


Abstract
Bacteria and archaea acquire resistance to foreign genetic elements by integrating fragments of foreign DNA into CRISPR (clustered regularly interspaced short palindromic repeats) loci. In Escherichia coli, CRISPR-derived RNAs (crRNAs) assemble with Cas proteins into a multi-subunit surveillance complex called Cascade (CRISPR-associated complex for antiviral defense). Cascade recognizes DNA targets via protein-mediated recognition of a protospacer adjacent motif and complementary base pairing between the crRNA spacer and the DNA target. Previously determined structures of Cascade showed that the crRNA is stretched along an oligomeric protein assembly, leading us to ask how crRNA length impacts the assembly and function of this complex. We found that extending the spacer portion of the crRNA resulted in larger Cascade complexes with altered stoichiometry and preserved in vitro binding affinity for target DNA. Longer spacers also preserved the in vivo ability of Cascade to repress target gene expression and to recruit the Cas3 endonuclease for target degradation. Finally, longer spacers exhibited enhanced silencing at particular target locations and were sensitive to mismatches within the extended region. These findings demonstrate the flexibility of the Type I-E CRISPR machinery and suggest that spacer length can be modified to fine-tune Cascade activity.
INTRODUCTION
CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) systems are widespread and diverse adaptive immune systems in bacteria and archaea (1,2). These systems identify and degrade foreign genetic material associated with plasmids and bacteriophages through three distinct stages termed acquisition, expression and interference (3–5). During acquisition, small pieces of foreign DNA are inserted as new spacers into the CRISPR array composed of alternating repeats and spacers. During expression, the CRISPR array is processed into mature CRISPR RNAs (crRNAs) each composed of a spacer and flanking portions of the repeat, yielding an RNA–protein complex composed of the crRNA and Cas effector proteins. During interference, the complex binds foreign genetic material complementary to the spacer portion of the crRNA, thereby activating cis or trans-acting nucleases that cleave the bound target.
Despite their common function in adaptive immunity, CRISPR-Cas systems are phylogenetically and functionally diverse, where the latest classifications define two classes, six types and nineteen subtypes (2,6). Class 1 systems rely on multi-subunit surveillance complexes while Class 2 systems rely on a single effector protein. The two main classes are subdivided into types and subtypes that further delineate the particular form and function of each system. The phylogenetic distribution of these systems vary widely, with the Class 1 Type I system as the most abundant and widespread in both bacteria and archaea (2). This type is defined by the signature cas3 gene encoding a protein with helicase and endonuclease activities (2,7–10).
Our current knowledge of Type I systems has largely stemmed from studies of the Type I-E system in Escherichia coli K-12. In E. coli, the Cas3 helicase-nuclease is recruited to DNA targets bound by a multi–subunit complex termed the CRISPR-associated complex for antiviral defense (Cascade) (9,11). Cascade is a 405 kDa complex composed of an uneven stoichiometry of five different Cas proteins (Cse11Cse22Cas76Cas51Cas6e1) and a 61-nt crRNA (12). This complex forms a seahorse-shaped architecture with subunits that represent the head (Cas6e), backbone (Cas7), belly (Cse2) and tail (Cse1, Cas5). Cascade engages foreign DNA by searching for a sequence called a protospacer-adjacent motif (PAM) (13,14). PAM recognition is thought to distort the DNA in a way that facilitates crRNA-guided interrogation of the flanking DNA sequence for complementarity with the crRNA spacer (9,12,14–18). Target binding triggers a conformational change that recruits Cas3, which degrades the non-target strand in the 3′-to-5′ direction (7,11,19).
The Type I-E system from E. coli has proven to be a versatile tool for programmable gene silencing and DNA destruction (20). Gene silencing relies on the capacity of Cascade to stably bind without cleaving target DNA in the absence of Cas3 (21,22). Directing Cascade to bind a promoter repressed expression of the downstream gene up to ∼1000-fold, whereas directing Cascade to the coding region led to more modest, strand-dependent silencing (21,22). Cascade has also been used in the presence of Cas3 to target and degrade the bacterial genome in a sequence-specific manner, spurring applications in sequence-specific antimicrobials, genome editing and biocontainment (23–25).
Studies to understand and exploit the Type I-E system from E. coli have relied on a 32-nt spacer. This length aligns with the extremely narrow range observed in naturally occurring CRISPR arrays across E. coli strains (26). Crystal structures of Cascade suggest that the RNA serves as a scaffold for the oligomerization of the six Cas7 proteins forming the backbone of the complex (15,16,27). Based on this insight, we speculated that we could control Cascade assembly by varying the length of the crRNA spacer and that these engineered complexes may represent a novel method for controlling target recognition. We found that extending the spacer resulted in larger Cascade complexes with altered protein subunit stoichiometry. Using a combination of in vitro binding studies with in vivo gene silencing and targeted killing assays, we demonstrate that these enlarged complexes preserve function in target binding, transcriptional repression and DNA degradation. We also found that longer spacers can enhance gene silencing depending on the targeted site. Finally, we showed that mismatches in the extended region disrupted DNA binding, gene silencing and targeted killing. These findings reveal that Cascade assembly is dependent on spacer length and that this length may be used as a method for fine-tuning the activity and specificity of Cascade.
MATERIALS AND METHODS
Strains and plasmid construction
See Supplementary Table S1 for a list of all E. coli K-12 strains used in this work. To generate BW25113 Δcas3::cat lacZ+, the lacZ gene from MG1655 was transferred by P1 transduction into BW25113 Δcas3::cat. Cells were selected on M9 agar plates containing chloramphenicol and 0.2% D-lactose as the sole carbon source. Next, the cat cassette from BW25113 Δcas3::cat lacZ+ was excised using the pCP20 plasmid as described previously (28) to generate BW25113 Δcas3 lacZ+.
See Supplementary Table S2 for a list of all plasmids used in this work. To generate cBAD33, the pBAD33 plasmid was digested with NsiI and SacI to remove araC through the araB promoter. Oligonucleotides encoding the J23108 promoter (pBAD33.J23108.fwd2/ pBAD33.J23108.rev2) were 5′ phosphorylated with T4 polynucleotide kinase and annealed by slowly cooling from 95 to 25°C. The resulting dsDNA was ligated with T4 DNA ligase into the digested pBAD33 and transformed into NovaBlue electrocompetent cells. The pCas3 plasmid was generated by PCR-amplifying the cas3 gene from E. coli MG1655 genomic DNA using primers to introduce a strong ribosome binding site, an upstream KpnI restriction site and a downstream XbaI restriction site (pCas3.for/pCas3.rev). The cas3 amplicon and cBad33 vector were digested with KpnI and XbaI, ligated together with T4 DNA ligase and transformed into NovaBlue electrocompetent cells.
The GFP reporter plasmids were based on the pUA66 plasmid (29). To generate pUA66lacZ-NT3PAM-mutant from pUA66lacZ, mutagenic primers (NT3-PAM.Q5.for/NT3-PAM.Q5.rev) were used with the Q5® Site-Directed Mutagenesis kit from New England Biolabs (NEB).
The crRNA expression plasmids, pcrRNA.ind and pcrRNA.con, were based on previous work (21). To insert new repeat-spacer pairs into pcrRNA.con or pcrRNA.ind, either plasmid first digested with KpnI and XhoI. Then, oligonucleotides encoding the palindromic repeat and crRNA spacers were annealed, 5′ phosphorylated with T4 polynucleotide kinase, ligated into the digested vector with T4 DNA ligase and transformed into TOP10 electrocompetent cells. All plasmid cloning was verified by Sanger sequencing. See Supplementary Table S3 for a list of all oligonucleotides used in this work. All oligonucleotides were chemically synthesized by Integrated DNA Technologies (IDT) or Eurofins Genomics. All enzymes were purchased from New England Biolabs (NEB).
Cascade expression and purification
Cascade and Cascade variants were expressed and purified using previously described methods (12,17). Briefly, crRNAs and E. coli K-12 Cas proteins were co-expressed in E. coli BL21 (DE3) cells from three different expression vectors: a pCDF vector containing Cse2 fused to an N-terminal Strep tag, Cas7, Cas5 and Cas6; a pRSF vector containing Cse1; and either pcrRNA.con or pcrRNA.ind vectors (described above) containing the CRISPR with a normal (32-nt) or extended spacer sequence (e.g. 32 +6, 32 +12). Cells were grown at 37°C in LB-media under antibiotic selection to an OD600nm of 0.5, then were induced with a final concentration of 0.2 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and 0.02% L-arabinose. After induction, cells were cultured overnight at 16°C, pelleted by centrifugation (5,000 g for 10 min), suspended in lysis buffer (100 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM tris(2-carboxyethyl)phosphine (TCEP) and 5% glycerol) and frozen at −80°C. Cells were lysed by sonication and lysates were clarified by centrifugation (22,000 g for 30 min). Cascade and Cascade variants self-assembled in vivo and were affinity purified on StrepTrap HP resin (GE) with the N-terminal Strep-II tag on Cse2. Cascade was eluted off StrepTrap HP resin with lysis buffer supplemented with 2.5 mM desthiobiotin. Cascade was concentrated and then further purified by gel filtration chromatography using a 26/60 Superdex 200 (GE Healthcare) equilibrated with 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM TCEP and 5% glycerol. The elution profile was visualized at 280 and 260 nm wavelengths to identify changes in complex size.
To identify size differences between Cascade variants, 600 μl of each complex was purified by gel filtration chromatography using a 16/60 Superdex 200 (GE Healthcare) equilibrated with 100 mM HEPES pH 7.0. Eluted complexes were concentrated, mixed with 5× loading buffer (100 mM HEPES pH 7.5, 375 mM NaCl, 50% glycerol) and loaded onto a 5% Blue Native Polyacrylamide gel and run at 85 V for 2 h (30). To analyze protein subunit abundance, 20 μg of each Cascade variant was loaded onto a 10% denaturing sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and run at 150 V for 45 min. To determine crRNA length, RNA was isolated by phenol/chloroform extraction and loaded onto a TBE (Tris Borate EDTA), 7M-Urea, 14% polyacrylamide gel and ran at 190 V for 30 min. Proteins were stained with Coomassie dye and RNA with SYBR Gold (Thermo Fisher Scientific).
SDS-PAGE gel densitometry and estimation of subunit stoichiometry
Coomassie stained SDS-PAGE gels were analyzed with GelQuant.Net (biochemlabsolutions.com). Band intensities were quantified with local background correction. Band intensity fractions were determined with the equation:
 |
Stoichiometry was estimated using the equation:
 |
Average stoichiometry and standard deviations were calculated from five different SDS-PAGE gels stained with Coomassie Blue.
Protein and native mass spectrometry
Cascade was buffer-exchanged into 100 mM ammonium acetate, pH 7 (Sigma) by repeated washing over 3 kDa molecular weight cutoff spin filters (Pall Corporation). A total of 2 μM Cascade was injected using gold-coated borosilicate glass capillaries and analyzed on a SYNAPT G2-Si electrospray time-of-flight instrument (Waters) in positive mode. The system was calibrated with sodium iodide dissolved in solution of 2-propanol and water (Waters). Gold-coated borosilicate glass capillaries were prepared as previously reported (31) with the following changes: Borosilicate glass capillaries, 1.2/0.68 OD/ID mm (World Precision Instruments) were pulled on model P-97 (Sutter Instrument Company) in one step with heat 540, pull 90, velocity 50 and time 150. Solutions were sprayed at a rate of 90 nl/min. Capillaries were coated with two layers (inner: Cr, 3 nm to increase coat resistance and prolong spray time; outer: Au, 60 nm to provide conductivity) using an AMod Evaporator System.
To assure optimal instrument performance in high mass-to-charge range, several adjustments to a previously published protocol (32) were made. To minimize complex dissociation and maximize ion transmission, source temperature was set to 30°C, capillary voltage to 1.7 kV and cone gas to 40 L/h. Sampling cone and source offset were set to 40 and 50 V, respectively. Inlet pressure was fixed at 3 mbar. To improve desolvation/declustering, trap bias voltage was adjusted to 16 V and argon pressure in the collision cell (trap) was 7 ml/min. Transfer collision energy was kept at constant level of 5 V while trap energy varied between 10–200 V. To determine accurate protein subunit masses, Cascade complexes were denatured by dilution in a 50:50 solution of 1% formic acid (Sigma) and acetonitrile. Collected spectra of Cascade complexes in native and denatured conditions were processed and analyzed in MassLynx software version 4.1 (Waters).
Electrophoretic mobility shift assays (EMSA)
Oligonucleotides (Operon) were 5′-end labeled with γ32P-ATP (PerkinElmer) using T4 polynucleotide kinase (NEB), and purified by phenol/chloroform extraction followed by MicroSpin G-25 column (GE Healthcare) filtration. Labeled oligonucleotides were hybridized with >5-fold molar excess of complementary strand in hybridization buffer (20mM HEPES pH 7.5, 75mM NaCl, 2mM EDTA, 10% glycerol and 0.01% bromophenol blue) by incubating at 95°C for 5 min and gradually cooling to 25°C in a thermocycler. DNA duplexes were gel purified, ethanol precipitated and recovered in hybridization buffer.
Varying concentrations of Cascade or Cascade variants were incubated with 32P labeled oligonucleotides in hybridization buffer with 1mM TCEP for 15 min at 37°C. Reactions were loaded onto a 6% native polyacrylamide gel, and run for 3 h at 150 V at 4°C. After electrophoresis, gels were dried, exposed to phosphor storage screens and scanned with a Typhoon (GE Healthcare) phosphorimager. Bound and unbound DNA fractions were quantified using ImageQuant software (GE Healthcare), and the fractions of bound oligonucleotides were plotted against total Cascade concentration. The data were fit by least-squares analysis using a standard binding isotherm:
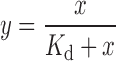 |
where, y is the fraction of bound DNA, x is the concentration of the Cascade complex and Kd is the apparent dissociation constant.
Growth conditions
All strains were cultured at 37°C and 250 RPM in up to 5 ml of LB medium (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl) or M9 minimal medium (1× M9 salts, 2 mM MgSO4, 0.1 mM CaCl2, 10 μg/ml thiamine hydrochloride) containing 0.4% glycerol and 0.2% casamino acids. All strains were plated on LB agar (LB medium with 1.2% agar) or on M9 agar with lactose (M9 medium with 1.2% agar and 0.2% D-lactose) in 100 × 15 mm polystyrene petri dishes. To maintain any plasmids, cells were cultured in liquid medium or on agar plates containing appropriate antibiotics at the following concentrations: 50 μg/ml of ampicillin, 34 μg/ml of chloramphenicol, 50 μg/ml of kanamycin.
Spacer design
See Supplementary Table S4 for a list of all of protospacers targeted in this work and Supplementary Table S5 for pUA66 promoter sequences. Protospacers were selected by identifying a PAM located at the 5′ end of the protospacer strand matching the spacer for the Type I-E system in E. coli (33). The AAG, AGG and AAC PAMs were used in this work. A non-functional ACG PAM shown previously to not support Cascade-based repression was used as a negative control (34). The 26–56 nts immediately downstream of the PAM were then used as the spacer. The cloning scheme required fixing the final 2 nts of the spacer to TC, as described previously (23).
Transformation assays
The transformation assay was conducted similar to previous work (23). Briefly, E. coli BW25113 Δcas3 lacZ+ harboring pCas3 were cultured overnight in LB medium. Cultures were back-diluted 1:250 into 25 ml of LB medium and grown to an ABS600 of 0.6–0.8 as quantified with a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific). The cells were then washed in ice-cold 10% glycerol and concentrated by a factor of ∼100. A total of 50 μl of the concentrated cells were transformed with 50 ng of plasmid DNA using a MicroPulser electroporator (Bio-Rad). Plasmids encoding the constitutively expressed spacers (pcrRNA.con-XX, where XX is replaced with the spacer name) were used for this assay. Transformed cells were recovered in 300 μl SOC medium for 1 h at 37°C. After the recovery period, the cells were diluted up to factors of 105 and 200 μl of the dilution were plated on LB agar with appropriate antibiotics.
Flow cytometry analysis
Flow cytometry was conducted similar to previous work (21). Briefly, cells grown overnight in M9 minimal medium with 0.2% casamino acids and 0.4% glycerol were back-diluted to an ABS600 of 0.01 into M9 minimal medium containing 0.2% casamino acids, 0.4% glycerol and appropriate inducers. For the BW25113 Δcas3::cat cells containing arabinose-inducible plasmids (pcrRNA.ind-XX, where XX is replaced with the spacer name) and pUA66-lacZ, 0.2% L-arabinose and 0.1 mM IPTG were added as inducers. For the MG1655 Δcas3::cat cells harboring constitutively-expressed pcrRNA.con-xylA-s1 with pUA66-xylA, the only inducer required was 0.2% D-xylose. Upon reaching an ABS600 of ∼0.2 after 3–4 h of growth, the cultures were diluted 1:100 in phosphate buffered saline and run on an Accuri C6 Flow Cytometer (Becton Dickinson) equipped with CFlow plate sampler, a 488-nm laser and a 530 ± 15 nm bandpass filter. Events reflecting cells were gated based on forward scatter (FSC-H) and side scatter (SSC-H) with respective lower cutoffs of 14 000 and 600 to reduce the measurement of particulates. The gate was set using E. coli cells stained with the DRAQ5 dye (Thermo Fisher Scientific). The fluorescence of the gated cells was then measured in FL1-H. At least 30,000 events were analyzed for each sample.
Statistical analyses
All statistical analyses were conducted using a one-tailed Student's t-test with unequal variance. Statistical analyses for the apparent dissociation constants (Kd) assumed that the variance is geometrically distributed, resulting in standard errors of measurement reported as fractions of the geometric mean.
RESULTS
Spacer length determines Cascade size and composition
Previously determined structures of Cascade revealed a seahorse-shaped complex composed of a Cas6e head, a backbone composed of six Cas7 subunits, a belly consisting of two Cse2 subunits and a tail composed of one Cse1 and one Cas5 (Figure 1A and Supplementary Figure S1A) (15–17,27). The crRNA is an integral component of this complex that is stretched from head to tail, where each of the Cas7 backbone subunits interacts with 6 nts of the crRNA spacer sequence. Furthermore, each Cse2 subunit makes direct contacts with two Cas7 subunits (35). Based on this structural information, we hypothesized that the composition and stoichiometry of Cascade might be affected by spacer length. We speculated that extending the spacer would result in an additional Cas7 subunit for every 6 nts of spacer extension and an additional Cse2 subunit for an extension of 12 nts (Figure 1A and Supplementary Figure S1A). To test this hypothesis, we purified Cascade expressed with a CRISPR array containing a naturally occurring spacer of 32 nts (+0) or a spacer extended to 38 (+6) or 44 (+12) nts. Analytical gel filtration and Blue native PAGE of Cascade complexes assembled using the extended crRNAs revealed increases in size that directly correlated with spacer length (Supplementary Figure S1B). Protein and RNA subunits of each complex were resolved using denaturing PAGE (Figure 1B). As expected, the larger complexes contained crRNAs that were ∼6 or ∼12 nts longer than the crRNA associated with wild-type Cascade. The extended complexes contained all five of the Cas protein subunits, where densitometry analysis of the denaturing PAGE gels suggested changes in protein subunit stoichiometry in line with our structural predictions (Supplementary Figure S1C).
Figure 1.

Escherichia coli Type I-E Cascades with extended spacers form stable complexes with altered stoichiometry. (A) Native mass spectra of Cascade complexes containing crRNA with spacers that are either 32 (+0), 38 (+6) or 44 nts (+12) in length and schematic representations of the corresponding Cascade complexes. Extending the crRNA spacer by 6 or 12 nts (light blue) results in complexes that are ∼41 kDa or ∼105 kDa larger than wild-type. The additional mass is consistent with the mass of the extended crRNA (light blue), Cas7 (orange) and in the case of the +12 complex an additional Cse2 subunit (purple). (B) SYBR-stained denaturing PAGE of crRNAs purified from Cascade complexes containing a 61-nt crRNA with a 32-nt spacer (+0), a 67-nt crRNA with a 38-nt spacer (+6), or a 73-nt crRNA with a 44-nt spacer (+12) (top) and SDS-PAGE stained with Coomassie blue (bottom). (C) Cascade complexes with a 32-nt spacer (+0, left) or a 44-nt spacer (+12, right) bind DNA targets with high affinity. Shown below each gel is the apparent dissociation constant (Kd), reported as the geometric mean and S.E.M. for three independent experiments. Experimental data and curve fits for the individual experiments are shown in Supplementary Figure S2.
To determine the subunit stoichiometry of the wild-type and engineered Cascade complexes, we performed native mass spectrometry to measure intact masses of the +0, +6 and +12 complexes (Figure 1A and Supplementary Table S6). The mass of the wild-type (+0) complex was 405,238.2 ± 45.8 Da, while the +6 and +12 complexes had masses of 446,655.1 ± 209.9 Da and 510,127.2 ± 48.9 Da, respectively. The +6 complex was ∼41 kDa larger than the wild-type (+0) complex, which is consistent with the addition of a Cas7 subunit (∼40 kDa) plus the mass of an additional 6 nts to the crRNA (∼1.6 kDa). The addition of 12 nts resulted in a complex ∼105 kDa larger than the wild-type complex, which matches the predicted addition of two Cas7 subunits (∼80 kDa), one Cse2 subunit (∼21 kDa) and 12 nts to the crRNA (∼3.5 kDa). Collectively, these results provide evidence that additional Cas7 subunits are incorporated into Cascade when the spacer is extended in 6-nt increments, and additional Cse2 subunits are incorporated in 12-nt increments.
To determine how spacer length impacts target binding, we measured the binding affinity of Cascade with a wild-type spacer (+0) or with a spacer extended by 12 nts (+12) to a dsDNA target. For two different spacers (s1 and p1), the +0 and +12 complexes bound target DNA with high affinity (Figure 1C and Supplementary Figure S2) (12,35). The apparent dissociation constant was marginally lower for the +12 complex around the level of significance (P = 0.062 for s1, P = 0.041 for p1, n = 3) (Supplementary Figure S2), suggesting that the spacer extension at least maintained affinity for the target.
Extended spacers permit transcriptional silencing and DNA interference
Our data demonstrated that Cascade accommodates extended spacers in vitro, although it remained unclear how longer spacers would impact Cascade activity in vivo. To determine the in vivo effect of elongated spacers on Cascade function, we used a previously developed gene repression assay (21,22). This system relies on an E. coli strain (BW25113 Δcas3::cat) in which cas3 was deleted and a constitutive promoter was introduced upstream of the cse1-cse2-cas7-cas5-cas6e operon (21). We transformed this strain with an L-arabinose-inducible plasmid encoding a designed CRISPR array and the pUA66-lacZ reporter plasmid encoding the green fluorescent protein (gfp) gene downstream of the lacZ promoter (Figure 2A). The spacers were designed to be 32 nts (+0) or 44 nts (+12) and target locations within the promoter or either strand of the transcribed region. For all sites targeted, the +12 spacers exhibited similar or improved silencing of GFP compared to the +0 spacers, indicating that extended Cascade complexes are capable of repressing expression of a target gene (Figure 2B). We observed the strongest silencing when targeting the promoter, intermediate silencing when targeting the sense strand of the transcribed region and weak silencing when targeting the antisense strand of the transcribed region (Figure 2C) in line with previous work (21).
Figure 2.

Extended Cascade assemblies repress gene expression and impair transformation efficiency in vivo. (A) Cascade complexes were designed to target sequences in the pUA66-lacZ reporter. Blue bars above or below the reporter represent spacers that match the top and bottom strands, respectively. Locations include the promoter (p) as well as the sense (s) and antisense (as) strand of lacZ. Black dots represent PAMs. Protospacer and PAM sequences can be found in Supplementary Table S4. (B) A schematic illustrating how Cascade binding may block gene transcription. (C) Impact of extending the spacer by 0 (dark blue) or 12 (light blue) nts on GFP silencing by Cascade. Fold-repression is calculated in comparison to a non-targeting control. (D) A schematic illustrating how the addition of Cas3 results in autoimmunity and reduced colonies after transformation of the plasmid containing Cas3. (E) Impact of extending the spacer by 0 (dark blue) or 12 (light blue) nts on DNA interference by Cascade and Cas3 is indicated (bottom). The transformation fold-reduction is calculated in comparison to a spacer-free control. The as2 and s2 spacers were not tested because the associated protospacers were not present in the genomic lacZ locus. Values represent the mean and S.E.M. of at least three measurements starting from independent colonies.
We next asked how extended crRNAs impact the ability of Cascade to recruit Cas3 and elicit targeted DNA degradation (Figure 2D). Hybridization of the crRNA spacer sequence to the complementary strand of a dsDNA target forms a displaced R-loop that is critical for Cas3 recruitment and DNA target degradation (12,36). To measure Cas3 activity, we constitutively expressed cas3 from a plasmid and measured the transformation efficiency of plasmids encoding genome-targeting crRNAs with 32-nt (+0) or 44-nt (+12) spacers. Previous work has shown that CRISPR-mediated genome targeting is lethal, resulting in greatly reduced transformation efficiencies in comparison to a non-targeting control (23,37). We used the lacZ-targeting spacers from the transcriptional silencing assay (p1, p2, as1 and s1), which required restoring the genomic lacZ locus by P1 transduction to generate BW25113 Δcas3 lacZ+. The transformation assays showed that all spacers resulted in ∼103–104 reduction in transformation efficiency when compared to a spacer-free control (Figure 2E). Therefore, extended Cascade complexes can recruit and activate Cas3 for target destruction, demonstrating that extended Cascade complexes can direct CRISPR-mediated interference.
Extended spacers can improve Cascade-mediated silencing
Extending the spacer markedly improved Cascade-mediated silencing for the s1 spacer complementary to the sense strand upstream of the ribosomal binding site of gfp (from 3- to 20-fold) (Figure 2C). To determine the relationship between the length of s1 and silencing activity, we designed spacers modified by multiples of six based on the interval of Cas7 binding (Figure 3A and Supplementary Figure S1A) (15). Silencing generally improved with each additional 6 nts at the s1 site (Figure 3B), while no trend was observed when extending a promoter-targeting spacer (p1) or an antisense-targeting spacer (as2) (Supplementary Figure S3). Extended spacers that deviated from multiples of six (i.e. +8, +10) also showed improvement in silencing efficiency compared to wild-type, whereas spacers shortened by six nucleotides lost silencing activity (Figure 3B).
Figure 3.

Spacer sequence extension can improve Cascade-mediated gene silencing. (A) Target locations within close proximity to the s1 spacer. The 32-nt spacer (dark blue) was extended by lengths divisible by six (light blue) or not divisible by six (purple). The 32-nt spacer for s1 was also shortened by 6 nts (dark blue). Black bars represent the PAM. Sequences match the bottom strand of the reporter construct. (B) Gene silencing resulting from Cascade complexes with spacers s1, s3 and s4 of varying sizes. The gray line indicates a fold-repression value of 1 reflecting no change in GFP fluorescence. (C) Targeting the sense strand of the untranslated region of the xylA promoter. The black dot indicates the PAM. To evaluate GFP silencing, the pUA66-xylA reporter construct was transformed into MG1655Δcas3::cat along with a plasmid encoding a constitutively expressed CRISPR array containing the xylA-s1 spacer. (D) Impact of extending the xylA-targeting spacer by +0 nts (dark blue) or +12 (light blue) nts on GFP silencing by Cascade. Fold-repression is calculated in comparison to a non-targeting control. (E) Evaluating the basis of spacer length-dependent silencing with Cascade. Testing a non-functional PAM required mutating the PAM of the s1 protospacer in the reporter construct. (F) Potential RNA-based antisense regulation was evaluated in the absence (No Cascade) or presence (Non-functional PAM) of Cascade. Values represent the mean and S.E.M. of at least three measurements starting from independent colonies.
Interestingly, enhanced silencing appears to be related to target location, as two spacers (s3 and s4) with targets near s1 improved silencing when extended (Figure 3B). This phenomenon was not limited to the lacZ promoter, as we observed enhanced silencing when targeting a similar location within the xylA promoter (Figure 3C and D), demonstrating that elongated Cascade complexes can be utilized to control silencing in different genetic contexts. To our knowledge, this serves as the first instance of quantitatively enhancing CRISPR-mediated silencing by increasing spacer length (38).
Enhanced silencing was only associated with spacers targeting the sense strand upstream of the coding region. This orientation could allow the crRNA to base pair with the transcribed gfp mRNA, potentially resulting in mRNA destabilization and translational inhibition; accordingly, longer crRNAs may be more efficient at base pairing for antisense regulation. To test whether the CRISPR array acts through antisense regulation (i.e. crRNA-guided binding of complementary mRNA), we assessed GFP silencing in the absence of the Cascade proteins. However, GFP silencing by the +0 and the +12 spacers was negligible (Figure 3F). To test whether enhanced silencing could be attributed to Cascade binding the gfp mRNA, we mutated the PAM to disrupt dsDNA binding but preserve PAM-independent RNA binding (Figure 3E) (17,39). Again, the +0 and +12 spacers yielded negligible GFP silencing (Figure 3F). Therefore, antisense regulation by the free crRNA or Cascade-bound crRNA cannot explain enhanced silencing with longer spacers.
Mismatches between the DNA target and the extended spacer impair Cascade activity
We finally asked whether base pairing through the extended region of the longer crRNAs is important for Cascade binding and activity. One possibility is that a longer crRNA with a mutated extended region would still be capable of base pairing through the first 32 nts of the natural spacer. However, recent work has shown that base pairing at the 3′ end of a normal spacer contributes to Cascade binding (36,40–42). To initially explore the importance of the extended spacer region, we measured the in vitro binding affinity of purified Cascade complexes in which the extended region of the p1 +12 spacer was mutated to disrupt base-pairing potential with the target (Supplementary Figure S4). Mutations that perturb base pairing in the extended region of the crRNA significantly lowered the apparent dissociation constant (P = 0.004, n = 3) (Figure 4A and Supplementary Figure S2), indicating that the extended region is important for Cascade to bind DNA in vitro. These mutations also required higher concentrations of Cascade to achieve saturated binding of the DNA target (Figure 4A and Supplementary Figure S2), suggesting that higher Cascade concentrations interfered with target binding.
Figure 4.

Mismatches between spacer extensions and DNA targets impair Cascade activity. (A) Schematics of Cascade complexes are depicted (top) with regular spacer length colored dark blue, complementary extensions colored in light blue and mismatching extensions in red. Representative gel images from electrophoretic mobility shift assays for each of the depicted complexes are shown. Shown below each gel is the apparent dissociation constant (Kd), reported as the geometric mean and S.E.M. for three independent experiments. Experimental data and curve fits for the individual experiments are shown in Supplementary Figure S2. (B) Impact of mutating the 3′ end of the spacer on gene silencing (left) and DNA interference (right) for the extended spacer variant (top) and the wild type length (bottom). The number in parentheses indicates the number of mutations made to the 3′ end of the s1 +0 or s1 +12 spacer. See Supplementary Figure S5 for the sequence of each mutated spacer. (C) Silencing efficiency plotted by the mismatch percentage of the total spacer length for the p1 spacer. Dark blue circles refer to the +0 spacer. Light blue circles refer to the +12 spacer. Values represent the mean and S.E.M. of at least three measurements starting from independent colonies.
To determine how mutations in the extended region impact transcriptional repression by Cascade, we tested different numbers of mismatches introduced at the 3′ end of the +12 spacer (Supplementary Figure S4). As a basis of comparison, we made similar mutations to the +0 spacer. The p1 spacer targeting the lacZ promoter was selected because the +0 and +12 versions of the spacer yielded similar levels of gene repression (Figure 2C). We found that gene silencing for either spacer was reduced with each additional mutation (Figure 4B). While the +0 spacer was more susceptible to the number of mutations, the susceptibilities were similar when accounting for mutations as a fraction of the total spacer length (Figure 4C). We also observed similar susceptibilities for the +12 variant of the s1 spacer (Supplementary Figure S5), suggesting that the mutational susceptibilities applied to different protospacer sequences. These results demonstrate that the extended portion of the spacer is critical for Cascade-mediated gene repression in vivo.
To assess how mutations in the extended spacer impact DNA interference, we tested the +0 and +12 variants of the p1 spacer with the same set of mutations at the 3′ end in the presence of Cas3 expression (Figure 4B). We found that more mismatches at the 3′ end led to more transformants, paralleling the trend observed with the transcriptional silencing assay. However, the +12 spacer maintained the lower transformation efficiency for up to six mismatches while the +0 spacer showed gradually improved transformation efficiencies with additional mismatches. Thus, extended and regular spacers exhibit different propensities for DNA interference and escape based on the number of mismatches at the 3′ end.
DISCUSSION
We found that extending the crRNA spacer altered the size and composition of the I-E Cascade complex from E. coli. Our in vitro data indicated that Cascade incorporates an extra Cas7 subunit with every 6 nts added to the crRNA spacer and an extra Cse2 subunit with every 12 nts added to the spacer. Furthermore, our in vivo repression data demonstrated that the altered complex maintained stable DNA binding and Cas3-mediated DNA degradation, where both functions required base pairing through the extended region of the spacer. Given that these functions require the wild-type complex to undergo large conformational rearrangements (16,27,35), our results suggest that the additional subunits still permit the necessary conformational changes to stably bind DNA and recruit Cas3 for target degradation.
We also found that longer spacers exhibited enhanced silencing when targeting the DNA sense strand between the promoter and the coding regions of lacZ and xylA. This trend suggests that improved silencing with extended spacers may be a general phenomenon. We were able to eliminate antisense interactions as possible explanations, though pinpointing the responsible mechanism will require further studies. Nevertheless, our results offer the possibility of tuning the extent of silencing simply by adjusting the number of nucleotides in the spacer. While we extended the spacer by up to 80% of its wild-type length (+24 nts), the Type I-E Cascade may be able to accommodate even longer spacers.
Despite the ability of the Type I-E Cascade to tolerate extended spacers, the naturally occurring spacer lengths for Type I-E systems are largely fixed (26). The natural selection of 32-nt spacers may reflect the mechanism of protospacer acquisition, which consistently integrates spacers of defined length. Accordingly, overexpression of the universal Cas1 and Cas2 acquisition proteins in E. coli leads to the incorporation of ∼32-nt spacers into the CRISPR locus (43–46).
The unique relationship between spacer length and complex size may be applicable across Class 1 systems based on the general assembly of their effector complexes (47). Evidence for this comes from Type III systems, which naturally generate different crRNA lengths and complex sizes. For example, the Type III-A system of Streptococcus thermophilus processes crRNAs to two distinct lengths where the longer crRNA results in a larger Csm effector complex (48). Similarly, the Type III-B systems from Thermus thermophilus and Pyrococcus furiosus have been shown to bind crRNAs of multiple lengths that differ by 6 nts (49–51). These insights from multiple species and CRISPR-Cas subtypes suggest that the assemblies of multi-subunit Class 1 complexes are templated by the length of the crRNA. In contrast, the single protein effectors of Class 2 systems appear to maintain a crRNA spacer that is not amenable to extension (52–57).
In summary, our work provides further insight into the flexibility and extent to which crRNAs from Type I-E CRISPR-Cas systems can be modified. The length of Type I-E crRNAs is not fixed and can be substantially extended, altering Cascade stoichiometry while maintaining in vivo functionality. These observations present additional opportunities to explore how other Class 1 systems respond to alterations to the crRNAs. Lastly, we uncovered a unique situation that offers the possibility of designing tunable transcriptional regulators whose design is dependent solely on the length of the crRNA. These insights are expected to inform our understanding of CRISPR biology and how these adaptive immune systems can be altered and improved toward applications in medicine and biotechnology.
Supplementary Material
Acknowledgments
We thank Rodolphe Barrangou for critical discussions and Charlotte Uetrecht for technical insights that were critical for measuring the intact masses of Cascade.
Authors’ contributions: M.L.L and C.L.B. conceived the project. R.N.J. and W.L. performed the in vitro experiments. M.T-L and B.B. performed the mass spectrometry. M.L.L., S.R.D. and K.R.M. performed the in vivo work. M.L.L., R.N.J., B.W. and C.L.B. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [CBET-1403135 to C.L.B., IIP-1549648 to C.L.B., EPS-110134 to B.W.]; National Institutes of Health [F32GM108436 to R.N.J., P20GM103500 to B.W., R01GM108888 to B.W., 5P20RR02437 to B.B.]; Department of Energy [DE-SC0012518 to B.B.]; Kenan Institute of Engineering, Technology, and Science (to C.L.B.); M.J. Murdock Charitable Trust (to B.W., B.B.); Amgen young investigator award (to B.W.); Montana State University Agricultural Experimental Station (to B.W.). Funding for open access charge: NSF [CBET-1403135].
Conflict of interest statement. C.L.B. is a co-founder of Locus Biosciences and serves on its scientific advisory board.
REFERENCES
- 1.Van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H., et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Oost J., Jore M.M., Westra E.R., Lundgren M., Brouns S.J.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Bhaya D., Davison M., Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 5.Wiedenheft B., Sternberg S.H., Doudna J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 6.Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K., et al. Discovery and functional characterization of diverse Class 2 CRISPR-Cas Systems. Mol. Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinkunas T., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011;30:1335–1342. doi: 10.1038/emboj.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huo Y., Nam K.H., Ding F., Lee H., Wu L., Xiao Y., Farchione M.D., Zhou S., Rajashankar K., Kurinov I., et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014;21:771–777. doi: 10.1038/nsmb.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westra E.R., van Erp P.B.G., Künne T., Wong S.P., Staals R.H.J., Seegers C.L.C., Bollen S., Jore M.M., Semenova E., Severinov K., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson R.N., Lavin M., Carter J., Wiedenheft B. Fitting CRISPR-associated Cas3 into the helicase family tree. Curr. Opin. Struct. Biol. 2014;24:106–114. doi: 10.1016/j.sbi.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulepati S., Bailey S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J. Biol. Chem. 2013;288:22184–22192. doi: 10.1074/jbc.M113.472233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jore M.M., Lundgren M., van Duijn E., Bultema J.B., Westra E.R., Waghmare S.P., Wiedenheft B., Pul U., Wurm R., Wagner R., et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 13.Redding S., Sternberg S.H., Marshall M., Gibb B., Bhat P., Guegler C.K., Wiedenheft B., Doudna J.A., Greene E.C. Surveillance and processing of foreign DNA by the Escherichia coli CRISPR-Cas system. Cell. 2015;163:854–865. doi: 10.1016/j.cell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes R.P., Xiao Y., Ding F., van Erp P.B.G., Rajashankar K., Bailey S., Wiedenheft B., Ke A. Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature. 2016;530:499–503. doi: 10.1038/nature16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson R.N., Golden S.M., van Erp P.B.G., Carter J., Westra E.R., Brouns S.J.J., van der Oost J., Terwilliger T.C., Read R.J., Wiedenheft B. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science. 2014;345:1473–1479. doi: 10.1126/science.1256328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao H., Sheng G., Wang J., Wang M., Bunkoczi G., Gong W., Wei Z., Wang Y. Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature. 2014;515:147–150. doi: 10.1038/nature13733. [DOI] [PubMed] [Google Scholar]
- 17.Wiedenheft B., Lander G.C., Zhou K., Jore M.M., Brouns S.J.J., van der Oost J., Doudna J.A., Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinkunas T., Gasiunas G., Waghmare S.P., Dickman M.J., Barrangou R., Horvath P., Siksnys V. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013;32:385–394. doi: 10.1038/emboj.2012.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo M.L., Leenay R.T., Beisel C.L. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol. Bioeng. 2016;113:930–943. doi: 10.1002/bit.25851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo M.L., Mullis A.S., Leenay R.T., Beisel C.L. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2015;43:674–681. doi: 10.1093/nar/gku971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rath D., Amlinger L., Hoekzema M., Devulapally P.R., Lundgren M. Efficient programmable gene silencing by Cascade. Nucleic Acids Res. 2015;43:237–246. doi: 10.1093/nar/gku1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomaa A.A., Klumpe H.E., Luo M.L., Selle K., Barrangou R., Beisel C.L. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. MBio. 2014;5 doi: 10.1128/mBio.00928-13. doi:10.1128/mBio.00928-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caliando B.J., Voigt C.A. Targeted DNA degradation using a CRISPR device stably carried in the host genome. Nat. Commun. 2015;6:6989. doi: 10.1038/ncomms7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Pan S., Zhang Y., Ren M., Feng M., Peng N., Chen L., Liang Y.X., She Q. Harnessing Type I and Type III CRISPR-Cas systems for genome editing. Nucleic Acids Res. 2016;44:e34. doi: 10.1093/nar/gkv1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Díez-Villaseñor C., Almendros C., García-Martínez J., Mojica F.J.M. Diversity of CRISPR loci in Escherichia coli. Microbiology. 2010;156:1351–1361. doi: 10.1099/mic.0.036046-0. [DOI] [PubMed] [Google Scholar]
- 27.Mulepati S., Héroux A., Bailey S. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014;345:1479–1484. doi: 10.1126/science.1256996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cherepanov P.P., Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 29.Zaslaver A., Bren A., Ronen M., Itzkovitz S., Kikoin I., Shavit S., Liebermeister W., Surette M.G., Alon U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 30.Wittig I., Braun H.-P., Schägger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 31.Kirshenbaum N., Michaelevski I., Sharon M. Analyzing large protein complexes by structural mass spectrometry. J. Vis. Exp. 2010;40:e1954. doi: 10.3791/1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michaelevski I., Kirshenbaum N., Sharon M. T-wave ion mobility-mass spectrometry: basic experimental procedures for protein complex analysis. J. Vis. Exp. 2010;41:e1985. doi: 10.3791/1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westra E.R., Semenova E., Datsenko K.A., Jackson R.N., Wiedenheft B., Severinov K., Brouns S.J.J. Type I-E CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013;9:e1003742. doi: 10.1371/journal.pgen.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leenay R.T., Maksimchuk K.R., Slotkowski R.A., Agrawal R.N., Gomaa A.A., Briner A.E., Barrangou R., Beisel C.L. Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol. Cell. 2016;62:137–147. doi: 10.1016/j.molcel.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Erp P.B.G., Jackson R.N., Carter J., Golden S.M., Bailey S., Wiedenheft B. Mechanism of CRISPR-RNA guided recognition of DNA targets in Escherichia coli. Nucleic Acids Res. 2015;43:8381–8391. doi: 10.1093/nar/gkv793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rutkauskas M., Sinkunas T., Songailiene I., Tikhomirova M.S., Siksnys V., Seidel R. Directional R-Loop formation by the CRISPR-Cas surveillance complex Cascade provides efficient off-target site rejection. Cell Rep. 2015;10:1534–1543. doi: 10.1016/j.celrep.2015.01.067. [DOI] [PubMed] [Google Scholar]
- 37.Vercoe R.B., Chang J.T., Dy R.L., Taylor C., Gristwood T., Clulow J.S., Richter C., Przybilski R., Pitman A.R., Fineran P.C. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 2013;9:e1003454. doi: 10.1371/journal.pgen.1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mougiakos I., Bosma E.F., de Vos W.M., van Kranenburg R., van der Oost J. Next generation prokaryotic engineering: the CRISPR-Cas toolkit. Trends Biotechnol. 2016 doi: 10.1016/j.tibtech.2016.02.004. doi:10.1016/j.tibtech.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Sashital D.G., Wiedenheft B., Doudna J.A. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell. 2012;46:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blosser T.R., Loeff L., Westra E.R., Vlot M., Künne T., Sobota M., Dekker C., Brouns S.J.J., Joo C. Two distinct DNA binding modes guide dual roles of a CRISPR-Cas protein complex. Mol. Cell. 2015;58:60–70. doi: 10.1016/j.molcel.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szczelkun M.D., Tikhomirova M.S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., Seidel R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. U.S.A. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fineran P.C., Gerritzen M.J.H., Suárez-Diez M., Künne T., Boekhorst J., van Hijum S.A.F.T., Staals R.H.J., Brouns S.J.J. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E1629–E1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yosef I., Goren M.G., Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arslan Z., Hermanns V., Wurm R., Wagner R., Pul Ü. Detection and characterization of spacer integration intermediates in type I-E CRISPR-Cas system. Nucleic Acids Res. 2014;42:7884–7893. doi: 10.1093/nar/gku510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nuñez J.K., Lee A.S.Y., Engelman A., Doudna J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–198. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nuñez J.K., Kranzusch P.J., Noeske J., Wright A. V, Davies C.W., Doudna J.A. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson R.N., Wiedenheft B. A conserved structural chassis for mounting versatile CRISPR RNA-guided immune responses. Mol. Cell. 2015;58:722–728. doi: 10.1016/j.molcel.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas C., Nwokeoji A.O., Dickman M.J., Horvath P., Siksnys V. Programmable RNA shredding by the Type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol. Cell. 2014;56:506–517. doi: 10.1016/j.molcel.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 49.Staals R.H.J., Agari Y., Maki-Yonekura S., Zhu Y., Taylor D.W., van Duijn E., Barendregt A., Vlot M., Koehorst J.J., Sakamoto K., et al. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of Thermus thermophilus. Mol. Cell. 2013;52:135–145. doi: 10.1016/j.molcel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hale C., Kleppe K., Terns R.M., Terns M.P. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA. 2008;14:2572–2579. doi: 10.1261/rna.1246808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ran F.A., Hsu P.D., Lin C.-Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shechner D.M., Hacisuleyman E., Younger S.T., Rinn J.L. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods. 2015;12:664–670. doi: 10.1038/nmeth.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dahlman J.E., Abudayyeh O.O., Joung J., Gootenberg J.S., Zhang F., Konermann S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015;33:1159–1161. doi: 10.1038/nbt.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zalatan J.G., Lee M.E., Almeida R., Gilbert L.A., Whitehead E.H., La Russa M., Tsai J.C., Weissman J.S., Dueber J.E., Qi L.S., et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.