

Abstract

Rhodacyclopentanones derived from carbonylative C–C activation of cyclopropyl ureas can be “captured” by pendant nucleophiles prior to “collapse” to 1,3-diazepanes. The choice of N-substituent on the cyclopropane unit controls the oxidation level of the product, such that C4–C5 unsaturated or saturated systems can be accessed selectively.
The Biginelli multicomponent reaction provides high versatility for the synthesis of 6-ring cyclic ureas and is used widely in diversity oriented synthesis.1 However, equally direct and flexible entries to homologated systems have not been reported despite the biological significance of the 1,3-diazepane scaffold (Scheme 1A). This is the core motif in a family of highly potent HIV protease inhibitors, as exemplified by DMP 450.2 Other important 1,3-diazepanes include the structurally intriguing β-lactamase inhibitor MK-7655 developed by Merck,3 and the mast cell inhibitory alkaloid (+)-monanchorin.4 The lack of general methods for accessing 1,3-diazepanes reflects wider difficulties in preparing medium ring systems containing multiple heteroatoms.5 Consequently, modular catalytic methodologies that address this issue are likely to be of interest to the pharmaceutical sector.6
Scheme 1.
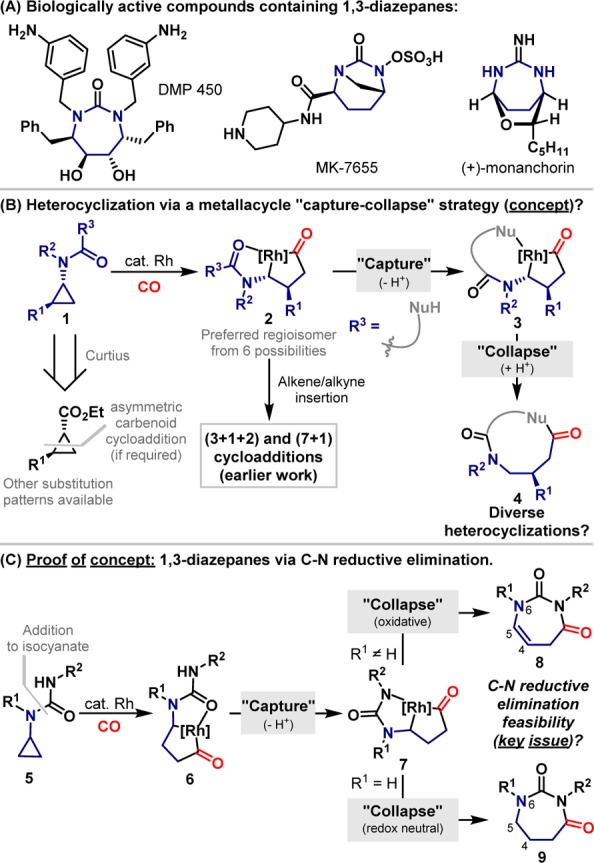
Our laboratory has developed a cycloaddition strategy that relies on N-directing group controlled insertion of Rh and CO into the proximal C–C bond of aminocyclopropanes 1 (Scheme 1B).7 The resulting rhodacyclopentanones 2 engage pendant alkynes or alkenes to provide (3 + 1 + 2)7a,7b,7d or (7 + 1)7c cycloaddition products. This approach harnesses the strain embedded within readily prepared (and enantiopure) aminocyclopropanes to provide byproduct-free access to complex N-heterocyclic ring systems. In seeking to expand further the scope of this catalysis platform, we considered whether rhodacyclopentanones 2 might be susceptible to attack by pendant nucleophiles. If successful, this would provide medium rings 4 via the intermediacy of kinetically accessible bicycles 3, with a key issue being the scope of the C–Nu reductive elimination step, a process only known for C–O bond formation.8 Such catalytic metallacycle “capture–collapse” sequences have the potential to generate a wide range of challenging rings containing multiple heteroatoms. In this report we outline our proof-of-concept studies toward this broad goal by demonstrating that readily prepared urea-based systems 5 can be converted directly to substituted 1,3-diazepanes via previously unknown C–N reductive elimination from rhodacyclopentanones 7 (Scheme 1C). We also show that the oxidation level of the product (8 vs 9) can be controlled by the choice of R1-substituent, thereby providing valuable additional flexibility to the methodology.
Initial studies examined a range of Rh-catalysts for the carbonylative cyclization of 5a (Table 1). Under 1 atm of CO, we found that a cationic Rh(I)-system derived from [Rh(cod)2]BARF and triphenylphosphine provided oxidative product 8a in 82% yield and 20:1 selectivity over the alternate C4–C5 saturated variant 9a. Notably, neutral Rh(I)-complexes were completely ineffective and higher CO pressures (e.g., 5 atm) offered no benefits. The presence of an acid cocatalyst (PhCO2H) was found to have a significant effect, providing approximately 20% enhancements to the yields of cyclizations described throughout this study (vide infra). For clarity, the numbering system used in Scheme 1C is retained throughout subsequent discussion: 5 = cyclopropyl substrate; 8 = C4–C5 unsaturated product; 9 = C4–C5 saturated product; letters = structural variant.
Table 1. Oxidative Carbonylative Heterocyclizationsa.


The ratio of 8a–i:9a–i is given in parentheses (determined by 1H NMR analysis of crude material).
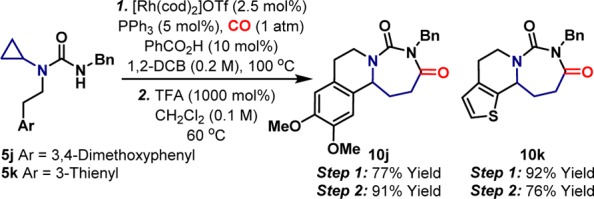
The scope of the process is outlined in Table 1. In the majority of cases cyclization was efficient, generating the target C4–C5 unsaturated systems with high selectivity (5:1 to 24:1) over their saturated congeners. The protocol tolerates a wide range of aryl or alkyl substituents at R1. Even potentially sensitive functionality survives, such as the ester of 5h and the cyclopropylmethyl group of 5i; the latter highlights the exquisite selectivity of the initial C–C activation step. The tolerance of the process to the R2 group shows greater variance: alkyl groups, including sterically encumbered variants (e.g., 5d), performed well but N-aryl groups were less effective, perhaps due to the lower nucleophilicity of the nitrogen center. The ability to access unsaturated products efficiently opens up further opportunities for methodology design (Scheme 2). For example, 5j/k, which are equipped with pendant (hetero)aryl groups underwent Rh-catalyzed heterocyclization and subsequent iminium ion triggered ring closure to provide complex tricyclic systems 10j/k, as confirmed by single crystal X-ray diffraction.
Scheme 2. Serial Rh-Catalyzed and Brønsted Acid Promoted Cyclizations.
The processes outlined in Table 1 provide efficient oxidative access to systems possessing C4–C5 unsaturation. If redox neutral processes could be promoted, then complementary access to C4–C5 saturated systems would also be possible, allowing an unusual oxidation level divergent entry to the target diazepanes. In the event, we observed that high selectivity for this pathway can be achieved using systems where R1 = H (Table 2). Under conditions similar to those outlined in Table 2, cyclization of 5l–r, which encompasses a range of alkyl or aryl R2-substituents, occurred efficiently to provide targets 9l–r with 3:1 to 12:1 selectivity over the corresponding C4–C5 unsaturated variants (8l–r).9 The efficient cyclization of tert-butyl substrate 5o is significant, as this result supports a carbonyl directed C–C activation pathway (5 to 6 to 7, Scheme 1C). In principle, N-directed C–C bond activation could lead directly to intermediates 7; however, steric hindrance would likely render such an initiation mode inefficient for 5o.
Table 2. Redox Neutral Carbonylative Heterocyclizations.


The ratio of 9l–r:8l–r is given in parentheses (determined by 1H NMR analysis of crude material).
Dioxane was used as solvent.
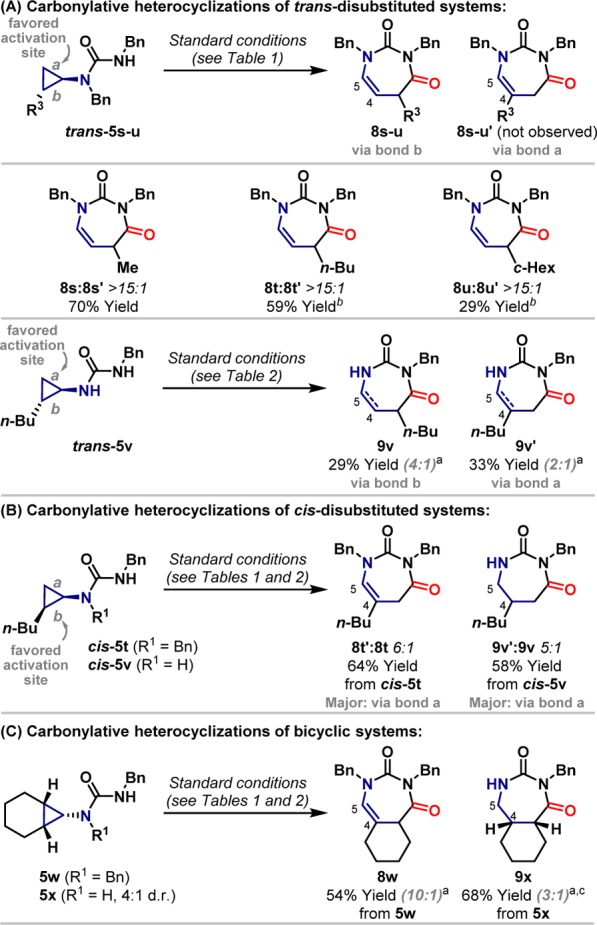
Adaption of the approaches outlined in Tables 1 and 2 to systems with substitution on the cyclopropane component revealed interesting and insightful results (vide infra). Trans-1,2-disubstituted systems 5s–u provided highly selective access to C4–C5 unsaturated ring systems; the observed regioisomers 8s–u (>15:1 r.r.) are derived from C–C activation of the more hindered bond b.10Previous studies involving isolable metallacycles have confirmed that this is the disfavored C–C activation pathway.7c As expected based on the results in Table 2, cyclization of 5v, which lacks an N-benzyl substituent on the aminocyclopropane unit, favored the formation of C4–C5 saturated products (5:2 saturated/unsaturated). In this case, however, C–C activation regioselectivity was low, providing an approximately 1:1 ratio of C3:C4 substituted systems 9v and 9v′, which are derived from cleavage of bond b vs bond a, respectively.11 We have previously established that cis-disubstituted aminocyclopropanes preferentially undergo directed C–C bond activation at the more hindered but more electron-rich bond b.7bHowever, cyclization of cis-5t and cis-5v delivered selectively regioisomers 8t′ and 9v′, derived from C–C activation of less hindered bond a. As expected, in both cases the oxidation level of the product was controlled by the presence or absence of an R1-substituent. Bicyclic systems 5w and 5x, which are not subject to directed C–C activation regiocontrol issues, cyclized without incident to deliver expected products 8w and 9x in an oxidation level divergent manner.
The results for cis- and trans-disubstituted cyclopropane systems (Scheme 3A/B) provide important insights into the mechanism of the processes described here. In these cases, product regioselectivities are (in general) opposite to what would be expected based on stoichiometric rhodacyclopentanone formations using carbamate protected aminocyclopropanes (trans-cyclopropanes: activation of bond a expected, products from activation of bond b observed; cis-cyclopropanes: activation of bond b expected, products from activation of bond a observed).7b,7c,12 Initial studies to rationalize this discrepancy confirmed that N,N,N′-trisubstituted urea directing groups provide the same C–C activation regioselectivities as carbamate variants (Scheme 4A). This was done by exposing diastereomeric systems trans-5s and cis-5s to the reaction conditions in the absence of CO; decomposition products generated from the putative rhodacyclobutane intermediate (not depicted) indicate the kinetically favored site of C–C oxidative addition.13Trans-disubstituted cyclopropane trans-5s generated selectively alkene 11b, which is derived from C–C activation of bond a, whereas cis-disubstituted system cis-5s provided predominantly 11a (11:1 11a:11b), via preferential activation of bond b. Thus, in both cases, C–C activation regioselectivity is in line with previous stoichiometric studies7b,7c but opposite to that observed in Scheme 3A/B.
Scheme 3.
Scheme 4.
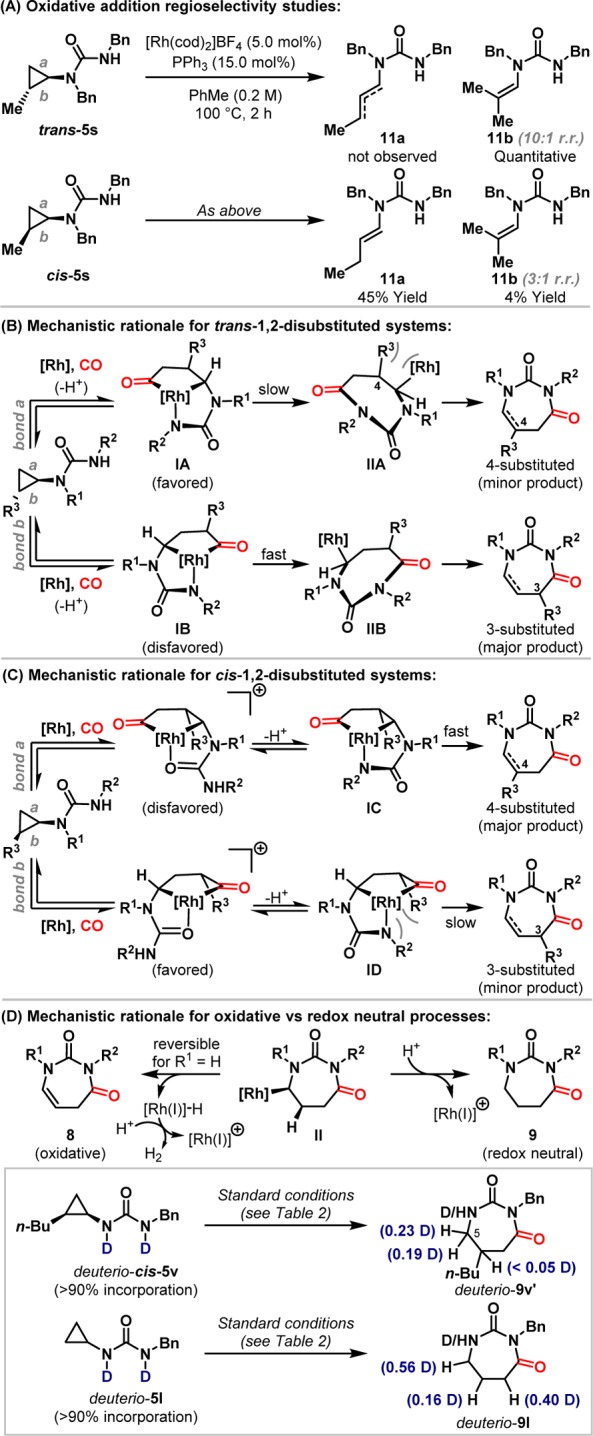
We have shown in earlier work that directed rhodacyclopentanone formation is (highly) reversible when cationic Rh-systems are employed.7b,7c As such, product regioselectivity can be controlled by the facility of processes other than the initial directed C–C activation step. This type of Curtin–Hammett scenario (i.e., reversible formation of 7 from 5, see Scheme 1C) underpins proposed mechanisms for the processes described here. For trans-disubstituted systems, rhodacyclopentanone formation via cleavage of bond a is preferred leading to metallacycle IA (Scheme 4B). However, subsequent C–N reductive elimination to IIA is slow because of the developing steric clash between the Rh-center and the R3-substituent. Accordingly, reprotonation of nitrogen, retro-carbonylation, and C–C reductive elimination regenerate the cyclopropane and enable equilibration to disfavored metallacycle IB. Here, C–N reductive elimination is likely more facile because the Rh-center of IIB is further from the R3-substituent (cf. IIA, 1,2- vs 1,3-relationship); consequently, the C3-substituted product is generated selectively. For trans-5v, where R1 = H, steric constraints for IA to IIA may, to a certain extent, be alleviated, such that progression to the 4-substituted product becomes competitive and low regioselectivity is observed.14 A similar but distinct selectivity model is proposed for cis-disubstituted systems (Scheme 4C). Here, rhodacyclopentanone formation via bond b is favored leading to metallacycle ID; however, C–N reductive elimination from this intermediate may be slow due to developing steric clashes between the N–R2 group and the R3-substituent. As before, reversible metallacycle formation allows equilibration to IC, where this steric impediment is alleviated en route to the C4-substituted product. Thus, we suggest that for both systems the unexpected product regioselectivities are determined by the facility of C–N reductive elimination rather than C–C activation.15
Why does the presence or absence of an R1-substituent control selectivity for C4–C5 unsaturated or saturated products (8 vs 9) (Scheme 4D)? Preliminary studies indicate that the conversion of II to 8 occurs in both cases, but this is reversible for R1 = H allowing eventual protodemetalation to 9. Cyclization of deuterio-cis-5v provided deuterio-9v′, where deuterium transfer from the urea to both diastereotopic C5 positions supports reversible β-hydride elimination and alkene dissociation (to 8)16 in advance of irreversible protodemetalation (to 9); incomplete deuterium transfer may be due to exchange with protic impurities (e.g., H2O) prior to C–H formation. For systems lacking a C4-substituent, isomerization along the ring occurs prior to protodemetalation; cyclization of deuterio-5l generated deuterio-9l, where deuterium incorporation was observed at C3, C4, and C5. Heterocyclization to 8 is oxidative, and we speculate that the active Rh(I) species is regenerated by protonation triggered reductive elimination of dihydrogen; this process is known for related systems.8,17 Presumably, the PhCO2H additive acts as a proton reservoir for both pathways, which otherwise would be reliant solely on the proton released by conversion of 6 to 7 (see Scheme 1C).
In summary, we demonstrate an approach to substituted 1,3-diazepanes as proof-of-concept for a general metallacycle “capture–collapse” strategy. We anticipate that the strategy will allow the generation of a range of medium ring systems containing multiple heteroatoms; studies toward this broad goal are underway.18 The findings described here enhance significantly the scope of the catalysis platform outlined in Scheme 1B (1 to 2), opening up numerous avenues for further exploration, while at the same time adding to the wider and emerging area of rhodacyclopentanone-based catalysis.7,8,19
Acknowledgments
European Research Council via the EU’s Horizon 2020 Programme (ERC grant 639594 CatHet). Bristol Chemical Synthesis Centre for Doctoral Training (EPSRC grant EP/G036764/1) for a studentship (N.G.M). University of Bristol X-ray crystallographic service for analysis of trans-5s, 10j, and 10k. Royal Society for a University Research Fellowship (J.F.B.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07046.
Author Contributions
† N.G.McC. and S.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Kappe C. O. Acc. Chem. Res. 2000, 33, 879. 10.1021/ar000048h. [DOI] [PubMed] [Google Scholar]; b Biggs-Houck J. E.; Younai A.; Shaw J. T. Curr. Opin. Chem. Biol. 2010, 14, 371. 10.1016/j.cbpa.2010.03.003. [DOI] [PubMed] [Google Scholar]
- a Lam P. Y. S.; Jadhav P. K.; Eyermann C. J.; Hodge C. N.; Ru Y.; Bacheler L. T.; Meek J. L.; Otto M. J.; Rayner M. M.; Wong Y. N.; Chang C.-H.; Weber P. C.; Jackson D. A.; Sharpe T. R.; Erickson-Viitanen S. Science 1994, 263, 380. 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- Blizzard T. A.; Chen H.; Kim S.; Wu J.; Bodner R.; Gude C.; Imbriglio J.; Young K.; Park Y.-W.; Ogawa A.; Raghoobar S.; Hairston N.; Painter R. E.; Wisniewski D.; Scapin G.; Fitzgerald P.; Sharma N.; Lu J.; Ha S.; Hermes J.; Hammond M. L. Bioorg. Med. Chem. Lett. 2014, 24, 780. 10.1016/j.bmcl.2013.12.101. [DOI] [PubMed] [Google Scholar]
- Meragelman K. M.; McKee T. C.; McMahon J. B. J. Nat. Prod. 2004, 67, 1165. 10.1021/np030434i. [DOI] [PubMed] [Google Scholar]
- Selected methodologies to 1,3-diazepanes:; a Anumandla D.; Littlefield R.; Jeffrey C. S. Org. Lett. 2014, 16, 5112. 10.1021/ol502460j. [DOI] [PubMed] [Google Scholar]; b Kim M.; Gajulapati K.; Kim C.; Jung H. Y.; Goo J.; Lee K.; Kaur N.; Kang H. J.; Chung S. J.; Choi Y. Chem. Commun. 2012, 48, 11443. 10.1039/c2cc35484e. [DOI] [PubMed] [Google Scholar]; c Dutta S.; Higginson C. J.; Ho B. T.; Rynearson K. D.; Dibrov S. M.; Hermann T. Org. Lett. 2010, 12, 360. 10.1021/ol9026914. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhou H.-B.; Alper H. J. Org. Chem. 2003, 68, 3439. 10.1021/jo020526x. [DOI] [PubMed] [Google Scholar]; e Hylton K.-G.; Main A. D.; McElwee-White L. J. Org. Chem. 2003, 68, 1615. 10.1021/jo026816v. [DOI] [PubMed] [Google Scholar]; f McReynolds M. D.; Sprott K. T.; Hanson P. R. Org. Lett. 2002, 4, 4673. 10.1021/ol027074v. [DOI] [PubMed] [Google Scholar]
- Bauer R. A.; Wenderski T. A.; Tan D. S. Nat. Chem. Biol. 2012, 9, 21. 10.1038/nchembio.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shaw M. H.; Melikhova E. Y.; Kloer D. P.; Whittingham W. G.; Bower J. F. J. Am. Chem. Soc. 2013, 135, 4992. 10.1021/ja401936c. [DOI] [PubMed] [Google Scholar]; b Shaw M. H.; McCreanor N. G.; Whittingham W. G.; Bower J. F. J. Am. Chem. Soc. 2015, 137, 463. 10.1021/ja511335v. [DOI] [PubMed] [Google Scholar]; c Shaw M. H.; Croft R. A.; Whittingham W. G.; Bower J. F. J. Am. Chem. Soc. 2015, 137, 8054. 10.1021/jacs.5b05215. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shaw M. H.; Whittingham W. G.; Bower J. F. Tetrahedron 2016, 72, 2731. 10.1016/j.tet.2015.08.052. [DOI] [Google Scholar]
- Murakami M.; Tsuruta T.; Ito Y. Angew. Chem., Int. Ed. 2000, 39, 2484.. [DOI] [PubMed] [Google Scholar]
- A one-pot synthesis of 9l (57% yield, 6:1 saturated:unsaturated) from cyclopropylamine is given in the Supporting Information (SI).
- Cyclization of (S,S)-trans-5s (>98% ee) provided (S)-8s in >98% ee (see the SI).
- Replacement of the n-butyl group with methyl or cyclohexyl substituents gave analogous results (see the SI).
- The relative stereochemistry of 1,2-disubstituted cyclopropane substrates was determined using a combination of X-ray crystallographic analysis (trans-5s) and NOE experiments.
- Similar results were obtained in the presence of PhCO2H (15 mol%). The SI details analogous experiments for carbamate protected systems, confirming that C–C activation selectivity is the same in the absence and presence of CO.
- For the conversion of trans-5v to 8v′, β-hydride elimination presumably occurs via N6-H.
- Alternate explanations cannot be discounted on the basis of available data. For example, the steric effects of the R3-group may result in slow formation of ID, rather than slow reductive elimination from this intermediate.
- The suggestion that the alkene dissociates assumes stereospecific protodemetalation.
- Gridnev I. D.; Higashi N.; Asakura K.; Imamoto T. J. Am. Chem. Soc. 2000, 122, 7183. 10.1021/ja000813n. [DOI] [Google Scholar]
- By the strictest (IUPAC) definition, 7-ring systems are not classed as medium rings. However, the chemistry described here is an important conceptual stepping stone towards this goal because conventional 7-ring cyclizations are, in terms of kinetics, significantly more demanding than 5- or 6-ring variants:; Illuminati G.; Mandolini L.; Masci B. J. Am. Chem. Soc. 1975, 97, 4960. 10.1021/ja00850a032. [DOI] [Google Scholar]
- a Review: Shaw M. H.; Bower J. F.. Chem. Commun. 2016, DOI: 10.1039/c6cc04359c. Selected recent contributions: [DOI] [Google Scholar]; b Zhou X.; Dong G. J. Am. Chem. Soc. 2015, 137, 13715. 10.1021/jacs.5b09799. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ko H. M.; Dong G. Nat. Chem. 2014, 6, 739. 10.1038/nchem.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Souillart L.; Parker E.; Cramer N. Angew. Chem., Int. Ed. 2014, 53, 3001. 10.1002/anie.201311009. [DOI] [PubMed] [Google Scholar]; e Souillart L.; Cramer N. Angew. Chem., Int. Ed. 2014, 53, 9640. 10.1002/anie.201405834. [DOI] [PubMed] [Google Scholar]; f Matsuda T.; Tsuboi T.; Murakami M. J. Am. Chem. Soc. 2007, 129, 12596. 10.1021/ja0732779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.