

Here, Molleston et al. find that signals from viral infections repurpose TRAMP complex components to a cytoplasmic surveillance role where they selectively engage viral RNAs for degradation to restrict a broad range of viruses.
Keywords: RNA degradation, TRAMP, exosome, antiviral, arbovirus, intrinsic immunity
Abstract
RNA degradation is tightly regulated to selectively target aberrant RNAs, including viral RNA, but this regulation is incompletely understood. Through RNAi screening in Drosophila cells, we identified the 3′-to-5′ RNA exosome and two components of the exosome cofactor TRAMP (Trf4/5–Air1/2–Mtr4 polyadenylation) complex, dMtr4 and dZcchc7, as antiviral against a panel of RNA viruses. We extended our studies to human orthologs and found that the exosome as well as TRAMP components hMTR4 and hZCCHC7 are antiviral. While hMTR4 and hZCCHC7 are normally nuclear, infection by cytoplasmic RNA viruses induces their export, forming a cytoplasmic complex that specifically recognizes and induces degradation of viral mRNAs. Furthermore, the 3′ untranslated region (UTR) of bunyaviral mRNA is sufficient to confer virus-induced exosomal degradation. Altogether, our results reveal that signals from viral infection repurpose TRAMP components to a cytoplasmic surveillance role where they selectively engage viral RNAs for degradation to restrict a broad range of viruses.
RNA decay is tightly regulated to ensure cellular homeostasis. This requires specific recognition and targeting by RNA exonucleases (Garneau et al. 2007). The 5′ RNA degradation machinery uses decapping enzymes to remove the 5′ cap, allowing 5′-to-3′ exonucleases to degrade target RNA (Hsu and Stevens 1993; Muhlrad et al. 1994). Degradation from the 3′ end is largely mediated by the RNA exosome, a highly conserved multisubunit complex generally consisting of nine core factors that form a barrel structure (Schneider and Tollervey 2013). RNAs are inserted into this barrel and subsequently degraded by the two associated 3′-to-5′ exonucleases Rrp6 and Dis3. Exosomal degradation has roles in normal RNA biogenesis and turnover as well as surveillance of aberrant RNAs, including misfolded or mismodified tRNAs and mRNAs with defective polyadenylation (Allmang et al. 1999; Kadaba 2004; Milligan et al. 2005). The exosome has also been implicated in regulation of RNA regulons, functionally related sets of mRNAs that are tightly coregulated through common sequences in their 3′ untranslated regions (UTRs) (Chen et al. 2001; Mukherjee et al. 2002; Keene 2007; Singer et al. 2012; Blackinton and Keene 2014).
Specificity for exosomal degradation is provided by RNA-binding cofactor complexes anchored by DExD/H-box helicases that directly associate with the target RNA and the exosome, inserting these RNAs for decay (Houseley and Tollervey 2009). The best-characterized cofactors are the yeast TRAMP (Trf4/5–Air1/2–Mtr4 polyadenylation) and Ski (Superkiller) complexes, which have known roles in the nucleus and cytoplasm, respectively (Brown et al. 2000; LaCava et al. 2005; Vanácová et al. 2005; Wyers et al. 2005). In yeast, the TRAMP complex facilitates nuclear surveillance of improperly processed mRNAs, rRNAs, snRNAs, snoRNAs, and tRNAs, while the Ski complex is involved in mRNA turnover, nonsense-mediated decay, and nonstop decay (Anderson and Parker 1998; Kadaba 2004; Milligan et al. 2005; Wyers et al. 2005; Houseley et al. 2006). In human cells, the nuclear surveillance roles are only beginning to be elucidated but appear to be further subdivided into two Mtr4-anchored complexes: Human TRAMP is restricted to the nucleolus and engages rRNA precursors, while the NEXT (nuclear exosome targeting) complex, which is restricted to the nucleoplasm, binds mRNAs, snRNAs, snoRNAs, and promoter upstream transcripts (PROMPTs) (Lubas et al. 2011, 2015; Andersen et al. 2013). The full spectrum of RNA targets of these cofactor complexes and the exosome remains unknown.
Viral RNAs, much like aberrant cellular transcripts, differ from normal RNAs. These differences can include the presence of dsRNA structures, 5′ triphosphates, and short or absent poly-A tails (Barbalat et al. 2011; Moon and Wilusz 2013). From mammals to arthropods, RNA-binding proteins can recognize these foreign RNA motifs and trigger a range of antiviral responses. Diverse helicases recognize viral RNAs; mammalian RIG-I and MDA5 recognize 5′ triphosphates and long dsRNAs, respectively, to induce antiviral transcriptional responses (Hornung et al. 2006; Kato et al. 2006; Pichlmair et al. 2006), and DDX17 recognizes bunyaviral RNA stem–loops to restrict infection in both Drosophila and human cells (Moy et al. 2014a). Other RNA-binding proteins have been implicated in the recognition or restriction of viral RNAs, suggesting that there are additional players to be discovered (Guo et al. 2004; Miyashita et al. 2011; Dong et al. 2016).
Emerging evidence suggests that viral RNAs are targeted for decay. RNASEL, which is induced by interferon signaling, nonspecifically degrades both viral and cellular RNA as a step toward cell death (Hassel et al. 1993; Castelli et al. 1997; Brennan-Laun et al. 2014). In contrast, recent studies have shown that 5′ exonucleases and decapping machinery selectively target flaviviruses and bunyaviruses, respectively (Hopkins et al. 2013, 2015; Moon and Wilusz 2013). The 3′-to-5′ RNA exosome and its associated cofactor complexes have been implicated in some immune functions but are less understood. Early work showed that the yeast Ski complex restricts dsRNA viruses, although the mechanism is not clearly understood (Masison et al. 1995; Anderson and Parker 1998; Benard et al. 1998). More recently, the human Ski component hSKIV2L has been shown to regulate endogenous RIG-I RNA targets to prevent autoimmunity; however, it likely does so independently of the canonical Ski complex and the exosome (Eckard et al. 2014). Additionally, other mammalian antiviral RNA-binding proteins (ZAP, DDX60, and AID) have been shown to bind the exosome and, in some cases, depend on the exosome for their antiviral function; however, none of these factors has been shown to induce exosome-mediated degradation of viral RNAs (Guo et al. 2007; Miyashita et al. 2011; Liang et al. 2015). Furthermore, no studies have explored potential roles of the nuclear exosome cofactors in viral infection.
We performed a series of RNAi screens to identify previously unknown RNA processing factors that restrict RNA viruses. First, we screened a library of 177 genes implicated in RNA biology in Drosophila cells (Zhou et al. 2008) against two disparate arthropod-borne RNA viruses (arboviruses): vesicular stomatitis virus (VSV) and Sindbis virus (SINV). VSV is a negative sense rhabdovirus that causes oral and skin lesions in livestock and an influenza-like fever in humans (Letchworth et al. 1999). SINV, a positive sense alphavirus, causes chronic polyarthritic disease (Kurkela et al. 2005). Second, we mined our previously published genome-wide RNAi screen against Rift Valley fever virus (RVFV), a trisegmented negative sense bunyavirus that causes a febrile illness with 1%–3% mortality in humans as well as abortions and juvenile mortality in livestock (Bird et al. 2009; Hopkins et al. 2013). The screens converged on the identification of the two exosome-associated exonucleases Rrp6 and Dis3 as antiviral against all three viruses in insect cells. Since the exonucleases may have functions outside of the RNA exosome (Schneider et al. 2007; Callahan and Butler 2008; Kiss and Andrulis 2011), we tested the role of two core exosome structural components, Rrp4 and Rrp41, and found that these also restrict infection. Since the exosome does not target RNAs directly but uses cofactor complexes, we screened orthologs of three major cofactor complexes—Ski, NEXT, and TRAMP—for their roles in viral infection. These studies revealed that only the TRAMP-associated helicase dMtr4 [l(2)35Df] and TRAMP-associated zinc finger RNA-binding protein dZcchc7 (CG9715) are antiviral in flies. We extended our studies to human cells, where we found that the broadly antiviral role for the RNA exosome and TRAMP orthologs hMTR4 and hZCCHC7 is conserved. Mechanistically, we found that infection with these cytoplasmic viruses induces the export of hMTR4 and hZCCHC7, which are nuclear in uninfected cells, to the cytoplasm, where they form a complex with the exosome as well as viral RNAs. Furthermore, we found that viral RNAs are shortened at the 3′ end and are stabilized by disruption of the exosome or the RNA-binding protein hZCCHC7. Additionally, we found that the RVFV mRNA 3′ UTR confers exosomal regulation. These findings show that a virus-induced cytoplasmic TRAMP-like complex specifically targets viral RNAs for exosome-mediated degradation to attenuate infection.
Results
RNAi screen identifies the RNA exosome as antiviral in Drosophila cells
We previously identified an antiviral role in Drosophila for dArs2 and the nuclear cap-binding complex through RNA silencing (Sabin et al. 2009). We hypothesized that additional genes involved in RNA metabolism and degradation have antiviral roles against RNA viruses, so we conducted a targeted RNAi screen against a panel of 177 genes with previously characterized roles in RNA metabolism in Drosophila cells, including dArs2 (Zhou et al. 2008). In order to identify broadly antiviral genes, this gene set was screened against two disparate arthropod-borne viruses: VSV and SINV. Drosophila DL1 cells were treated with previously validated dsRNAs targeting each gene in the panel, and knockdown was allowed to proceed for 3 d, after which cells were infected with GFP-expressing VSV or SINV, and percent infection was quantified by automated fluorescence microscopy. Each screen was performed in duplicate, and genes with robust Z-scores of ≥2 for percent infection in both replicates (P < 0.001) were considered antiviral hits. Using these metrics, the positive control dArs2 as well as 25 other genes were antiviral against both viruses (Supplemental Table S1). One of these hits, dRrp6, is a catalytic 3′–5′ exonuclease associated with the RNA exosome and the only member of the RNA exosome in the 177-gene screening set (Fig. 1A,B). We compared these data with our published genome-wide RVFV screen and found that another exosome-associated exonuclease, dDis3, was a validated antiviral hit (Hopkins et al. 2013). The identification of exosome components in multiple screens suggested a broadly antiviral role, and thus we focused on the exosome for further study.
Figure 1.
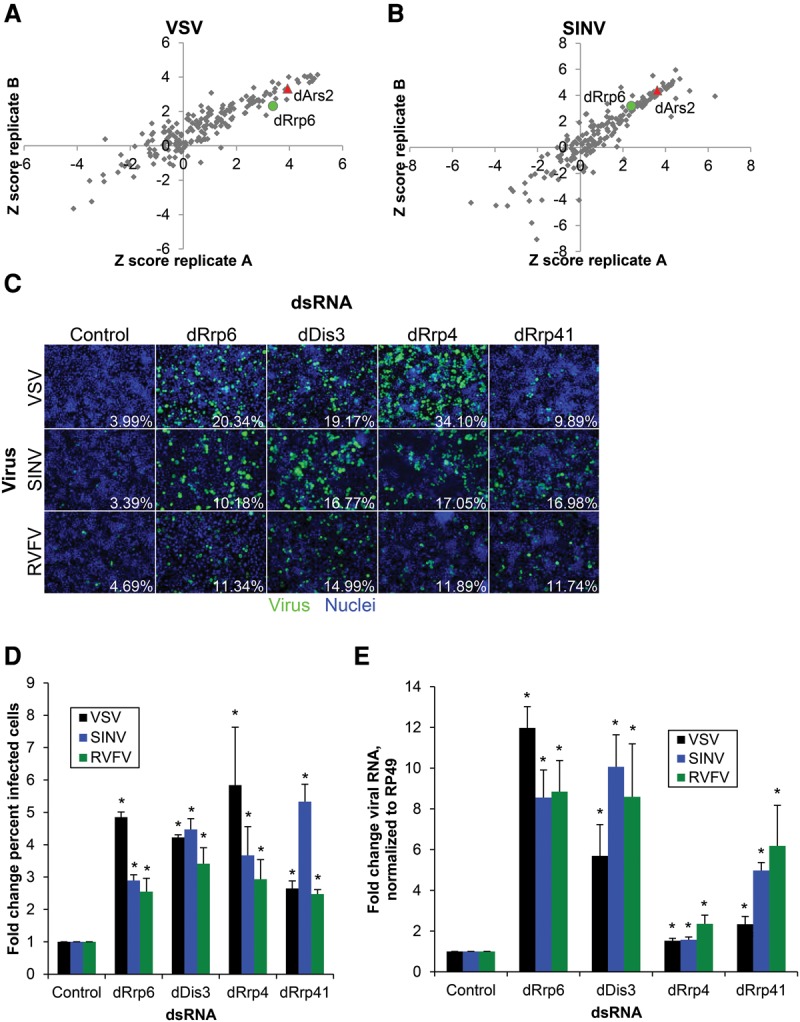
The RNA exosome is broadly antiviral in Drosophila cells. A panel of 177 genes with roles in RNA biology was depleted by RNAi in DL1 cells for 3 d and infected with VSV-GFP (A) (multiplicity of infection [MOI] = 0.1, 24 h) or SINV-GFP (B) (MOI = 2.5, 36 h) and screened by immunofluorescence measuring the percentage of infected cells. Robust Z-scores are shown for two replicates. These screens identified the positive control dArs2 (red) and the exosome component dRrp6 (green). (C) DL1 cells were treated with dsRNAs targeting the indicated genes or negative control dsRNA targeting β-galactosidase; infected with VSV-GFP (MOI = 0.1, 24 h), SINV-GFP (MOI = 2.5, 36 h), or RVFV (MOI = 0.1, 30 h); and subsequently processed for automated immunofluorescence microscopy for GFP or RVFV nucleocapsid (N). Representative images are shown with quantification of the percentage of infected cells. (D) Mean ± SEM of at least three experiments as shown in B, normalized to control. Mean percent infection in control cells was 5.40% (VSV), 3.80% (SINV), and 5.62% (RVFV). (*) P < 0.05 compared with control by Student's t-test. (E) Cells were infected as above and processed for RT-qPCR for VSV N, SINV Nsp1, or RVFV N compared with the housekeeping gene Rp49. Mean ± SEM normalized to control is shown. n ≥ 3. (*) P < 0.05, compared with control by Student's t-test.
Since these exonucleases can potentially function outside of the canonical exosome (Schneider et al. 2007; Callahan and Butler 2008; Kiss and Andrulis 2011), we tested two core components of the exosome: dRrp4 and dRrp41. In addition, we validated our screening results using independent dsRNAs against dRrp6 and dDis3. Knockdown of dRrp4 and dRrp6 was verified by immunoblot of ectopically expressed tagged proteins (Hessle et al. 2009), as antibodies against the endogenous proteins are not commercially available (Supplemental Fig. S1A). Depletion of each of these genes had little impact on cell viability as measured by cell number (Supplemental Fig. S1B) but led to significantly increased infection of VSV, SINV, and RVFV as measured by both immunofluorescence microscopy (Fig. 1C,D) and RT-qPCR (Fig. 1E). The effect of the exonucleases dRrp6 and dDis3 on viral RNA was stronger than that of the structural genes dRrp4 and dRrp41, suggesting the possibility that the exonucleolytic activity of the exosome is limiting. Taken together, these studies suggest that the RNA exosome complex restricts a broad range of RNA viruses in Drosophila cells.
Orthologs of TRAMP complex components are antiviral in Drosophila
The exosome is dependent on RNA-binding cofactor complexes to bring RNA targets to the exosome for degradation. The best-characterized of these are the Ski, TRAMP, and NEXT complexes (Lubas et al. 2011; Schneider and Tollervey 2013). The Ski complex is cytoplasmic and consists of a DExH/D-box helicase (Ski2), a tetratricopeptide repeat-containing protein (Ski3), and a WD repeat-containing protein (Ski8), all of which have Drosophila orthologs (Brown et al. 2000; Orban and Izaurralde 2005). Depletion of the three Ski components (dSki2/tst, dSki3/CG8777, and dSki8/CG3909) had no significant effect on viral infection (Fig. 2B; Supplemental Fig. S2A)
Figure 2.
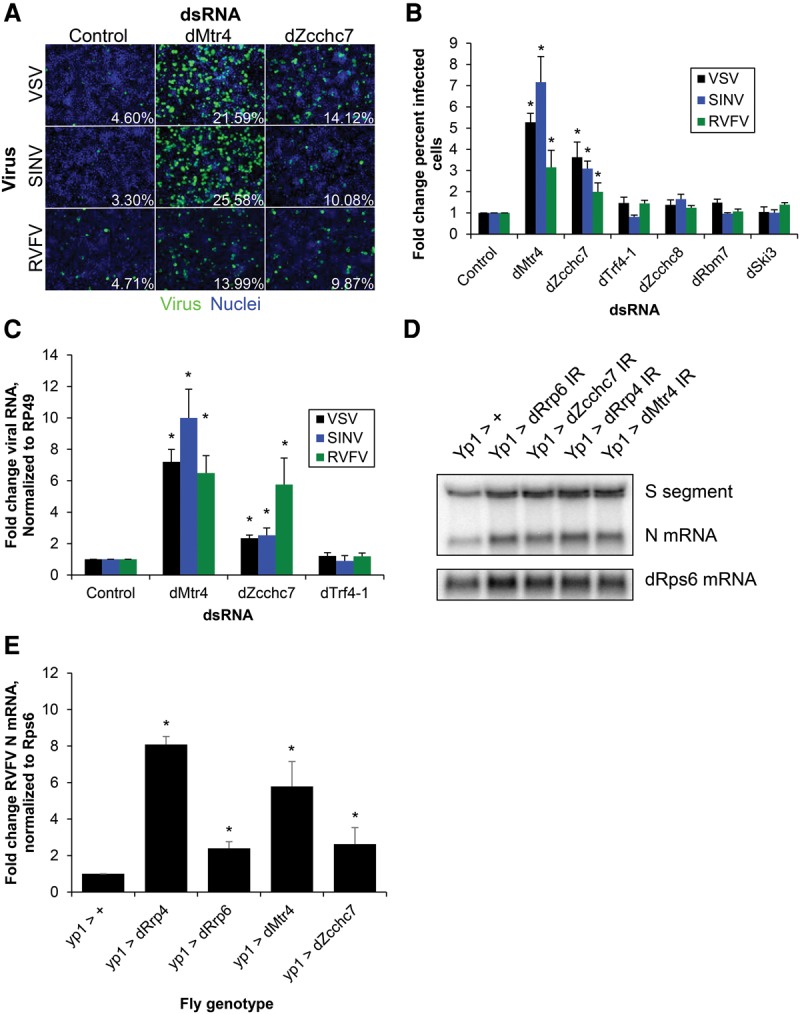
TRAMP orthologs dMtr4 and dZcchc7 are antiviral in Drosophila. (A) DL1 cells were treated with dsRNAs targeting the indicated genes or negative control dsRNA targeting β-galactosidase; infected with VSV-GFP (MOI = 0.1, 24 h), SINV-GFP (MOI = 2.5, 36 h), or RVFV (MOI = 0.1, 30 h); and subsequently processed for automated immunofluorescence microscopy for GFP or RVFV N. Representative images are shown with quantification of the percentage of infected cells. (B) Mean ± SEM of at least three experiments as shown in A, normalized to control. (*) P < 0.05, compared with control by Student's t-test. (C) Cells were infected as above and processed for RT-qPCR for VSV N, SINV Nsp1, or RVFV N compared with the housekeeping gene Rp49. Mean ± SEM normalized to control is shown. n ≥ 3. (*) P < 0.05, compared with control by Student's t-test. (D) Adult flies depleted of exosome or TRAMP genes in the fat body (YP1-Gal4>IR) or controls (YP1-Gal4>+) were challenged with RVFV for 6 d and then processed for Northern blot. A probe that identified the S segment genome/anti-genome and the N mRNA was used. The housekeeping gene RpS6 was used as a loading control. (E) Quantification of RVFV N mRNA from three or more experiments as shown in D. Mean ± SEM normalized to control. (*) P < 0.05, compared with control by Student's t-test.
The TRAMP complex, which is nuclear in yeast and nucleolar in humans, consists of a DExH/D-box helicase, Mtr4; a poly-A polymerase, Trf4/5 (Trf4-1 is the active TRAMP polymerase in Drosophila); and a zinc finger RNA-binding protein, Air1/2 (LaCava et al. 2005; Houseley and Tollervey 2008; Nakamura et al. 2008; Fasken et al. 2011; Lubas et al. 2011). The Drosophila ortholog of Air1/2 is not well characterized: Previous BLAST searches with Saccharomyces cerevisiae Air1p and the human Air ortholog hZCCHC7 found that CG9715 is the closest Drosophila ortholog, which we refer to here as dZcchc7 (Fasken et al. 2011; Lubas et al. 2011). Mining of our recent RVFV genome-wide RNAi screen revealed that dZcchc7 was also a validated antiviral gene (Hopkins et al. 2013).
In humans, Mtr4 forms an additional nuclear complex called the NEXT complex, which is excluded from nucleoli (Lubas et al. 2011). This complex is composed of hMTR4, hZCCHC8 (a zinc finger RNA-binding protein similar to hZCCHC7), and hRBM7 (an RNA-binding motif-containing protein) (Guo et al. 2003; Gustafson et al. 2005).
We tested each of the Drosophila TRAMP orthologs [dMtr4/l(2)35Df, dTrf4-1, and dZcchc7/CG9715] as well as the NEXT orthologs (dZcchc8/CG4622 and dRbm7/CG11454) for their roles in antiviral defense. While depletion of the TRAMP component dTrf4-1 or the NEXT components dZcchc8 and dRbm7 did not increase infection, depletion of dMtr4 or dZcchc7 significantly increased infection of VSV, SINV, and RVFV by both microscopy (Fig. 2A,B) and RT-qPCR (Fig. 2C). Knockdown of dMtr4 was confirmed by immunoblot of tagged overexpressed dMtr4 (Supplemental Fig. S2B) and RT-qPCR of endogenous transcripts for the other genes (Supplemental Fig. S2C). Altogether, this demonstrates that the helicase (dMtr4) and RNA-binding zinc finger (dZcchc7) TRAMP orthologs have a role in the control of viral infection in Drosophila cells.
The exosome and TRAMP orthologs restrict RVFV infection of adult flies
By taking advantage of genome-wide in vivo RNAi transgenic libraries, we tested the roles of exosome and TRAMP genes during infection of adult animals. Because these genes are essential, in vivo RNAi was performed in a nonessential organ, the fat body, which is the primary target of RVFV infection in adult flies (Moy et al. 2014b). We expressed inverted repeats targeting dRrp4, dRrp6, dMtr4, or dZcchc7 in the female fat body (Vidal et al. 2001) and challenged these flies with RVFV for 6 d, after which infection was assessed by Northern blot. We found that depletion of all four genes resulted in a significant increase in RVFV replication compared with control (Fig. 2D,E). Flies with exosome components depleted in the fat body had survival similar to that of control flies, suggesting that increased viral replication is not due to generalized frailty (Supplemental Fig. S2D). These data suggest that the RNA exosome and TRAMP orthologs dMtr4 and dZcchc7 are antiviral both in cell culture and at the organismal level.
The antiviral role of the exosome, Mtr4, and Zcchc7 is conserved from flies to humans
The RNA exosome and TRAMP have conserved roles from yeast to flies to humans (Houseley and Tollervey 2009; Schneider and Tollervey 2013). While some studies have explored the human RNA exosome, the human TRAMP components were only recently identified, and few RNA targets have been characterized (Fasken et al. 2011; Lubas et al. 2011). We tested whether the exosome and TRAMP genes hMTR4 and hZCCHC7 also restrict viral infection in human cells. We depleted the human exosome exonucleases hRRP6 and hDIS3, the core exosome subunits hRRP4 and hRRP41, and the TRAMP components hMTR4 and hZCCHC7 using siRNAs in human osteosarcoma cells (U2OS), which are permissive to infection by a large number of viruses, including VSV, SINV, and RVFV (Moy et al. 2014a). Knockdown was confirmed by immunoblot for genes with a commercial antibody that we could validate or RT-qPCR for the remaining genes (Supplemental Fig. S3A,B). Quantification of cell number revealed only modest effects on cell number under these conditions (Supplemental Fig. S3C). Cells depleted of the exosome, hMTR4, or hZCCHC7 and infected with VSV, SINV, or RVFV showed increased viral RNA levels as measured by RT-qPCR (Fig. 3A) and increased viral protein by immunoblot (Fig. 3B–D). Together, these data suggest that the RNA exosome and TRAMP components are antiviral in human cells against diverse RNA viruses.
Figure 3.
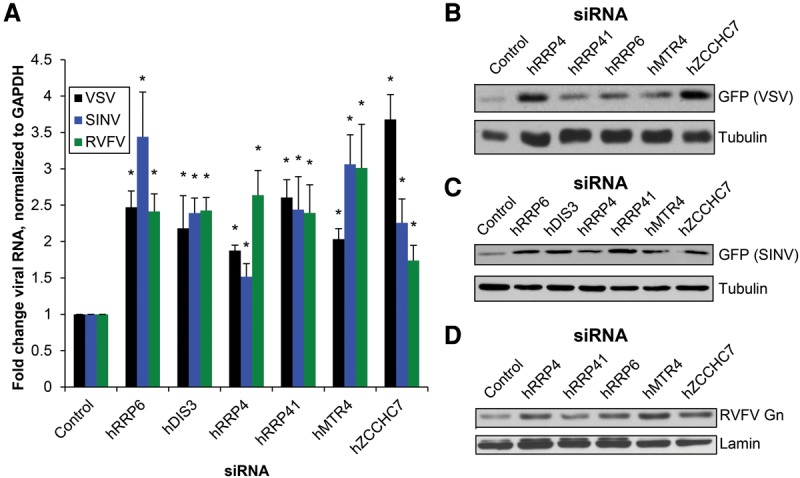
The RNA exosome and TRAMP orthologs are antiviral in human cells. (A) U2OS cells were transfected with the indicated siRNAs; infected with VSV-GFP (MOI = 0.05, 14 h), SINV-GFP (MOI = 1, 16 h), or RVFV (MOI = 0.03, 18 h); and subsequently processed for RT-qPCR for VSV N, SINV Nsp1, or RVFV N compared with the housekeeping gene GAPDH. Mean ± SEM is shown normalized to control. n ≥ 3. (*) P < 0.05, compared with control by Student's t-test. (B) Cells were transfected with the indicated siRNAs, infected with VSV-GFP (MOI = 0.05, 14 h), and then processed for GFP immunoblot. A representative blot is shown. n ≥ 3. (C) Cells were transfected with the indicated siRNAs, infected with SINV-GFP (MOI = 1, 8 h), and then processed for GFP immunoblot. A representative blot is shown. n ≥ 3. (D) Cells were transfected with the indicated siRNAs, infected with RVFV (MOI = 0.3, 18 h), and then processed for RVFV Gn glycoprotein immunoblot. A representative blot is shown. n ≥ 3.
MTR4 and ZCCHC7 form a cytoplasmic complex with the exosome upon viral infection
While the RNA exosome is present in the nucleus and cytoplasm, the human TRAMP complex is thought to be exclusively nucleolar (Fasken et al. 2011; Lubas et al. 2011). However, VSV, SINV, and RVFV are all cytoplasmic RNA viruses that are not thought to transport their RNAs into the nucleus (Strauss and Strauss 1994; Letchworth et al. 1999; Moon and Wilusz 2013). Therefore, we hypothesized that if the TRAMP components were directly targeting viral RNA, they would have to relocalize to the cytoplasm during infection. We validated an antibody against hZCCHC7 by both immunoblot and immunofluorescence (Supplemental Figs. S3B, S4A). While we validated an hMTR4 antibody by immunoblot (Supplemental Fig. S3B), it did not recognize hMTR4 by immunofluorescence (data not shown). Next, U2OS cells were either mock-infected or infected with RVFV or SINV, and hZCCHC7 localization was monitored. While hZCCHC7 was exclusively nucleolar in uninfected cells, in RVFV- or SINV-infected cells, hZCCHC7 accumulated in cytoplasmic punctae (Fig. 4A–D; monochrome images in Supplemental Fig. S4B,C). Furthermore, the average number of cytoplasmic punctae per infected cell was similar between RVFV and SINV infection (Supplemental Fig. S4D).
Figure 4.
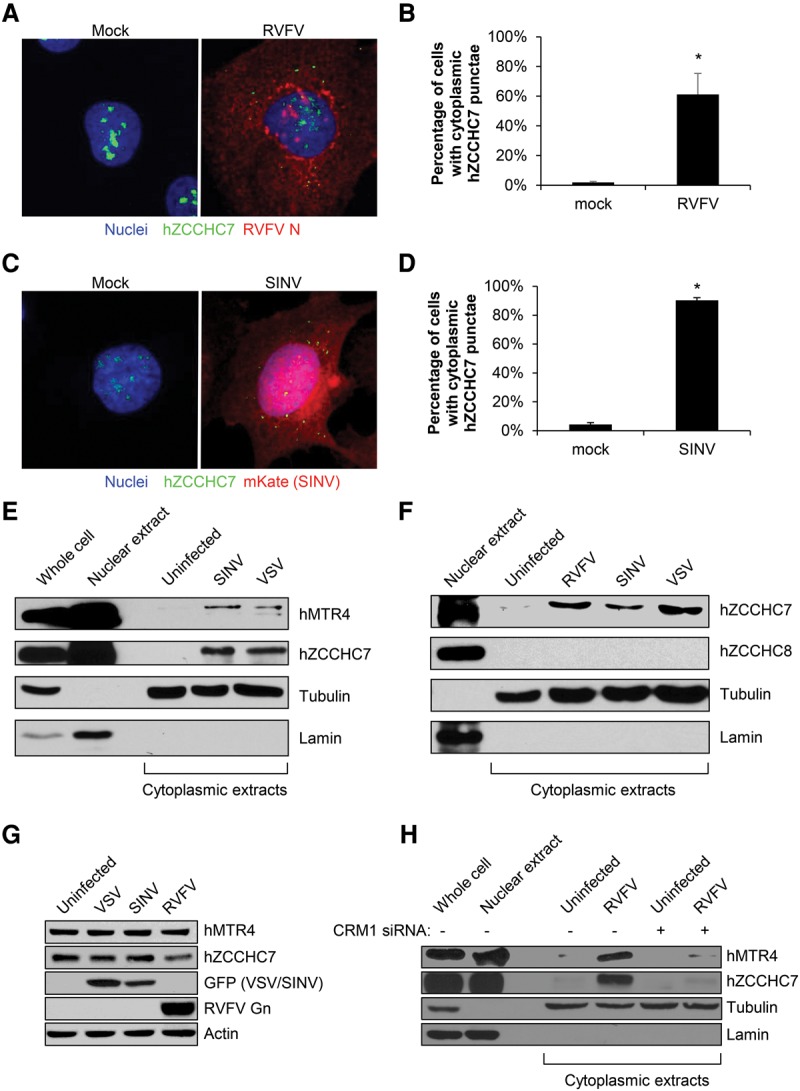
Human MTR4 and ZCCHC7 are exported to the cytoplasm upon viral infection. (A) U2OS cells were infected with RVFV (MOI = 10, 12 h) or mock-infected and processed for immunofluorescence microscopy for hZCCHC7 (green), RVFV N (red), and nuclei (blue). (B) Quantification of the percentage of cells with cytoplasmic hZCCHC7 punctae in mock- or RVFV-infected cells in at least three experiments as in A. Mean ± SEM is shown. (*) P < 0.05, compared with mock by Student's t-test. (C) U2OS cells were infected with SINV-mKate (MOI = 10, 5 h) or mock-infected and processed for immunofluorescence microscopy for hZCCHC7 (green), mKate (red), and nuclei (blue). (D) Quantification of the percentage of cells with cytoplasmic hZCCHC7 punctae in mock- or SINV-infected cells in at least three experiments as in C. Mean ± SEM is shown. (*) P < 0.05, compared with mock by Student's t-test. (E) U2OS cells were infected with SINV-GFP or VSV-GFP (MOI = 10, 8 h), subjected to nuclear/cytoplasmic fractionation and immunoblot, and probed for the nuclear protein lamin and the cytoplasmic protein tubulin to verify extract purity along with hMTR4 and hZCCHC7. A representative blot is shown. n ≥ 3. (F) U2OS cells were infected with RVFV (MOI = 10, 12 h), SINV-GFP (MOI = 10, 8 h), or VSV-GFP (MOI = 10, 8 h); subjected to nuclear/cytoplasmic fractionation and immunoblot; and probed for the nuclear protein lamin and the cytoplasmic protein tubulin to verify extract purity along with hZCCHC7 and hZCCHC8. A representative blot is shown. n ≥ 3. (G) U2OS cells were infected with VSV-GFP (MOI = 10, 8 h), SINV-GFP (MOI = 10, 8 h), or RVFV (MOI = 10, 12 h), and whole-cell lysates were processed for immunoblot. A representative image is shown. n = 2. (H) U2OS cells were transfected with siRNA specific to CRM1 or control, infected with RVFV (MOI = 10, 12 h), subjected to nuclear/cytoplasmic fractionation and immunoblot, and probed for the nuclear protein Lamin and the cytoplasmic protein tubulin to verify extract purity along with hMTR4 and hZCCHC7. A representative image is shown. n ≥ 3.
Next, we set out to explore the localization of these factors using biochemical fractionation. Nuclear and cytoplasmic lysates were isolated, and the purity of fractionation was established by monitoring the nuclear protein lamin and the cytoplasmic protein tubulin. As expected, hMTR4 and hZCCHC7 were detected in the nuclear but not cytoplasmic fraction of uninfected cells (Fig. 4E). However, upon infection with VSV or SINV, both hMTR4 and hZCCHC7 accumulated in the cytoplasm. We also examined localization of the NEXT component hZCCHC8, since it is similar in structure to hZCCHC7 and also associates with hMTR4 in the nucleus (Lubas et al. 2011). In contrast to hMTR4 and hZCCHC7, hZCCHC8 was exclusively nuclear in both uninfected and infected cells (Fig. 4F; Supplemental Fig. S4E), suggesting that there is specificity in the proteins relocalized to the cytoplasm.
To determine whether the accumulation of these TRAMP components in the cytoplasm was due to their export from the nucleus or increased synthesis, we first examined whether viral infection altered overall levels of these proteins and observed no increase upon infection with VSV, SINV, or RVFV (Fig. 4G). Next, we assessed whether cytoplasmic accumulation was dependent on nuclear export by testing whether the major nuclear export protein CRM1, which we found previously to be broadly antiviral (Yasunaga et al. 2014), was required for virus-induced cytoplasmic accumulation. We validated that siRNA treatment efficiently depleted CRM1 (Supplemental Fig. S4F). Next, CRM1-depleted or control cells were infected with RVFV, and the nuclear and cytoplasmic fractions were isolated. As with VSV and SINV, RVFV infection led to the accumulation of hMTR4 and hZCCHC7 in the cytoplasm (Fig. 4H). Furthermore, this virus-induced increase was lost upon CRM1 depletion. These data suggest that signals from viral infection induce the export of these antiviral proteins from the nucleus to the cytoplasm.
The TRAMP components hMTR4 and hZCCHC7 function in the nucleolus as a complex with the exosome (Lubas et al. 2011). We reasoned that in order to target viral RNAs for exosomal degradation, these proteins must both relocalize and form a complex in the cytoplasm. To test this hypothesis, we first expressed Flag-tagged hMTR4 (Lubas et al. 2011) or a vector control and confirmed expression by immunoblot (Supplemental Fig. S5A). Next, transfected cells were infected with RVFV or mock-infected, and anti-Flag immunoprecipitations were performed. As expected, hMTR4-Flag immunoprecipitated hZCCHC7 and the exosomal protein hRRP6 in both infected and uninfected cells in whole-cell lysates (Fig. 5A). In contrast, cytoplasmic hMTR4-Flag precipitated hZCCHC7 and hRRP6 only in RVFV-infected cells (Fig. 5B). While the specificity of hZCCHC7 coimmunoprecipitation during infection can be explained by its export to the cytoplasm, hRRP6 was coimmunoprecipitated only during infection despite having a cytoplasmic localization in uninfected cells as well. Coimmunoprecipitation of these factors was unchanged upon RNase A treatment (data not shown), suggesting that these interactions are not RNA-dependent. Taken together, our data indicate that hZCCHC7 and hMTR4 are exported and form a complex with the exosome in the cytoplasm upon viral infection.
Figure 5.
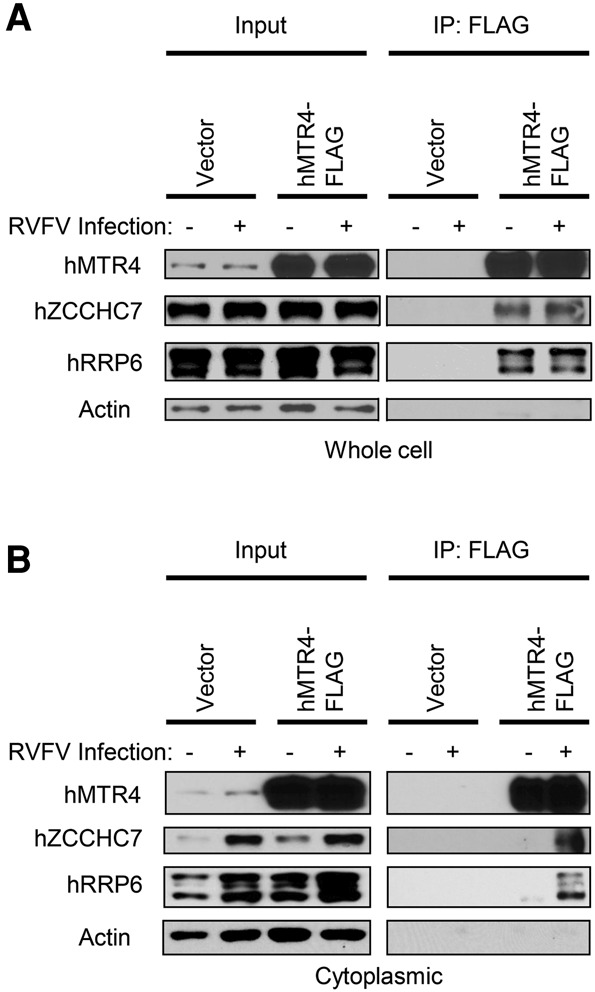
Human MTR4 and ZCCHC7 form a cytoplasmic complex with the exosome upon infection. U2OS cells were transfected with an hMTR4-Flag expression vector or empty vector and infected with RVFV (MOI = 10, 12 h) or mock-infected, and then either whole-cell lysates (A) or cytoplasmic fractions (B) were processed for coimmunoprecipitation. Four percent input was loaded for the hMTR4 immunoblot, and 8% input was loaded for other proteins. Actin was used as a loading control. Representative blots are shown. n ≥ 3.
ZCCHC7 specifically binds RVFV mRNA and SINV RNA
In yeast, the zinc finger-containing Air proteins are thought to confer RNA-binding specificity to TRAMP, which in turn delivers its RNA cargo to the exosome for degradation (Schmidt et al. 2012). This led us to hypothesize that hZCCHC7, as the human Air ortholog, may be the specificity factor that binds viral RNAs to target them to the exosome. To examine this possibility, we performed RNA immunoprecipitation (RNA-IP) with hZCCHC7. We transfected Flag-tagged hZCCHC7 (Lubas et al. 2011) or control vector and verified expression and cytoplasmic localization of hZCCHC7-Flag during infection by immunoblot (Supplemental Fig. S5A). Next, transfected cells were infected with RVFV or SINV, cytoplasmic fractions were collected (input), and a fraction was subjected to Flag-immunoprecipitation followed by RT-qPCR. RNA quantification was normalized to vector control for both input and Flag immunoprecipitation to demonstrate that ectopic expression of hZCCHC7 did not increase the input levels and to remove any signal from nonspecific RNA binding to beads or the Flag antibody. We found that RVFV nucleocapsid (N) and nonstructural (NSs) mRNAs but not the S segment genome or anti-genome from which they were transcribed were selectively and significantly bound by hZCCHC7 (Fig. 6A; Supplemental Fig. S5B, schematic of RNAs). This selective binding is not explained by RNA abundance, as Northern blot analysis revealed that N mRNA levels are lower than those of S segment genome/anti-genome RNA in both Drosophila and humans (Fig. 2D; Moy et al. 2014b). An endogenous mRNA, hDCP2 (the levels of which are unaffected by RVFV infection) (Hopkins et al. 2015), was not significantly bound. Similar results were found for SINV infection, in which SINV genomic and subgenomic RNAs (which function as mRNAs) were significantly bound by hZCCHC7 (Fig. 6B). These data suggest that hZCCHC7 selectively binds viral mRNAs in the cytoplasm during infection.
Figure 6.

Viral mRNA is bound by hZCCHC7. (A,B) U2OS cells were transfected with an hZCCHC7-Flag expression vector or empty vector, infected with RVFV (A) (MOI = 10, 12 h) or SINV-GFP (B) (MOI = 10, 8 h), and then fractionated. Cytoplasmic extracts were collected (input), and a fraction was subjected to Flag immunoprecipitation and processed for RT-qPCR. RNA quantification was normalized to vector control input or Flag immunoprecipitation. Fold change in hZCCHC7-bound RNA normalized to vector-bound RNA is presented. hDCP2 was used as an endogenous mRNA control, as it is not known to be regulated during RVFV or SINV infection. Mean ± SEM shown. (*) P < 0.05 by Student's t-test.
RVFV mRNA, but not genomic or anti-genomic RNA, is shortened at the 3′ end
The exosome processively degrades RNA 3′ to 5′; however, RNAs partially degraded by the exosome can leave 3′ truncated degradation intermediates (Eckwahl et al. 2015). Since we found that hZCCHC7 selectively bound RVFV mRNA, but not RVFV genomic or anti-genomic RNA, we hypothesized that the mRNA would be truncated at the 3′ end. To test this, 3′ rapid amplification of cDNA ends (RACE) was performed, and individual RNAs were sequenced. Briefly, RNA was harvested from RVFV-infected cells, and a linker was ligated to the 3′ end of the RNA. RT–PCR was then performed using a linker-specific reverse primer and a virus-specific forward primer targeting S segment genomic RNA, anti-genomic RNA, or N mRNA. PCR products were cloned, and individual clones were sequenced to identify the proportion of full-length clones. We classified reads as full length or shortened based on the full-length sequences of the genomic segment as well as the transcription termination site that defines the 3′ end of the N mRNA, N coding sequence (ORF), and 3′ UTR (Ikegami et al. 2007). 3′ RACE revealed that while most sequence reads from genomic and anti-genomic S segment RNA clones were full length, the majority of N mRNA reads were shortened at the 3′ end (Fig. 7A). A variety of truncated N mRNA sequences were found with both intact and disrupted N coding sequence (Fig. 7B; Supplemental Tables S2–S4, full sequence alignments). This suggests that the viral mRNA, but not genomic or anti-genomic RNA, is subject to 3′ degradation.
Figure 7.
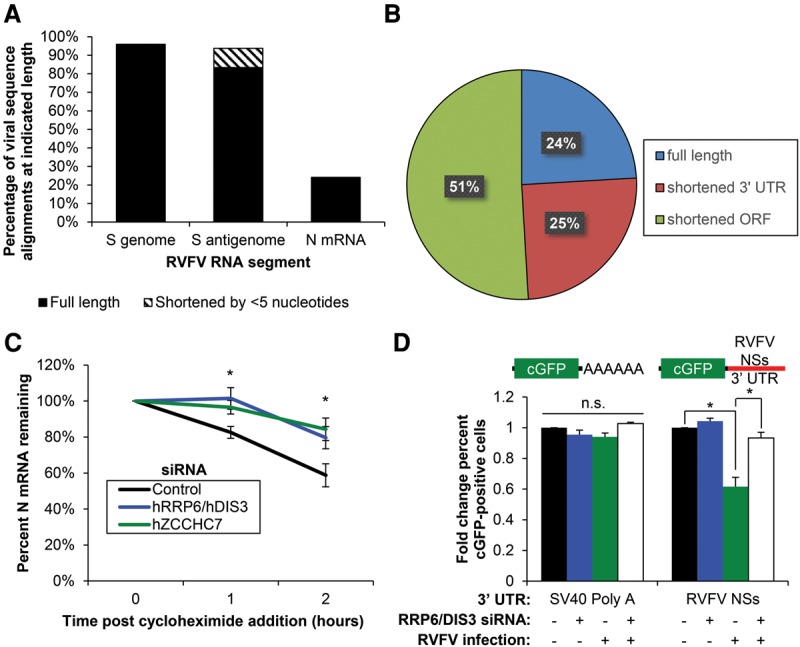
The RNA exosome and hZCCHC7 target viral mRNAs for decay. (A) U2OS cells were infected with RVFV (MOI = 0.3, 18 h), and 3′ RACE was performed using primers that detect the RVFV small segment RNAs as indicated. Sequenced reads were aligned to RVFV, classified as full length or slightly shortened (<5 nucleotides), and plotted. Pooled data from three biological replicates are shown. (B) Pie chart of N mRNA reads from A that are full length (blue), encode a full-length ORF but truncated 3′ UTR (red), or encode a truncated ORF are shown (green). (C) U2OS cells were transfected with the indicated siRNAs and infected with RVFV (MOI = 1, 12 h). Infected cells were treated with 50 µg/mL cycloheximide and processed for RT-qPCR at the indicated time points. Mean ± SEM is shown normalized to hDCP2. n ≥ 3. (*) P < 0.05, compared with control by Student's t-test. (D) U2OS cells stably expressing cGFP reporters with the indicated 3′ UTRs were transfected with siRNAs targeting hRRP6 and hDIS3 or control and either uninfected or infected with RVFV (MOI = 10, 18 h). Cells were subsequently processed for automated immunofluorescence microscopy. Mean ± SEM is shown normalized to mock-infected control. n ≥ 3. (*) P < 0.05, compared with control by Student's t-test.
RVFV mRNA stability is dependent on the exosome and ZCCHC7
Bunyaviruses such as RVFV are unique in that mRNA transcription but not the replication of the genome requires concomitant protein translation (Barr 2007). Cycloheximide, which inhibits translational elongation, can therefore be used to block new mRNA synthesis, allowing us to assess the rate of decay of previously transcribed RVFV mRNA (Hopkins et al. 2013). To determine whether the stability of RVFV mRNA is exosome- and hZCCHC7-dependent, we depleted the two exosome exonucleases hRRP6 and hDIS3 or hZCCHC7 by siRNA in U2OS cells. Cells were infected with RVFV (multiplicity of infection [MOI] = 1, 12 h) and treated with cycloheximide for 0, 1, or 2 h, and RNA was processed for RT-qPCR. As previously observed (Hopkins et al. 2013), we found that RVFV mRNA significantly decayed after addition of cycloheximide in control cells (Fig. 7C), but RVFV genome/anti-genome RNA did not (Supplemental Fig. S5C). Furthermore, we observed that depletion of hRRP6/hDIS3 or hZCCHC7 significantly reduced this decay, suggesting that RVFV mRNA is destabilized by the RNA exosome and hZCCHC7.
The RVFV NSs 3′ UTR is specifically regulated by the exosome during infection
We set out to determine whether there are specific signals in the RVFV mRNA that direct exosomal degradation. Since cellular mRNAs are often targeted to the exosome through signals at the 3′ end, including hypoadenylation, 3′ extension, and AU-rich elements in the 3′ UTR (Chen et al. 2001; Milligan et al. 2005; Lubas et al. 2015), we hypothesized that the 3′ UTR of a RVFV mRNA would be sufficient to render an mRNA susceptible to control by the exosome. To test this, we used a reporter system in which the RVFV NSs 3′ UTR is cloned downstream from a cGFP ORF. Since the mRNAs of RVFV are not polyadenylated, we generated the exact 3′ end by cloning the mascRNA (MALAT1-associated small cytoplasmic RNA) sequence downstream from the 3′ UTR, which is processed by endogenous RNase P, leaving the mature viral 3′ end (Supplemental Fig. S6A–D; Wilusz et al. 2012). U2OS cells stably expressing this reporter or a control reporter with the SV40 polyadenylation signal cloned downstream from cGFP were transfected with siRNAs targeting hRRP6 and hDIS3 or control and either mock-infected or infected with RVFV (MOI = 10, 18 h). Automated fluorescence microscopy was used to calculate the percentage of cells expressing cGFP. We found that while the NSs 3′ UTR reporter was unaffected by exosome depletion in uninfected cells, RVFV infection caused a significant reduction in reporter signal that was rescued to uninfected control levels by exosome depletion (Fig. 7D). In contrast, the SV40 poly-A reporter was unaffected by either exosome depletion or viral infection. This demonstrates that, upon viral infection, the exosome specifically regulates the RVFV 3′ UTR but not a conventional polyadenylated mRNA.
Discussion
Increasing evidence suggests that the RNA recognition and decay machinery plays an important role in the control of viral infection. Through RNAi screening, we found that the RNA exosome and two components of the exosomal cofactor TRAMP complex were antiviral against diverse RNA viruses from insects to humans.
Since the TRAMP complex normally binds specific RNAs for exosome-dependent processing, we hypothesized that the antiviral TRAMP components would recognize viral RNAs and recruit the exosome to degrade them. However, human TRAMP is thought to reside exclusively in the nucleolus (Fasken et al. 2011; Lubas et al. 2011); therefore, we tested whether these antiviral TRAMP components translocate upon infection to the cytoplasm, where the viral RNAs are located. Indeed, we found that hMTR4 and hZCCHC7 are dependent on the nuclear export protein CRM1 for their cytoplasmic accumulation and associate with each other and the exosome in the cytoplasm during infection. In contrast, the related NEXT complex component hZCCHC8 remained exclusively nuclear during infection, suggesting that the export of antiviral TRAMP components to the cytoplasm is specific. Furthermore, immunofluorescence analysis revealed infection-induced hZCCHC7 cytoplasmic punctae. The RNA exosome and some cofactors have been found previously in cytoplasmic granules distinct from P bodies or stress granules (Sheth and Parker 2003; Graham et al. 2006; Lin et al. 2007). These data suggest that hZCCHC7 and hMTR4 accumulate in exosome granules during viral infection to facilitate the specific degradation of viral RNA.
The virus-induced translocation of antiviral factors from the nucleus to the cytoplasm is an emerging theme; we previously found that two additional RNA-binding proteins, DDX17 and Drosha, are exported during infection for their roles in antiviral defense (Moy et al. 2014a; Shapiro et al. 2014). Furthermore, we found CRM1 to be broadly antiviral in both Drosophila and human cells (Yasunaga et al. 2014), suggesting that this is at least in part related to the requirement for the export of antiviral RNA-binding proteins. The sensors necessary for export of these effectors of antiviral defense are not yet well understood; we hypothesize that detection of virus-specific signals such as dsRNA by sensors like RIG-I, MDA5, or PKR may trigger export of hMTR4 and hZCCHC7. Indeed, PKR sensing of viral RNA has been shown to induce the formation of antiviral stress granules in the cytoplasm, opening the possibility that a similar process may induce hZCCHC7 granules (Onomoto et al. 2012).
Next, we investigated whether this exported complex binds viral RNA. We found that RVFV and SINV mRNA are bound by hZCCHC7, but RVFV genome RNA, RVFV anti-genome RNA, and an endogenous mRNA are not. This specificity may be explained at least in part by accessibility; bunyaviral genome and anti-genome RNAs are coated by N and as such may not be accessible to cellular degradation machinery, while mRNA must be accessible in order to be translated and thus is vulnerable to RNases (Kolakofsky and Hacker 1991; Strauss and Strauss 1994). Furthermore, 3′ RACE revealed that while RVFV S segment genome and anti-genome RNAs were largely full length, the majority of RVFV N mRNA was shortened at the 3′ end. Although RVFV N mRNA has been studied using 3′ RACE, previous studies sequenced only pooled and size-selected RACE products, which would mask shortened ends (Albarino et al. 2007; Lara et al. 2011). This led us to test whether the exosome impacts the stability of viral mRNAs. Using cycloheximide to disrupt protein translation and thus RVFV mRNA transcription (Barr 2007), we found that RVFV mRNA decay is exosome- and hZCCHC7-dependent and that the genomic RNA is not subject to this targeting.
This specificity suggests that there may be signals or sequences in the viral mRNAs that direct their decay. Indeed, the exosome degrades several classes of mRNAs based on signals in their 3′ UTRs. Therefore, we tested whether the 3′ UTR of RVFV mRNA confers this specificity and found that the RVFV NSs 3′ UTR directs exosome-dependent decay only during RVFV infection. This regulated decay of viral mRNAs is reminiscent of other cohorts of mRNAs, known as RNA regulons, which are coregulated through signals in their 3′ UTRs such as AU-rich elements or C-rich motifs; regulation of these RNAs is also exosome-dependent (Chen et al. 2001; Mukherjee et al. 2002; Keene 2007; Singer et al. 2012; Blackinton and Keene 2014).
We characterized a new role for an RNA-binding exosome cofactor complex that is regulated by infection to specifically target viral RNAs for exosome-dependent degradation. This may be part of a larger spectrum of exosomal cofactors activated during infection. The antiviral RNA-binding proteins DDX17, DDX60, ZAP, and AID have all been found to coimmunoprecipitate with the exosome (Guo et al. 2007; Lubas et al. 2011; Miyashita et al. 2011; Moy et al. 2014a; Liang et al. 2015). Furthermore, both ZAP and AID restrict viral infection only if the exosome is intact. Further studies are needed to determine whether these complexes drive exosomal degradation of viral RNAs. Nevertheless, taken together with the present study, these data suggest that viral infection may induce a panel of RNA-binding cofactor complexes to target viral RNAs for selective exosome-dependent decay.
Materials and methods
Cells, viruses, antibodies, and reagents
Drosophila DL1 cells and human U2OS cells were grown and maintained as previously described (Cherry and Perrimon 2004; Moser et al. 2012). VSV-eGFP (gift from J. Rose) was grown in BHK cells as described (Ramsburg et al. 2005). SINV-GFP (gift from R. Hardy) and SINV-mKate (gift from M. Heise) were grown in C636 cells as described (Burnham et al. 2007). An attenuated strain of RVFV (MP-12) was grown in Vero cells as described (Filone et al. 2010). Viral titers were calculated by plaque assay on BHK cells. Antibodies are described in Supplemental Table S5. Fluorescent secondary antibodies were obtained from Invitrogen, and HRP-conjugated antibodies were from Amersham. Other chemicals were purchased from Sigma.
Drosophila RNAi
dsRNAs were generated as described (Boutros et al. 2004). Knockdowns for RNAi screening were performed in 384-well plates prearrayed with 0.25 µg of dsRNA per well, as described (Zhou et al. 2008; Hopkins et al. 2013). Briefly, to knock down genes using RNAi, DL1 cells were passaged into serum-free medium and seeded into plates containing dsRNAs targeting the indicated genes or β-galactosidase as a nontargeting control. Cells were serum-starved for 1 h, after which complete medium was added, and cells were incubated for 3 d. Knockdown was validated by cotransfection of dsRNAs with plasmids expressing dRrp4-V5, dRrp6-V5, or dMtr4-Flag using Effectene transfection reagent (Qiagen) (Hessle et al. 2009). Plasmids are detailed in Supplemental Table S6.
Mammalian RNAi
Silencer Select siRNAs were obtained from Ambion and transfected into U2OS cells using HiPerFect (Qiagen) as per the manufacturer's protocol. Cells were incubated for 3 d. The specific siRNAs used are in Supplemental Table S7; siRNAs for each gene were mixed and used together. Silencer Select Negative Control #2 (Ambion) was used as a nontargeting control.
Viral infections
Three days after RNAi, cells were infected with the indicated viruses. MOI was calculated based on viral titers on BHK cells. For DL1 cells, VSV-GFP (MOI = 0.1) was processed at 24 h post-infection (hpi). SINV-GFP (MOI = 2.5) and RVFV (MOI = 0.1) were spinoculated at 1200 rpm for 2 h and processed at 36 and 30 hpi, respectively. For infectivity studies in U2OS cells, VSV-GFP (MOI = 0.05), SINV-GFP (MOI = 1), and RVFV (MOI = 0.03) were added to cells in complete medium for 14, 16, and 18 h, respectively, for RNA or 14, 8, and 18 h, respectively, for protein. For immunofluorescence localization studies, U2OS cells were infected with RVFV or SINV-mKate (MOI = 10) for 12 and 5 h, respectively. For fractionation and immunoprecipitation, U2OS cells were infected with VSV-GFP (MOI = 10, 8 h), SINV-GFP (MOI = 10, 8 h), or RVFV (MOI = 10, 12 h).
Immunofluorescence
Cells were processed as previously described (Shelly et al. 2009). Cells were imaged with an ImageXpress Micro automated microscope. At least four sites in each of three wells were imaged per condition per experiment, and MetaXpress cell scoring was used to calculate the number of cells and the percentage of infection. For protein relocalization studies, U2OS cells grown on coverslips were imaged with a Leica DMI 4000 B fluorescent microscope. MetaXpress software was used to quantify cytoplasmic hZCCHC7 punctae between 0.8 and 2 µm in size in mock-infected and infected cells (infection was verified by immunofluorescence for each cell to be quantified), with at least 25 cells quantified per condition. All experiments were performed at least three times.
RNA quantification
Total RNA was extracted, and Northern blotting or RT-qPCR were performed as previously described (Cherry 2005; Xu et al. 2012). Primer sequences are described in Supplemental Table S8.
Adult fly infections
Transgenic flies for in vivo RNAi were obtained from the Vienna Drosophila RNAi Center or Bloomington Drosophila Stock Center and crossed to YP1-GAL4 (fly strains are listed in Supplemental Table S9). Four-day-old to 7-d-old flies were challenged with RVFV (Cherry and Perrimon 2004), and 15 flies per condition were processed for RNA 6 d after infection as previously described (Xu et al. 2012).
Nuclear/cytoplasmic fractionation
Cells were lysed in buffer A (30 mM Hepes at pH 7.4, 2 mM MgOAc, 0.1% NP40) supplemented with 5 mM DTT, PMSF, and protease inhibitors (25× Roche complete tablets) by pipetting three times through a 26-gauge needle. Nuclei were pelleted for 20 min at 500 g. Cytoplasmic supernatant was removed, and the nuclear pellet was rinsed twice with buffer A and then lysed with sonication in RIPA buffer (50 mM Tris-HCl at pH 7.4, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 50 mM NaF) supplemented with PMSF and protease inhibitors. Lysates were analyzed by immunoblot. Each experiment was repeated at least three times, and representative blots are shown.
Immunoprecipitation
U2OS cells were transfected with doxycycline-inducible hMTR4-Flag, hZCCHC7-Flag, or empty vector control using Xtremegene 9 (Roche) (Lubas et al. 2011). Plasmids are detailed in Supplemental Table S6. Plasmid expression was induced at 24 h with 1 µg/mL doxycycline, and cells were infected at 48 h with RVFV (MOI = 10) for 12 h. Cytoplasmic extracts or whole-cell lysates were immunoprecipitated with anti-Flag M2 magnetic beads (Sigma). For analysis of proteins, beads were treated with 100 μg/mL RNase A or untreated, washed with buffer A, and analyzed by immunoblot. For analysis of bound RNA, beads were washed in buffer A supplemented with 150 mM NaCl and 0.5% NP40 and analyzed by RT-qPCR for efficient expression of the construct and target RNA binding. Each experiment was repeated at least three times, and representative blots are shown.
3′ RACE
3′ RACE was performed as previously described (Wilusz et al. 2008). U2OS cells were infected with RVFV (MOI = 0.3) for 18 h. Total RNA was harvested and treated with CIP (New England Biolabs) to remove terminal phosphates, ligated to miRNA linker #3 (IDT), and reverse-transcribed using a linker-specific primer. RACE PCR was performed against S segment genome, anti-genome, or N mRNA (see the Supplemental Material for primers), and products were cloned using TOPO-TA (Invitrogen). Individual colonies were screened for insert with colony hybridization Southern blot and sequenced. Sequences were classified based on previous studies of RVFV transcriptional termination (Ikegami et al. 2007).
RNA stability assay
Three days after RNAi, cells were infected with RVFV (MOI = 1) for 12 h. Cells were treated with 50 µg/mL cycloheximide for 0, 1, or 2 h, after which cells were processed for RT-qPCR. DCP2 was used as a control.
GFP 3′ UTR reporters
To generate plasmids encoding cGFP 3′ UTR reporters, the previously described pCRII-TOPO CMV-cGFP-SV40 poly-A sense plasmid (Wilusz et al. 2012) was cleaved by the NotI restriction enzyme to remove the SV40 polyadenylation signal. The 3′ UTR of RVFV NSs, RNase P cleavage site, and mascRNA sequences flanked by NotI sites (sequence: GCGGCCGCAGGTTAAGGCTGCCCCACCCCCCACCCCCTAATCCCGACCGTAACCCCAACTCCCCTTCCCCCCAACCCCCTGGGACGCTGGTGGCTGGCACTCCTGGTTTCCAGGACGGGGTTCAAGTCCCTGCGGTGTCTTTGCTTGCGGCCGC) were then inserted downstream from the cGFP ORF. The original plasmid expressing cGFP with a downstream SV40 polyadenylation signal was used as a control. Proper processing of cGFP mRNA and mascRNA was validated by Northern blot and small RNA Northern blot, respectively. Expression of cGFP protein was validated by Western blot. Stable cell lines were generated and used for immunofluorescence experiments.
Supplementary Material
Acknowledgments
We thank E. Klinman, D. Klinman, and J.P. Molleston for critical reading of the manuscript; R. Doms for RVFV antibodies; J. Rose for VSV-eGFP; R. Hardy for SlNV-GFP; M. Heise for SINV-mKate; Vienna Drosophila RNAi Center and The Bloomington Stock Center for transgenic flies; and the Cherry laboratory for advice. This work was supported by National Institute of Health grants R01AI074951, U54AI057168, and R01AI095500 to S.C.; R00GM104166 to J.E.W.; and T32AI007324 to J.M.M. S.C. is a recipient of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award. J.E.W. is a Rita Allen Foundation Scholar.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.284604.116.
References
- Albarino CG, Bird BH, Nichol ST. 2007. A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever sicilian, and Toscana viruses. J Virol 81: 5246–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. 1999. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J 18: 5399–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. 2013. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol 20: 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JS, Parker RP. 1998. The 3′' to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J 17: 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. 2011. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29: 185–214. [DOI] [PubMed] [Google Scholar]
- Barr JN. 2007. Bunyavirus mRNA synthesis is coupled to translation to prevent premature transcription termination. RNA 13: 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard L, Carroll K, Valle RCP, Wickner RB. 1998. Ski6p is a homolog of RNA-processing enzymes that affects translation of non-poly(A) mRNAs and 60S ribosomal subunit biogenesis. Mol Cell Biol 18: 2688–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird BH, Ksiazek TG, Nichol ST, Maclachlan NJ. 2009. Rift Valley fever virus. J Am Vet Med Assoc 234: 883–893. [DOI] [PubMed] [Google Scholar]
- Blackinton JG, Keene JD. 2014. Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin Cell Dev Biol 34: 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N. 2004. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303: 832–835. [DOI] [PubMed] [Google Scholar]
- Brennan-Laun SE, Ezelle HJ, Li X-L, Hassel BA. 2014. RNase-L control of cellular mRNAs: roles in biologic functions and mechanisms of substrate targeting. J Interferon Cytokine Res 34: 275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Bai X, Johnson AW. 2000. The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6: 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham AJ, Gong L, Hardy RW. 2007. Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 367: 212–221. [DOI] [PubMed] [Google Scholar]
- Callahan KP, Butler JS. 2008. Evidence for core exosome independent function of the nuclear exoribonuclease Rrp6p. Nucleic Acids Res 36: 6645–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli JC, Hassel BA, Wood KA, Li XL, Amemiya K, Dalakas MC, Torrence PF, Youle RJ. 1997. A study of the interferon antiviral mechanism: apoptosis activation by the 2-5A system. J Exp Med 186: 967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107: 451–464. [DOI] [PubMed] [Google Scholar]
- Cherry S. 2005. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev 19: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S, Perrimon N. 2004. Entry is a rate-limiting step for viral infection in a Drosophila melanogaster model of pathogenesis. Nat Immunol 5: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Ye W, Yang J, Han P, Wang Y, Ye C, Weng D, Zhang F, Xu Z, Lei Y. 2016. DDX21 translocates from nucleus to cytoplasm and stimulates the innate immune response due to dengue virus infection. Biochem Biophys Res Commun 473: 648–653. [DOI] [PubMed] [Google Scholar]
- Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, Stetson DB. 2014. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol 15: 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckwahl MJ, Sim S, Smith D, Telesnitsky A, Wolin SL. 2015. A retrovirus packages nascent host noncoding RNAs from a novel surveillance pathway. Genes Dev 29: 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasken MB, Leung SW, Banerjee A, Kodani MO, Chavez R, Bowman EA, Purohit MK, Rubinson ME, Rubinson EH, Corbett AH. 2011. Air1 zinc knuckles 4 and 5 and a conserved IWRXY motif are critical for the function and integrity of the Trf4/5–Air1/2–Mtr4 polyadenylation (TRAMP) RNA quality control complex. J Biol Chem 286: 37429–37445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filone CM, Hanna SL, Caino MC, Bambina S, Doms RW, Cherry S. 2010. Rift Valley fever virus infection of human cells and insect hosts is promoted by protein kinase Cε. PLoS One 5: e15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126. [DOI] [PubMed] [Google Scholar]
- Graham AC, Kiss DL, Andrulis ED. 2006. Differential distribution of exosome subunits at the nuclear lamina and in cytoplasmic foci. Mol Biol Cell 17: 1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TB, Boros LG, Chan KC, Hikim APS, Hudson AP, Swerdloff RS, Mitchell AP, Salameh WA. 2003. Spermatogenetic expression of RNA-binding motif protein 7, a protein that interacts with splicing factors. J Androl 24: 204–214. [DOI] [PubMed] [Google Scholar]
- Guo X, Carroll J-WN, Macdonald MR, Goff SP, Gao G. 2004. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J Virol 78: 12781–12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Ma J, Sun J, Gao G. 2007. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc Natl Acad Sci 104: 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson MP, Welcker M, Hwang HC, Clurman BE. 2005. Zcchc8 is a glycogen synthase kinase-3 substrate that interacts with RNA-binding proteins. Biochem Biophys Res Commun 338: 1359–1367. [DOI] [PubMed] [Google Scholar]
- Hassel BA, Zhou A, Sotomayor C, Maran A, Silverman RH. 1993. A dominant negative mutant of 2-5A-dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J 12: 3297–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessle V, Björk P, Sokolowski M, González de Valdivia E, Silverstein R, Artemenko K, Tyagi A, Maddalo G, Ilag L, Helbig R, et al. 2009. The exosome associates cotranscriptionally with the nascent pre-mRNP through interactions with heterogeneous nuclear ribonucleoproteins. Mol Biol Cell 20: 3459–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins KC, McLane LM, Maqbool T, Panda D, Gordesky-Gold B, Cherry S. 2013. A genome-wide RNAi screen reveals that mRNA decapping restricts bunyaviral replication by limiting the pools of Dcp2-accessible targets for cap-snatching. Genes Dev 27: 1511–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins KC, Tartell MA, Herrmann C, Hackett BA, Taschuk F, Panda D, Menghani SV, Sabin LR, Cherry S. 2015. Virus-induced translational arrest through 4EBP1/2-dependent decay of 5′-TOP mRNAs restricts viral infection. Proc Natl Acad Sci 112: 201418805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K-K, Schlee M, et al. 2006. 5′-triphosphate RNA is the ligand for RIG-I. Science 314: 994–997. [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D. 2008. The nuclear RNA surveillance machinery: the link between ncRNAs and genome structure in budding yeast? Biochim Biophys Acta 1779: 239–246. [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D. 2009. The many pathways of RNA degradation. Cell 136: 763–776. [DOI] [PubMed] [Google Scholar]
- Houseley J, LaCava J, Tollervey D. 2006. RNA-quality control by the exosome. Nat Rev Mol Cell Biol 7: 529–539. [DOI] [PubMed] [Google Scholar]
- Hsu CL, Stevens A. 1993. Yeast cells lacking 5′→3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol Cell Biol 13: 4826–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S. 2007. Characterization of Rift Valley fever virus transcriptional terminations. J Virol 81: 8421–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba S. 2004. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev 18: 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441: 101–105. [DOI] [PubMed] [Google Scholar]
- Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8: 533–543. [DOI] [PubMed] [Google Scholar]
- Kiss DL, Andrulis ED. 2011. The exozyme model: a continuum of functionally distinct complexes. RNA 17: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakofsky D, Hacker D. 1991. Bunyavirus RNA synthesis: genome transcription and replication. Curr Top Microbiol Immunol 169: 143–159. [DOI] [PubMed] [Google Scholar]
- Kurkela S, Manni T, Myllynen J, Vaheri A, Vapalahti O. 2005. Clinical and laboratory manifestations of Sindbis virus infection: prospective study, Finland, 2002–2003. J Infect Dis 191: 1820–1829. [DOI] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. 2005. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121: 713–724. [DOI] [PubMed] [Google Scholar]
- Lara E, Billecocq A, Leger P, Bouloy M. 2011. Characterization of wild-type and alternate transcription termination signals in the Rift Valley fever virus genome. J Virol 85: 12134–12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letchworth GJ, Rodriguez LL, Del Cbarrera J. 1999. Vesicular stomatitis. Vet J 157: 239–260. [DOI] [PubMed] [Google Scholar]
- Liang G, Liu G, Kitamura K, Wang Z, Chowdhury S, Monjurul AM, Wakae K, Koura M, Shimadu M, Kinoshita K, et al. 2015. TGF-β suppression of HBV RNA through AID-dependent recruitment of an RNA exosome complex. PLoS Pathog 11: e1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W-J, Duffy A, Chen C-Y. 2007. Localization of AU-rich element-containing mRNA in cytoplasmic granules containing exosome subunits. J Biol Chem 282: 19958–19968. [DOI] [PubMed] [Google Scholar]
- Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. 2011. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 43: 624–637. [DOI] [PubMed] [Google Scholar]
- Lubas M, Andersen PR, Schein A, Dziembowski A, Kudla G, Jensen TH. 2015. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep 10: 178–192. [DOI] [PubMed] [Google Scholar]
- Masison DC, Blanc A, Ribas JC, Carroll K, Sonenberg N, Wickner RB. 1995. Decoying the cap− mRNA degradation system by a double-stranded RNA virus and poly(A)– mRNA surveillance by a yeast antiviral system. Mol Cell Biol 15: 2763–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan L, Torchet C, Allmang C, Shipman T, Tollervey D. 2005. A nuclear surveillance pathway for mRNAs with defective polyadenylation. Mol Cell Biol 25: 9996–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita M, Oshiumi H, Matsumoto M, Seya T. 2011. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol 31: 3802–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SL, Wilusz J. 2013. Cytoplasmic viruses: rage against the (cellular RNA decay) machine. PLoS Pathog 9: e1003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser TS, Schieffer D, Cherry S. 2012. AMP-activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog 8: e1002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy RH, Cole BS, Yasunaga A, Gold B, Shankarling G, Varble A, Molleston JM, tenOever BR, Lynch KW, Cherry S. 2014a. Stem-loop recognition by DDX17 facilitates miRNA processing and antiviral defense. Cell 158: 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano MV, Yagi Y, Fitzgerald KA, Stanger BZ, Soldan SS, et al. 2014b. Antiviral autophagy restricts rift valley fever virus infection and is conserved from flies to mammals. Immunity 40: 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Decker CJ, Parker R. 1994. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′→3′ digestion of the transcript. Genes Dev 8: 855–866. [DOI] [PubMed] [Google Scholar]
- Mukherjee D, Gao M, O'Connor JP, Raijmakers R, Pruijn G, Lutz CS, Wilusz J. 2002. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J 21: 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R, Takeuchi R, Takata K-I, Shimanouchi K, Abe Y, Kanai Y, Ruike T, Ihara A, Sakaguchi K. 2008. TRF4 is involved in polyadenylation of snRNAs in Drosophila melanogaster. Mol Cell Biol 28: 6620–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onomoto K, Jogi M, Yoo J-S, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, et al. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7: e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orban TI, Izaurralde E. 2005. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 11: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314: 997–1001. [DOI] [PubMed] [Google Scholar]
- Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. 2005. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol 79: 15043–15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin LR, Zhou R, Gruber JJ, Lukinova N, Bambina S, Berman A, Lau C-K, Thompson CB, Cherry S. 2009. Ars2 regulates both miRNA- and siRNA- dependent silencing and suppresses RNA virus infection in Drosophila. Cell 138: 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Xu Z, Mathews DH, Butler JS. 2012. Air proteins control differential TRAMP substrate specificity for nuclear RNA surveillance. RNA 18: 1934–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Tollervey D. 2013. Threading the barrel of the RNA exosome. Trends Biochem Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Anderson JT, Tollervey D. 2007. The exosome subunit Rrp44 plays a direct role in RNA substrate recognition. Mol Cell 27: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS, Schmid S, Aguado LC, Sabin LR, Yasunaga A, Shim JV, Sachs D, Cherry S, tenOever BR. 2014. Drosha as an interferon-independent antiviral factor. Proc Natl Acad Sci 111: 7108–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. 2009. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30: 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300: 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S, Zhao R, Barsotti AM, Ouwehand A, Fazollahi M, Coutavas E, Breuhahn K, Neumann O, Longerich T, Pusterla T, et al. 2012. Nuclear pore component Nup98 is a potential tumor suppressor and regulates posttranscriptional expression of select p53 target genes. Mol Cell 48: 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58: 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanácová S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. 2005. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol 3: e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B. 2001. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-κB-dependent innate immune responses. Genes Dev 15: 1900–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, Freier SM, Spector DL. 2008. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135: 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, JnBaptiste CK, Lu LY, Kuhn C-D, Joshua-Tor L, Sharp PA. 2012. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26: 2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle J-C, Dufour M-E, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B. 2005. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737. [DOI] [PubMed] [Google Scholar]
- Xu J, Grant G, Sabin LR, Gordesky-Gold B, Yasunaga A, Tudor M, Cherry S. 2012. Transcriptional pausing controls a rapid antiviral innate immune response in Drosophila. Cell Host Microbe 12: 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga A, Hanna SL, Li J, Cho H, Rose PP, Spiridigliozzi A, Gold B, Diamond MS, Cherry S. 2014. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathog 10: e1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Hotta I, Denli AM, Hong P, Perrimon N, Hannon GJ. 2008. Comparative analysis of argonaute-dependent small RNA pathways in Drosophila. Mol Cell 32: 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.