

Abstract
The replication licensing factor CDC6 recruits the MCM2-7 replicative helicase to the replication origin, where MCM2-7 is activated to initiate DNA replication. MCM2-7 is activated by both the CDC7-Dbf4 kinase and cyclin-dependent kinase and via interactions with CDC45 and go-ichi-ni-san complex (GINS) to form the CDC45·MCM2-7·GINS (CMG) helicase complex. TIMELESS (TIM) is important for the subsequent coupling of CMG activity to DNA polymerases for efficient DNA synthesis. However, the mechanism by which TIM regulates CMG activity for proper replication fork progression remains unclear. Here we show that TIM interacts with MCM2-7 prior to the initiation of DNA replication. TIM depletion in various human cell lines results in the accumulation of aberrant CMG helicase complexes on chromatin. Importantly, the presence of these abnormal CMG helicase complexes is not restricted to cells undergoing DNA synthesis. Furthermore, even though these aberrant CMG complexes interact with the DNA polymerases on human chromatin, these complexes are not phosphorylated properly by cyclin-dependent kinase/CDC7-Dbf4 kinase and exhibit reduced DNA unwinding activity. This phenomenon coincides with a significant accumulation of the p27 and p21 replication inhibitors, reduced chromatin association of CDC6 and cyclin E, and a delay in S phase entry. Our results provide the first evidence that TIM is required for the correct chromatin association of the CMG complex to allow efficient DNA replication.
Keywords: cell cycle, cyclin-dependent kinase (CDK), DNA binding protein, DNA helicase, DNA replication, protein complex
Introduction
During cell growth, faithful DNA duplication is essential for correctly transmitting genetic material to daughter cells during cell division, and defects in this process can result in genome instability, tumorigenesis, or cell death. During DNA replication, double-stranded DNA must be unwound by the MCM2-7a hexameric helicase to generate ssDNA,2 which is then used as the template for DNA synthesis by DNA polymerase. However, even though there is an abundance of MCM2-7 protein on the chromatin throughout the cell cycle, most of the MCM2-7 is kept inactive, and only a small population is recruited and associated with active replicons to participate in DNA synthesis (1–3). During early G1 phase, the MCM2-7 protein is recruited to the active replication origins, where it forms part of the pre-replication complex, and its recruitment requires the CDC6 and CDT1 licensing factors (4, 5). To ensure the accurate timing of replication initiation, the activity of the chromatin-bound MCM2-7 protein is tightly regulated both by post-translational modifications and protein-protein interactions. Specifically, the catalytic activity of the MCM2-7 helicase is suppressed during G1 phase and can only be activated both via the interaction with GINS and CDC45, which forms the CMG complex, and via phosphorylation by DDK and CDK during replication initiation (6, 7).
Mammalian TIM is an essential protein for embryonic development (8). TIM and its interacting protein, TIPIN, are highly conserved replication factors that directly interact with the MCM2-7 helicase (9). TIM and TIPIN form a stable heterodimer, and this interaction is crucial for the stability and nuclear localization of both proteins in vivo (10–12). They are the mammalian homologs of Saccharomyces cerevisiae Tof1 and Csm3, respectively (13, 14). Tof1 and Csm3 are part of the replication progression complex that couples DNA unwinding and DNA synthesis activities and stabilizes replication forks at pause sites (15–18). Tof1 also plays a role in activating the DNA damage response pathway during S phase (19, 20). The functions of Tof1 and Csm3 are conserved in their vertebrate homologs, TIM and TIPIN (21, 22). For example, when cells encounter DNA damage during S phase, TIM-TIPIN dimers promote phosphorylation of CHK1, which activates the intra-S phase checkpoint response and arrests replication forks. In the absence of TIM-TIPIN, cells continue to synthesize damaged DNA, leading to catastrophic consequences, as demonstrated by increased cell death (21, 22). In undamaged cells, TIM dysfunction decreases the rate of replication fork progression and uncouples the DNA polymerase and MCM2-7 helicase activity (21). TIM-TIPIN also facilitates the loading of cohesin subunits to establish sister chromatid cohesions (23, 24). The role of TIM-TIPIN in cohesion establishment is consistent with the discovery of Csm3 and Tof1 mutations in genetic screens for chromosome segregation defects (14, 25).
Here we report a novel function of human TIM for the correct association of the CMG complex on chromatin. We found that TIM-TIPIN interacts with MCM2-7 not only during S phase but also throughout the whole cell cycle. Human cell lines treated with TIM siRNAs contain elevated amounts of the p21 and p27 replication inhibitors, and this phenotype coincides with a delay in S phase entry and decreased association of CDC6 and cyclin E with chromatin. As a consequence, there is reduced recruitment of MCM2-7 to the active replication origin. Unexpectedly, despite the inefficient recruitment of MCM2-7 to the active replication origin during G1 phase in TIM-deficient cells, the levels of chromatin-bound CMG complexes remain unchanged, and the presence of these CMG complexes on the chromatin is no longer restricted to S phase. Although these CMG complexes interact with DNA polymerases, the MCM4 subunit has an altered phosphorylation pattern at the DDK- and CDK-dependent PG sites, which are important for efficient DNA replication (26, 27). Our data unveil a novel role for TIM in preventing the accumulation of aberrant CMG complexes on the chromatin outside of S phase. We propose that the presence of these non-S phase CMG complexes with altered post-translational modifications acts as a false negative feedback signal to prevent CDC6 and cyclin E from binding to DNA, thereby hindering DNA replication in TIM-deficient cells.
Results
TIM Deficiency Leads to Inefficient S Phase Entry
Mammalian TIM is a component of the replication fork progression complex and is required for the efficient progression of replication forks during S phase (21, 22, 28). In addition, TIM promotes the sister chromatid cohesion necessary for proper chromosomal segregation during mitosis (23, 24). Reduced levels of cohesin complexes during early G1 phase can also lead to slow replication progression and can lengthen S phase by limiting the number of replication origins that fire (29). It is therefore expected that TIM deficiency would lead to the accumulation of S phase cells. To test this, we depleted TIM using two separate siRNAs in HEK293 cells (Fig. 1A, left panel) and human osteosarcoma U2OS cells (Fig. 1B, left panel). Consistent with a published report (11), we observed a destabilization of TIPIN in TIM knockdown cells using either of the TIM siRNAs (Fig. 1, A and B, left panels, second blots). The percentage of the asynchronized cell population undergoing DNA synthesis was monitored by measuring DNA content (Fig. 1, A and B, right panels, x axis) and BrdU incorporation by pulse-labeling the cells with BrdU prior to analysis (Fig. 1, A and B, right panels, y axis). Unexpectedly, BrdU incorporation analysis of the TIM siRNA-treated HEK293 (Fig. 1A, right panel) and U2OS (Fig. 1B, right panel) cells showed that the S phase population was reduced compared with the control cells.
FIGURE 1.

TIM knockdown cells show delayed S phase entry. A and B, left panels, the protein levels of TIM, TIPIN, p21, and p27 were analyzed by SDS-PAGE and Western blotting of whole cell extracts (WCE). HEK293 (A) and U2OS (B) cells were treated with either control or TIM siRNAs. Actin was used as the loading control. Right panels, flow cytometry to determine cell cycle distribution of asynchronized HEK293 (A) and U2OS (B) cells treated with control or TIM siRNAs. The cells were labeled with BrdU and co-stained for BrdU incorporation (y axis) and DNA content (propidium iodide, x axis). Percentages of cells in S phase are shown in black, and the differences in the percentages between control and TIM knockdown cells are shown in red. C, flow cytometry analysis of HEK293 cells treated with control or TIM siRNAs after release from nocodazole blocking at the indicated time points. 2C and 4C represent cells containing one or two copies of each chromosome, respectively. All data are representative of a minimum of two independent experiments. All Western blots in each subfigure were from the same lysate or experiment.
An accelerated replication fork progression rate could lead to a shorter S phase and, hence, a reduction in the S phase population. Alternatively, a decrease in the number of cells undergoing DNA synthesis may result from inefficient S phase entry. To test these possibilities, we evaluated the cell cycle progression of HEK293 cells that were transfected with either control or TIM siRNAs for 48 h and then synchronized to G2/M phase by nocodazole treatment for an additional 20 h. After nocodazole release, cell cycle progression was monitored by flow cytometry. Within the first 4–6 h after release, the number of G1 phase cells increased in both the control and TIM siRNA knockdown cultures (Fig. 1C, first and second columns). 12 h after nocodazole release, the majority of the control knockdown cells showed positive BrdU staining (Fig. 1C, fourth column). However, the initiation of DNA synthesis was delayed at 12 h after nocodazole release in the majority of TIM-depleted cells that had completed mitosis and entered G1 phase. Overall, these cell cycle analyses revealed an unexpected defect in S phase entry caused by TIM depletion.
High Levels of the Replication Inhibitors p21 and p27 Are Present in TIM-depleted Cells without Evidence of DNA Damage
Consistent with the observation of a reduced S phase population, elevated levels of p21 and p27 were present in TIM knockdown HEK293 and U2OS cells compared with control cells (Fig. 1, A and B, left panels, third and fourth blots). Expression of the SV40 large T antigen has been reported to stimulate the initiation of host DNA replication and reduce the expression levels of p21 and p27 (30, 31). However, HEK293T cells, which contain the SV40 large T antigen, also accumulated p21 and p27 when TIM was depleted (Fig. 2A, third and fourth panels). p21 and p27 block DNA replication by inactivating CDK2-cyclin E, which is important for G1/S phase transition (32). For cells to enter S phase, p21 and p27 must be ubiquitinated and degraded during late G1 phase to allow the activation of CDK2-cyclin E for replication initiation. Therefore, we asked whether the delay in S phase entry in TIM knockdown cells was associated with the persistent presence of p21 and p27. Indeed, when we synchronized HEK293T cells with nocodazole to various cell cycle stages (Fig. 2B), we found that, unlike in control knockdown cells, both p21 and p27 failed to be degraded in TIM-depleted cells 11 h after nocodazole release, when most of the control knockdown cells were in S phase (Fig. 2, B and C).
FIGURE 2.

TIM-deficient cells accumulate high levels of replication inhibitors without accumulating DNA damage. A, the protein levels of TIM, TIPIN, p21, and p27 were analyzed by SDS-PAGE and Western blotting of WCE from HEK293T cells, which contain the SV40 large T antigen, treated with either control or TIM siRNAs. Actin was used as the loading control. B, flow cytometry of HEK293T control (left panel) and TIM knockdown (right panel) cells after release from nocodazole block at the indicated time points. C, the protein levels of TIM, p21, p27, SKP2, and cyclin A were analyzed on Western blots of WCEs that were prepared from the synchronized control and TIM knockdown HEK293T cells shown in B. Actin was used as the loading control for SDS-PAGE. noc, nocodazole. D, the protein levels of TIM, CHK1, and p-CHK1 were analyzed by Western blotting using WCE prepared from control and TIM knockdown HEK293T cells with or without UV irradiation. E, quantification of the percentage of HEK293T cells (>200 cells/sample) containing ssDNA, based on positive staining of pulse-labeled BrdU under non-denaturing conditions 2, 6, and 13 h after release from nocodazole treatment. Error bars represent S.D. F—I, flow cytometry (left panels) and representative immunofluorescent staining of the pulse-labeled BrdU (right panels) in control siRNA-treated HEK293T cells (F and G) and TIM siRNA-treated cells (H and I) shown in E. J, the protein levels of TIM, SKP2, and p27 were analyzed by SDS-PAGE and Western blotting of WCE from HEK293T, U2OS, or HT0180 cells treated with either control or TIM siRNAs. Actin was used as the loading control. 2C and 4C represents cell containing one or two copies of each chromosome, respectively. All data are representative of a minimum of two independent experiments. All Western blots in each subfigure were from the same lysate or experiment.
TIM is important for replication fork stability and efficient DNA replication progression (21, 22). Therefore, we tested the possibility that the delay in S phase entry observed in TIM-depleted G1 cells was due to the presence of unreplicated or damaged DNA inherited from the previous cell cycle. Because DNA damage activates the ATR-CHK1-dependent checkpoint response during G1 phase (33, 34), we analyzed the level of p-CHK1 in TIM knockdown cells. Consistent with a previous report that TIM facilitates efficient CHK1 activation (35), we found that UV irradiation induced CHK1 phosphorylation in both control and TIM knockdown cells. However, the amount of UV-induced p-CHK1 was reduced in TIM knockdown cells (Fig. 2D). As anticipated, TIM depletion alone did not activate CHK1 in the absence of UV treatment (Fig. 2D).
To further confirm that TIM depletion does not stimulate DNA damage, we asked whether ssDNA accumulates during G1 phase. This ssDNA accumulation can occur because of either unreplicated or unresolved damaged DNA inherited from the previous cell cycle in TIM-depleted cells. To test this hypothesis, we carried out immunofluorescence microscopy using an anti-BrdU antibody to detect ssDNA foci formed under non-denaturing conditions in cells precultured with BrdU for 18 h. Under non-denaturing conditions, only the BrdU at ssDNA regions is recognized by the anti-BrdU antibody. ssDNA foci were normally visible only in cells synchronized to S phase (13 h) but not in those synchronized to G1 phase (6 h; Fig. 2, E–G). Even though ssDNA-positive cells were also observed during G1 phase when TIM was depleted (Fig. 2H), the number of ssDNA-positive G1 cells remained low (<7%). These results indicate that the inefficient S phase entry seen in the majority of the TIM-depleted cells (Fig. 1C) is not due to the presence of unreplicated or damaged DNA. In addition, even though at 13 h post nocodazole release ssDNA foci were detected in TIM-depleted cells, the number of ssDNA-positive cells (Fig. 2E) and the number of ssDNA foci per cell were reduced in these cells compared with control knockdown cells (Fig. 2, compare I with G). Together, these results are consistent with reduced efficiency of DNA replication initiation rather than DNA damage.
Reduced Chromatin Association of CDC6 and Cyclin E in TIM-depleted Cells
We further determined the cause for the persistent presence of p21 and p27 in TIM-depleted cells. The degradation of p21/p27 during late G1 phase is mediated by SCFSKP2-dependent ubiquitination (36). We found that the SKP2 protein level was reduced in HEK293T, U2OS, and HT1080 cells (Fig. 2J). This reduction is most likely due to the fact that even though SKP2 was visible during M phase, it failed to accumulate during G1 and S phases in TIM knockdown cells (Fig. 2C, fourth panel). APC/C is known to degrade SKP2 (37). However, the level of cyclin A, which is an APC/C substrate (38), remained similar to the level in control cells (Fig. 2C, fifth panel). Therefore, the low level of SKP2 in the absence of TIM is unlikely to result from increased APC/C activity.
In addition to APC/C-dependent degradation of SKP2, the cellular level of SKP2 is also controlled by E2F-dependent transcription (39–41). Prior to replication initiation, E2F activity is suppressed by the binding of Rb at promoters (42–45). During late G1 phase, cyclin E-CDK2 is recruited to the chromatin to phosphorylate Rb and disrupt the Rb-E2F interaction at promoters, allowing the activation of E2F-dependent transcription (42, 46). Therefore, we analyzed the levels of chromatin-bound cyclin E and found that the amount of cyclin E on the chromatin was substantially reduced in TIM knockdown cells (Fig. 3A). Furthermore, the level of chromatin-bound CDC6, which is required for chromatin recruitment and the activation of cyclin E-CDK2 activity (47–49), was also reduced in TIM knockdown cells (Fig. 3A).
FIGURE 3.

Reduced chromatin recruitment of CDC6 and cyclin E in TIM knockdown cells. A, Western blotting analyses of CB ORC2, CDC6, cyclin E, MCM2-7 (represented by MCM7), CDC45, and GINS (represented by SLD5 and PSF2) in the soluble chromatin fractions isolated from HEK293, HEK293T, and U2OS cells treated either with control or TIM siRNAs. Histone H3 was used as the loading control. B, Western blotting analyses of CDC6, MCM2-7 (represented by MCM7) and GINS (represented by SLD5) in the cytoplasmic (Cyt), nucleoplasmic (Nuc), and CB fractions prepared from HEK293T cells treated with control or CDC6 siRNA. C, Western blotting analysis of MCM5, ORC2, and active RNAPII isolated from ChIP in formaldehyde-cross-linked control and TIM knockdown cells. D and E, real-time PCR analysis of the relative abundance of MCM5, ORC2, and RNAPII bound to the lamin B2 origin in HEK293 cells (D) and HEK293T cells (E) 6 h after nocodazole release, when cells were predominantly in G1 phase. ChIP using IgG alone was used as the background level, and the relative abundance of MCM5, ORC2, and RNAPII shown was after subtracting the background signal from IgG ChIP. Each value represents the mean ± S.D. calculated from triplicate PCRs per one representative experiment. All data are representative of a minimum of two independent experiments. All Western blots in each subfigure were from the same lysate or experiment.
The chromatin-bound CDC6 is necessary for the recruitment of MCM2-7 to the active origins during G1 phase and the subsequent interaction with CDC45 and GINS to form the CMG complex and initiate DNA replication (4, 6, 7). Therefore, in CDC6 siRNA knockdown cells, chromatin recruitment of the components of the CMG complex, such as SLD5 and MCM7, was reduced (Fig. 3B). Because the amount of chromatin-bound CDC6 was reduced in TIM-depleted cells, we expected that the recruitment of MCM2-7 helicase to the active origin would be compromised in TIM knockdown cells. Indeed, the amount of MCM5 found at the active lamin B2 origin was reduced in TIM knockdown cells compared with control knockdown cells synchronized to G1 phase (Fig. 3, C–E). On the other hand, there was no significant change in the amount of origin-bound ORC (Fig. 3C-E, ORC2), whose association with the origin is independent of CDC6 (50). In summary, the reduced levels of chromatin-bound CDC6 and cyclin E are consistent with a low level of SKP2, reduced recruitment of MCM2-7 helicase to the replication origin, the accumulation of p21/p27, and the inefficient S phase entry observed in TIM knockdown cells.
CMG Helicase Complexes Accumulate on Human Chromatin in Non-S Phase, TIM-depleted Cells
Surprisingly, even though the levels of the chromatin-bound CDC6 and the origin-bound MCM2-7 in TIM knockdown cells were reduced, we found that components of the CMG complex, including the GINS (SLD5 and PSF2) and CDC45, were still found on the chromatin (Fig. 3, A, fifth through seventh panels, and B). To further determine whether these chromatin-bound GINS and CDC45 proteins were part of CMG complexes, we purified the CMG complexes using FLAG-CDC45 from U2OS (Fig. 4A) and HEK293 (Fig. 4B) cells that were treated with either control or TIM siRNA. Unexpectedly, the protein-protein interactions among the components of the CMG complex in TIM knockdown cells were still detected (Fig. 4, A and B, compare the first and second columns). Given that CDC6 is required to establish the pre-replication complexes and subsequently assemble the CMG complexes in normal cells (5), we tested whether depleting CDC6 in TIM knockdown cells would eliminate these CMG complexes. When the cells were treated with CDC6 siRNA for 60 h, we found that all of the CMG complexes were abolished in these cells (Fig. 4, A and B, compare the first and third columns). However, depletion of CDC6 in TIM knockdown cells (e.g. TIM-CDC6 double knockdown) failed to eliminate the chromatin-bound CMG complexes (Fig. 4, A and B, compare the third and fourth columns). To exclude the possibility that the chromatin-bound CMG complexes in the CDC6-TIM double knockdown cells were due to an increase in the S phase population compared with CDC6-deficient cells, we analyzed the cell cycle distribution of HEK293 knockdown cells by flow cytometry. We found that the S phase population was decreased in TIM, CDC6, and TIM-CDC6 knockdown cells compared with control cells (Fig. 4C). In addition, even though CMG complexes were formed in TIM-CDC6 knockdown cells, these cells still showed severely disrupted cell cycle distributions similar to CDC6 knockdown cells (Fig. 4C). To further confirm that the accumulation of these CMG complexes on the chromatin were caused by TIM deficiency, we exogenously expressed an siRNA-resistant TIM construct in CDC6-TIM double knockdown cells. We found that the exogenously expressed TIM proteins stabilized endogenous TIPIN (Fig. 4B, second panel, right lane) and suppressed the CMG complexes (Fig. 4B, bottom four panels, compare the fifth and sixth lanes).
FIGURE 4.

TIM knockdown cells accumulate aberrant CMG helicase complexes. A, top panels, protein levels of TIM, CDC6, and MCM7 were analyzed on Western blots of WCE prepared from U2OS cells stably expressing FLAG-CDC45 and treated with control, TIM, CDC6, or TIM-CDC6 (double knockdown) siRNAs. Bottom panels, Western blotting analyses of FLAG-CDC45 complexes immunopurified (IP) from soluble chromatin fractions isolated from the corresponding knockdown cells to detect the presence of CDC45 (FLAG), MCM2-7 (MCM2 and MCM7), and GINS (SLD5 and PSF2). B, Western blotting analyses of the protein levels of TIM, TIPIN, and CDC6 in WCE (top panels), and the presence of MCM7, SLD5, and MCM10 in the FLAG-CDC45 prepared from HEK293 cells stably expressing FLAG-CDC45 (bottom panels), as performed in (A), with the exception that a lane containing WCE and FLAG-CDC45 IP prepared from TIM-CDC6 double knockdown cells complemented with a siRNA-resistant TIM expression construct was also included. C, flow cytometry analysis of HEK293 cells treated with control, TIM, CDC6, or CDC6-TIM double siRNAs. Percentages of cells in S phase are shown in black, and the differences in the percentages between control and knockdown cells are shown in red. All data are representative of a minimum of two independent experiments. All Western blots in each subfigure were from the same lysate or experiment.
Because the accumulation of CMG complexes in TIM knockdown cells cannot be eliminated by CDC6 depletion, we next asked whether the chromatin association of these CMG complexes still takes place only in S phase. When we purified chromatin-bound FLAG-CDC45 from control knockdown HEK293T cells synchronized to either early G1 (4 h) or S phase (12 h) after nocodazole release (Fig. 5A), we found that, as expected, MCM2-7 and GINS were co-purified with CDC45 during S phase but not during early G1 phase (Fig. 5B). However, CMG complexes were detected in an abundant amount in both G1 phase and S phase cells, when TIM was depleted (Fig. 5, A and B), suggesting that the presence of CMG is no longer restricted to S phase when TIM is absent. Previously, we showed that human RECQ4 DNA helicase forms a chromatin-specific complex with MCM10, MCM2-7, CDC45, GINS, and TIM-TIPIN during S phase to promote efficient DNA replication initiation (51). Here we found that the interaction of RECQ4 and MCM10 with CMG also failed to be eliminated by CDC6 depletion in TIM knockdown cells (Figs. 4B and 5, C and D). Importantly, in control knockdown cells, RECQ4 only interacted with CMG in cells harvested 11 h after nocodazole release, when a substantial portion of the cells were in S phase (Fig. 5, E, bottom panel, and F, fifth lane). However, in TIM knockdown cells, we found that an increasing amount of CMG was co-purified with FLAG-RECQ4 from the human chromatin as early as 2 h after nocodazole release, when cells were undergoing mitosis (Fig. 5, E, top panel, and F, second lane). Together, these results provide evidence that, in TIM knockdown cells, the CMG complexes are present on the chromatin even when they are not undergoing DNA synthesis.
FIGURE 5.

Aberrant CMG helicase complexes accumulate on the chromatin of non-S phase cells depleted with TIM. A, flow cytometry of control cells (left panel) and TIM knockdown HEK293T cells (right panel) stably expressing FLAG-CDC45 at different time points after release from nocodazole block. B, Western blotting analysis of FLAG-CDC45 complexes immunopurified from the soluble chromatin fractions of control and TIM knockdown cells synchronized at different cell cycle stages, as described in A, to detect the presence of CDC45 (FLAG), TIM, MCM2-7 (MCM5 and MCM7), and GINS (SLD5 and PSF2). Noc, nocodazole. C, the protein levels of TIM, CDC6, and SLD5 were analyzed by Western blotting using WCE prepared from control, TIM knockdown, CDC6 knockdown, and TIM-CDC6 double knockdown HEK293T cells expressing FLAG-RECQ4. D, Western blotting analysis of FLAG-RECQ4 complexes immunopurified from the soluble chromatin fractions of the corresponding knockdown cells described in C using antibodies against FLAG, MCM10, MCM7, CDC45, and SLD5. E, flow cytometry of control cells (left panel) and TIM knockdown cells (right panel) stably expressing FLAG-RECQ4 at multiple time points after release from nocodazole block. F, Western blotting analysis of FLAG-RECQ4 complexes immunopurified from the soluble chromatin fractions of control and TIM knockdown cells synchronized at various cell cycle stages, as described in E, to detect the presence of RECQ4 (FLAG), TIPIN, MCM10, MCM2-7 (MCM7), CDC45 and GINS (SLD5 and PSF2). G, Western blotting analysis of FLAG-RECQ4 complexes immunopurified from the chromatin fractions prepared from control cells and TIM siRNA knockdown cells using anti-FLAG antibody. H, helicase activity of equal amounts of FLAG-RECQ4 complex purified from control cells or TIM knockdown HEK293T cells assayed using 32P-labeled splayed-arm substrates. The 32P-labeled ssDNA products were visualized by autoradiography following neutral PAGE. All data are representative of a minimum of three independent experiments. All Western blots in each subfigure were from the same lysate or experiment. 2C and 4C represents cell containing one or two copies of each chromosome, respectively.
The CMG Complexes in TIM-depleted Cells Exhibit Altered Patterns of MCM4 Phosphorylation
Despite the increased amount of the RECQ4-CMG helicase complex (Fig. 5F), TIM knockdown cells showed inefficient DNA synthesis. We hypothesized that the RECQ4-CMG replicative helicase complexes that accumulate in TIM knockdown cells could be catalytically defective. Indeed, when the helicase activity of equal amounts of CMG-containing FLAG-RECQ4 helicase complex purified from the TIM knockdown and control cells was compared (Fig. 5G), the amount of single-stranded dissociation product in the presence of the TIM knockdown RECQ4-CMG complex was significantly reduced compared with that of the control cell complex (Fig. 5H).
We next wished to determine the cause of the decreased catalytic activity of the CMG-containing helicase complex in TIM knockdown cells. It has been established that both CDK and DDK are required for the assembly and activation of the CMG complex at active replication origins and that MCM4 is a direct target of DDK and CDK during replication initiation (27, 52–55). Indeed, chromatin-bound MCM4 was present in multiple modified forms that exhibited slower mobility on SDS-PAGE, and the presence of this modified MCM4 protein was abolished by treatment with inhibitors of either the CDC7 component of DDK or CDK (Fig. 6A). Given that, in the absence of TIM, the interactions of CDC45 and GINS with the MCM2-7 helicase are no longer restricted to S phase (Fig. 5, B and F), we next determined whether the CMG complexes that formed in TIM knockdown cells contained the expected post-translational modifications. First, we measured the levels of phosphorylated MCM4 by SDS-PAGE mobility shift in the purified chromatin-bound FLAG-MCM7 complex. We found a reduction in phosphorylated MCM4 when TIM was depleted (Fig. 6B, fourth panel). We used an antibody against phosphorylated Ser/Thr residues to further evaluate the presence of phosphorylated MCM4. Consistent with the observed change in MCM4 mobility, we also observed reduced levels of phosphorylated polypeptides with SDS-PAGE mobility between 100 and 150 kDa, where phosphorylated MCM4 was found in these purified complexes (Fig. 6B, fifth panel).
FIGURE 6.

The aberrant CMG helicase complexes in TIM knockdown cells exhibit altered posttranslational modifications. A, Western blotting analysis of chromatin-bound FLAG-MCM4 purified from HEK293T cells treated with DMSO, CDC7 inhibitor (PHA 767491), or CDK inhibitor (roscovitine). B, Western blotting analysis of TIM, MCM2, MCM4, and phosphorylated polypeptides in chromatin-bound FLAG-MCM7 complexes purified from control and TIM knockdown cells. C, Western blotting analysis of MCM4, MCM7, and MCM2 of the chromatin-bound FLAG-MCM7 complex purified from control or TIM knockdown HEK293T cells treated with DMSO or APH. D, amino acid sequence of the first 150 residues of human MCM4. The three intrinsic DDK target sites (S/T-D/E) are underlined, and the six PG sites (S/T-S/T-P) are shown in bold. E, Western blotting analysis of chromatin-bound FLAG-MCM4 WT, the phosphomimetic DDK mutant (3E-D/E), and the phosphomimetic PG mutant (6E-EP) purified from control and TIM knockdown cells. F, Western blotting analysis of chromatin-bound FLAG-MCM4 WT and phosphodefective PG mutant (6A-AP) purified from control and TIM knockdown cells. G, Western blotting analysis of MCM4 in purified FLAG-CDC45 or FLAG-MCM7 complexes treated with or without λ phosphatase (λ PPase). FLAG-CDC45 and FLAG-MCM7 were detected using an anti-FLAG antibody. H, Western blotting analysis of chromatin-bound FLAG-CDC45 complexes purified from HEK293 cells (left two lanes) or U2OS cells (right two lanes) treated with control or TIM siRNA. I, Western blotting analysis of chromatin-bound FLAG-CDC45 complexes purified from control or TIM siRNA-treated HEK293T cells. The purified complexes were analyzed using antibodies to detect MCM2-7 (MCM4, MCM2), GINS (SLD5), CDC45 (FLAG), and DNA polymerase δ (Pol δ). All data are representative of a minimum of three independent experiments. All Western blots in each subfigure were from the same lysate or experiment.
We next tested whether the altered phosphorylation pattern of the chromatin-bound MCM4 is due to the accumulation of stalled replication forks. It has been shown that replication fork arrest by hydroxyurea or APH leads to ATR-induced MCM4 hyperphosphorylation as part of the replication block checkpoint response (56). Indeed, we observed an increase in hyperphosphorylated MCM4 in APH-treated cells (Fig. 6C, compare the first and third lanes). However, this APH-induced hyperphosphorylation of MCM4 is distinct from the MCM4 phosphorylation pattern found in TIM knockdown cells (Fig. 5C, compare the second and third lanes). This APH-induced MCM4 hyperphosphorylation is not dependent on TIM, as this phenomenon was also observed in TIM knockdown cells treated with APH (Fig. 6C, fourth lane). This result indicates that the reduced phosphorylation of MCM4 on chromatin in the absence of TIM is not likely to be a consequence of replication fork arrest.
In yeast, multiple DDK-dependent phosphorylation sites, which are important for DNA replication, have been identified within the N terminus of MCM4 (27). In humans, the MCM4 N terminus contains three intrinsic DDK target sites (Ser or Thr followed by Asp or Glu (Ser/Thr-Asp/Glu)), and they are Ser26, Ser131, and Ser141 (Fig. 6D, underlined residues). We found that a phosphomimetic mutant containing Ser-to-Glu mutations at the three intrinsic DDK sites (Fig. 6E, 3 E-D/E) failed to rescue the alternation of MCM4 phosphorylation levels in TIM knockdown cells (Fig. 6E, compare the four left lanes), suggesting that the phosphorylation of these intrinsic DDK target sites is not affected by TIM. In addition to the intrinsic DDK target sites, DDK can also target the first Ser/Thr of the MCM4 PG sites (phosphorylated Ser/Thr-Ser/Thr-Pro) when the second Ser/Thr is already phosphorylated by CDK (26, 27, 57). Human MCM4 contains six PG sites within the N terminus: Ser2, Ser6, Ser31, Thr53, Ser70 and Ser87 (Fig. 6C, residues in bold). We found that phosphomimetic mutations at these PG sites (e.g. Fig. 6E, 6E-EP) or phosphodefective mutations (e.g. Fig. 6F, 6A-AP) eliminated the difference in MCM4 mobility on SDS-PAGE between control and TIM knockdown cells, suggesting that the chromatin-bound MCM2-7 helicase in the absence of TIM may not contain properly phosphorylated PG sites.
The PG phosphorylation events on MCM4, which are necessary for CMG assembly and DNA synthesis, are tightly regulated during the cell cycle (26, 27). Restricting CDC45 and GINS to only interacting with the PG-phosphorylated MCM2-7 helicase ensures the correct timing of CMG activation and replication initiation in normal cells. Indeed, CDC45 normally interacts only with the MCM2-7 helicase, which contains hyperphosphorylated MCM4. This hyperphosphorylated MCM4 can be converted to unphosphorylated MCM4 by λ phosphatase, as demonstrated by FLAG-CDC45 immunoprecipitation (Fig. 6G, compare the first and third lanes). Because there is an excessive amount of inactive MCM2-7 protein on the chromatin (1, 2), the majority of the chromatin-bound MCM2-7, which was purified using FLAG-MCM7, was not hyperphosphorylated (Fig. 6G, second lane). Hence, the CDC45-associated, hyperphosphorylated MCM2-7 was a minor species of the total MCM2-7 helicases (Fig. 6G, compare the first and second lanes). When we analyzed the phosphorylation states of the CMG complexes purified from HEK293T, U2OS, and HEK293 cells stably expressing FLAG-CDC45, we found that the complexes purified from TIM knockdown cells contained differently modified forms of MCM4 with faster mobility (Fig. 6, H and I). Even though these CMG complexes were capable of interacting with DNA polymerases such as Pol δ (Fig. 6I, fifth panel), these CMG helicase complexes were not likely to support efficient DNA replication because of their altered phosphorylation states. Although we cannot exclude the possibility that other post-translational modifications on CMG complexes may be affected by TIM depletion, our data indicate that TIM is at least important for preventing the accumulation of CMG complexes that are not phosphorylated correctly at the PG sites.
The Interaction of TIM with MCM2-7 Is Independent of the Cell Cycle
The ability of TIM to prevent the accumulation of the aberrant CMG complexes in non-S phase cells suggests that TIM may associate with the MCM2-7 helicase independent of DNA synthesis. However, TIM is only known to be a component of the replication fork progression complex (22). Therefore, we looked at the kinetics of the interaction between TIM and MCM2-7 throughout the cell cycle. First, we found that the stable interaction of TIM-TIPIN with MCM2-7 takes place on the chromatin (Fig. 7A). Interestingly, when we examined a HEK293T cell line stably expressing FLAG-MCM7, we found that, although CDC45 and GINS primarily associate with the MCM2-7 helicase during S phase (11 h), TIM-TIPIN interacts with MCM2-7 on the chromatin throughout the cell cycle (Fig. 7, B and C). On the other hand, TIM was primarily enriched in the FLAG-CDC45 complex purified from S phase cells (11 h) compared with the other phases (Fig. 7, D and E), and this result further supports the interaction of TIM with MCM2-7 prior to the interaction between MCM2-7 and CDC45 to form CMG. Consistent with this finding, we showed that CDC6 was also co-purified with TIM in the CB fraction (Fig. 7A). In addition, MCM2-7 could still be co-purified with TIM and TIPIN in the absence of CDC6, RECQ4, or MCM10 (Fig. 7, F and G), all of which are proteins that interact with MCM2-7 during G1 phase and G1/S transition to facilitate CMG assembly in human cells (51, 58). Therefore, we concluded that the interaction of TIM-TIPIN with MCM2-7 is independent of the cell cycle.
FIGURE 7.
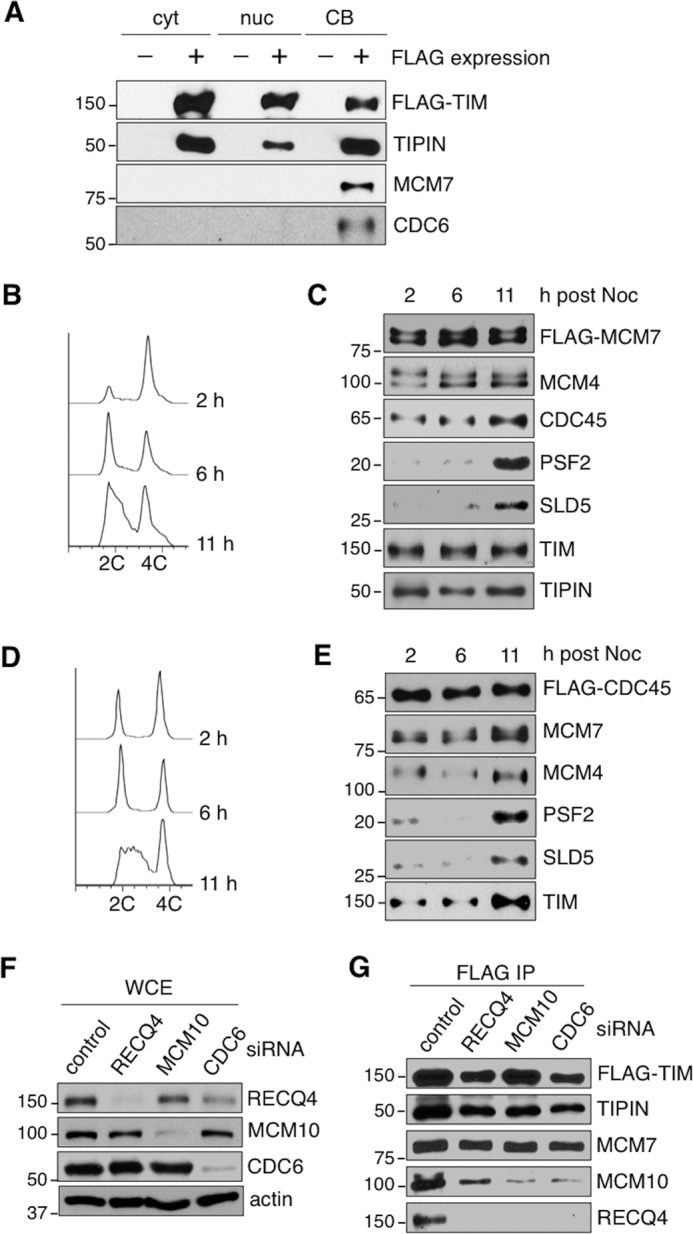
TIM-TIPIN associates with MCM2-7 prior to DNA replication initiation. A, Western blotting analysis for the presence of TIM, TIPIN, MCM2-7 (MCM7), and CDC6 in FLAG complexes purified from the cytoplasmic (cyt), nucleoplasmic (nuc), and CB fractions prepared from HEK293T cells transfected with a control vector or a vector expressing FLAG-TIM. B, flow cytometry of HEK293T cells stably expressing FLAG-MCM7 at the indicated time points after release from nocodazole block. C, Western blotting analysis of FLAG-MCM7 complexes immunopurified with anti-FLAG M2-agarose from soluble chromatin-bound fractions of HEK293T cells synchronized at different time points after nocodazole (Noc) release as described in B. The blots were probed with the indicated antibodies to detect the presence of MCM2-7 (MCM4 and FLAG), GINS (PSF2 and SLD5), CDC45, TIM, and TIPIN. D, flow cytometry of HEK293T cells stably expressing FLAG-CDC45 at the indicated time points after release from nocodazole block. E, Western blotting analysis of the FLAG-CDC45 complex immunopurified with anti-FLAG M2-agarose from soluble chromatin-bound fractions of HEK293T cells synchronized at different time points after nocodazole release as described in D. The purified complexes were analyzed with the indicated antibodies to detect the presence of MCM2-7 (MCM4 and MCM7), GINS (PSF2 and SLD5), and TIM. F, the protein levels of RECQ4, MCM10, and CDC6 were analyzed by Western blotting using WCE prepared from control, RECQ4, MCM10, and CDC6 knockdown cells stably expressing FLAG-TIM. G, Western blotting analysis of FLAG-TIM complexes immunopurified from the soluble chromatin fractions of the corresponding knockdown cells described in A to detect the presence of TIM (FLAG), TIPIN, MCM2-7 (MCM7), MCM10, and RECQ4. All data are representatives of a minimum of two independent experiments. All Western blots in each subfigure were from the same lysate or experiment. 2C and 4C represents cell containing one or two copies of each chromosome, respectively.
Discussion
This is the first report that demonstrates a function for human TIM in ensuring the correct chromatin association of the CMG complexes during the cell cycle (Fig. 8). During the normal cell cycle, CMG is only present on the chromatin during S phase to initiate DNA replication and allow cells to progress through S phase (5). We showed that TIM and TIPIN stably interact with the MCM2-7 helicase throughout the cell cycle (Fig. 7) and that TIM is important for preventing aberrant CMG complexes from accumulating on the chromatin when cells are not in S phase (Fig. 5, B and F). As a consequence, in the absence of TIM, aberrant CMG complexes containing MCM4, with altered phosphorylation at PG sites (Fig. 6), accumulate on the chromatin even in non-S phase cells (Fig. 8). Despite their abundance in TIM knockdown cells, these abnormal CMG complexes are likely to be catalytically altered (Fig. 5H).
FIGURE 8.

Model for TIM-TIPIN function in facilitating efficient DNA replication initiation by suppressing chromatin accumulation of aberrant CMG complexes. In normal cells, TIM-TIPIN is known to travel with the CMG complex to maintain proper replication fork progression (left). However, TIM-TIPIN also interacts with MCM2-7 outside of S phase. We suggest that, after completion of DNA synthesis, the interaction of TIM-TIPIN with MCM2-7 may be responsible for the disassembly and dissociation of the CMG from the chromatin prior to mitosis or entry to the next round of the cell cycle. In the absence of TIM, aberrant CMG complexes are present on the chromatin during G1 phase (right). The presence of these abnormal CMG complexes may provide false signals to prevent CDC6-dependent CMG assembly at active origins and initiate DNA replication.
What is the molecular mechanism by which TIM prevents the accumulation of aberrant CMG complexes in non-S phase cells? After DNA synthesis is completed, CMG is disassembled by CDC48-dependent ubiquitination (59). It is possible that TIM has a role in the disassembly or dissociation of the CMG complexes from the chromatin by facilitating or preserving this modification (Fig. 8). In TIM-deficient cells, the persistent chromatin association of the CMG complexes after DNA replication may continue to the next cell cycle, leading to the appearance of these CMG complexes on the chromatin prior to the next round of DNA replication. Because CDC6 is not known to be involved in the disassembly of the CMG complex, CDC6 depletion fails to eliminate these aberrant CMG complexes accumulated on the DNA in TIM knockdown cells (Fig. 4). Even though these CMG complexes remain on the DNA, they might still be targeted for dephosphorylation, leading to the altered phosphorylation pattern observed on the CMG complexes (Fig. 6). Alternatively, we cannot exclude the possibility that, in the absence of TIM, the accumulation of the CMG complex in the non-S phase cells may be a consequence of aberrant MCM2-7 chromatin loading or CMG assembly. In cells, CDC6 has been implicated in loading MCM2-7 at both active origins and the excess dormant origins, and this may be achieved by CDC6 continuously loading MCM2-7 at the active origin and releasing the loaded MCM2-7 to allow the helicase to slide away from the active origin (60, 61). However, in vitro, purified MCM2-7 helicase alone is capable of binding to DNA (53, 62). Furthermore, interactions among MCM2-7, CDC45, and GINS can form in the absence of CDC6 in vitro using recombinant proteins (53, 63). Most likely, negative regulatory mechanisms to minimize CDC6-independent DNA binding of MCM2-7 and CMG assembly may exist in cells. Indeed, multiple phosphorylation events that inhibit MCM2-7 binding to DNA have been reported (64, 65). In addition, in yeast, the SLD2 and SLD3 initiation factors interact with MCM2-7 helicase to block its interaction with CDC45 and GINS, and phosphorylation of SLD2 by CDK removes the block, thereby allowing the CMG complex to assemble (63, 66, 67). It is possible that TIM interacts with MCM2-7 prior to S phase to ensure that the MCM2-7 helicase can only be loaded onto the DNA via a CDC6-dependent pathway to form proper CMG complexes. In addition, mammalian TIM-TIPIN can interact with the MCM3, 4, 5, 6, and 7 subunits (9), whereas CDC45 and GINS interact with MCM2, 3, and 5 (68). It is plausible that TIM-TIPIN behaves similarly to yeast SLD2 and SLD3 and that the interactions of MCM2-7 with TIM-TIPIN and CDC45/GINS are normally mutually exclusive but can simultaneously interact with the MCM2-7 helicase after post-translational modification when cells enter S phase. Hence, in the absence of TIM, the interactions of MCM2-7 with CDC45 and GINS are not regulated and may form prior to DNA synthesis (Fig. 8). If so, it will be interesting to determine whether the interface of the MCM2-7 helicase that interacts with TIM-TIPIN can be modified at active replicons by CDK or DDK in a CDC6-dependent manner to allow the simultaneous interaction of CDC45 and GINS with MCM2-7 when TIM-TIPIN is present.
We further demonstrated that, in addition to preventing the accumulation of aberrant CMG complexes in non-S-phase cells, TIM contributes to the stable chromatin association of CDC6 and cyclin E (Fig. 3A). We propose the following possible mechanism by which TIM promotes the association of CDC6 with chromatin. CDC6 binding to DNA is tightly regulated to avoid re-replication during each round of the cell cycle. Possible mechanisms to prevent CDC6 from binding to DNA after replication initiation include nuclear export and cyclin A-dependent phosphorylation (4). It is possible that the assembly of CMG complexes may also provide a signal to release CDC6 from DNA for nuclear export to avoid re-replication. If so, the presence of the aberrant CMG complexes on the chromatin during G1 phase could provide false signals that either prevent CDC6 from binding to chromatin or weaken its DNA binding (Fig. 8).
When cells enter G1 phase of the cell cycle, CDC6 is recruited by ORC to the origin of replication (69), and this is followed by the chromatin recruitment of the MCM2-7 helicase and CDK2-cyclin E (Fig. 8) (70, 71). The chromatin binding and activation of CDK2-cyclin E provide a positive feedback loop that further enriches CDC6 on the DNA. This enrichment of CDC6 on the DNA can be achieved by protecting CDC6 from APC/C-dependent degradation (72) and activating the E2F transcription factor (73), which further amplifies the expression of CDC6 (74, 75). E2F-dependent transcription is also necessary for accumulation of SKP2, a component of the SCFSKP2 ubiquitin ligase, which promotes replication initiation by degrading p27 and p21 (36) during late G1 phase (39–41, 76). The reduced chromatin loading of CDC6 observed in TIM knockdown cells may explain why these cells also exhibited decreased levels of DNA-bound cyclin E (Fig. 3A), reduced recruitment of MCM2-7 helicase to the active replication origin (Figs. 3D and 8), reduced MCM4 phosphorylation (Fig. 6), low levels of SKP2 in G1 and S phases (Fig. 2C), high levels of p21 and p27 throughout the cell cycle (Figs. 1, A and B, and 2A), and a delay in S phase entry (Figs. 1C and 2B). The presence of these altered CMG helicase complexes may also explain the slow rate of replication fork progression that is observed in TIM knockdown cells when the cells start DNA synthesis (12).
Experimental Procedures
Plasmids
The pCMV-FLAG-RECQ4 plasmid was constructed as described previously (51). The MCM4, MCM7, and CDC45 cDNAs were purchased from Open Biosystems. The MCM4 and MCM7 cDNAs were subcloned into the Nde1 and EcoR1 sites of the pCMV-FLAG vector. The CDC45 cDNA was subcloned into the NdeI and XhoI sites of the pCMV-FLAG vector. The primer sequences were as follows: MCM7FW, 5′-GGA ATT CCA TAT GGC ACT GAA GGA CTA CGC-3′; MCM7RV, 5′-GGT GCA GGA AGA GCG ACA AAA GTG ATC CGT GTC-3′; MCM4FW, 5′-GGA ATT CCA TAT GTC CCC GGC GTC G-3′; MCM4RV, 5′-CCG GAA TTC GAG CAA GCG CAC GGT CTT CC-3′; CDC45FW, 5′-GGA ATT CCA TAT GTT CGT GTC CGA TTT CCG-3′; and CDC45RV, 5′-CCG CTC GAG CTA GGA CAG GAG GGA AAT AAG-3′. Mutations of putative phosphorylation sites in MCM4 were generated using the QuikChange® multisite-directed mutagenesis kit (Agilent Technologies). MCM4–3E represents mutants in which the Ser residues at positions Ser26, Ser131, and Ser142 were mutated to Glu. MCM4–6EE contains mutations in which the Ser and Thr residues at positions Ser2Ser3, Ser6Thr7, Ser31Ser32, Thr53Ser54, Ser70Ser71, and Ser87Ser88 were mutated to Glu. The primers used for mutagenesis were as follows: S26E, 5′-GCC CAG ACG CCT CGG GAG GAG GAT GCC AGG TCA-3′; S131E, 5′-GCA AGT GGA TCT GCA GGA GGA CGG GGC AGC AGC AG-3′; S142E, 5′-GCA GCA GAA GAT ATA GTG GCA GAG GAG CAG TCT CTA GGC CAA AAA-3′; Ser2Ser3Ser6Thr7Glu, 5′-GAT GAT AAA CAT ATG GAG G AG CCG GCG GAG GAG CCG AGC CGC CG-3′; Ser31Ser32Glu, 5′-GGA GTG AGG ATG CCA GGG AGG AGC CCT CTC AGA GAC GTA G-3′; Thr53Ser54Glu, 5′-GTT GCA GCC GAT GCC AGA GGA GCC TGG AGT GGA CCT G-3′; Ser70Ser71Glu, 5′-GCG CAG GAC GTG CTG TTT GAG GAG CCT CCC CAA ATG CAT TCT-3′; and Ser87Ser88Glu, 5′-CTC TTG ACT TTG ATG TTG AGG AGC CAC TGA CAT ACG GCA C-3′. To generate the pCMV-TIM plasmid, the TIM cDNA was PCR-amplified from a HeLa cDNA library using TIMFW (5′-CCG CGA AGC TTA TGG ACT TGC ACA TGA TGA ACT G-3′) and TIMRV (5′-CCG GGT ACC TCA GTC ATC CTC ATC ATC CTC AAT C-3′) and cloned into the HindIII and KpnI sites of the pCMV vector. pCMV-TIM-R1, encoding wild-type TIM, was rendered resistant to TIM siRNA #1 by introducing eight silent mutations at TIM codons 799–807 using the primer TIMSIR (5′-GGA AGA ACA CAG CTG TAG TCC GGG AAA TGA CGG AAG GTT ACG GCT CCC TGG ATG AC-3′).
Antibodies
Mouse anti-CDC6 (sc-9964), goat anti-actin (sc-1616), rabbit anti-p27 (sc-528), rabbit anti-H3 (sc-10809), rabbit anti-DNA pol δ (sc10784), mouse anti-β-tubulin (sc-5274), and rabbit anti-CDC45 (sc-20685) were purchased from Santa Cruz Biotechnology. Mouse anti-p21 (ab16767), mouse anti-Cyclin E (ab3927), mouse anti-Cyclin A (ab16726), mouse anti-RNAPII Ser(P)5 (4H8), and rabbit anti-MCM7 (ab52489) were purchased from Abcam. Goat anti-MCM2 (A300-122A), rabbit anti-TIM (A300-961A-1), rabbit anti-TIPIN (A301-474A), rabbit anti-MCM4 (A300-193A), rabbit anti-MCM5 (A300-195A), rabbit anti-Skp2 (A302-436A), and rabbit anti-ORC2 (A302-734A) were purchased from Bethyl Laboratories. Mouse anti-Ser/Thr(P) (61548) was purchased from BD Biosciences. Rabbit anti-RECQ4 was generated as described previously against residues 71–80 of human RECQ4 (51, 77). Rabbit anti-MCM10 (12251-1-AP) and rabbit anti-Psf2 (16247-1-AP) were purchased from ProteinTech. Rabbit anti-FLAG (F7425) was purchased from Sigma. Rabbit anti-Sld5 (2-79.00.02) was purchased from Strategic Diagnostics. Rat anti-BrdU (MCA2060T) was purchased from AbD Serotec. Goat anti-rat DyLight 488-conjugated (112-485-167) and donkey anti-rat IgG conjugated with Rhodamine (712-026-150) were purchased from Jackson ImmunoResearch Laboratories. Rabbit anti-phospho-CHK1 (p-CHK1 Ser345, 133D3, 2348) and rabbit anti-CHK1 (2345) were purchased from Cell Signaling Technology. Mouse anti-ORC2 (M055-3) was purchased from MBL.
Cell Culture, siRNA, Cell Fractionation, and Immunopurification
The human cell lines HEK293T and HT1080 were generous gifts from Dr. Stephen West (The Francis Crick Institute). The human cell lines HEK293 and U2OS were obtained from Dr. Jeremy Stark (City of Hope). The cells were cultured in DMEM supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 incubator. Plasmid transfection, chromatin fractionation, and immunopurification were performed as described previously (51). The siRNA sequences for RECQ4, MCM10, and CDC6 were also described previously (51). The TIM stealth siRNAs #1 (5′-GGUUCGAGAGAUGACUGAGG GCUAU-3′ (11)) and #2 (5′-UCCAGGGUAGCUU AGUCCUUUCAAA-3′) were purchased from Invitrogen. The siRNAs were transfected into the cells using DharmaFECT 1 transfection reagent (Thermo Scientific) according to the protocol of the manufacturer. For TIM siRNA or TIM-CDC6 double siRNA knockdown, the cells were treated with 25 nm siRNA for 24 h followed by a second 25 nm siRNA treatment. The cells were harvested 36 h after the second siRNA transfection. For cell cycle synchronization, the cells were cultured with nocodazole-containing medium 24 h after the second siRNA transfection. For UV treatment, cells in log phase were irradiated with 30 J/m2 UV light and harvested after 1 h of incubation. For APH treatment, the cells were cultured with 1 μg/ml of APH for 24 h prior to harvest. For CDC7 and CDK inhibitor treatments, PHA 767491 (5 μm, Santa Cruz Biotechnology) and roscovitine (20 μm, Sigma) were added to the tissue culture media to a final concentration as indicated for 24 h prior to harvest.
Cell Cycle and ssDNA Analyses
The cells were synchronized at G2/M phase with 50 ng/ml nocodazole in complete medium for 20 h. The cells were released by washing twice with complete DMEM. For DNA content analysis, the cells were stained with propidium iodide (50 μg/ml) and analyzed in a CyAn ADP Analyzer (Beckman Coulter). For the BrdU incorporation assay, the cells were pulse-labeled with 10 μm BrdU (Sigma) for 30 min. The BrdU-positive cells were detected using a rat anti-BrdU antibody followed by a goat anti-rat DyLight 488-conjugated secondary antibody. The cell cycle profile distributions were determined with the FlowJo software package (Tree Star Inc.). ssDNA detection was performed as described previously (78). Briefly, the siRNA-transfected cells were grown on coverslips and labeled with 10 μm BrdU for 18 h, followed by nocodazole synchronization for 18 h. After nocodazole release, the cells were fixed with 3% paraformaldehyde containing 0.2% Triton X-100 for 15 min, followed by blocking with blocking buffer (3% BSA and 0.5% Tween 20 in PBS) for 30 min. The coverslips were then incubated overnight with a rat anti-BrdU antibody (1:500), diluted in blocking buffer overnight at 4 °C, washed with PBS, stained with anti-rat IgG conjugated with Rhodamine (1:1000) for 2 h at room temperature, washed with PBS, stained with DAPI, and mounted in anti-fade mounting medium.
Phosphatase Treatment
The co-purified FLAG-CDC45 and FLAG-MCM7 complexes were eluted with 3× FLAG peptide in Elution-A buffer (10 mm HEPES (pH 7.9), 0.2 m NaCl, 0.2 mm EDTA, 0.05% Triton X-100, and 10% glycerol). The eluates were then dialyzed against dialysis buffer (10 mm Tris (pH 7.5), 100 mm NaCl, 0.2 mm EDTA, and 10% glycerol). An aliquot was incubated in 30 μl of the buffer supplied by the manufacturer with or without 400 U of λ protein phosphatase (New England Biolabs). The samples were separated by SDS-PAGE and detected on Western blots using an anti-MCM4 antibody.
Helicase Assay
FLAG-RECQ4 co-purified complexes were eluted with 3× FLAG peptide (Sigma) in Elution-A buffer (10 mm HEPES (pH 7.9), 0.2 m NaCl, 0.2 mm EDTA, 0.05% Triton X-100, and 10% glycerol). The eluates were used to perform helicase assays as described previously (51).
ChIP and Real-time PCR
The cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and the reaction was stopped by adding glycine to a final concentration of 0.125 m for 5 min at room temperature. The fixed cells were rinsed twice with PBS and lysed with cytoplasmic lysis buffer (10 mm Tris (pH 8.0), 0.34 m sucrose, 3 mm CaCl2, 2 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, and 0.5% Nonidet P-40) plus protease inhibitors. Lysates were pelleted by centrifugation at 5000 rpm for 5 min at 4 °C. The pellets were resuspended in ChIP lysis buffer (1.0% SDS, 10 mm EDTA, and 50 mm Tris (pH 8.0)) plus protease inhibitors, and chromatin was sheared by sonication to generate DNA fragments of <1 kb. Chromatin was diluted 10 times in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris (pH 8.0), and 167 mm NaCl) plus protease inhibitor and precleared with protein A/G beads (Thermo Scientific) for 1 h at 4 °C. Antibodies (ORC2, MCM5, and RNA polymerase II) were used for immunoprecipitation. A rabbit IgG (Santa Cruz Biotechnology) was used as a negative control. ChIP complexes were collected with protein A/G beads and washed sequentially twice with low-salt buffer (0.1% SDS, 1.0% Triton X-100, 2 mm EDTA, 20 mm Tris (pH 8.0), and 0.15 m NaCl), high-salt buffer (0.1% SDS, 1.0% Triton X-100, 2 mm EDTA, 20 mm Tris (pH 8.0), and 0.5 m NaCl), LiCl buffer (0.25 m LiCl, 1.0% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, and 10 mm Tris (pH 8.0)), and Tris-EDTA buffer. The ChIP complexes were eluted with 300 μl of elution buffer (0.1 m sodium bicarbonate and 1.0% SDS) at room temperature for 15 min. The chromatin was reverse cross-linked by adding 20 μl of 5 m NaCl and incubated at 65 °C overnight. The DNA was digested with RNase A and proteinase K and purified by phenol-chloroform and ethanol precipitation. Real-time PCR was performed to amplify the human Lamin B2 origin region (B48) (79) with an ABI 7500 fast system using SYBR Green fluorescence. Enrichment was calculated using the comparative Ct method.
Author Contributions
X. X. conducted most of the experiments and analyzed the results. J. T. W. contributed to the analysis of CMG complex formation. M. L. contributed to the cell cycle analysis. Y. L. conceived the idea for the project, analyzed the results with X. X., and wrote the paper.
Acknowledgments
The research reported in this publication included work performed in the Analytical Cytometry and Drug Discovery and Structural Biology Cores supported by the NCI, National Institutes of Health under Award P30CA33572. We thank Dr. Nancy Linford for comments and expert editing of the manuscript.
This work was supported by National Institutes of Health Grant R01 CA151245 to (Y. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ssDNA
- single-stranded DNA
- GINS
- go-ichi-ni-san complex
- DDK
- CDC7-Dbf4 kinase
- CDK
- cyclin-dependent kinase
- TIM
- TIMELESS
- TIPIN
- TIM-interacting protein
- CMG
- CDC45·MCM2-7·GINS complex
- PG
- phosphorylation-generated
- ATR
- ataxia telangiectasia- and Rad3-related protein
- APC/C
- anaphase-promoting complex/cyclosome
- Rb
- retinoblastoma protein
- ORC
- origin recognition complex
- APH
- aphidicolin
- WCE
- whole-cell extract(s)
- RNAPII
- RNA polymerase II
- IP
- immunopurification.
References
- 1. Dimitrova D. S., Todorov I. T., Melendy T., and Gilbert D. M. (1999) Mcm2, but not RPA, is a component of the mammalian early G1-phase prereplication complex. J. Cell Biol. 146, 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Edwards M. C., Tutter A. V., Cvetic C., Gilbert C. H., Prokhorova T. A., and Walter J. C. (2002) MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 277, 33049–33057 [DOI] [PubMed] [Google Scholar]
- 3. Woodward A. M., Göhler T., Luciani M. G., Oehlmann M., Ge X., Gartner A., Jackson D. A., and Blow J. J. (2006) Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 173, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim J., and Kipreos E. T. (2008) Control of the Cdc6 replication licensing factor in metazoa: the role of nuclear export and the CUL4 ubiquitin ligase. Cell Cycle 7, 146–150 [DOI] [PubMed] [Google Scholar]
- 5. Labib K. (2010) How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 24, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aparicio T., Ibarra A., and Méndez J. (2006) Cdc45-MCM-GINS, a new power player for DNA replication. Cell Div. 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masai H., You Z., and Arai K. (2005) Control of DNA replication: regulation and activation of eukaryotic replicative helicase, MCM. IUBMB Life 57, 323–335 [DOI] [PubMed] [Google Scholar]
- 8. Gotter A. L., Manganaro T., Weaver D. R., Kolakowski L. F. Jr, Possidente B., Sriram S., MacLaughlin D. T., and Reppert S. M. (2000) A time-less function for mouse timeless. Nat. Neurosci. 3, 755–756 [DOI] [PubMed] [Google Scholar]
- 9. Numata Y., Ishihara S., Hasegawa N., Nozaki N., and Ishimi Y. (2010) Interaction of human MCM2-7 proteins with TIM, TIPIN and Rb. J. Biochem. 147, 917–927 [DOI] [PubMed] [Google Scholar]
- 10. Gotter A. L. (2003) Tipin, a novel timeless-interacting protein, is developmentally co-expressed with timeless and disrupts its self-association. J. Mol. Biol. 331, 167–176 [DOI] [PubMed] [Google Scholar]
- 11. Chou D. M., and Elledge S. J. (2006) Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. U.S.A. 103, 18143–18147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Unsal-Kaçmaz K., Chastain P. D., Qu P. P., Minoo P., Cordeiro-Stone M., Sancar A., and Kaufmann W. K. (2007) The human Tim/Tipin complex coordinates an intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 27, 3131–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park H., and Sternglanz R. (1999) Identification and characterization of the genes for two topoisomerase I-interacting proteins from Saccharomyces cerevisiae. Yeast 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 14. Rabitsch K. P., Tóth A., Gálová M., Schleiffer A., Schaffner G., Aigner E., Rupp C., Penkner A. M., Moreno-Borchart A. C., Primig M., Esposito R. E., Klein F., Knop M., and Nasmyth K. (2001) A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr. Biol. 11, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 15. Nedelcheva M. N., Roguev A., Dolapchiev L. B., Shevchenko A., Taskov H. B., Shevchenko A., Stewart A. F., and Stoynov S. S. (2005) Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J. Mol. Biol. 347, 509–521 [DOI] [PubMed] [Google Scholar]
- 16. Calzada A., Hodgson B., Kanemaki M., Bueno A., and Labib K. (2005) Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 19, 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gambus A., Jones R. C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R. D., and Labib K. (2006) GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 8, 358–366 [DOI] [PubMed] [Google Scholar]
- 18. Bando M., Katou Y., Komata M., Tanaka H., Itoh T., Sutani T., and Shirahige K. (2009) Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J. Biol. Chem. 284, 34355–34365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foss E. J. (2001) Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics 157, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katou Y., Kanoh Y., Bando M., Noguchi H., Tanaka H., Ashikari T., Sugimoto K., and Shirahige K. (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424, 1078–1083 [DOI] [PubMed] [Google Scholar]
- 21. Smith K. D., Fu M. A., and Brown E. J. (2009) Tim-Tipin dysfunction creates an indispensable reliance on the ATR-Chk1 pathway for continued DNA synthesis. J. Cell Biol. 187, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McFarlane R. J., Mian S., and Dalgaard J. Z. (2010) The many facets of the Tim-Tipin protein families' roles in chromosome biology. Cell Cycle 9, 700–705 [DOI] [PubMed] [Google Scholar]
- 23. Leman A. R., Noguchi C., Lee C. Y., and Noguchi E. (2010) Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 123, 660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith-Roe S. L., Patel S. S., Simpson D. A., Zhou Y. C., Rao S., Ibrahim J. G., Kaiser-Rogers K. A., Cordeiro-Stone M., and Kaufmann W. K. (2011) Timeless functions independently of the Tim-Tipin complex to promote sister chromatid cohesion in normal human fibroblasts. Cell Cycle 10, 1618–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayer M. L., Pot I., Chang M., Xu H., Aneliunas V., Kwok T., Newitt R., Aebersold R., Boone C., Brown G. W., and Hieter P. (2004) Identification of protein complexes required for efficient sister chromatid cohesion. Mol. Biol. Cell 15, 1736–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masai H., Taniyama C., Ogino K., Matsui E., Kakusho N., Matsumoto S., Kim J. M., Ishii A., Tanaka T., Kobayashi T., Tamai K., Ohtani K., and Arai K. (2006) Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281, 39249–39261 [DOI] [PubMed] [Google Scholar]
- 27. Randell J. C., Fan A., Chan C., Francis L. I., Heller R. C., Galani K., and Bell S. P. (2010) Mec1 is one of multiple kinases that prime the Mcm2-7 helicase for phosphorylation by Cdc7. Mol. Cell 40, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshizawa-Sugata N., and Masai H. (2007) Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 282, 2729–2740 [DOI] [PubMed] [Google Scholar]
- 29. Guillou E., Ibarra A., Coulon V., Casado-Vela J., Rico D., Casal I., Schwob E., Losada A., and Méndez J. (2010) Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 24, 2812–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Porrás A., Gaillard S., and Rundell K. (1999) The simian virus 40 small-t and large-T antigens jointly regulate cell cycle reentry in human fibroblasts. J. Virol. 73, 3102–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caracciolo V., Reiss K., Khalili K., De Falco G., and Giordano A. (2006) Role of the interaction between large T antigen and Rb family members in the oncogenicity of JC virus. Oncogene 25, 5294–5301 [DOI] [PubMed] [Google Scholar]
- 32. Sherr C. J., and Roberts J. M. (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 33. Gamper A. M., Rofougaran R., Watkins S. C., Greenberger J. S., Beumer J. H., and Bakkenist C. J. (2013) ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 41, 10334–10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Averbeck N. B., Ringel O., Herrlitz M., Jakob B., Durante M., and Taucher-Scholz G. (2014) DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell Cycle 13, 2509–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kemp M. G., Akan Z., Yilmaz S., Grillo M., Smith-Roe S. L., Kang T. H., Cordeiro-Stone M., Kaufmann W. K., Abraham R. T., Sancar A., and Unsal-Kaçmaz K. (2010) Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem. 285, 16562–16571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu Z., and Hunter T. (2010) Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 9, 2342–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bashir T., Dorrello N. V., Amador V., Guardavaccaro D., and Pagano M. (2004) Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428, 190–193 [DOI] [PubMed] [Google Scholar]
- 38. Geley S., Kramer E., Gieffers C., Gannon J., Peters J. M., and Hunt T. (2001) Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ji P., Jiang H., Rekhtman K., Bloom J., Ichetovkin M., Pagano M., and Zhu L. (2004) An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 16, 47–58 [DOI] [PubMed] [Google Scholar]
- 40. Binné U. K., Classon M. K., Dick F. A., Wei W., Rape M., Kaelin W. G. Jr, Näär A. M., and Dyson N. J. (2007) Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat. Cell Biol. 9, 225–232 [DOI] [PubMed] [Google Scholar]
- 41. Yung Y., Walker J. L., Roberts J. M., and Assoian R. K. (2007) A Skp2 autoinduction loop and restriction point control. J. Cell Biol. 178, 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chau B. N., and Wang J. Y. (2003) Coordinated regulation of life and death by RB. Nat. Rev. Cancer 3, 130–138 [DOI] [PubMed] [Google Scholar]
- 43. Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 44. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 45. Lim S., and Kaldis P. (2013) Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093 [DOI] [PubMed] [Google Scholar]
- 46. Harbour J. W., Luo R. X., Dei Santi A., Postigo A. A., and Dean D. C. (1999) Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859–869 [DOI] [PubMed] [Google Scholar]
- 47. Biggar K. K., and Storey K. B. (2009) Perspectives in cell cycle regulation: lessons from an anoxic vertebrate. Curr. Genomics 10, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Furstenthal L., Kaiser B. K., Swanson C., and Jackson P. K. (2001) Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J. Cell Biol. 152, 1267–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lunn C. L., Chrivia J. C., and Baldassare J. J. (2010) Activation of Cdk2/Cyclin E complexes is dependent on the origin of replication licensing factor Cdc6 in mammalian cells. Cell Cycle 9, 4533–4541 [DOI] [PubMed] [Google Scholar]
- 50. Bell S. P., and Stillman B. (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357, 128–134 [DOI] [PubMed] [Google Scholar]
- 51. Xu X., Rochette P. J., Feyissa E. A., Su T. V., and Liu Y. (2009) MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 28, 3005–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heller R. C., Kang S., Lam W. M., Chen S., Chan C. S., and Bell S. P. (2011) Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ilves I., Petojevic T., Pesavento J. J., and Botchan M. R. (2010) Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 37, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sheu Y. J., and Stillman B. (2006) Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sheu Y. J., and Stillman B. (2010) The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ishimi Y., Komamura-Kohno Y., Kwon H. J., Yamada K., and Nakanishi M. (2003) Identification of MCM4 as a target of the DNA replication block checkpoint system. J. Biol. Chem. 278, 24644–24650 [DOI] [PubMed] [Google Scholar]
- 57. Komamura-Kohno Y., Karasawa-Shimizu K., Saitoh T., Sato M., Hanaoka F., Tanaka S., and Ishimi Y. (2006) Site-specific phosphorylation of MCM4 during the cell cycle in mammalian cells. FEBS J. 273, 1224–1239 [DOI] [PubMed] [Google Scholar]
- 58. Im J. S., Ki S. H., Farina A., Jung D. S., Hurwitz J., and Lee J. K. (2009) Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 15628–15632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maric M., Maculins T., De Piccoli G., and Labib K. (2014) Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346, 1253596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Powell S. K., MacAlpine H. K., Prinz J. A., Li Y., Belsky J. A., and MacAlpine D. M. (2015) Dynamic loading and redistribution of the Mcm2-7 helicase complex through the cell cycle. EMBO J. 34, 531–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Remus D., Beuron F., Tolun G., Griffith J. D., Morris E. P., and Diffley J. F. (2009) Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 139, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bochman M. L., and Schwacha A. (2008) The Mcm2-7 complex has in vitro helicase activity. Mol. Cell 31, 287–293 [DOI] [PubMed] [Google Scholar]
- 63. Bruck I., Kanter D. M., and Kaplan D. L. (2011) Enabling association of the GINS protein tetramer with the mini chromosome maintenance (Mcm)2-7 protein complex by phosphorylated Sld2 protein and single-stranded origin DNA. J. Biol. Chem. 286, 36414–36426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moritani M., and Ishimi Y. (2013) Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J. Biochem. 154, 363–372 [DOI] [PubMed] [Google Scholar]
- 65. Montagnoli A., Valsasina B., Brotherton D., Troiani S., Rainoldi S., Tenca P., Molinari A., and Santocanale C. (2006) Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 281, 10281–10290 [DOI] [PubMed] [Google Scholar]
- 66. Bruck I., and Kaplan D. L. (2011) Origin single-stranded DNA releases Sld3 from Mcm2-7, allowing GINS to bind Mcm2-7. J. Biol. Chem. 286, 18602–18613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bruck I., and Kaplan D. L. (2011) GINS and Sld3 compete with one another for Mcm2-7 and Cdc45 binding. J. Biol. Chem. 286, 14157–14167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Costa A., Ilves I., Tamberg N., Petojevic T., Nogales E., Botchan M. R., and Berger J. M. (2011) The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 18, 471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coleman T. R., Carpenter P. B., and Dunphy W. G. (1996) The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell 87, 53–63 [DOI] [PubMed] [Google Scholar]
- 70. Tanaka T., Knapp D., and Nasmyth K. (1997) Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell 90, 649–660 [DOI] [PubMed] [Google Scholar]
- 71. Furstenthal L., Swanson C., Kaiser B. K., Eldridge A. G., and Jackson P. K. (2001) Triggering ubiquitination of a CDK inhibitor at origins of DNA replication. Nat. Cell Biol. 3, 715–722 [DOI] [PubMed] [Google Scholar]
- 72. Mailand N., and Diffley J. F. (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 122, 915–926 [DOI] [PubMed] [Google Scholar]
- 73. Morris L., Allen K. E., and La Thangue N. B. (2000) Regulation of E2F transcription by cyclin E-Cdk2 kinase mediated through p300/CBP co-activators. Nat. Cell Biol. 2, 232–239 [DOI] [PubMed] [Google Scholar]
- 74. Hateboer G., Wobst A., Petersen B. O., Le Cam L., Vigo E., Sardet C., and Helin K. (1998) Cell cycle-regulated expression of mammalian CDC6 is dependent on E2F. Mol. Cell. Biol. 18, 6679–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ohtani K., Tsujimoto A., Ikeda M., and Nakamura M. (1998) Regulation of cell growth-dependent expression of mammalian CDC6 gene by the cell cycle transcription factor E2F. Oncogene 17, 1777–1785 [DOI] [PubMed] [Google Scholar]
- 76. Hsu J. Y., Reimann J. D., Sørensen C. S., Lukas J., and Jackson P. K. (2002) E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 4, 358–366 [DOI] [PubMed] [Google Scholar]
- 77. Wang J. T., Xu X., Alontaga A. Y., Chen Y., and Liu Y. (2014) Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 7, 848–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Raderschall E., Golub E. I., and Haaf T. (1999) Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 96, 1921–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Thangavel S., Mendoza-Maldonado R., Tissino E., Sidorova J. M., Yin J., Wang W., Monnat R. J. Jr, Falaschi A., and Vindigni A. (2010) Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol. Cell. Biol. 30, 1382–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]