulmonary compliance and resistance are not attenuated in CD73-KO mice. A–D: effects of H1N1 influenza A/WSN/33 infection for 6 days on static lung compliance (CST, ml/cmH2O × 10), dynamic lung compliance (CDYN, ml/cmH2O × 10), baseline total lung resistance (RBASAL, cmH2O·s·ml−1), and maximal lung resistance following nebulization of 50 mg/ml methacholine (RMAX, cmH2O·s·ml−1). #P < 0.001 vs. WT at the same time point. Values are means ± SE (n = 15 per mouse strain).
Basal airway resistance did not differ between uninfected WT and CD73-KO mice (Fig. 4C). Basal airway resistance increased in both mouse strains at 6 dpi. However, basal airway resistance was significantly higher in CD73-KO mice than WT controls at this time point. Relative to uninfected animals, exposure to escalating doses of the muscarinic agonist methacholine resulted in a significant increase in maximal airway resistance at 6 dpi in WT mice, which is indicative of airway hyperresponsiveness to bronchoconstrictors (Fig. 4D). Airway hyperresponsiveness was not attenuated in infected CD73-KO mice.
Neutrophil counts and protein levels are reduced in BALF from influenza A virus-infected CD73-KO mice.
Infection with influenza A virus resulted in significant increases in BALF alveolar macrophage, neutrophil, and lymphocyte counts at 6 dpi in WT and CD73-KO mice (Fig. 5, A–C). Total BALF cell counts did not differ significantly between WT controls (7.1 ± 0.2 × 106/ml) and CD73-KO mice (6.5 ± 0.5 × 106/ml) at 6 dpi. Similarly, there was no difference in BALF alveolar macrophage and lymphocyte content between strains. However, BALF from CD73-KO mice contained significantly fewer neutrophils than BALF from WT controls at this time point. Finally, influenza A virus infection resulted in a 12-fold increase in BALF protein at 6 dpi in WT controls, but this increase was greatly attenuated in CD73-KO mice (Fig. 5D).
Fig. 5.

Neutrophil counts and protein levels are reduced in bronchoalveolar lavage fluid (BALF) from influenza A virus-infected CD73-KO mice. A–D: effects of H1N1 influenza A/WSN/33 infection for 6 days on BALF alveolar macrophages (AMs, n = 7–9 per mouse strain), BALF neutrophils (PMNs, n = 7–9 per mouse strain), BALF lymphocytes (Lymphs, n = 7–9 per mouse strain), and BALF protein (μg/ml, n = 13 and 6 for WT and CD73-KO mice, respectively). *P < 0.05; #P < 0.001 vs. WT mice. Values are means ± SE.
BALF cytokine and chemokine responses to influenza A virus infection are significantly altered in CD73-KO mice.
Prior to infection, no inflammatory mediators could be detected in BALF from WT or CD73-KO mice (data not shown). BALF from WT and CD73-KO mice contained comparable levels of the cytokines IFN-γ, IL-10, and TGF-β at 6 dpi (Table 1). Similarly, there were no differences in BALF CCL-2/monocyte chemotactic protein-1 and CCL-5/RANTES chemokine content between mouse strains at this time point. However, BALF from CD73-KO mice contained significantly higher levels of the cytokine IL-6 and the chemokine CXCL-10/IP-10 than BALF from WT controls at 6 dpi. In contrast, BALF neutrophil chemoattractant CXCL-1/KC content was significantly lower in CD73-KO mice than WT controls at this time point. Finally, BALF from CD73-KO mice contained significantly more IL-12 and less IL-1β and TNF-α than BALF from WT controls at 6 dpi, but levels of all three cytokines were very low, so these differences are unlikely to be of biological importance (data not shown).
Table 1.
BALF cytokine and chemokine responses to influenza A virus infection in CD73-KO and APCP-treated WT mice at 6 dpi
| IFN-γ | IL-6 | IL-10 | IP-10 | KC | MCP-1 | RANTES | TGF-β | |
|---|---|---|---|---|---|---|---|---|
| WT | 5,850 ± 600 | 480 ± 40 | 820 ± 70 | 2,920 ± 240 | 170 ± 20 | 2,320 ± 140 | 310 ± 30 | 620 ± 80 |
| CD73-KO | 6,220 ± 1,000 | 1,450 ± 190# | 600 ± 100 | 5,510 ± 550** | 60 ± 10# | 2,600 ± 70 | 250 ± 40 | 460 ± 100 |
| APCP-Tx | 6,880 ± 550 | 1,930 ± 310# | 520 ± 100* | ND | 120 ± 10* | ND | 300 ± 20 | 1,800 ± 250#¶ |
Values (means ± SE) are pg/ml. BALF, bronchoalveolar lavage fluid; CD73-KO, C57BL/6-congenic CD73-knockout mice (n = 5–7); WT, wild-type, untreated C57BL/6 mice (n = 6–10); APCP, adenosine 5′-(α,β-methylene) diphosphate; dpi, days postinfection; APCP-Tx, APCP-treated WT C57BL/6 mice (n = 4–5); IP-10, IFN-γ−induced protein 10; KC, keratinocyte chemoattractant; MCP-1, monocyte chemotactic protein 1; RANTES, regulated on activation, normal T cell expressed and secreted; TGF-β, transforming growth factor-β; ΝD, not done.
P < 0.05,
P < 0.005,
P < 0.001 vs. WT.
P < 0.001 vs. CD73-KO.
Importantly, BALF adenosine levels in CD73-KO mice (0.94 ± 0.1) did not differ from WT controls (0.89 ± 0.04) at 6 dpi (n = 3 per group). BALF adenosine levels in WT mice were consistent with our previous data (3).
Treatment of WT mice with the CD73 inhibitor APCP reduces influenza-induced hypoxemia and BALF leukocyte counts.
Treatment of uninfected WT mice with APCP by oral gavage for 6 days had no effect on body weight or carotid arterial O2 saturation at any treatment time point (data not shown). APCP gavage also did not alter the rate and extent of postinfection weight loss in influenza-infected WT mice (Fig. 6A). However, unlike the situation in infected, saline-treated WT mice and CD73-KO mice, hypoxemia was somewhat attenuated in APCP-treated infected WT animals at 6 dpi (Fig. 6B). In fact, carotid arterial saturation was significantly higher in APCP-treated WT mice than CD73-KO animals at 6 dpi (P < 0.005). Nevertheless, APCP treatment had no effect on severity of pulmonary edema at 6 dpi (Fig. 6C). Interestingly, treatment with APCP, but not saline, also resulted in significant reductions in BALF alveolar macrophages from WT animals at 6 dpi, which we did not observe in CD73-KO mice at this time point (Fig. 6D). Indeed, BALF alveolar macrophage counts were significantly lower in APCP-treated WT mice than CD73-KO animals at 6 dpi (P < 0.005). However, as in CD73-KO mice, APCP treatment of WT mice significantly reduced BALF neutrophil counts at 6 dpi. Consequently, total BALF cell counts were significantly lower in APCP-treated WT mice (4.2 ± 0.5 × 106/ml) than in untreated WT controls (7.1 ± 0.2 × 106/ml) and saline-treated WT mice (7.2 ± 1.1 × 106/ml) at 6 dpi (P < 0.005). However, BALF adenosine and protein content did not differ between untreated, saline-treated, and APCP-treated WT mice at 6 dpi (Fig. 6, E and F). Similarly, APCP treatment had no effect on lung homogenate viral titers at 6 dpi (data not shown).
Fig. 6.

Treatment of WT mice with the CD73 inhibitor adenosine 5'-(α,β-methylene) diphosphate (APCP) reduces influenza-induced hypoxemia and BALF leukocyte counts. A–F: effects of treatment of H1N1 influenza A/WSN/33-infected mice with 200 μl of saline or 200 μl of saline containing 20 mg/kg APCP by oral gavage (every other day) on body weight (%change from day 0, n > 15 per treatment group) and carotid arterial O2 saturation (n > 15 per treatment group) at 0–6 dpi and lung water content (wet-to-dry weight ratio, n = 7–9 per treatment group), BALF alveolar macrophages and neutrophils (AMs and PMNs, n = 5–9 per treatment group), BALF adenosine (n = 3–5 per treatment group), and BALF protein (n = 3–5 per treatment group) at 6 dpi. UNTx, untreated C57BL/6 mice; saline-Tx, saline-treated C57BL/6 mice; APCP-Tx, APCP-treated C57BL/6 mice. *P < 0.05 vs. UNTx. Values are means ± SE.
BALF cytokine and chemokine responses to influenza A virus infection are significantly altered in APCP-treated WT mice.
BALF cytokine and chemokine responses to influenza A virus infection in APCP-treated WT mice were generally comparable in magnitude to those in CD73-KO mice at 6 dpi (Table 1). Amounts of IFN-γ and RANTES in BALF from APCP-treated WT mice were comparable to those in untreated WT controls, but levels of IL-6 were significantly higher and CXCL-1/KC content was significantly lower in BALF from APCP-treated WT mice. However, unlike the situation in CD73-KO mice, APCP treatment also resulted in a significant reduction in BALF IL-10 and an increased amount of TGF-β relative to untreated WT controls at 6 dpi. In fact, BALF TGF-β, but not IL-10, was significantly higher in APCP-treated WT mice than CD73-KO mice at this time point. As in CD73-KO mice, BALF from APCP-treated WT mice contained low levels of IL-12, IL-1β, and TNF-α at 6 dpi (data not shown).
TNAP expression and activity are not higher in CD73-KO mouse lung.
Because there may be functional redundancy between CD73 and TNAPs, we measured whole lung homogenate TNAP expression and activity at 6 dpi. We found no differences in tnap gene expression (Fig. 7A), TNAP protein expression (Fig. 7, B and C), or TNAP enzymatic activity (Fig. 7D) between WT and CD73-KO mice at this time point.
Fig. 7.

Tissue-nonspecific alkaline phosphatase (TNAP) expression and activity is not higher in CD73-KO mouse lung. A–D: effects of H1N1 influenza A/WSN/33 infection for 6 days on whole lung homogenate tnap gene expression (fold change relative to uninfected mice), whole lung homogenate TNAP and GAPDH protein expression by Western blot analysis (no adjustments were made to original images other than conversion from color to gray scale), whole lung homogenate TNAP protein expression by densitometry of Western blot bands (normalized to GAPDH), and whole lung homogenate TNAP enzymatic activity (U/l). Values are means ± SE (n = 3 per group).
DISCUSSION
Extracellular nucleotides and nucleosides are important signaling molecules in the lung (21). Nucleotide and nucleoside concentrations in the alveolar lining fluid are controlled by a complex network of surface ectonucleotidases, and different cell types express different combinations of these enzymes (27). Previously, we observed that H1N1 influenza A virus induces rapid, nucleotide-mediated impairment of alveolar fluid clearance and showed that this effect was partially due to adenosine/A1-AdoR-mediated stimulation of CFTR-mediated Cl− secretion into the bronchoalveolar space (38). We also showed that influenza-induced ALI was highly attenuated in A1-AdoR-knockout mice, which indicates that ALI development is also adenosine-dependent (3). Because it has been proposed that AMP hydrolysis by CD73 is rate-limiting for generation of adenosine in the normal lung, we hypothesized that ALI would not develop, or at least be attenuated, in CD73-KO mice. To our surprise, we found that indexes of ALI severity were either unaltered or modestly increased in CD73-KO mice relative to WT controls. Similarly, pulmonary edema was not attenuated in WT mice treated with the CD73 inhibitor APCP, although hypoxemia was paradoxically somewhat less severe in these animals. Nevertheless, the magnitude and character of BALF cellular and humoral innate immune responses to infection differed quite markedly between CD73-KO mice, APCP-treated WT mice, and untreated WT controls. Overall, therefore, our results indicate that CD73 is not necessary for development of influenza-induced ALI. However, they also show that CD73 activity is required for development of a normal innate immune response following influenza A virus infection.
Our prior studies indicated that activation of A1-AdoRs by adenosine contributes substantially to development of ALI in influenza-infected mice. Hence, we were surprised to find that infection-induced bradycardia, viral replication, and bronchoconstriction were actually modestly increased in CD73-KO mice and APCP-treated WT animals. Moreover, neither cd73 gene deletion nor the CD73 inhibitor APCP reduced lung water content. Interestingly, however, while infection-induced hypoxemia was not attenuated in CD73-KO mice, APCP treatment attenuated hypoxemia by ∼25% in infected mice at 6 dpi. However, we found a similar disconnect between these two indexes of ALI severity in A1-AdoR-knockout mice: pulmonary edema was only modestly attenuated in infected A1-AdoR-knockout mice at 6 dpi, yet arterial O2 saturation was significantly increased relative to WT controls (3). We cannot provide a physiological explanation for the beneficial effect of APCP on arterial oxygenation. It does not appear to be related to differences in cardiovascular function between APCP-treated WT mice and CD73-KO animals, since infection-induced bradycardia was more severe in APCP-treated WT mice (data not shown). It is possible that attenuation of hypoxemia in APCP-treated WT mice reflects a fundamental difference between the effects of life-long CD73 knockout and short-term CD73 inhibition. There may be intrinsic compensatory mechanisms to maintain normal adenosine metabolism in CD73-KO mice that do not have time to develop following acute treatment with APCP. Nevertheless, our results emphasize that determination of ALI severity in influenza-infected mice cannot be based on measurements of a single outcome parameter, even if that parameter is of clinical relevance (25).
We found previously that influenza infection resulted in twofold increases in BALF ATP and adenosine levels at 6 dpi (2, 3). Our current data indicate that this cannot be ascribed to an increase in whole lung cd73 gene expression or CD73 enzymatic activity. If anything, cd73 gene expression was reduced and enzymatic activity was highly variable at this time point. We hypothesize that the absence of a direct correlation between cd73 mRNA and enzymatic activity may reflect either epigenetic regulation of CD73 protein expression or persistence of active CD73 protein in the cell even as gene expression begins to decline. Moreover, BALF adenosine content did not differ between WT and CD73-KO mice at 6 dpi. Similarly, we found that APCP treatment did not reduce BALF adenosine content in WT mice. Taken together, these findings would suggest that increased adenosine generation in the infected lung is CD73-independent. CD73 is described as being the rate-limiting enzyme for generation of adenosine in the lung at baseline (14). However, studies in polarized primary cultures of human bronchial epithelial cells indicate that TNAPs can compete with CD73 for the conversion of AMP to adenosine (28). Importantly, TNAPs and CD73 are expressed in different anatomic regions of the lung: the ectonucleotidases are prominent in upper airways, while TNAPs are more concentrated in distal airways and lung parenchyma (28). TNAPs have lower overall affinity for AMP than CD73. However, unlike CD73, they can also dephosphorylate ATP and ADP (6). It has been proposed that high-affinity, low-capacity metabolic enzymes such as CD73 regulate extracellular adenosine levels under normal conditions, while low-affinity, high-capacity enzymes such as TNAPs may play a more important role in situations of massive nucleotide release (27). We showed previously that influenza induces release of large amounts of ATP into the distal (bronchoalveolar) compartment of the lung (2), which is also the primary site of histopathological lesions in our infection model (1, 38). In the current study we found that TNAP was comparably expressed in the lungs of WT and CD73-KO mice at 6 dpi and that its enzymatic activity did not differ between strains. Taken together, our data suggest that there is functional redundancy between CD73 and TNAPs and that TNAP-mediated hydrolysis of ATP released by bronchoalveolar epithelial cells in response to influenza infection is more important for adenosine generation in the distal lung than is hydrolysis of AMP by CD73, which may be more significant in the upper airways. However, confirmation of this hypothesis would require cell type-specific knockout of CD73, and such experiments are beyond the scope of the current study.
Although ALI severity did not differ significantly between WT, CD73-KO, and APCP-treated WT mice, we did find some differences in the immune response to infection between these three experimental groups. Analysis of BALF indicated that the composition of the inflammatory leukocyte infiltrate in the infected lung was greatly affected by CD73 expression and activity. In particular, the neutrophil response to infection was attenuated in both CD73-KO and APCP-treated WT mice and to a similar extent. This effect was associated with comparable reductions in BALF levels of the neutrophil chemoattractant CXCL-1/KC in both experimental groups, which may itself result from adenosine-mediated inhibition of endothelial CXCL-1/KC production (5). However, our prior data indicate that activation of A1-AdoRs by adenosine may be more important than CXCL-1/KC for neutrophil recruitment to the lungs of influenza-infected mice. CD73-dependent generation of adenosine at the leading edge of migrating neutrophils has been shown to promote their forward movement (21). Hence, lack of CD73 expression or activity in neutrophils could directly impede their migration into the inflamed lung in influenza-infected mice, irrespective of BALF CXCL-1/KC content. On the other hand, earlier reports showed that accumulation of neutrophils in the lung in response to inhaled LPS is enhanced in CD73-KO mice, suggesting that the requirement for CD73 may be pathogen-specific (29).
As with BALF neutrophil content, we found differences in BALF alveolar macrophage counts and cytokine/chemokine profiles between WT, CD73-KO, and APCP-treated WT mice, as well as differences between the latter two groups. BALF from both CD73-KO and APCP-treated WT mice contained significantly more IL-6 than BALF from untreated WT animals. However, BALF from APCP-treated WT mice, but not CD73-KO animals, also contained significantly fewer alveolar macrophages, less IL-10, and more TGF-β. The underlying cause of these disparities remains unclear, but, as noted above, they may reflect a fundamental difference between the effects of life-long CD73 knockout and short-term CD73 inhibition on TNAP activity.
We found previously that development of pulmonary edema in H1N1 influenza A/WSN/33 virus-infected mice was accompanied by an early decrease in alveolar fluid clearance but that alveolar barrier function did not decline until later time points (38). In contrast, Gotts et al. (16) found that alveolar barrier breakdown preceded development of pulmonary edema in H1N1 influenza A/PR/8 virus-infected mice. One unexpected finding in the current study was that BALF protein was lower in CD73-KO mice than untreated WT controls, despite comparable pulmonary edema severity. This was not the case in APCP-treated WT mice. The lower BALF protein level in CD73-KO mice indicates that alveolar barrier integrity is better maintained in these mice than in APCP-treated WT animals. Tate et al. (31) showed that alveolar macrophages are barrier-protective in influenza, and we found a reduction in BALF macrophages in APCP-treated WT mice, but not in CD73-KO animals. Hence, there is a correlation between BALF protein content and BALF alveolar macrophage counts in our model. Therefore, we hypothesize that attenuation of barrier dysfunction in influenza-infected CD73-KO mice is due to the preservation of a normal alveolar macrophage response to infection in this strain.
Davies et al. (11) reported that exposure to hyperoxia resulted in lower BALF adenosine and greater pulmonary edema in CD73-KO mice than WT controls but did not alter leukocyte infiltration. These effects were attributed to loss of barrier-protective A2B-AdoR activation. Conversely, treatment with the A2B-AdoR agonist BAY 60-6583 increased alveolar fluid clearance and attenuated pulmonary edema in endotoxin-induced ALI (18). However, we did not find an effect of A2B-AdoR blockade on alveolar fluid clearance inhibition in influenza-infected mice (38). Moreover, we found no differences in BALF adenosine content or pulmonary edema severity between CD73-KO and WT mice following influenza infection, but we did see reduced BALF neutrophils. These results are strikingly similar to our previous findings in influenza-infected A1-AdoR-KO mice (3). We propose that, in our model, generation of adenosine by TNAP compensates for lack of CD73 activity in CD73-KO mice. However, Davies et al. did not measure TNAP expression or activity, and so it is possible that hyperoxia also causes TNAP downregulation or inhibition. In that case, there would be no compensatory mechanism for adenosine generation in the BALF in the absence of CD73.
In conclusion, we found that, despite being both ATP- and adenosine-dependent, development of ALI in influenza-infected mice is independent of conversion of ATP to adenosine by CD73 but may, instead, be TNAP-mediated. Taken together, the findings from this and our prior studies indicate that while strategies to inhibit de novo pyrimidine synthesis and/or A1-AdoR activation are likely to ameliorate influenza-induced ALI, the same cannot be said for CD73 inhibition (2, 3). However, the current study did reveal a previously unrecognized role for CD73 in shaping the innate immune response to influenza A virus infection.
GRANTS
This work was supported by the C. Glenn Barber Fund and National Heart, Lung, and Blood Institute Grant R01 HL-102469.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
F.A., P.S.W., and I.C.D. developed the concept and designed the research; F.A. and P.S.W. performed the experiments; F.A., P.S.W., and I.C.D. analyzed the data; F.A., P.S.W., and I.C.D. interpreted the results of the experiments; F.A. and I.C.D. prepared the figures; F.A., P.S.W., and I.C.D. edited and revised the manuscript; I.C.D. drafted the manuscript; I.C.D. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Lisa Joseph for excellent technical support.
Current affiliation of F. Aeffner: Flagship Biosciences Inc., Westminster, CO.
REFERENCES
- 1.Aeffner F, Abdulrahman B, Hickman-Davis JM, Janssen PM, Amer A, Bedwell DM, Sorscher EJ, Davis IC. Heterozygosity for the F508del mutation in the cystic fibrosis transmembrane conductance regulator anion channel attenuates influenza severity. J Infect Dis 208: 780–789, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aeffner F, Bratasz A, Flaño E, Powell KA, Davis IC. Post-infection A77-1726 treatment improves cardiopulmonary function in H1N1 influenza-infected mice. Am J Respir Cell Mol Biol 47: 543–551, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol 88: 10214–10227, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Askovich PS, Sanders CJ, Rosenberger CM, Diercks AH, Dash P, Navarro G, Vogel P, Doherty PC, Thomas PG, Aderem A. Differential host response, rather than early viral replication efficiency, correlates with pathogenicity caused by influenza viruses. PLos One 8: e74863, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouma M, van der Wildenburg F, Buurman W. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am J Physiol Cell Physiol 271: C522–C529, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Buchet R, Millán JL, Magne D. Multisystemic functions of alkaline phosphatases. In: Phosphatase Modulators, edited by Millán JL. New York: Humana, 2013, p. 27–51. [DOI] [PubMed] [Google Scholar]
- 7.Caruso M, Alamo A, Crisafulli E, Raciti C, Fisichella A, Polosa R. Adenosine signaling pathways as potential therapeutic targets in respiratory disease. Expert Opin Ther Targets 17: 761–772, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Chan KM, Delfert D, Junger KD. A direct colorimetric assay for Ca2+-stimulated ATPase activity. Anal Biochem 157: 375–380, 1986. [DOI] [PubMed] [Google Scholar]
- 9.Clark N, Lynch J. Influenza: epidemiology, clinical features, therapy, prevention. Semin Respir Crit Care Med 32: 373–392, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal 2: 351–360, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies J, Karmouty-Quintana H, Le TT, Chen N, Weng T, Luo F, Molina J, Moorthy B, Blackburn MR. Adenosine promotes vascular barrier function in hyperoxic lung injury. Physiol Rep 2: e12155, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis IC, Sullender WM, Hickman-Davis JM, Lindsey JR, Matalon S. Nucleotide-mediated inhibition of alveolar fluid clearance in BALB/c mice after respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol 286: L112–L120, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Davis IC, Xu A, Gao Z, Hickman-Davis JM, Factor P, Sullender WM, Matalon S. Respiratory syncytial virus induces insensitivity to β-adrenergic agonists in mouse lung epithelium in vivo. Am J Physiol Lung Cell Mol Physiol 293: L281–L289, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunwiddie TV, Diao L, Proctor WR. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci 17: 7673–7682, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. The role of interferon in influenza virus tissue tropism. J Virol 72: 8550–8558, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gotts JE, Abbott J, Matthay MA. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am J Physiol Lung Cell Mol Physiol 307: L395–L406, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamilton BE, Miniño AM, Martin JA, Kochanek KD, Strobino DM, Guyer B. Annual summary of vital statistics: 2005. Pediatrics 119: 345–360, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar epithelial A2B-adenosine receptors in pulmonary protection during acute lung injury. J Immunol 195: 1815–1824, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofer CC, Woods PS, Davis IC. Infection of mice with influenza A/WSN/33 (H1N1) virus alters alveolar type II cell phenotype. Am J Physiol Lung Cell Mol Physiol 308: L628–L638, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irvin CG, Bates JH. Measuring the lung function in the mouse: the challenge of size. Respir Res 4: 4–13, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 11: 201–212, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreindler JL, Shapiro SD. Lung turns to AA (adenosine analogues) to dry out. Nat Med 13: 406–408, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Kunzelmann K, Konig J, Sun J, Markovich D, King NJ, Karupiah G, Young JA, Cook DI. Acute effects of parainfluenza virus on epithelial electrolyte transport. J Biol Chem 279: 48760–48766, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Kunzelmann K, Sun J, Meanger J, King NJ, Cook DI. Inhibition of airway Na+ transport by respiratory syncytial virus. J Virol 81: 3714–3720, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM, Acute Lung Injury in Animals Study Group. An official American Thoracic Society Workshop Report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills JH, Thompson LF, Mueller C, Waickman AT, Jalkanen S, Niemela J, Airas L, Bynoe MS. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 105: 9325–9330, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picher M. Mechanisms regulating airway nucleotides. Subcell Biochem 55: 17–49, 2011. [DOI] [PubMed] [Google Scholar]
- 28.Picher M, Burch LH, Hirsh AJ, Spychala J, Boucher RC. Ecto 5'-nucleotidase and nonspecific alkaline phosphatase. Two AMP-hydrolyzing ectoenzymes with distinct roles in human airways. J Biol Chem 278: 13468–13479, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J 23: 473–482, 2009. [DOI] [PubMed] [Google Scholar]
- 30.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110: 993–1002, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tate MD, Pickett DL, Van Rooijen N, Brooks AG, Reading PC. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol 84: 7569–7580, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taubenberger JK, Morens DM. Influenza: the once and future pandemic. Public Health Rep 125 Suppl 3: 16–26, 2010. [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 200: 1395–1405, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong HH, Long JP, Li D, DeMaria TF. Alteration of gene expression in human middle ear epithelial cells induced by influenza A virus and its implication for the pathogenesis of otitis media. Microb Pathog 37: 193–204, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Traylor ZP, Aeffner F, Davis IC. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza Other Respir Viruses 7: 472–479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster RG, Govorkova EA. Continuing challenges in influenza. Ann NY Acad Sci 1323: 115–139, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wink MR, Lenz G, Braganhol E, Tamajusuku AS, Schwartsmann G, Sarkis JJ, Battastini AM. Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett 198: 211–218, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, Durbin JE, Davis IC. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med 178: 969–976, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem Mol Biol 49: 473–497, 2014. [DOI] [PubMed] [Google Scholar]
- 40.Zimmermann H, Zebisch M, Sträter N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8: 437–502, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]