

Abstract
We here reported the mitochondrial (mt) genome of one of the heterotrophic microeukaryotes related to cryptophytes, Palpitomonas bilix. The P. bilix mt genome was found to be a linear molecule composed of “single copy region” (∼16 kb) and repeat regions (∼30 kb) arranged in an inverse manner at both ends of the genome. Linear mt genomes with large inverted repeats are known for three distantly related eukaryotes (including P. bilix), suggesting that this particular mt genome structure has emerged at least three times in the eukaryotic tree of life. The P. bilix mt genome contains 47 protein-coding genes including ccmA, ccmB, ccmC, and ccmF, which encode protein subunits involved in the system for cytochrome c maturation inherited from a bacterium (System I). We present data indicating that the phylogenetic relatives of P. bilix, namely, cryptophytes, goniomonads, and kathablepharids, utilize an alternative system for cytochrome c maturation, which has most likely emerged during the evolution of eukaryotes (System III). To explain the distribution of Systems I and III in P. bilix and its phylogenetic relatives, two scenarios are possible: (i) System I was replaced by System III on the branch leading to the common ancestor of cryptophytes, goniomonads, and kathablepharids, and (ii) the two systems co-existed in their common ancestor, and lost differentially among the four descendants.
Keywords: mitochondrial genome, Cryptista, inverted repeats, cytochrome c maturation
Introduction
The mitochondrion is an organelle that is a remnant of the endosymbiotic α-proteobacterium captured by the common ancestor of eukaryotes, and still retains its own genome of bacterial origin (Gray 1993). Despite the single origin of mitochondria, mitochondrial (mt) genomes are largely divergent in the aspects of gene content and genome structure (Gray 1999; Gray et al. 2004). The most gene-rich mt genome known to date is that of the jakobid Andalucia godoyi containing 100 genes (Lang et al. 1997; Burger et al. 2013). In contrast, apicomplexan parasites (e.g., a malaria parasite Plasmodium falciparum) and their photosynthetic relatives (e.g., Chromera velia) have the least gene-rich mt genomes containing only 3–5 genes (Hikosaka et al. 2013; Flegontov et al. 2015). Mitochondrial genome structure also varies among eukaryotes. Besides the “typical” single-circular form represented by the human mt genome, single-linear, multi-linear, and multi-circular forms have been reported amongst phylogenetically diverse eukaryotes [reviewed in Smith and Keeling (2015)].
Cryptophytes, one of major photosynthetic microeukaryotic lineages, have a clear phylogenetic affinity to heterotrophic microeukaryotes, namely, goniomonads, kathablepharids, and Palpitomonas bilix. Furthermore, three heterotrophic groups, such as Picozoa represented by Picomonas judraskeda (Not et al. 2007; Seenivasan et al. 2013), Telonemia represented by Telonema subtilis (Yabuki et al. 2013), and Endohelea represented by Microheliella maris (Yabuki et al. 2012), were proposed to be parts of the monophyletic assemblage containing cryptophytes, which is designated as Cryptista (Cavalier-Smith et al. 2015). Large-scale transcriptomic data of cryptophytes, goniomonads, and P. bilix are available in the Marine Microbial Eukaryote Transcriptome Sequencing Project databases (Keeling et al. 2014). For the cryptophyte Guillardia theta, the nuclear genome has been completed by Curtis et al. (2012). In contrast, no complete mt genome of cryptists is available, except those of two cryptophytes (Hauth et al. 2005; Kim et al. 2008).
In this study, we sequenced the complete mt genome of P. bilix as the first step towards understanding the mt genome evolution in Cryptista. The mt genome of P. bilix was found to be a linear molecule with a tripartite structure—“single copy region” (∼16 kb) and two identical copies of an approximately 30 kb-region arranged inversely (∼76 kb in total). In terms of gene content, the P. bilix mt genome was found to be unique among members of Cryptista in containing ccmA, ccmB, ccmC, and ccmF genes encoding protein subunits involved in cytochrome c maturation in mitochondria. Using comparative genomic approaches, we here discuss (i) the evolution of mt genome with the tripartite structure, and (ii) that of cytochrome c maturation system in the clade of cryptophytes, goniomonads, kathablepharids, and P. bilix.
Materials and Methods
Cell Culture
P. bilix cells were cultured in the seawater-based URO medium (http://mcc.nies.go.jp/02medium.html#uro; last accessed September 12, 2016) containing 10% of the YT medium (http://mcc.nies.go.jp/02medium.html#YT; last accessed September 12, 2016) at 20 °C as described in Yabuki et al. (2010). After the density of the P. bilix cells reached approximately 105 cells/mL, the cells were harvested for DNA extraction and preparation of the plugs for pulsed-field gel electrophoresis (PFGE).
DNA Extraction, Sequencing, and In Silico Reconstruction of the mt Genome
Total DNA was extracted from the harvested P. bilix cells by the standard phenol/chloroform-based method. About 100 μg of total DNA was used for preparation of a 350 bp-, paired-end library with the TruSeq Nano DNA LT Sample Prep Kit (Illumina). The library was subjected to the Illumina HiSeq2000 sequence system at Hokkaido System Science Co., Ltd (Sapporo, Japan). We obtained ∼346 million of paired-end 100-base reads (∼35 Gbp in total). Of these, 10 million reads were assembled by SPAdes (Nurk et al. 2013). We subjected the resultant contigs to TBLASTN to identify potential mt genome fragments. The queries used in the TBLASTN search were the putative amino acid (aa) sequences encoded in the genes identified in the 12-kb mt genome fragment of the kathablepharid Leucocryptos marina (Nishimura et al. 2012), and those identified in the complete mt genomes of two cryptophytes, Rhodomonas salina (Hauth et al. 2005) and Hemiselmis andersenii (Kim et al. 2008). We retrieved the contigs, which matched with the queries at E-values of less than 10−10, as the candidates for mt genome fragments. The contig sequences retrieved by the first BLAST analysis were then subjected to BLASTN against NCBI nr database to exclude bacterial contamination. We discarded the contigs that matched to known bacterial sequences with identity greater than 90% in the second BLAST analysis. The procedure described above selected only two contigs, one was ∼62 kb and the other was ∼14 kb in size, as candidates for the P. bilix mt genome fragments. The two contigs appeared to be parts of a single, continuous DNA molecule, as we successfully amplified the DNA fragment bridging the two contigs by PCR with a set of primers designed based on the contig sequences (data not shown).
Genome Annotation
Within the putative mt genome, open reading frames (ORFs) encoding proteins longer than 50 aa residues were identified by using Artemis (Carver et al. 2012). Functions of the putative proteins were predicted by subjecting the identified ORFs to BLASTX against NCBI nr database. Transfer RNA genes were predicted with the tRNAscan-SE Search Server (http://trna.ucsc.edu/tRNAscan-SE/citation.html; last accessed September 12, 2016). Genes encoding rRNAs (rns, rnl, and rrn5) and RNase P RNA were searched using MFannot (http://megasun.bch.umontreal.ca/RNAweasel/; last accessed September 12, 2016). The gene encoding transfer-messenger RNA (tmRNA) was detected by using Infernal ver. 1.1.1 (Eddy and Durbin 1994) with a covariance model built from the tmRNA sequences identified in mt genomes (Burger et al. 2013; Hafez et al. 2013). The mt genome sequence with the annotation was deposited in the DNA Data Bank of Japan (GenBank/EmBL/DDBJ; accession no. AP017433).
Gel Electrophoreses
The overall structure and length of the P. bilix mt genome were initially predicted from the analyses of Illumina sequence reads (see above), and then were confirmed by a combination of PFGE and Southern blot analyses. The confluently grown P. bilix cells in 500 mL culture were used for preparing the plugs for PFGE as described by Eschbach et al. (1991) and Tanifuji et al. (2006). The harvested cells were rapidly embedded in 1% Certified Low Melt Agarose (Bio-Rad Laboratories) with envelope buffer (10 mM Tris-HCl pH 8.0, 100 mM EDTA, 200 mM NaCl). The agarose was poured into Disposable Plug Mold (Bio-Rad Laboratories) and solidified at 4 °C. The solidified agarose gel (plug) was incubated in N-lauroylsarcosine-Na buffer (10 mM Tris-HCl pH 8.0, 400 mM EDTA, 1% N-lauroylsarcosine-Na) with 5 mg proteinase K (3.5 U/mg; Sigma-Aldrich) for 10 days at 50 °C. The plug was then transferred to SDS buffer (50 mM Tris-HCl pH 8.0, 100 mM EDTA, 1% SDS) with 5 μL of proteinase K (100 μg/μL) and incubated for 10 days at 50 °C. The plug was washed with TE buffer before PFGE.
To digest the mt genome by XbaI, the plug was soaked in 300 μL of XbaI reaction mixture containing 25 units of XbaI (TaKaRa) at 37 °C for 12 h. We then added 25 units of XbaI to the reaction mixture, and continued the digestion for 12 h. The plugs with and without XbaI digestion were subjected to PFGE using 1% agarose gel (prepared with 0.5 × TBE buffer), which was run in 0.5 × TBE buffer at 14 °C. CHEF-DR II Pulsed-Field Electrophoresis System (Bio-Rad Laboratories) was used for the PFGE in this study. The PFGE ran for 16 h at a voltage of 5.1 V/cm with switching time set to 2.5–25 s. The agarose gel was then subjected to Southern blot analysis (see below).
For rough estimation of the 5′ and 3′ terminal regions of the linear mt genome (see below for details), 3 μg of total DNA extracted from the P. bilix cells was digested by EcoRI. We also prepared the DNA sample digested by SalI. The two digested DNA samples were subjected to standard gel electrophoresis using 1% agarose gel (prepared with 1 × TAE buffer), and run in 1 × TAE buffer at room temperature. The gel electrophoresis was run for 4.5 h at 50V. The agarose gel was then subjected to Southern blot analysis (see below).
Southern Blot Analyses
The DNA probe for Southern blot analyses was set within atp1 (see below). To prepare the template DNA for digoxigenin (DIG) probe synthesis, we firstly amplified a partial region of atp1 from P. bilix total DNA by PCR with a set of primers (Forward: 5′-TTGACCACGACCAATCGGTAC-3′, and Reverse: 5′-GTAGGAGATGGTATTGCTCGTG-3′), and cloned into pGEM-T Easy vector (Promega). The DIG probe was then synthesized using PCR DIG Probe Synthesis Kit (Roche Applied Science) with the cloned atp1 amplicon as the template and the primer set described above. This DNA probe was used for all the Southern blot analyses conducted in this study (see below). The hybridization reaction was performed at 37 °C for 16 h in DIG Easy Hyb (Roche Applied Science) with the DIG probe (Final concentration: 2 μL DIG probe/1 mL DIG Easy Hyb). The DNA separated by agarose gel electrophoresis was transferred to a positively charged nylon membrane (Amersham Hybond–N+, GE Healthcare) as described in DIG Application Manual for filter hybridization (Roche Applied Science). The membrane was washed twice for 5 min at room temperature in 2 × SSC, 0.1% SDS, and then for 15 min at 65 °C in 0.2 × SSC, 0.1% SDS. The signal from the DIG probe was detected using the Ready-to-use CDP-Star (Roche Applied Science). The detailed processes for hybridization and probe detection were followed as described in DIG Application Manual for filter hybridization (Roche Applied Science).
Results and Discussion
Genome Structure
The analysis of Illumina HiSeq 2000 reads, coupled with PCR experiments, recovered a single, linear contig for the P. bilix mt genome, which is 76,874 bp in size (fig. 1A). The G + C content of this contig is 27.6%. This “76 Kb-contig” appeared to bear large repeats at both ends, which are arranged in an inverse manner. Each inverted repeat is ∼30 Kb in size, and contains 25 ORFs and 18 structural RNA genes (fig. 1A and B). “Single copy region,” which is sandwiched between the two inverted repeats, is ∼16 kb in size, and contains 22 ORFs and 8 structural RNA genes (fig. 1A and B). We experimentally examined whether the P. bilix mt genome can be represented by 76 Kb-contig with respect to genome size and structure. First, the PFGE of undigested total DNA, followed by a Southern blot analysis, detected a single DNA fragment of ∼75.5 kb in size, consistent with the size of 76 kb-contig reconstructed from the sequence analysis described above (fig. 2A). Second, the mt genome fragments produced by XbaI digestion strongly suggested that the P. bilix mt genome comprise a single linear molecule. As a single XbaI site was found in the central (single copy) region, the linear mt genome is anticipated to be split into two fragments of ∼44 and ∼32 kb in size after XbaI digestion (fig. 2A). Alternatively, if the mt genome forms a circular or multimeric linear structure, a single 76 kb-fragment is anticipated. After XbaI digestion of the mt genome, two DNA fragments, which were estimated to be ∼45.5 and ∼36.8 kb in size, were detected (fig. 2A). Although the precise sizes of DNA fragments are difficult to be estimated based on this PFGE image, this result agrees with the monomeric linear genome structure shown in figure 2A.
Fig. 1.—
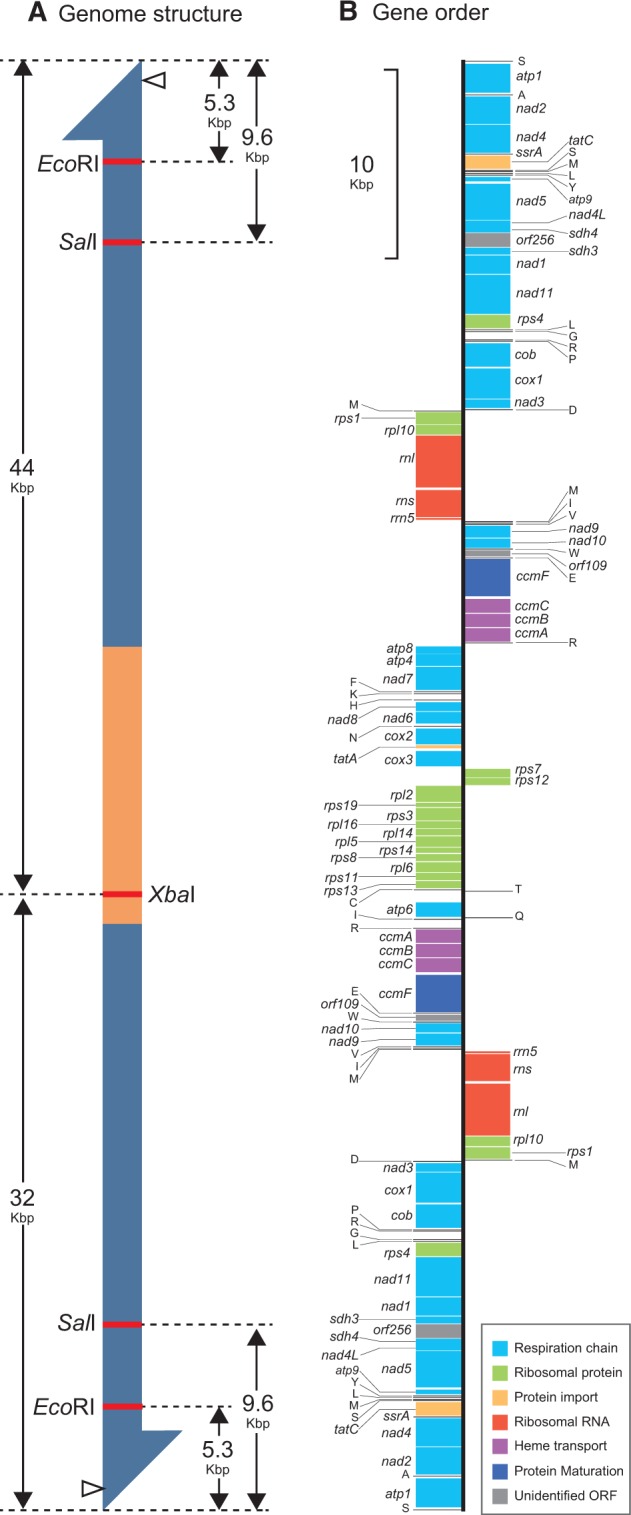
Palpitomonas bilix mitochondrial genome. (A) Putative mitochondrial (mt) genome structure. Inverted repeats are shown in blue, and the central portion of the mt genome (single copy region) is shown in yellow. The DIG probe was designed to hybridize atp1 gene region in inverted repeats (open arrowheads). According to the monomeric linear form of the P. bilix mt genome, XbaI digestion is anticipated to produce ∼44 kb- and ∼32 kb-fragments. Likewise, ∼5.3 kb- and ∼9.6 kb-fragments are likely to be detected by Southern blot analyses following EcoRI and SalI digestion, respectively. (B) Gene order. Protein-coding genes and ribosomal RNA genes are shown by boxes, and their putative functions are color-coded (see the inset). Transfer RNA and transfer-messenger RNA genes are shown by lines.
Fig. 2.—

Southern blot analyses of the Palpitomonas bilix mitochondrial genome. A DIG DNA probe set within atp1 gene region (fig. 1) was used in all the Southern blot analyses in this study. (A) Analysis of the DNA sample separated by pulsed-field gel electrophoresis (PFGE). The DNA samples with and without XbaI digestion were run in lanes 1 and 2, respectively. The sizes of the detected DNA fragments were found to be consistent with the predicted monomeric linear form of the mitochondrial (mt) genome (fig. 1A). Note that the precise sizes of the two DNA fragments were difficult to estimate from the PFGE image. (B). Analysis of the DNA samples digested by EcoRI and SalI. Both restriction enzymes generated the DNA fragments, the sizes of which were found to be similar to those expected from the mt genome contig reconstructed from Illumina pair-end short reads (fig. 1A).
To assess whether 76 kb-contig covers the ends of the mt genome sufficiently, we more carefully evaluated the lengths of the mt genome fragments generated by EcoRI and SalI digestions. Each repeat region bears a single EcoRI site, which is ∼5.3 kb distant from the ends of the contig (fig. 1A). If both contig ends bear sufficiently long unassembled regions (e.g., telomeric repeats), the probe for Southern blotting, which is located at the edge of the inverted repeat region, will detect DNA fragments longer than ∼5.3 kb after EcoRI digestion. Similarly, if a large portion of the mt genome was missed at both ends of 76 kb-contig, SalI digestion was anticipated to produce DNA fragments longer than ∼9.6 kb (fig. 1A). Nevertheless, both EcoRI and SalI digestions were found to produce ∼5.31 and ∼9.51 kb fragments, respectively (fig. 2B). Thus, 76 kb-contig most likely covers almost entire mt genome, albeit we cannot exclude the possibility of short telomeric repeats not being assembled into the contig.
Linear mt genomes have been reported from phylogenetically diverse eukaryotes (Dinouël et al. 1993; Vahrenholz et al. 1993; Takano et al. 1994; Fan and Lee 2002; de Graaf et al. 2009; Swart et al. 2012; Hikosaka et al. 2013). Prior to this study on the P. bilix mt genome, there were only two known linear mt genomes bearing large inverted repeats containing multiple genes—the stramenopile Proteromonas (Pr.) lacertae (Pérez-Brocal et al. 2010) and Acavomonas peruviana which branches at the base of the clade of apicomplexans, dinoflagellates, and their relatives (Janouškovec et al. 2013; Tikhonenkov et al. 2014). As P. bilix, Pr. lacertae, and A. peruviana are distantly related to each other in the tree of eukaryotes, the linear mt genomes with large inverted repeats were established on the separate branches leading to the aforementioned species.
The linear mt genome of A. peruviana possesses telomere-like tandem repeats at both ends (Janouškovec et al. 2013), which are likely critical to maintain chromosome ends. On the other hand, for the linear mt genomes of P. bilix and Pr. laceratae, no repetitive sequence has been detected (Pérez-Brocal et al. 2010; this study). Thus, the P. bilix and Pr. lacertae mt genomes require alternative mechanisms to maintain their chromosome ends. For instance, the undetermined portion of the P. bilix and/or Pr. lacertae mt genome ends may have hairpin structures as observed in yeast mt genomes (Dinouël et al. 1993). It is also possible that P. bilix and/or Pr. lacertae possesses a novel mechanism for maintaining the ends of the linear mt genome.
Gene Content
The experimental data described in the previous section strongly suggest that 76 kb-contig most likely covers almost entire mt genome of P. bilix. Henceforth here, we regard 76 kb-contig as the P. bilix mt genome. In this genome, we identified genes encoding 47 protein species, 26 tRNA species, one tmRNA (ssrA), and three rRNA species (rnl, rns, and rrn5), which occupy approximately 91% of the genome. No gene encoding RNase P RNA was found. Among the 47 putative proteins, 23 are parts of the respiratory chain complexes I–V (highlighted in light blue in fig. 1B), 16 are ribosomal proteins (highlighted in green), and six are involved in protein transport, protein maturation, or heme transport (highlighted in orange, dark blue or purple). Only two out of the 47 putative proteins were functionally unassignable based on sequence similarity (highlighted in grey in fig. 1B). The tRNA genes are sufficient to translate all aa codons (supplementary table S1, Supplementary Material online). To the best of our knowledge, the gene encoding tmRNA has been found only in the mt genomes of jakobids and oomycetes (Burger et al. 2013; Hafez et al. 2013), except the P. bilix mt genome (this study). Similar to most mitochondrial tmRNA (mt-tmRNA) previously identified, the putative P. bilix mt-tmRNA was predicted to lack the mRNA domain, and have the CCA sequence added to the 3′-terminus post-transcriptionally (supplementary figs. S1A and B, Supplementary Material online). The following genes were found to overlap with each other; (i) nad3 and trnD, (ii) rps1 and rpl10, (iii) trnM and trnI, (iv) trnI and trnV, and (v) rpl2 and rps19. Neither RNA editing, intron nor a deviant genetic code was detected. We found potential Shine-Dalgarno (SD) sequences (5′-A/GAAGG-3′), which can assist mRNAs to recruit the ribosomes in the prokaryotic translation system, in the 5′ upstream regions of 15 out of the 47 ORFs (supplementary fig. S2, Supplementary Material online). The 5′ upstream regions including potential SD sequences were found to be complementary to the 3′ terminus of small subunit rRNA sequence (supplementary fig. S2, Supplementary Material online).
In recent phylogenomic analyses, P. bilix and cryptophytes, together with kathablepharids and goniomonads, constantly form a clade with high statistical support (Yabuki et al. 2014; Cavalier-Smith et al. 2015). Reflecting the phylogenetic affinity between P. bilix and cryptophytes, the gene content in the P. bilix mt genome was found to be similar to those in the mt genomes of two cryptophytes, Rhodomonas salina and Hemiselmis andersenii (Hauth et al. 2005; Kim et al. 2008; Note that no complete mt genome is available for kathablepharids or goniomonads in publicly accessible databases). In our comparison, we considered only functionally assignable protein-coding genes. As shown in figure 3, the gene content of the two cryptophyte mt genomes appeared to be identical, except rpl31 is absent in the R. salina mt genome. The P. bilix mt genome possesses four genes encoding proteins for cytochrome c maturation (ccmA, ccmB, ccmC, and ccmF) and two ribosomal proteins (rpl2 and rpl10), which are absent in the two cryptophyte mt genomes (fig. 3). On the other hand, two genes encoding ribosomal proteins (rps2 and rpl31) are present in the H. andersenii mt genome, but absent in the P. bilix mt genome (fig. 3). No rpl31 transcript, but the rps2 transcript was identified in the transcriptomic data of P. bilix deposited in the Marine Microbial Eukaryote Transcriptome Sequencing Project (Database accession, MMTESP0780; protein ID, CAMPEP_0113873348) by BLAST, suggesting that rps2 is encoded in the nuclear genome. In P. bilix, RPS2 is likely to be imported post-translationally into the mitochondrion, as a mitochondria targeting signal was predicted in its N-terminal region by TargetP 1.1 Server (Emanuelsson et al. 2007; http://www.cbs.dtu.dk/services/TargetP/; last accessed September 12, 2016). As the genome data is not available, we could not conclude whether rpl31 is genuinely absent in P. bilix.
Fig. 3.—

Venn diagram to compare the gene content among the mitochondrial genomes of Palpitomonas bilix and two cryptophytes (Rhodomonas salina and Hemiselmis andersenii). Note that tatA, rps1, and atp4 are not annotated in the GenBank file of the R. salina mitochondrial (mt) genome (GenBank accession no. NC_002572), but listed in the genome map (fig. 1) in Hauth et al. (2005). Only functionally assignable and vertically inherited protein-coding genes are included in this diagram. Functionally unassignable genes in the three mt genomes are indicated in parentheses.
Prior to this study, at least one of ccm genes involved in cytochrome c maturation system has been identified in mt genomes of ciliates, malawimonads, jakobids, streptophytes, red algae, heteroloboseans, and a collodictyonid (Pritchard et al. 1990; Lang et al. 1997; Unseld et al. 1997; Ohta et al. 1998; Fritz-Laylin et al. 2011; Kamikawa et al. 2016; See GenBank accession nos. NC_026311 and NC_002553 for the two malawimonad mt genomes). Amongst the mt genomes containing ccm genes, the set of ccmA, ccmB, ccmC, and ccmF has been found solely in the mt genomes of the red alga Cyanidioschyzon merolae (Ohta et al. 1998), jakobids (Lang et al. 1997; Burger et al. 2013), and P. bilix (this study; see figs. 2A and 3). Furthermore, the operon of the four ccm genes was retained only in the mt genomes of jakobids and P. bilix.
Evolution of Cytochrome c Maturation in Cryptista
Cytochromes of c-type bind to heme through two cysteine residues (at a conserved CXXCH motif) and transfer electrons between the respiratory chains in the mitochondria (Sanders et al. 2010). Due to the importance of heme binding in cytochrome c, all eukaryotes with canonical (aerobic) mitochondria require a pathway for cytochrome c maturation (Giegé et al. 2008). There are three evolutionarily distinct pathways for covalent attachment of heme to apocytochrome c in mitochondria, namely System I, System III, and an uncharacterized pathway in euglenozoans, which is sometimes referred to as System V (Allen et al. 2008). System I apparently shares the evolutionary origin with the bacterial system, of which subunits are encoded by the ccm operon (ccmA-B-C-D-E-F-G-H). Some eukaryotes (e.g., land plants) are believed to utilize System I, as the protein subunits required for System I have been found in both mt and nuclear genomes (Unseld et al. 1997; Meyer et al. 2005; Rayapuram et al. 2007; Hamel et al 2009). System III, a key component of which is holocytochrome c synthase (HCCS) (alternatively known as heme lyase), has been found exclusively in eukaryotes (Allen et al. 2008; Babbitt et al. 2015). Although euglenozoans utilize cytochrome c in their mitochondria, neither System I nor System III have been found in this lineage, implying the presence of a unique system (System V) for cytochrome c maturation (Allen et al. 2004; Allen 2011).
The phylogenetic distribution of System I and that of System III are mutually exclusive amongst aerobic eukaryotes, and, to the best of our knowledge, no organism harboring both evolutionarily distinct systems for cytochrome c maturation has been identified (Allen et al. 2008). It has been speculated that the entire System I, which includes membrane proteins, cannot be nucleus-encoded (Allen et al. 2008). For instance, if ccmC and ccmF were nucleus-encoded and their gene products—inner mitochondrial membrane (IMM)-associated proteins containing multiple transmembrane helices—were synthesized in the cytosol, it would be difficult to transport these proteins into the mitochondrion and place them within the IMM. Indeed, in all of the eukaryotes utilizing System I known so far, at least one subunit was found to be mitochondrially encoded (Allen et al. 2008). Thus, eukaryotes with the mt genomes carrying no ccm gene are anticipated to utilize nucleus-encoded HCCS for cytochrome c maturation (i.e. System III)
P. bilix apparently uses System I for cytochrome c maturation, as the four ccm genes were found in the mt genome (fig. 3), and no HCCS transcript was detected in the transcriptomic data of this organism deposited in the Marine Microbial Eukaryote Transcriptome Sequencing Project. In contrast, no ccm gene was found in the R. salina or H. andersenii mt genome, suggesting that cryptophytes utilize System III for cytochrome c maturation. Indeed, the transcripts for HCCS were detected in the transcriptomic data of multiple cryptophyte species including R. salina and H. andersenii (summarized in table 1). Likewise, HCCS transcripts were detected in the transcriptomic data of the kathablepharid Roombia sp. NY0200, Goniomonas pacifica, and Goniomonas sp. strain m, suggesting that both kathablepharids and goniomonads utilize System III as well (table 1).
Table 1.
Holocytochrome c Synthase (HCCS) Detected in the Transcriptomic Data of Cryptist Members
| Organism | Transcriptome project | Accession nos for the transcripts encoding HCCS | |
|---|---|---|---|
| Cryptophytes | Cryptomonas curvata CCAP979/52 | MMETSP1050 | CAMPEP_0172183370, CAMPEP_0172202336 |
| Cryptomonas paramecium CCAP977/2a | MMETSP0038 | CAMPEP_0113663230, CAMPEP_0113669566 | |
| Geminigera cryophila CCMP2564 | MMETSP0799 | CAMPEP_0173105750, CAMPEP_0179442806, CAMPEP_0179461534* | |
| Geminigera sp. | MMETSP1102 | CAMPEP_0173080548*, CAMPEP_0173119622* | |
| Hemiselmis andersenii CCMP439 | MMETSP1041 | CAMPEP_0172005192*, CAMPEP_0172040676* | |
| Hemiselmis andersenii CCMP1180 | MMETSP1042 | CAMPEP_0169556210* | |
| Hemiselmis andersenii CCMP441 | MMETSP1043 | CAMPEP_0173412080, CAMPEP_0172005192, CAMPEP_0172089158*, CAMPEP_0172061530* | |
| Hemiselmis rufescens PCC563 | MMETSP1357 | CAMPEP_0173471772, CAMPEP_0173442208 | |
| Hemiselmis tepida CCMP443 | MMETSP1355 | CAMPEP_0174942424 | |
| Hemiselmis virescens PCC157 | MMETPS1356 | CAMPEP_0173394540 | |
| Proteomonas sulcata CCMP704 | MMETSP1049 | CAMPEP_018431988 | |
| Rhodomonas abbreviata | MMETSP1101 | CAMPEP_0181296906, CAMPEP_0181288406, CAMPEP_0181343696 | |
| Rhodomonas lens | MMETSP0484 | CAMPEP_0177719224, CAMPEP_0177727028* | |
| Rhodomonas salina CCMP 1319 | MMETSP1047 | CAMPEP_0172101522 | |
| Undescribed cryptophyte species CCMP2293 | MMETSP0987 | CAMPEP_0180215772, CAMPEP_0180369356*, CAMPEP_0180305760*, CAMPEP_0180144656* | |
| Goniomonads | Goniomonas pacifica CCMP1869 | MMETSP0108 | CAMPEP_0175913492 |
| Goniomonas sp. Strain m | MMETSP0114 | CAMPEP_0114548122, CAMPEP_0114556322 | |
| Kathablepharid | Roombia sp. NY0200 | In-house | LC148861, LC148862 |
Note.—The accession numbers of crypotophyte and goniomonad HCCS transcripts are of the Marine Microbial Eukaryote Transcriptome Sequence Project (http://marinemicroeukaryotes.org/project_organisms; last accessed September 12, 2016). The sequences marked with asterisks were omitted from the HCCS phylogenetic tree presented in Fig. S3. For Roombia sp. NY0200, two distinct HCCS transcripts were identified in our in-house transcriptomic data and deposited in DDBJ/EMBL/GenBank under the accession nos. listed in the table.
If Systems I and III cannot co-exist in a single cell (Allen et al. 2008), the evolution of cytochrome c maturation among P. bilix, kathablepharids, goniomonads, and cryptophytes (so-called “core cryptists”) is not complicated. By superimposing the distribution of System I/System III on the relationship amongst core cryptists, we can assume that (i) the common ancestor of core cryptists used System I, and (ii) after the separation of P. bilix and the common ancestor of kathablepharids, goniomonads, and cryptophytes, the switch from System I to System III took place on the branch leading to the latter (“Scenario 1” in fig. 4A). It is straightforward to speculate that the system for cytochrome c maturation in the ancestor of core cryptists remains in P. bilix. This scenario demands the HCCS genes found in kathablepharids, goniomonads, and cryptophytes to be of a lateral origin (or origins). Unfortunately, the phylogenetic relationship amongst HCCS aa sequences was little resolved, providing no clue for the origin of the HCCS genes in core cryptists (supplementary fig. S3, Supplementary Material online).
Fig. 4.—
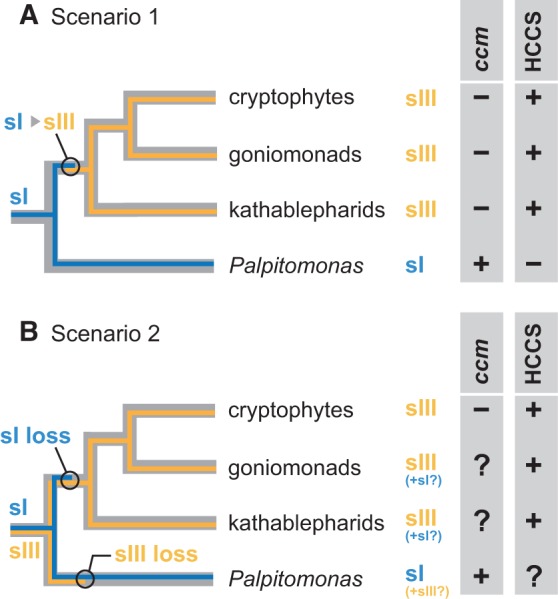
Scenarios for the evolution of cytochrome c maturation system in Palpitomonas bilix, kathablepharids, goniomonads, and cryptophytes. The phylogenetic relationship amongst the four lineages/species is based on Yabuki et al. (2014). (A) Scenario 1 assuming no co-existence of Systems I and III. Under this assumption, the distribution of Systems I and III can be predicted as follows. Although no nuclear genome is available for Palpitomonas bilix, we can conclude that this organism lacks a HCCS gene for System III, as its mitochondrial (mt) genome contains ccm genes for System I. Likewise, although no mt genome data is available for goniomonads or kathablepharids, we can conclude that the two lineages lack ccm genes for System I, as HCCS genes were identified in their transcriptomic data. By combining both nuclear and mt genome data of cryptophytes, this lineage most probably possesses a single system for cytochrome c maturation (i.e., System III). For each of the four lineages/species, the presence/absence (+/−) of ccm genes and a HCCS gene is indicated. This scenario assumes (i) the ancestral cell used the bacterial system (i.e., System I; designated as “sI”), and (ii) the switch from System I to System III (designated as “sIII”) occurred in the common ancestor of kathablepharids, goniomonads, and cryptophytes. (B) Scenario 2. This scenario assumes (i) the ancestral cell possessed the two evolutionarily distinct systems, and (ii) System I or System III was differentially lost on the branch leading to P. bilix and that leading to the common ancestor of kathablepharids, goniomonads, and cryptophytes. If the co-existence of Systems I and III is possible in a single cell, some uncertainties about the distribution of Systems I and III remain (represented by question marks). As no nuclear genome is available for P. bilix, it is uncertain whether a HCCS gene (System III) is absent in this organism. Likewise, no complete mt genome is available for kathablepharids or goniomonads, and the presence of ccm genes for System I in either (or both) lineages remains unclear.
In the literature, no organism has been known to possess both Systems I and III so far. However, in the strict sense, the possibility of the two systems co-existing in a single cell cannot be ruled out. If the co-existence of Systems I and III is possible, alternative scenarios are conceivable and one of them is presented in figure 4B. This particular scenario assumes that the ancestor of core cryptists possessed both Systems I and III, and the two distinct systems were differentially lost on the branch leading to P. bilix and that leading to the common ancestor of kathablepharids, goniomonads, and cryptophytes. Nevertheless, the hypothesis described above would give P. bilix a somewhat uncertain status; the presence of a HCCS gene (i.e., System III) cannot be excluded, as no genome data is currently available for P. bilix. Likewise, HCCS genes were identified in the transcriptomic data of kathablepharids and goniomonads (table 1), but the presence of ccm genes (i.e., System I) cannot be excluded until their mt genomes are completed. At this moment, we are certain only about a single system for cytochrome c maturation (System III) in cryptophytes, as both their nuclear and mt genome data are available (Hauth et al. 2005; Kim et al. 2008; Curtis et al. 2012).
We need to revisit the evolution of cytochrome c maturation in core cryptists when both nuclear and mt genome data become available for P. bilix, kathablepharids, and goniomonads. It is also important to characterize cytochrome c maturation in Picozoa, Telonemia, and Endohelea, which were nominated as the phylogenetic relatives of core cryptists (Cavalier-Smith et al. 2015). In particular, the endohelean Microheliella maris is critical to infer the early evolution of cytochrome c maturation in Cryptista, as the Bayesian multigene phylogenetic analyses constantly placed this microeukaryote at the base of the clade of core cryptists including P. bilix (Cavalier-Smith et al. 2015).
Supplementary Material
Supplementary table S1 and figures S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported in part by grants from the Japanese Society for Promotion of Sciences (JSPS) awarded to R. K. (15H05606 and 15H14591), G. T. (26840123), T. H. (23117005 and 15H05231), and Y. I. (23117006 and 16H04826). A. Y. and Y. N. were supported by research fellowships from the JSPS for Young Scientists (no. 201242 and 236484 for A. Y., and no. 25789 for Y. N.). R. K. was supported by a grant from the Institute for Fermentation, Osaka, Japan.
Literature Cited
- Allen JW, Ginger ML, Ferguson SJ. 2004. Maturation of the unusual single-cysteine (XXXCH) mitochondrial c-type cytochromes found in trypanosomatids must occur through a novel biogenesis pathway. Biochem. J. 383:537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JW, et al. 2008. Order within a mosaic distribution of mitochondrial c-type cytochrome biogenesis systems? FEBS J. 275:2385–2402. [DOI] [PubMed] [Google Scholar]
- Allen JW. 2011. Cytochrome c biogenesis in mitochondria—Systems III and V. FEBS J. 278:4198–4216. [DOI] [PubMed] [Google Scholar]
- Babbitt SE, Sutherland MC, Francisco BS, Mendez DL, Kranz RG. 2015. Mitochondrial cytochrome c biogenesis: no longer an enigma. Trends Biochem. Sci. 40:446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Forget L, Lang BF. 2013. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol. Evol. 5:418–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T, Chao EE, Lewis R. 2015. Multiple origins of Heliozoa from flagellate ancestors: new cryptist subphylum Corbihelia, superclass Corbistoma, and monophyly of Haptista, Cryptista, Hacrobia and Chromista. Mol. Phylogenet. Evol. 93:331–362. [DOI] [PubMed] [Google Scholar]
- Curtis BA, et al. 2012. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 492:59–65. [DOI] [PubMed] [Google Scholar]
- Dinouël N, et al. 1993. Linear mitochondrial DNAs of yeasts: closed-loop structure of the termini and possible linear-circular conversion mechanisms. Mol. Cell. Biol. 13:2315–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR, Durbin R. 1994. RNA sequence analysis using covariance models. Nucleic Acids Res. 22:2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. 2007. Locating proteins in the cell using TargetP, SignalP, and related tools. Nat. Protoc. 2:953–971. [DOI] [PubMed] [Google Scholar]
- Eschbach S, Hofmann CJ, Maier UG, Sitte P, Hansmann P. 1991. A eukaryotic genome of 660 kb: electrophoretic karyotype of nucleomorph and cell nucleus of the cryptomonad alga, Pyrenomonas salina. Nucleic Acids Res. 19:1779–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lee RW. 2002. Mitochondrial genome of the colorless green alga Polytomella parva: two linear DNA molecules with homologous inverted repeat termini. Mol. Biol. Evol. 19:999–1007. [DOI] [PubMed] [Google Scholar]
- Flegontov P, et al. 2015. Divergent mitochondrial respiratory chains in phototrophic relatives of apicomplexan parasites. Mol. Biol. Evol. 32: 1115–1131. [DOI] [PubMed] [Google Scholar]
- Fritz-Laylin L, Ginger M, Walsh C, Dawson S, Fulton C. 2011. The Naegleria genome: a free-living microbial eukaryote lends unique insights into core eukaryotic cell biology. Res. Microbiol. 162:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegé P, Grienenberger JM, Bonnard G. 2008. Cytochrome c biogenesis in mitochondria. Mitochondrion 8:61–73. [DOI] [PubMed] [Google Scholar]
- de Graaf RM, et al. 2009. The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genomics 10:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW. 1993. Origin and evolution of organelle genomes. Curr. Opin. Genet. Dev. 3:884–890. [DOI] [PubMed] [Google Scholar]
- Gray MW. 1999. Evolution of organellar genomes. Curr. Opin. Genet. Dev. 9:678–687. [DOI] [PubMed] [Google Scholar]
- Gray MW, Lang BF, Burger G. 2004. Mitochondria of protists. Annu. Rev. Genet. 38:477–524. [DOI] [PubMed] [Google Scholar]
- Hafez M, Burger G, Steinberg SV, Lang F. 2013. A second eukaryotic group with mitochondrion-encoded tmRNA. RNA Biol. 10: 1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel P, Corvest V, Giegé P, Bonnard G. 2009. Biochemical requirements for the maturation of mitochondrial c-type chtochromes. Biochim. Biophys. Acta. 1793:125–138. [DOI] [PubMed] [Google Scholar]
- Hauth AM, Maier UG, Lang BF, Burger G. 2005. The Rhodomonas salina mitochondrial genome: bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res. 33:4433–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka K, Kita K, Tanabe K. 2013. Diversity of mitochondrial genome structure in the phylum Apicomplexa. Mol. Biochem. Parasitol. 188: 26–33. [DOI] [PubMed] [Google Scholar]
- Janouškovec J, et al. 2013. Colponemids represent multiple ancient alveolate lineages. Curr. Biol. 23:2546–2552. [DOI] [PubMed] [Google Scholar]
- Kamikawa R, Shiratori T, Ishida K, Miyashita H, Roger AJ. 2016. Group II intron-mediated trans-splicing in the gene-rich mitochondrial genome of an enigmatic eukaryote, Diphylleia rotans. Genome Biol. Evol. 8:458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, et al. 2014. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12:e1001889.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, et al. 2008. Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Genomics 9:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, et al. 1997. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature 387:493–497. [DOI] [PubMed] [Google Scholar]
- Meyer EH, et al. 2005. AtCCMH, an essential component of the c-type cytochrome maturation pathway in Arabidopsis mitochondria, interacts with apocytochrome c. Proc. Natl. Acad. Sci. U S A. 102: 16113–16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Kamikawa R, Hashimoto T, Inagaki Y. 2012. Two independent lateral group I intron transfers in the katablepharid Leuocryptos marina. PLoS One 7:e37307.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Not F, et al. 2007. Picobiliphytes: a marine picoplanktonic algal grouped with unknown affinities to other eukaryotes. Science 315:253–255. [DOI] [PubMed] [Google Scholar]
- Nurk S, et al. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20:714–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta N, Sato N, Kuroiwa T. 1998. Structure and organaization of the mitochondrial genome of the unicellular red alga Cyanidioschyzon merolae deduced from the complete nucleotide sequence. Nucleic Acids Res. 26:5190–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Brocal V, Shahar-Golan R, Clark CG. 2010. A linear molecule with two large inverted repeats: the mitochondrial genome of the stramenopile Proteromonas lacertae. Genome Biol. Evol. 2:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard AE, et al. 1990. Nucleotide sequence of the mitochondrial genome of Paramecium. Nucleic Acids Res. 18:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayapuram N, Hagenmuller J, Grienenberger J-MM, Giegé P, Bonnard G. 2007. AtCCMA interacts with AtCcmB to form a novel mitochondrial ABC transporter involved in cytochrome c maturation in Arabidopsis. J. Biol. Chem. 282:21015–21023. [DOI] [PubMed] [Google Scholar]
- Sanders C, Turkarslan S, Lee DW, Daldal F. 2010. Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 18:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seenivasan R, Sausen N, Medlin LK, Melkonian M. 2013. Picomonas judraskeda gen. et sp. nov.: the first identified member of the Picozoa phylum nov., a widespread group of picoeukaryotes, formerly known as “picobiliphytes”. PLoS One 8:e59565.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ. 2015. Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. U S A. 112:10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, Nowacki M, Shum J, et al. 2012. The Oxytricha trifallax mitochondrial genome. Genome Biol. Evol. 4:136–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano H, Kawano S, Kuroiwa T. 1994. Complex terminal structure of a linear mitochondrial plasmid from Physarum polycephalum: three terminal inverted repeats and an ORF encoding DNA polymerase. Curr. Genet. 25:252–257. [DOI] [PubMed] [Google Scholar]
- Tanifuji G, Erata M, Ishida K, Onodera N, Hara Y. 2006. Diversity of secondary endosymbiont-derived actin-coding genes in cryptomonads and their evolutionary implications. J. Plant Res. 119:205–215. [DOI] [PubMed] [Google Scholar]
- Tikhonenkov DV, et al. 2014. Description of Colponema vietnamica sp. n. and Acavomonas peruviana n. gen. n. sp., two new alveolate phyla (Colponemidia nom. nov. and Acavomonidia nom. nov.) and their contributions to reconstructing the ancestral state of alveolates and eukaryotes. PLoS One 9:e95467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unseld M, Marienfeld JR, Brandt P, Brennicke A. 1997. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 15:57–61. [DOI] [PubMed] [Google Scholar]
- Vahrenholz C, Riemen G, Pratje E, Dujon B, Michaelis G. 1993. Mitochondrial DNA of Chlamydomonas reinhardtii: the structure of the ends of the linear 15.8-kb genome suggests mechanisms for DNA replication. Curr. Genet. 24:241–247. [DOI] [PubMed] [Google Scholar]
- Yabuki A, Inagaki Y, Ishida K. 2010. Palpitomonas bilix gen. et sp. nov.: a novel deep-branching heterotroph possibly related to Archaeplastida or Hacrobia. Protist 161:523–538. [DOI] [PubMed] [Google Scholar]
- Yabuki A, Chao EE, Ishida K, Cavalier-Smith T. 2012. Microheliella maris (Microhelida ord. n.), an ultrastructurally highly distinctive new axopodial protist species and genus, and the unity of phylum Heliozoa. Protist 163:356–388. [DOI] [PubMed] [Google Scholar]
- Yabuki A, Eikrem W, Takishita K, Patterson DJ. 2013. Fine structure of Telonema subtilis Griessmann, 1913: a flagellate with an unique cytoskeletal structure among eukayrotes. Protist 164:556–569. [DOI] [PubMed] [Google Scholar]
- Yabuki A, et al. 2014. Palpitomonas bilix represents a basal cryptist lineage: insight into the character evolution in Cryptista. Sci. Rep. 4:4641.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.