

Abstract
Tat interaction with astrocytes has been shown to be important for Tat neurotoxicity and HIV/neuroAIDS. We have recently shown that Tat expression leads to increased glial fibrillary acidic protein (GFAP) expression and aggregation and activation of unfolded protein response/endoplasmic reticulum (ER) stress in astrocytes and causes neurotoxicity. However, the exact molecular mechanism of astrocyte-mediated Tat neurotoxicity is not defined. In this study, we showed that neurotoxic factors other than Tat protein itself were present in the supernatant of Tat-expressing astrocytes. Two-dimensional gel electrophoresis and mass spectrometry revealed significantly elevated lysosomal hydrolytic enzymes and plasma membrane-associated proteins in the supernatant of Tat-expressing astrocytes. We confirmed that Tat expression and infection of pseudotyped HIV.GFP led to increased lysosomal exocytosis from mouse astrocytes and human astrocytes. We found that Tat-induced lysosomal exocytosis was tightly coupled to astrocyte-mediated Tat neurotoxicity. In addition, we demonstrated that Tat-induced lysosomal exocytosis was astrocyte-specific and required GFAP expression and was mediated by ER stress. Taken together, these results show for the first time that Tat promotes lysosomal exocytosis in astrocytes and causes neurotoxicity through GFAP activation and ER stress induction in astrocytes and suggest a common cascade through which aberrant astrocytosis/GFAP up-regulation potentiates neurotoxicity and contributes to neurodegenerative diseases.
Keywords: astrocyte, cathepsin B (CTSB), exocytosis, glial cell, human immunodeficiency virus (HIV), neuron
Introduction
HIV-1 infection of the CNS occurs within hours of viral acquisition in the periphery (1) and often leads to neurological symptoms that include motor and cognitive dysfunction (2). These symptoms have collectively been termed HIV/neuroAIDS. Currently, there is no specific treatment for neuroAIDS, mainly because of incomplete understanding of the mechanisms of neuronal damage induced by HIV. Introduction of combination antiretroviral therapy (cART)2 has increased the lifespan of HIV-infected individuals and, as a result, has changed the landscape of HIV/neuroAIDS, with increased prevalence of minor cognitive motor disorder (MCMD) (3, 4). It is clear that cART has failed to prevent or reverse MCMD at the present time (5). Thus, new approaches to prevention and treatment of MCMD are undoubtedly warranted and urgently needed.
The main neuropathologies associated with HIV MCMD include widespread astrocytosis, chronic neuroinflammation, and compromised neuronal integrity (4, 6). At the cellular level, the primary cell targets for HIV infection are macrophages/microglia and, to a lesser extent, astrocytes (7, 8). However, HIV does not infect neurons that are mostly affected in the CNS of HIV-infected individuals. Therefore, a number of indirect mechanisms have been proposed for HIV/neuroAIDS pathogenesis. The proposed mechanisms could generally be grouped into three main categories: improper immune activation of macrophages/microglia, soluble factors of both viral and host origins from macrophages/microglia and astrocytes, and astrocyte dysfunction. It is highly conceivable that all three categories of mechanisms may function in an integrated manner in the HIV/neuroAIDS pathogenesis.
One such example is the HIV-1 Tat protein, which has been shown to be a major factor in HIV/neuroAIDS pathogenesis through various mechanisms. Tat is one of the viral soluble factors secreted from HIV-infected microglia/macrophages and astrocytes in the CNS or HIV-infected macrophages/monocytes and CD4+ T lymphocytes outside the CNS (9). It is present in the brain of HIV-infected individuals with and without cART treatment (10). Direct Tat exposure causes acute neuronal damage both in vitro and in vivo (11, 12). Tat could also affect neuron survival at more physiologically relevant concentrations indirectly through increased recruitment of macrophages/monocytes and lymphocytes into the CNS (13–16) or through alterations in cytokine expression, excitatory properties, intracellular signaling, autophagy, and endolysosomal function (13, 16–19). In agreement with these findings, Tat expression in or injection into the CNS in the absence of HIV infection is sufficient to cause neuropathologies similar to those noted in the brain of AIDS patients (16). In addition, Tat could affect neuron survival through interaction with astrocytes and subsequent alterations of astrocyte function (see more below) (20–22).
HIV infection of astrocytes has been characterized to be restricted, namely only abundantly expressing HIV early gene products Tat, Nef, and Rev proteins (23, 24). Tat has indeed been detected in and secreted by astrocytes that are latently infected with HIV (9, 25). Tat alters astrocyte growth (21, 22, 26) and induces expression of cytokines and chemokines in astrocytes such as MCP-1, IL-1β, IL-6, RANTES, and CXCL10 (17, 27, 28), which could in turn recruit more macrophages/monocytes and lymphocytes into the CNS (13–16). Studies, including ours, have shown that astrocytes potentiate Tat neurotoxicity (16, 20, 22), probably through Tat-mediated transcriptional activation of glial fibrillary acidic protein (GFAP) expression (29–31). Of particular note is that Tat expression in astrocytes alone is sufficient to induce pathological changes such as reactive astrocytes or astrocytosis, loss of neuron axons and dendrites, and neurobehavioral deficits such as impaired motor and cognitive functions in the CNS reminiscent of HIV-associated MCMD (22, 29, 32, 33). These findings support the notion that astrocyte-mediated Tat neurotoxicity plays important roles in HIV/neuroAIDS. However, the exact underlying molecular mechanisms are not well understood.
In the study, we aimed to determine the neurotoxic factors responsible for astrocyte-mediated Tat neurotoxicity. Specifically, we compared the protein profile in the supernatants of Tat-expressing mouse primary astrocytes and wild-type mouse primary astrocytes. We demonstrated that Tat expression in astrocytes induced lysosomal exocytosis and, as a result, led to astrocyte-mediated Tat neurotoxicity. Moreover, Tat-induced GFAP expression and ER stress were directly involved in this process. These findings provide new important insights not only about the roles of this critical and pervasive protein Tat in HIV/neuroAIDS, particularly in the era of cART, but also about the general roles of astrocytosis/GFAP up-regulation in contributing to neurodegenerative diseases.
Results
Neurotoxic Factors Other than Tat for Astrocyte-mediated Tat Neurotoxicity
To identify neurotoxic factors that were present in the supernatant of Tat-expressing astrocytes, we first determined whether Tat was the sole contributor of astrocyte-mediated Tat neurotoxicity. We transfected human primary astrocytes with Tat-expressing plasmid, immunodepleted the soluble Tat protein in the supernatant secreted by Tat-expressing astrocytes, and determined the neurotoxicity of the supernatants. The immunodepletion efficiency was assessed using the LTR-driven luciferase reporter gene activity assay. Compared with the C3 control, the supernatant from Tat-transfected astrocytes gave rise to a higher Luc activity (Fig. 1A) and neurotoxicity (Fig. 1B). Immunodepletion of the supernatants with IgG had little effect on Tat activity on the LTR promoter and Tat neurotoxicity. In contrast, immunodepletion of the supernatants with an antibody against c-Myc, an epitope tag used to identify Tat protein, abrogated Tat activity on the LTR promoter but had little effect on Tat neurotoxicity.
FIGURE 1.
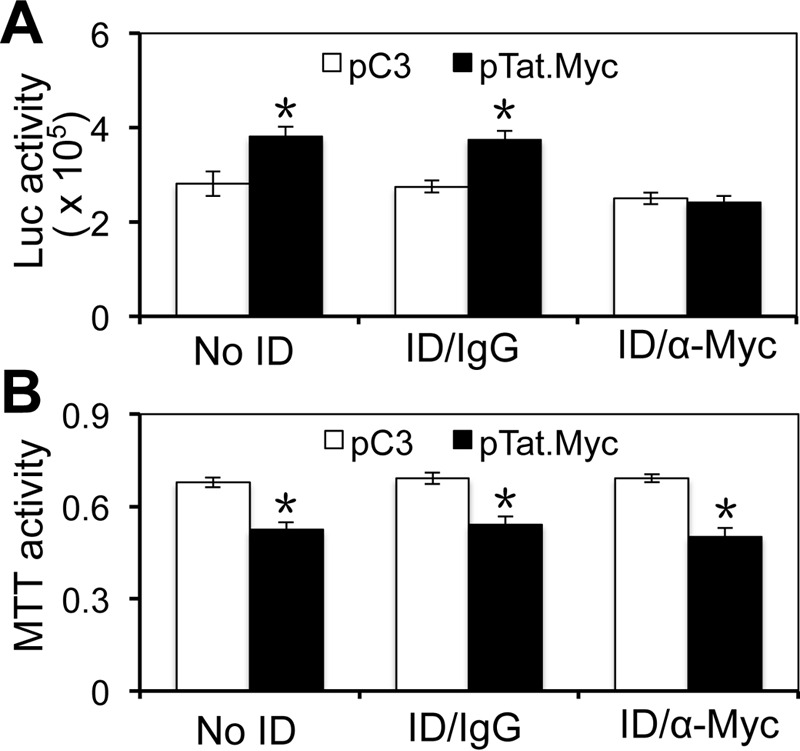
Neurotoxicity of Tat-immunodepleted supernatants from Tat-expressing astrocytes. U373.MG cells were transfected with pcDNA3 or pTat.Myc. Culture supernatants were harvested 72 h post-transfection and incubated with anti-Myc antibody for Tat immunodepletion (ID/α-Myc). Normal rabbit IgG was used as the control (ID/IgG). In addition, the supernatant without immunodepletion (No ID) was also included as a control. All supernatants were exposed to LTR-Luc-transfected 293T for their activity on the LTR promoter (A) or exposed to SH-SY5Y and evaluated for their effects on cell viability (B). Error bars, S.D.; *, p < 0.05.
Besides acute Tat neurotoxicity, Tat has been shown to induce production of TNF-α, free radicals, and glutamate in activated microglia/astrocytes and cause neurotoxicity (34–36). Thus, we next evaluated the role of each of those factors in astrocyte-mediated Tat neurotoxicity. We took advantage of the primary mouse astrocytes isolated from the doxycycline-inducible and astrocyte-specific mice (iTat) and TNF-α antagonist WP9QY peptide, nitrite oxide (NO) production inhibitor l-NAME, free radical scavenger superoxide dismutase (SOD), and MK-801 (glutamate receptor channel blocker) (37, 38). We collected the culture supernatant from Dox-induced iTat primary astrocytes, preincubated the supernatant with each of the above agents, and determined the neurotoxicity of the supernatant in mouse primary neurons. Compared with the control, treatment of l-NAME and SOD only showed slight but not statistically significant recovery of neuron survival. Meanwhile, WP9QY and MK-801 showed little effect. In contrast, heat-inactivated supernatant showed significant improvements in neuron survival (Fig. 2). These results together suggest that Tat protein itself or Tat-induced factors such as TNF-α, free radicals, and glutamate are not significant contributors to the neurotoxicity of the supernatants of Tat-expressing astrocytes.
FIGURE 2.

Relative contribution of several known factors to astrocyte-mediated Tat neurotoxicity. Wild-type primary astrocytes (WT) or iTat primary astrocytes were induced with Dox for 3 days. Supernatants were collected and incubated with PBS, WP9QY (5 and 10 μm), l-NAME (10 and 20 μm), SOD (10 and 20 μg/ml), or MK-801 (0.5 and 2 μm) or heat-inactivated. After the indicated treatment, supernatants were added into mouse primary neurons, cultured for 72 h, and evaluated for neurotoxicity using the MTT assay. WT supernatants were similarly treated and showed no neurotoxicity (data not shown). All comparisons were made against iTat treated with PBS. Error bars, S.D.; *, p < 0.05.
Tat Expression Induced Lysosomal Exocytosis from Astrocytes
To understand the molecular mechanisms of astrocyte-mediated Tat neurotoxicity, we collected the culture supernatants from WT or iTat astrocytes treated with Dox and subjected them to proteomic analysis. A total of 45 protein spots exhibited 1.5-fold or greater difference between WT supernatant and iTat supernatant (supplemental Fig. S1A). All of those 45 proteins were then recovered and sequenced by mass spectrometry. Only 39 proteins were identified with high confidence (supplemental Table S1). The DAVID pathway analysis identified two groups of proteins with the highest stringency: one associated with lysosomes and the other with cytoplasmic membrane-bound vesicles (supplemental Table S2). Of particular interest was detection of increased acid hydrolases, such as cathepsins A, B, D, and S and β-hexosaminidase, also known as N-acetyl-β-d-glucosaminidase (NAG), in the supernatant of Tat-expressing astrocytes over the WT control (supplemental Fig. S1B). These results indicate the possibility that Tat induces lysosomal exocytosis from astrocytes.
Constitutive lysosomal exocytosis is a continuous and slow process. Thus, to validate the proteomic results and to determine the above-mentioned possibility, we first performed the ionomycin-stimulated NAG release assay, which was developed and has been widely used as a standard indicator of lysosomal exocytosis (39), because ionomycin serves to catalyze the reaction, namely the rate of lysosomal exocytosis, but it does not alter the total number of lysosomal exocytosis events. Specifically, we compared the NAG release with Tat expression in astrocytes. iTat astrocytes were cultured in the presence or absence of Dox before they were subjected to the NAG release assay. There were significantly increased levels of NAG at 8 and 16 min from Dox-treated iTat astrocytes compared with those from iTat astrocytes treated without Dox (Fig. 3A). Next, we performed total internal reflection fluorescence microscopy (TIRF) to directly visualize and quantitate lysosomal exocytosis through the kinetics of ATP-laden vesicles and their fusion with the plasma membrane (40). WT and iTat astrocytes were treated with Dox for 3 days; labeled with quinacrine, a fluorescent marker of intracellular ATP vesicles co-localized with the lysosome-specific marker LAMP-1 (41, 42); and analyzed by TIRF. The overall lysosomal exocytosis in Dox-treated WT astrocytes and Dox-treated iTat astrocytes was recorded (supplemental Movies S1 and S2). Individual lysosomal exocytosis events in both Dox-treated WT and iTat astrocytes were counted manually based on the drastic drop of the fluorescence intensity within seconds shown in the histogram (Fig. 3B, top) and the images (Fig. 3B, bottom panel and insets). Compared with the Dox-treated WT astrocytes, Dox-treated iTat astrocytes exhibited 7 times more lysosomal exocytosis events (Fig. 3C). In addition, we also determined the relationship between Tat expression and HIV infection and lysosomal exocytosis in human astrocytes. Increased NAG was also detected in Tat-transfected U373.MG (Fig. 4A), VSV-G-pseudotyped HIV.GFP-infected U373.MG (Fig. 4B), and VSV-G-pseudotyped HIV.GFP-infected human primary fetal astrocytes (Fig. 4C). These results provide the direct evidence that Tat expression alone or in the context of HIV infection induced lysosomal exocytosis in astrocytes.
FIGURE 3.

Tat expression induced lysosomal exocytosis in astrocytes. A, iTat primary astrocytes were cultured in the absence (Dox−) or in the presence (Dox+) of 5 μg/ml Dox for 3 days. The cells were then processed for the NAG assay. The data are the mean ± S.D. of triplicate samples and are representative of three independent experiments. B and C, WT or iTat primary astrocytes were cultured in the presence of 5 μg/ml Dox for 3 days and then processed for TIRF. Each exocytosis event was counted manually (B, bottom panel and insets) based on the drastic drop of the fluorescence intensity within seconds shown in the histogram (B, top). One hundred quinacrine-labeled ATP-containing vesicles were randomly selected in each astrocyte. Three astrocytes from each sample were randomly selected for quantitation of lysosomal exocytosis events. The number of lysosomal exocytosis events is expressed as a percentage of the total number of vesicles (C). Error bars, S.D.; *, p < 0.05.
FIGURE 4.

Lysosomal exocytosis from Tat-expressing astrocytes and VSV-G-pseudotyped HIV.GFP-infected astrocytes. U373.MG cells were transfected with pcDNA3 (C3) or Tat.Myc (A) or infected with VSC-G-pseudotyped HIV.GFP (B), or human primary fetal astrocytes were infected with VSC-G-pseudotyped HIV.GFP (C). The cells were analyzed for NAG release 72 h post-transfection or 48 h post-infection using the NAG assay. Error bars, S.D.; *, p < 0.05.
To ascertain that Tat induced constitutive lysosomal exocytosis in Tat-expressing astrocytes, we treated WT or iTat astrocytes with Dox and performed immunofluorescence staining for cell surface LAMP-1, a marker for exocytosized lysosomes (43), in the absence of ionomycin stimulation. Compared with the WT astrocytes, iTat astrocytes had more LAMP-1-positive vesicles on their surface (Fig. 5A). Flow cytometry analysis confirmed that there was about 5% LAMP-1-positive staining on the cell surface of the WT astrocytes and about 15% LAMP-1-positive staining on the surface of iTat astrocytes (Fig. 5, B and C). These results showed that Tat expression also led to increased constitutive lysosomal exocytosis.
FIGURE 5.
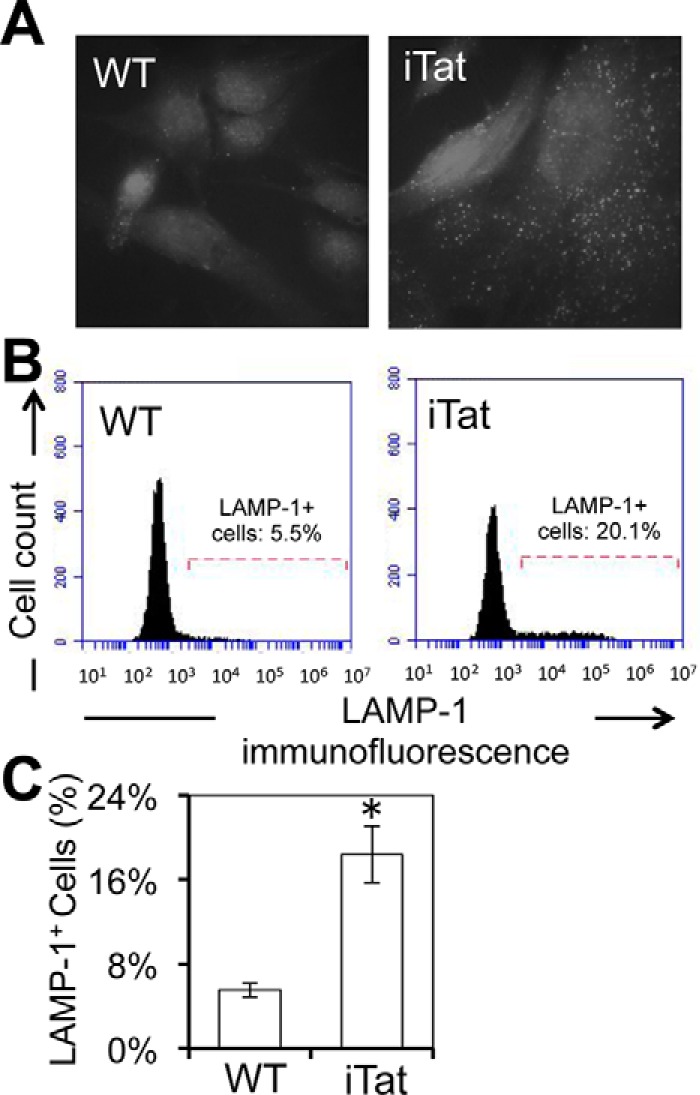
Lysosomal exocytosis from Tat-expressing astrocytes using cell surface LAMP-1 as a marker. WT and iTat primary astrocytes were cultured in the presence of 5 μg/ml Dox for 3 days, processed for cell surface LAMP-1 staining, and evaluated by confocal imaging for LAMP-1 staining (A) and by flow cytometry for quantitation of LAMP-1+ cell percentage (B). A and B, representative of three independent experiments; C, mean ± S.D. of LAMP-1+ cell percentages from three independent experiments. Error bars, S.D.; *, p < 0.05.
Tat-induced lysosomal exocytosis could simply result from Tat-induced increases of the total number of lysosomes. To address this possibility, we compared the total NAG level, measured by the NAG activity, in the supernatants and cell lysates. There were no significant differences in the total NAG activity between U373.MG cells that were transfected with Tat and C3 (supplemental Fig. S2A) or between iTat mouse astrocytes that were treated with and without Dox (supplemental Fig. S2B). In addition, there were no significant differences in the total NAG activity between human fetal astrocytes that were mock-infected and infected with VSV-G-pseudotyped HIV.GFP (supplemental Fig. S2C). Therefore, it did not seem likely that Tat expression altered the total number of lysosomes in astrocytes.
Lysosomal Exocytosis Contributed to Astrocyte-mediated Tat Neurotoxicity
To directly determine the relationship between lysosomal exocytosis and astrocyte-mediated Tat neurotoxicity, we took advantage of vacuolin-1, a specific inhibitor of lysosome exocytosis (44). iTat astrocytes were treated with or without Dox and then with 0, 1, 5, or 10 μm vacuolin-1, followed by the NAG assay for lysosomal exocytosis and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay for neurotoxicity. iTat astrocytes treated with Dox showed increased NAG release (Fig. 6A) and increased neurotoxicity (Fig. 6B) when compared with iTat astrocytes treated without Dox. Treatment of vacuolin-1 at 1 μm slightly decreased the NAG release and slightly decreased the neurotoxicity in Dox-treated iTat astrocytes. At both 5 and 10 μm, vacuolin-1 completely blocked the NAG release and, accordingly, completely blocked the neurotoxicity of Dox-treated iTat astrocytes. Meanwhile, treatment of vacuolin-1 at all three concentrations did not show any significant effects on the NAG release and neurotoxicity in iTat astrocytes treated without Dox. These results support the notion that lysosomal exocytosis is directly involved in astrocyte-mediated Tat neurotoxicity.
FIGURE 6.
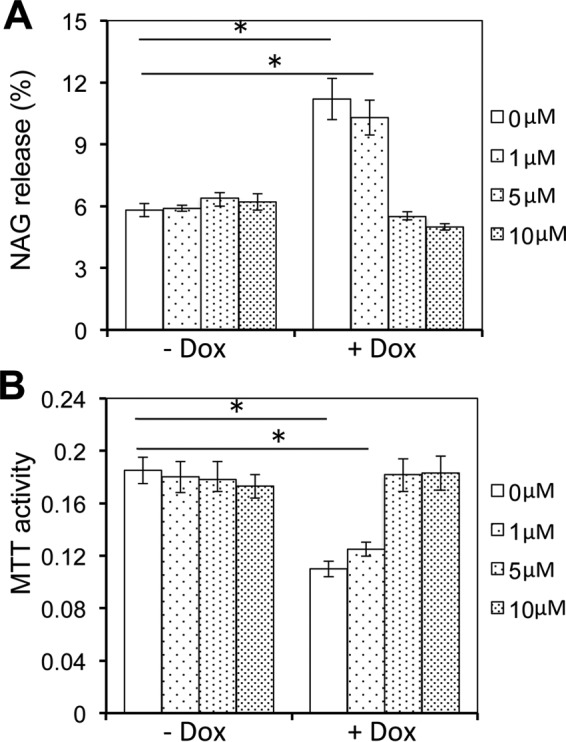
Inhibition of lysosomal exocytosis decreased astrocyte-mediated Tat neurotoxicity. iTat primary astrocytes were cultured in the absence or presence of 5 μg/ml Dox for 3 days and then in the presence of the indicated concentrations of vacuolin-1 for 1 h. The cells were then processed for the NAG assay (A). The culture supernatants from the vacuolin-1-treated cells were collected and used to determine the neurotoxicity in SH-SY5Y cells using the MTT assay (B). Error bars, S.D.; *, p < 0.05.
Lysosomal Enzyme Cathepsin B Contributed to Astrocyte-mediated Tat Neurotoxicity
Lysosomes contain a variety of hydrolytic enzymes that are able to break down proteins, nucleic acids, carbohydrates, and lipids. Our proteomics assay above revealed increased release of lysosomal enzyme cysteine proteases, such as cathepsins B, D, and S, in the supernatant of Tat-expressing astrocytes. Thus, we next determined the importance of cysteine proteases in astrocyte-mediated Tat neurotoxicity. We treated the supernatants of Tat-transfected human primary astrocytes with Z-FA-fmk, a potent irreversible inhibitor of cysteine proteases (45, 46), and determined the neurotoxicity of the supernatants. Compared with the control, the neurotoxicity of the Z-FA-fmk-treated supernatant from Tat-transfected human primary astrocytes exhibited significantly lower neurotoxicity (Fig. 7A). Similar results were observed with Tat-expressing mouse primary astrocytes (Fig. 7B). Cathepsin B has been shown to be a potential neurotoxic factor (47–49). Consistent with these findings, an increased level of cathepsin B was detected in the supernatants of Tat-expressing astrocytes by Western blotting (Fig. 7C). To further ascertain the role of cathepsin B in astrocyte-mediated Tat neurotoxicity, the supernatants were incubated with cathepsin B neutralization antibody and then assayed for their neurotoxicity. Anti-cathepsin B antibody treatment led to less neurotoxicity of the supernatants of Tat-expressing astrocytes (Fig. 7D), which appeared to be antibody concentration-dependent. These results suggest that that cysteine protease cathepsin B is a major factor for astrocyte-mediated indirect Tat neurotoxicity.
FIGURE 7.
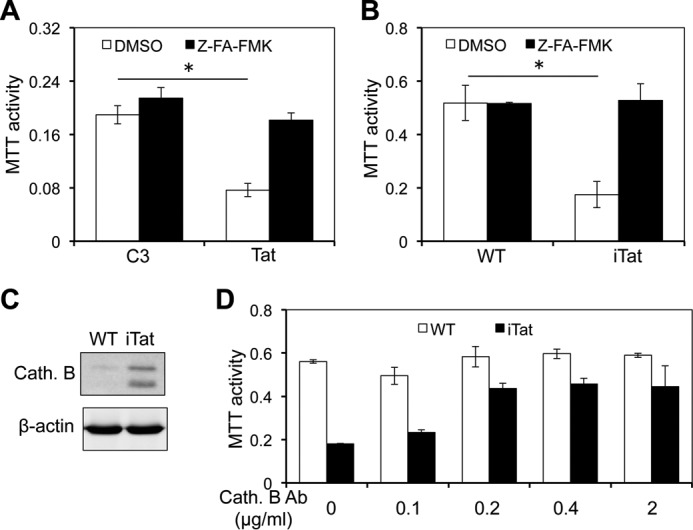
Cathepsin B inhibitor inhibited the neurotoxicity of the supernatants of Tat-expressing astrocytes. A, human primary fetal astrocytes were transfected with pcDNA3 (C3) or Tat, cultured for 72 h, and then treated with cathepsin B inhibitor Z-FA-fmk (100 μm) for 24 h. DMSO, the solvent of Z-FA-fmk, was included as a control. The supernatants were collected and used to determine the neurotoxicity in SH-SY5Y using the MTT assay. B–D, primary astrocytes were isolated from WT and iTat mice and cultured in the presence of Dox for 3 days and treated with cathepsin B inhibitor Z-FA-fmk as above. The supernatants were evaluated for their neurotoxicity as above (B). The cells were harvested for cell lysates. The cathepsin B (Cath. B) in both supernatants and cell lysates was determined by Western blotting (C). In addition, the supernatants were incubated with different concentrations of an anti-cathepsin B antibody (Cath. B) for 1 h and assayed for their neurotoxicity as above (D). Error bars, S.D.; *, p < 0.05.
Requirement of GFAP Expression and ER Stress for Tat-induced Lysosomal Exocytosis in Astrocytes
Next, we determined whether Tat induced lysosomal exocytosis in non-astrocytic cells. To this end, we transfected 293T human embryonic kidney cells and Huh7.5.1 human hepatoma cells with Tat and determined lysosomal exocytosis in those cells. Compared with respective controls and U373.MG (Fig. 4A), no significantly different NAG activity was detected at all time points tested in both 293T (Fig. 8A) and Huh7.5.1 (Fig. 8B). Interestingly, compared with human primary astrocytes (Fig. 1B), only the supernatant from Tat-transfected 293T showed neurotoxicity in human primary neurons (Fig. 8C), but not in SH-SY5Y human neuroblastoma cells (Fig. 8D). In contrast, the supernatant from Tat-transfected Huh7.5.1 cells did not show neurotoxicity in both human primary neurons and SH-SY5Y cells.
FIGURE 8.

Effects of Tat expression on lysosome exocytosis and neurotoxicity in 239T and Huh 7.5.1. 293T (A) and Huh 7.5.1 cells (B) were transfected with cDNA3 (C3) or Tat.Myc. The cells were analyzed for the NAG release using the NAG assay. The supernatants were collected for their neurotoxicity in human primary fetal neurons (C) and SH-SY5Y (D) using the MTT assay. Error bars, S.D.; *, p < 0.05.
In addition, we have shown that GFAP activation is important for astrocyte-mediated Tat neurotoxicity (22, 29–31). Thus, we next determined the role of GFAP expression in Tat-induced lysosomal exocytosis. We took advantage of the iTat/GFAP− mice, isolated primary astrocytes from those mice, treated them with Dox, and determined the NAG release from those cells. Consistent with previous findings, more NAG was released in iTat astrocytes treated with Dox than in those treated without Dox (Fig. 9A). However, there were no differences in the NAG release among iTat/GFAP− astrocytes treated with and without Dox and iTat astrocytes treated without Dox. Meanwhile, the NAG release was correlated with the neurotoxicity (Fig. 9B).
FIGURE 9.

GFAP knock-out abrogated Tat-induced lysosomal exocytosis in Tat-expressing astrocytes. Primary astrocytes were isolated from iTat and iTat/GFAP− mice and cultured in the absence or presence of 5 μg/ml Dox for 3 days. The cells were processed for the NAG assay (A). The supernatants were analyzed for their neurotoxicity using the MTT assay (B). Error bars, S.D.; *, p < 0.05.
Furthermore, we have recently shown in a separate accompanying article (84) that Tat-induced GFAP activation leads to unfolded protein response and endoplasmic reticulum stress. Thus, we also determined the relationship between Tat/GFAP expression, lysosomal exocytosis, and endoplasmic reticulum stress. To this end, we transfected U373.MG cells with Tat or GFAP expression plasmids; treated the cells with 4-phenylbutyrate (4-PBA), a chemical chaperone that has commonly been used to alleviate ER stress (50); and determined the NAG release. Tat expression and GFAP expression both led to increased NAG release, whereas 4-PBA treatment completely abrogated Tat- or GFAP-induced NAG release (Fig. 10A). Similar results were obtained in Dox-treated iTat astrocytes (Fig. 10B). Taken together, these results indicate that GFAP expression/endoplasmic reticulum stress were directly involved in Tat-induced lysosomal exocytosis and astrocyte-mediated Tat neurotoxicity.
FIGURE 10.
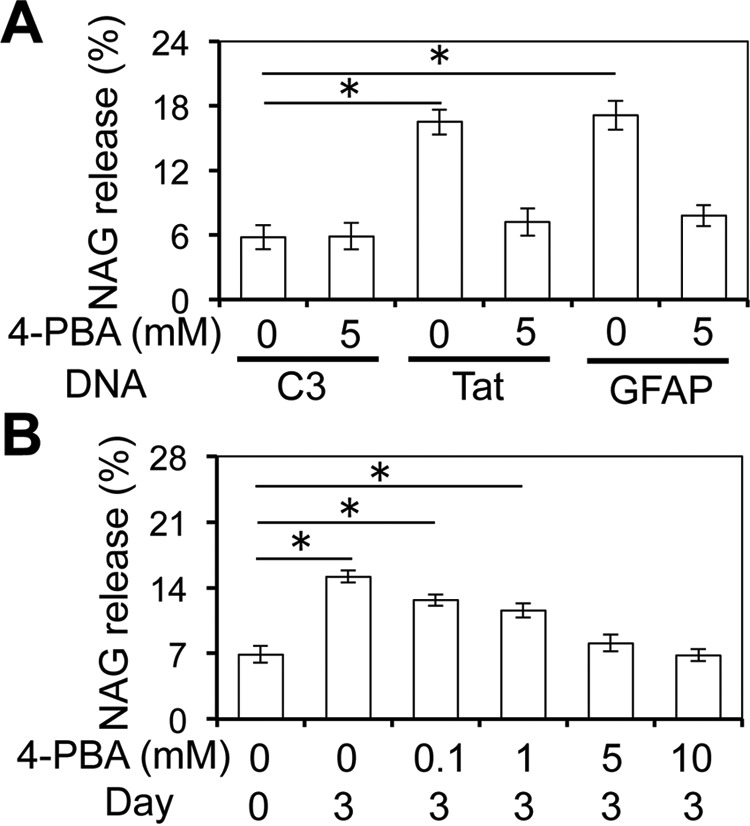
4-PBA inhibited lysosomal exocytosis in astrocytes. A, U373.MG were transfected with cDNA3, Tat, or GFAP expression plasmid, cultured for 48 h, and continued to culture in the absence or presence of 5 mm 4-PBA for 24 h. The cells were harvested and analyzed for the NAG release using the NAG assay. B, iTat primary astrocytes were cultured in the presence of 5 μg/ml Dox for 0 and 3 days and then in the presence of 0, 0.1, 1, 5, or 10 mm 4-PBA for 24 h. The cells were harvested and analyzed for the NAG release using the NAG assay. Error bars, S.D.; *, p < 0.05.
Discussion
In this study, we first showed that neurotoxic factors other than Tat protein itself were present in the supernatant of Tat-expressing astrocytes (Figs. 1 and 2). Proteomic analysis and subsequent studies revealed elevated levels of lysosomal hydrolytic enzymes though lysosomal exocytosis in Tat-expressing mouse primary astrocytes and human astrocytes and VSV-G-pseudotyped HIV.GFP-infected human astrocytes and contributed to astrocyte-mediated Tat neurotoxicity (Figs. 3, 4, and 6 and supplemental Fig. S1). Moreover, we determined that GFAP expression and endoplasmic reticulum stress were directly involved in Tat-induced lysosomal exocytosis and neurotoxicity (Figs. 8–10). These findings provide novel insights into astrocyte-mediated Tat neurotoxicity and support important roles of Tat in HIV/neuroAIDS, particularly in the era of cART, when Tat is still detected in the CNS of cART-treated individuals (51).
Tat is detected in the brain of HIV-1-infected patients at both the mRNA and protein level (10). It is secreted by HIV-1-infected cells within the brain, or it could be transported across the blood-brain barrier from the periphery (52). HIV-1 Tat causes neuron death by stimulating TNF-α release from astrocytes (34), which has been shown to be indirectly mediated by pro-inflammatory microglial activation (53). Similarly, Tat-induced oxidative stress, NO production, and MCP-1 could all be mediated by activation of microglia (35, 36). On the other hand, direct exposure of Tat to astrocytes can inhibit glutamate uptake by astrocytes, which results in increasing glutamate concentrations in the neuronal microenvironment and causes neuron death, whereas this process is under the control of TNF-α and Tat in a dose- and time-dependent manner (34, 54). These findings indicate that Tat-induced neuron death through TNF-α, NO, protein oxidation, and glutamate is complex and either requires other accessory cells or is highly context-dependent. The Tat expression level is extremely low in the iTat system, because it is under the control of the weak GFAP promoter (32). Thus, the likelihood of direct or acute Tat neurotoxicity in the setting is low.
We have recently shown that Tat induces UPR/ER stress in astrocytes (84). Protein aggregation-triggered UPR/ER stress is crucial in many biological responses and is generally considered an adaptive reaction of cells to environmental stress, serving as a survival signal (55). Astrocytes have the unique ability to tolerate and even proliferate under the conditions that lead to ER stress (56). However, little is known about how GFAP aggregate formation and ER stress in astrocytes affect neurons. Calcium storage is an important function of the ER (57), and ER stress often leads to calcium release (58). In astrocytes, increases in intracellular calcium trigger fusion of secretory vesicles with the plasma membrane, termed exocytosis, and result in release of cargoes from the vesicles, such as neurotransmitters, hormones, and enzymes (59–61). The majority of these secretory vesicles are lysosomes in cortical astrocytes that have Ca2+-dependent asynchronous exocytosis to modulate ATP release as a feedback modulation of synaptic functions (62). Lysosomes induce necrosis, autophagy, or apoptosis through hydrolytic enzymes or cathepsins within the vesicles (63, 64). Tat has been shown to induce calcium release from the ER (65, 66). In this study, we showed that Tat expression resulted in lysosomal exocytosis (Figs. 3 and 4 and supplemental Fig. S1), which in turn caused neurotoxicity (Figs. 5 and 7). Moreover, Tat-induced GFAP aggregation/endoplasmic reticulum stress has been directly linked to Tat-induced lysosomal exocytosis and astrocyte-mediated Tat neurotoxicity (Figs. 8–10). These findings support the following working model (Fig. 11): astrocyte uptake of Tat, secreted from HIV-infected macrophages/microglia, or Tat expression in HIV-infected astrocytes activates GFAP expression/astrocytosis; increased GFAP expression induces UPR/ER stress in astrocytes; induction of UPR/ER stress leads to calcium release from the ER storage into cytoplasm; elevated cytoplasmic calcium leads to lysosomal exocytosis from astrocytes; and released lysosomes and their components cause the neurotoxicity. It is important to point out that the effects of Tat-induced lysosomal exocytosis on astrocyte-mediated Tat neurotoxicity are modest but significant. Considering the vast number of astrocytes in the brain, one would expect the magnitude of the collective effects to be much higher in the context of HIV infection of the brain.
FIGURE 11.

Working model for astrocyte-mediated Tat neurotoxicity. Tat is secreted from HIV-infected macrophages/microglia (1); Tat is taken up into astrocytes or expressed in HIV-infected astrocytes (2); Tat activates GFAP expression (3); increased GFAP expression leads to formation of GFAP aggregation (4); GFAP aggregation triggers UPR/ER stress (5); ER stress leads to calcium release into the cytoplasm (6); increased intracellular calcium stimulates Ca2+-dependent lysosomal exocytosis (7) and release of lysosomal hydrolytic enzymes and cathepsins into the extracellular space (8); and lysosomal hydrolytic enzymes and cathepsins cause neurotoxicity (9).
The primary goal of this study was to elucidate the molecular mechanisms of astrocyte-mediated Tat neurotoxicity. We showed that lysosomal exocytosis was induced in astrocytes by HIV-1 Tat and contributed to astrocyte-mediated Tat neurotoxicity. Lysosome-induced necrosis and autophagy are considered to be two of the main pathways of lysosome-associated cell death, whereas recently, the lysosomal pathway of apoptosis caused by lysosomal protease has generally been accepted as an important model of lysosome-mediated cell death (63). Hydrolytic enzymes contained within lysosomes contribute to programmed cell death either by direct digestion of main cellular substrates or by induction in caspases of signaling pathways (64, 67), whereas lysosomal cathepsin-induced cell death has been identified primarily in pathological situations other than developmental cell death (68). For instance, cathepsin B contributes to astrocyte activation, malignant progression of glioma, and neuronal cell death (69–72). In our experiments, cysteine protease inhibitor Z-FA-fmk and cathepsin B neutralization both led to significantly lower neurotoxicity of the supernatants of Tat-expressing astrocytes (Fig. 5). It is likely that cathepsin D also contributes to astrocyte-mediated Tat neurotoxicity through lysosomal exocytosis. Cytosolic cathepsin D is elevated in neurotoxic conditions (73). Cathepsin D is released from toxicity-induced neurons and causes neuron death by activation of autophagy (73). Nevertheless, lysosomal cathepsins D and B have been shown to be neuroprotective (70, 74, 75). Thus, it is important to maintain the homeostasis of those enzymes or lysosomal exocytosis. Consistent with our findings, increased cathepsin B release has recently been linked to HIV infection-associated neuronal death and neurocognitive deficits (76, 77). Taken together, these findings suggest important roles of lysosomal exocytosis and cathepsins in neurodegenerative diseases, including HIV/neuroAIDS, and may provide new strategies for development of therapeutics for those diseases.
Materials and Methods
Animals and Tissues
C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Dox-inducible and astrocyte-specific HIV-1 Tat transgenic mice (iTat) were created in our laboratory as described previously (32). GFAP-null mice were generously provided by Dr. Albee Messing of University of Wisconsin (Madison, WI) (78). iTat/GFAP-null (iTat/GFAP−) mice were obtained by cross-breeding iTat mice with GFAP-null mice and characterized as described previously (79).
Cells and Cell Cultures
Human embryonic kidney 293T cells, human astrocytoma U373.MG cells, and human neuroblastoma SH-SY5Y cells were purchased from American Tissue Culture Collection (Manassas, VA). Human hepatoma Huh7.5.1 were obtained from Dr. Charles Rice (Rockefeller University, New York). 293T, U373.MG, and Huh7.5.1 cells were cultured in DMEM, whereas SH-SY5Y cells were cultured in 50% DMEM and 50% F-12 medium (Cellgro, Manassas, VA). Both culture media were supplemented with 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin. Mouse primary astrocytes were isolated from the brain of embryonic day 18.5 embryos and induced to express Tat as described previously (22). Human primary fetal astrocytes were prepared as before (80). Primary astrocytes were cultured in F12-K medium (Cellgro) containing 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin and passaged every 3–4 days.
Plasmids and Transfection
Plasmids pcDNA3 and pCMV-βgal were purchased from Clontech (Mountain View, CA). Plasmid pTat.Myc was constructed as described previously (81). Plasmid pLTR-Luc (donated by Drs. R. Jeeninga and B. Berkhout (82, 83)) was obtained through the National Institutes of Health AIDS Reagent Program. All cell transfections were performed using the standard calcium phosphate precipitation method.
Preparation of VSV-G-pseudotyped HIV.GFP Virus and Infection of Astrocytes
293T cells were transfected with pHIV.GFP plasmid and pHCMV-G plasmid using the standard calcium phosphate precipitation method. The culture supernatants were collected 48 h after the medium change, briefly centrifuged to remove cell debris, and used as the virus stock. U373.MG and human primary fetal astrocytes were infected with VSV-G-pseudotyped HIV.GFP viruses in the presence of 8 μg/ml Polybrene at 37 °C for 2 h. The cells were then thoroughly washed with culture medium to remove unbound viruses and cultured for an additional 48 h. The dosages of the viruses for infection were titrated to ensure minimal cell death and maximal infection efficiency.
Western Blotting
Cells were washed twice with ice-cold PBS and lysed in RIPA buffer (50 mm Tris·HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 2 mm PMSF, and 1× protease inhibitor mixture (Roche Applied Science)) on ice for 20 min for whole cell lysates. Protein concentration of the cell lysates was determined using a Bio-Rad DC protein assay kit (Bio-Rad). Cell lysates were electrophoretically separated by 10% SDS-polyacrylamide gel, followed by probing with appropriate primary and secondary antibodies, ECL detection, and imaging using a Bio-Rad ChemicDoc imaging system (Bio-Rad). For proteins in conditioned media, conditioned media (5 ml) were precipitated overnight with 9 volumes of 100% ethanol at −20 °C. The precipitates were collected by centrifugation at 13,000 rpm for 10 min, washed twice with 75% ethanol, and suspended in 50 μl of RIPA buffer for SDS-PAGE and ECL detection. Mouse monoclonal α-β-actin antibody was from Sigma, and rabbit polyclonal α-c-Myc and mouse anti-human cathepsin B antibody were from Santa Cruz Biotechnology, Inc. (Dallas, TX).
Immunodepletion
Cell culture supernatants were collected and incubated overnight in the presence of rabbit polyclonal α-c-Myc (1 μg/ml; Santa Cruz Biotechnology) at 4 °C with intermittent mixing. Normal rabbit IgG (Santa Cruz Biotechnology) was included as the control. Then protein A beads (20 μl/ml) were added and incubated at 4 °C for 2 h with intermittent mixing. The supernatants were removed from the beads by centrifugation and saved for further analysis.
Luciferase Activity Assay
Cells were washed twice with ice-cold PBS and refilled with 200 μl of 1× lysis buffer (Promega, Madison, WI) and then incubated at room temperature for 5 min. The clear supernatant was obtained by brief centrifugation. The luciferase reporter gene assay was done using a luciferase assay system (Promega, Madison, WI). The luciferase activity was quantitated using an Opticomp Luminometer (MGM Instruments, Hamden, CT).
MTT Assay
The neurotoxicity of the astrocyte culture supernatants was determined in SH-SY5Y unless stated otherwise using the MTT assay. Briefly, SH-SY5Y cells were plated in a 48-well plate at a density of 1 × 105 cells/well and cultured for 2 days. The cells were exposed to culture supernatants (2:1 ratio) collected from astrocytes and continued to culture for an additional 3 days. MTT (5 mg/ml) was added directly to the culture medium to a final concentration of 1 mg/ml, and the cultures were incubated at 37 °C for 4 h. The medium was then removed, and the purple crystal precipitates were dissolved in 200 μl of acid-isopropyl alcohol (44 ml of isopropyl alcohol plus 6 ml of 0.2 n HCl). Aliquots of the acid-isopropyl alcohol solvent were transferred into the wells of a 96-well plate, and the optical density was determined using a microplate reader at a test wavelength of 490 nm and a reference wavelength of 650 nm and then used to represent the relative cell viability. For treatment of the supernatants with cathepsin B inhibitor Z-AM-fmk, the supernatants were incubated with 100 μm Z-AM-fmk (46) (BD Pharmingen, San Jose, CA) for 1 h before being used for the neurotoxicity assay. For cathepsin B neutralization, the supernatants were collected, incubated with the indicated concentrations of mouse anti-human cathepsin B antibody (Santa Cruz Biotechnology) at room temperature for 1 h, and then assayed for their neurotoxicity.
Cell Surface Lysosome-associated Membrane Protein 1 (LAMP-1) Staining and Flow Cytometry
Cells were washed with ice-cold PBS once and incubated with rat anti-human LAMP-1 antibody (1:100, sc-19992, Santa Cruz Biotechnology) at 4 °C overnight and then with goat anti-rat Alexa Fluor 555 (1:200; Life Technologies, Inc.) at room temperature for 1 h. Extensive washes with PBS were performed between each step. Omission of the primary antibody in parallel staining was included as a control to ensure no nonspecific staining. Single cells suspensions were analyzed for LAMP-1-positive cells using flow cytometry.
Diazyme NAG Assay
Lysosomal exocytosis was determined using a fluorescence-based NAG microplate assay as described (39). Briefly, cells were plated in a 12-well plate at a density of 1 × 105 cells/well and cultured for 3 days with or without 5 μg/ml Dox. At the time of assay, the cells were washed once with PBS and cultured at 37 °C in 200 μl of PBS containing 1 mm CaCl2 and 10 μm ionomycin for 0, 2, 4, 8, or 16 min. At the end of each incubation time point, the incubation buffer was collected, and the cells were lysed in 1 ml of 1% Nonidet P-40 for cell lysates. Both the incubation buffer and the cleared cell lysates (175 μl each) were mixed with 25 μl of 6 mm 4-methyl-umbellyferyl-N-acetyl-β-d-glucosaminide (Sigma) in a 96-well plate and incubated at 37 °C for 15 min. The stop buffer (50 μl of 2 m Na2CO3 and 1.1 m glycine) was then added. The fluorescence intensity was determined at excitation of 365 nm and emission of 450 nm using a TECAN Infinite M200 microplate reader. The NAG activity in the incubation buffer was calculated as a percentage of the total NAG activity in both incubation buffer and cell lysates, and the percentage was used to represent the relative lysosomal exocytosis activity. To inhibit lysosomal exocytosis, astrocytes were treated with the indicated amount of vacuolin-1 (44) (Sigma-Aldrich) for 1 h before the cells were harvested for the NAG assay and the supernatants were added to neurons for the MTT assay. To inhibit the ER stress, astrocytes were treated with the indicated amounts of 4-PBA (50) (Sigma) for 1 h before the cells were harvested for the NAG assay.
TIRF
Lysosomal exocytosis was also directly visualized and quantitated using TIRF as described (40, 62) and performed at the core facility “Fluorescence Technologies and Nanomedicine” of the University of North Texas Health Science Center (Fort Worth, TX). Briefly, mouse primary astrocytes from WT or iTat mice were plated on a polylysine-coated square coverslip (20 mm; Corning) in a 6-well plate at a density of 1 × 104 cells/well and cultured for 3 days with or without 5 μg/ml Dox. At the time of the TIRF imaging, the cells were washed once with PBS and incubated with 10 μm quinacrine (Sigma) at room temperature for 10 min. The cells were then washed and incubated with 500 μl of PBS containing 1 mm CaCl2 and 1 μm ionomycin on the 37 °C heating stage of the TIRF microscope. The images were acquired at an interval of one image every 2 s for a total of 2 min. The TIRF angle was set up at 68º (with numerical aperture 1.45) to ensure to only capture vesicles docked at the plasma membrane. The samples were blinded throughout the TIRF analysis. The images were analyzed for lysosomal exocytosis events using SimplePCI version 6.0 software (Hamamatsu, Corp., Sewickley, PA). One hundred ATP-positive vesicles were randomly selected and monitored in each astrocyte. Three astrocytes from each sample were randomly selected for TIRF imaging. Each lysosomal exocytosis event was counted manually based on the drastic drop of the fluorescence intensity within seconds. The number of lysosomal exocytosis events was calculated as a percentage of the total number of monitored vesicles.
Data Analysis
All experiment data are the mean ± S.D. of three or more independent experiments and analyzed by two-tailed Student's t test. A p value of <0.05 was considered to be statistically significant and shown as an asterisk; a p value of <0.01 was considered to be statistically highly significant and shown as a double asterisk.
Author Contributions
Y. F. conducted and designed the experiments and prepared the data and the draft, and J. J. H. designed the experiments, prepared the data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Xiaoyu Luo for technical assistance and Dr. Pejman Rahimian and Amanda Whitmill for critical reading of the manuscript.
This work was supported by NINDS, National Institutes of Health Grants R01NS065785, R01094108, and R01MH092673 (to J. J. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1 and S2, Figs. S1 and S2, and Movies S1 and S2.
- cART
- combination antiretroviral therapy
- MCMD
- minor cognitive motor disorder
- RANTES
- regulated on activation normal T cell expressed and secreted
- GFAP
- glial fibrillary acidic protein
- LTR
- long terminal repeat
- l-NAME
- l-NG-nitroarginine methyl ester
- NAG
- N-acetyl-β-d-glucosaminidase
- TIRF
- total internal reflection fluorescence microscopy
- Dox
- doxycycline
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- 4-PBA
- 4-phenylbutyrate
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- SOD
- superoxide dismutase
- RIPA
- radioimmune precipitation assay.
References
- 1. An S. F., Groves M., Gray F., and Scaravilli F. (1999) Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J. Neuropathol. Exp. Neurol. 58, 1156–1162 [DOI] [PubMed] [Google Scholar]
- 2. Price R. W., and Brew B. J. (1988) The AIDS dementia complex. J. Infect. Dis. 158, 1079–1083 [DOI] [PubMed] [Google Scholar]
- 3. Ellis R. J., Deutsch R., Heaton R. K., Marcotte T. D., McCutchan J. A., Nelson J. A., Abramson I., Thal L. J., Atkinson J. H., Wallace M. R., and Grant I. (1997) Neurocognitive impairment is an independent risk factor for death in HIV infection. San Diego HIV Neurobehavioral Research Center Group. Arch. Neurol. 54, 416–424 [DOI] [PubMed] [Google Scholar]
- 4. Cohen R. A., and Gongvatana A. (2009) HIV-associated brain dysfunction in the era of HAART: reasons for hope, but continued concern. Neurology 73, 338–339 [DOI] [PubMed] [Google Scholar]
- 5. Ellis R., Langford D., and Masliah E. (2007) HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat. Rev. Neurosci. 8, 33–44 [DOI] [PubMed] [Google Scholar]
- 6. Del Valle L., and Piña-Oviedo S. (2006) HIV disorders of the brain: pathology and pathogenesis. Front. Biosci. 11, 718–732 [DOI] [PubMed] [Google Scholar]
- 7. Lee S. C., Hatch W. C., Liu W., Brosnan C. F., and Dickson D. W. (1993) Productive infection of human fetal microglia in vitro by HIV-1. Ann. N.Y. Acad. Sci. 693, 314–316 [DOI] [PubMed] [Google Scholar]
- 8. Saito Y., Sharer L. R., Epstein L. G., Michaels J., Mintz M., Louder M., Golding K., Cvetkovich T. A., and Blumberg B. M. (1994) Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology 44, 474–481 [DOI] [PubMed] [Google Scholar]
- 9. Ensoli B., Buonaguro L., Barillari G., Fiorelli V., Gendelman R., Morgan R. A., Wingfield P., and Gallo R. C. (1993) Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J. Virol. 67, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hudson L., Liu J., Nath A., Jones M., Raghavan R., Narayan O., Male D., and Everall I. (2000) Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J. Neurovirol. 6, 145–155 [DOI] [PubMed] [Google Scholar]
- 11. Nath A., Psooy K., Martin C., Knudsen B., Magnuson D. S., Haughey N., and Geiger J. D. (1996) Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J. Virol. 70, 1475–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sabatier J. M., Vives E., Mabrouk K., Benjouad A., Rochat H., Duval A., Hue B., and Bahraoui E. (1991) Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J. Virol. 65, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benelli R., Barbero A., Ferrini S., Scapini P., Cassatella M., Bussolino F., Tacchetti C., Noonan D. M., and Albini A. (2000) Human immunodeficiency virus transactivator protein (Tat) stimulates chemotaxis, calcium mobilization, and activation of human polymorphonuclear leukocytes: implications for Tat-mediated pathogenesis. J. Infect. Dis. 182, 1643–1651 [DOI] [PubMed] [Google Scholar]
- 14. Lafrenie R. M., Wahl L. M., Epstein J. S., Hewlett I. K., Yamada K. M., and Dhawan S. (1996) HIV-1-Tat protein promotes chemotaxis and invasive behavior by monocytes. J. Immunol. 157, 974–977 [PubMed] [Google Scholar]
- 15. Park I. W., Wang J. F., and Groopman J. E. (2001) HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 97, 352–358 [DOI] [PubMed] [Google Scholar]
- 16. Jones M., Olafson K., Del Bigio M. R., Peeling J., and Nath A. (1998) Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J. Neuropathol. Exp. Neurol. 57, 563–570 [DOI] [PubMed] [Google Scholar]
- 17. Conant K., Garzino-Demo A., Nath A., McArthur J. C., Halliday W., Power C., Gallo R. C., and Major E. O. (1998) Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc. Natl. Acad. Sci. U.S.A. 95, 3117–3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Magnuson D. S., Knudsen B. E., Geiger J. D., Brownstone R. M., and Nath A. (1995) Human immunodeficiency virus type 1 tat activates non-N-methyl-d-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann. Neurol. 37, 373–380 [DOI] [PubMed] [Google Scholar]
- 19. Hui L., Chen X., Haughey N. J., and Geiger J. D. (2012) Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro 4, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chauhan A., Turchan J., Pocernich C., Bruce-Keller A., Roth S., Butterfield D. A., Major E. O., and Nath A. (2003) Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J. Biol. Chem. 278, 13512–13519 [DOI] [PubMed] [Google Scholar]
- 21. Zhou B. Y., and He J. J. (2004) Proliferation inhibition of astrocytes, neurons, and non-glial cells by HIV-1 Tat protein. Neurosci. Lett. 359, 155–158 [DOI] [PubMed] [Google Scholar]
- 22. Zhou B. Y., Liu Y., Kim B. O., Xiao Y., and He J. J. (2004) Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol. Cell Neurosci. 27, 296–305 [DOI] [PubMed] [Google Scholar]
- 23. Ranki A., Nyberg M., Ovod V., Haltia M., Elovaara I., Raininko R., Haapasalo H., and Krohn K. (1995) Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS 9, 1001–1008 [DOI] [PubMed] [Google Scholar]
- 24. Brack-Werner R. (1999) Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS 13, 1–22 [DOI] [PubMed] [Google Scholar]
- 25. Atwood W. J., Tornatore C. S., Meyers K., and Major E. O. (1993) HIV-1 mRNA transcripts from persistently infected human fetal astrocytes. Ann. N.Y. Acad. Sci. 693, 324–325 [DOI] [PubMed] [Google Scholar]
- 26. Kolson D. L., Buchhalter J., Collman R., Hellmig B., Farrell C. F., Debouck C., and Gonzalez-Scarano F. (1993) HIV-1 Tat alters normal organization of neurons and astrocytes in primary rodent brain cell cultures: RGD sequence dependence. AIDS Res. Hum. Retroviruses 9, 677–685 [DOI] [PubMed] [Google Scholar]
- 27. El-Hage N., Gurwell J. A., Singh I. N., Knapp P. E., Nath A., and Hauser K. F. (2005) Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 50, 91–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Williams R., Yao H., Dhillon N. K., and Buch S. J. (2009) HIV-1 Tat co-operates with IFN-γ and TNF-α to increase CXCL10 in human astrocytes. PLoS One 4, e5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fan Y., Zou W., Green L. A., Kim B. O., and He J. J. (2011) Activation of Egr-1 expression in astrocytes by HIV-1 Tat: new insights into astrocyte-mediated Tat neurotoxicity. J. Neuroimmune Pharmacol. 6, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou W., Wang Z., Liu Y., Fan Y., Zhou B. Y., Yang X. F., and He J. J. (2010) Involvement of p300 in constitutive and HIV-1 Tat-activated expression of glial fibrillary acidic protein in astrocytes. Glia 58, 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan Y., Timani K. A., and He J. J. (2015) STAT3 and its phosphorylation are involved in HIV-1 Tat-induced transactivation of glial fibrillary acidic protein. Curr. HIV Res. 13, 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim B. O., Liu Y., Ruan Y., Xu Z. C., Schantz L., and He J. J. (2003) Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am. J. Pathol. 162, 1693–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fitting S., Ignatowska-Jankowska B. M., Bull C., Skoff R. P., Lichtman A. H., Wise L. E., Fox M. A., Su J., Medina A. E., Krahe T. E., Knapp P. E., Guido W., and Hauser K. F. (2013) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol. Psychiatry 73, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen P., Mayne M., Power C., and Nath A. (1997) The Tat protein of HIV-1 induces tumor necrosis factor-α production. Implications for HIV-1-associated neurological diseases. J. Biol. Chem. 272, 22385–22388 [DOI] [PubMed] [Google Scholar]
- 35. Turchan-Cholewo J., Dimayuga F. O., Gupta S., Keller J. N., Knapp P. E., Hauser K. F., and Bruce-Keller A. J. (2009) Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J. Neurochem. 108, 202–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boje K. M., and Arora P. K. (1992) Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 587, 250–256 [DOI] [PubMed] [Google Scholar]
- 37. Shute A. A., Cormier R. J., Moulder K. L., Benz A., Isenberg K. E., Zorumski C. F., and Mennerick S. (2005) Astrocytes exert a pro-apoptotic effect on neurons in postnatal hippocampal cultures. Neuroscience 131, 349–358 [DOI] [PubMed] [Google Scholar]
- 38. Papagapiou M. P., and Auer R. N. (1990) Regional neuroprotective effects of the NMDA receptor antagonist MK-801 (dizocilpine) in hypoglycemic brain damage. J. Cereb. Blood Flow Metab. 10, 270–276 [DOI] [PubMed] [Google Scholar]
- 39. Rodríguez A., Webster P., Ortego J., and Andrews N. W. (1997) Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 137, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Akopova I., Tatur S., Grygorczyk M., Luchowski R., Gryczynski I., Gryczynski Z., Borejdo J., and Grygorczyk R. (2012) Imaging exocytosis of ATP-containing vesicles with TIRF microscopy in lung epithelial A549 cells. Purinergic Signal. 8, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Parks A., Charest-Morin X., Boivin-Welch M., Bouthillier J., and Marceau F. (2015) Autophagic flux inhibition and lysosomogenesis ensuing cellular capture and retention of the cationic drug quinacrine in murine models. PeerJ 3, e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jung J., Uesugi N., Jeong N. Y., Park B. S., Konishi H., and Kiyama H. (2016) Increase of transcription factor EB (TFEB) and lysosomes in rat DRG neurons and their transportation to the central nerve terminal in dorsal horn after nerve injury. Neuroscience 313, 10–22 [DOI] [PubMed] [Google Scholar]
- 43. Bastow E. R., Last K., Golub S., Stow J. L., Stanley A. C., and Fosang A. J. (2012) Evidence for lysosomal exocytosis and release of aggrecan-degrading hydrolases from hypertrophic chondrocytes, in vitro and in vivo. Biol. Open 1, 318–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cerny J., Feng Y., Yu A., Miyake K., Borgonovo B., Klumperman J., Meldolesi J., McNeil P. L., and Kirchhausen T. (2004) The small chemical vacuolin-1 inhibits Ca2+-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 5, 883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schotte P., Schauvliege R., Janssens S., and Beyaert R. (2001) The cathepsin B inhibitor z-FA.fmk inhibits cytokine production in macrophages stimulated by lipopolysaccharide. J. Biol. Chem. 276, 21153–21157 [DOI] [PubMed] [Google Scholar]
- 46. Lawrence C. P., Kadioglu A., Yang A. L., Coward W. R., and Chow S. C. (2006) The cathepsin B inhibitor, z-FA-fmk, inhibits human T cell proliferation in vitro and modulates host response to pneumococcal infection in vivo. J. Immunol. 177, 3827–3836 [DOI] [PubMed] [Google Scholar]
- 47. Rodriguez-Franco E. J., Cantres-Rosario Y. M., Plaud-Valentin M., Romeu R., Rodríguez Y., Skolasky R., Meléndez V., Cadilla C. L., and Melendez L. M. (2012) Dysregulation of macrophage-secreted cathepsin B contributes to HIV-1-linked neuronal apoptosis. PLoS One 7, e36571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gan L., Ye S., Chu A., Anton K., Yi S., Vincent V. A., von Schack D., Chin D., Murray J., Lohr S., Patthy L., Gonzalez-Zulueta M., Nikolich K., and Urfer R. (2004) Identification of cathepsin B as a mediator of neuronal death induced by Aβ-activated microglial cells using a functional genomics approach. J. Biol. Chem. 279, 5565–5572 [DOI] [PubMed] [Google Scholar]
- 49. Sun L., Wu Z., Baba M., Peters C., Uchiyama Y., and Nakanishi H. (2010) Cathepsin B-dependent motor neuron death after nerve injury in the adult mouse. Biochem. Biophys. Res. Commun. 399, 391–395 [DOI] [PubMed] [Google Scholar]
- 50. Qi X., Hosoi T., Okuma Y., Kaneko M., and Nomura Y. (2004) Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol. Pharmacol. 66, 899–908 [DOI] [PubMed] [Google Scholar]
- 51. Johnson T. P., Patel K., Johnson K. R., Maric D., Calabresi P. A., Hasbun R., and Nath A. (2013) Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. U.S.A. 110, 13588–13593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Banks W. A., Robinson S. M., and Nath A. (2005) Permeability of the blood-brain barrier to HIV-1 Tat. Exp. Neurol. 193, 218–227 [DOI] [PubMed] [Google Scholar]
- 53. Jin J., Lam L., Sadic E., Fernandez F., Tan J., and Giunta B. (2012) HIV-1 Tat-induced microglial activation and neuronal damage is inhibited via CD45 modulation: a potential new treatment target for HAND. Am. J. Transl. Res. 4, 302–315 [PMC free article] [PubMed] [Google Scholar]
- 54. Fine S. M., Angel R. A., Perry S. W., Epstein L. G., Rothstein J. D., Dewhurst S., and Gelbard H. A. (1996) Tumor necrosis factor α inhibits glutamate uptake by primary human astrocytes: implications for pathogenesis of HIV-1 dementia. J. Biol. Chem. 271, 15303–15306 [DOI] [PubMed] [Google Scholar]
- 55. Høyer-Hansen M., and Jäättelä M. (2007) Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 14, 1576–1582 [DOI] [PubMed] [Google Scholar]
- 56. Saito A., Hino S., Murakami T., Kondo S., and Imaizumi K. (2007) A novel ER stress transducer, OASIS, expressed in astrocytes. Antioxid. Redox Signal. 9, 563–571 [DOI] [PubMed] [Google Scholar]
- 57. Harding H. P., Zhang Y., and Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 58. Hammadi M., Oulidi A., Gackière F., Katsogiannou M., Slomianny C., Roudbaraki M., Dewailly E., Delcourt P., Lepage G., Lotteau S., Ducreux S., Prevarskaya N., and Van Coppenolle F. (2013) Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J. 27, 1600–1609 [DOI] [PubMed] [Google Scholar]
- 59. Südhof T. C. (2012) Calcium control of neurotransmitter release. Cold Spring Harbor Perspect. Biol. 4, a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Buma P., and Roubos E. W. (1983) Calcium dynamics, exocytosis, and membrane turnover in the ovulation hormone-releasing caudo-dorsal cells of Lymnaea stagnalis. Cell Tissue Res. 233, 143–159 [DOI] [PubMed] [Google Scholar]
- 61. Raraty M., Ward J., Erdemli G., Vaillant C., Neoptolemos J. P., Sutton R., and Petersen O. H. (2000) Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 97, 13126–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang Z., Chen G., Zhou W., Song A., Xu T., Luo Q., Wang W., Gu X. S., and Duan S. (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 9, 945–953 [DOI] [PubMed] [Google Scholar]
- 63. Ferri K. F., and Kroemer G. (2001) Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3, E255–E263 [DOI] [PubMed] [Google Scholar]
- 64. Leist M., and Jäättelä M. (2001) Triggering of apoptosis by cathepsins. Cell Death Differ. 8, 324–326 [DOI] [PubMed] [Google Scholar]
- 65. Haughey N. J., and Mattson M. P. (2002) Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr. 31, S55–S61 [DOI] [PubMed] [Google Scholar]
- 66. Mayne M., Holden C. P., Nath A., and Geiger J. D. (2000) Release of calcium from inositol 1,4,5-trisphosphate receptor-regulated stores by HIV-1 Tat regulates TNF-α production in human macrophages. J. Immunol. 164, 6538–6542 [DOI] [PubMed] [Google Scholar]
- 67. Leist M., and Jäättelä M. (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2, 589–598 [DOI] [PubMed] [Google Scholar]
- 68. Los M., Wesselborg S., and Schulze-Osthoff K. (1999) The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity 10, 629–639 [DOI] [PubMed] [Google Scholar]
- 69. Houseweart M. K., Pennacchio L. A., Vilaythong A., Peters C., Noebels J. L., and Myers R. M. (2003) Cathepsin B but not cathepsins L or S contributes to the pathogenesis of Unverricht-Lundborg progressive myoclonus epilepsy (EPM1). J. Neurobiol. 56, 315–327 [DOI] [PubMed] [Google Scholar]
- 70. Lieuallen K., Pennacchio L. A., Park M., Myers R. M., and Lennon G. G. (2001) Cystatin B-deficient mice have increased expression of apoptosis and glial activation genes. Hum. Mol. Genet. 10, 1867–1871 [DOI] [PubMed] [Google Scholar]
- 71. Rempel S. A., Rosenblum M. L., Mikkelsen T., Yan P. S., Ellis K. D., Golembieski W. A., Sameni M., Rozhin J., Ziegler G., and Sloane B. F. (1994) Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 54, 6027–6031 [PubMed] [Google Scholar]
- 72. Tsuchiya K., Kohda Y., Yoshida M., Zhao L., Ueno T., Yamashita J., Yoshioka T., Kominami E., and Yamashima T. (1999) Postictal blockade of ischemic hippocampal neuronal death in primates using selective cathepsin inhibitors. Exp. Neurol. 155, 187–194 [DOI] [PubMed] [Google Scholar]
- 73. Amritraj A., Wang Y., Revett T. J., Vergote D., Westaway D., and Kar S. (2013) Role of cathepsin D in U18666A-induced neuronal cell death: potential implication in Niemann-Pick type C disease pathogenesis. J. Biol. Chem. 288, 3136–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liang Q., Ouyang X., Schneider L., and Zhang J. (2011) Reduction of mutant huntingtin accumulation and toxicity by lysosomal cathepsins D and B in neurons. Mol. Neurodegener. 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Koike M., Nakanishi H., Saftig P., Ezaki J., Isahara K., Ohsawa Y., Schulz-Schaeffer W., Watanabe T., Waguri S., Kametaka S., Shibata M., Yamamoto K., Kominami E., Peters C., von Figura K., and Uchiyama Y. (2000) Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 20, 6898–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cantres-Rosario Y., Plaud-Valentín M., Gerena Y., Skolasky R. L., Wojna V., and Meléndez L. M. (2013) Cathepsin B and cystatin B in HIV-seropositive women are associated with infection and HIV-1-associated neurocognitive disorders. AIDS 27, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rivera L. E., Colon K., Cantres-Rosario Y. M., Zenon F. M., and Melendez L. M. (2014) Macrophage derived cystatin B/cathepsin B in HIV replication and neuropathogenesis. Curr. HIV Res. 12, 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McCall M. A., Gregg R. G., Behringer R. R., Brenner M., Delaney C. L., Galbreath E. J., Zhang C. L., Pearce R. A., Chiu S. Y., and Messing A. (1996) Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc. Natl. Acad. Sci. U.S.A. 93, 6361–6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zou W., Kim B. O., Zhou B. Y., Liu Y., Messing A., and He J. J. (2007) Protection against human immunodeficiency virus type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving glial fibrillary acidic protein. Am. J. Pathol 171, 1923–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu Z., Zhao F., and He J. J. (2014) Hepatitis C virus (HCV) interaction with astrocytes: nonproductive infection and induction of IL-18. J. Neurovirol. 20, 278–293 [DOI] [PubMed] [Google Scholar]
- 81. Liu Y., Li J., Kim B. O., Pace B. S., and He J. J. (2002) HIV-1 Tat protein-mediated transactivation of the HIV-1 long terminal repeat promoter is potentiated by a novel nuclear Tat-interacting protein of 110 kDa, Tip110. J. Biol. Chem. 277, 23854–23863 [DOI] [PubMed] [Google Scholar]
- 82. Klaver B., and Berkhout B. (1994) Comparison of 5′ and 3′ long terminal repeat promoter function in human immunodeficiency virus. J. Virol. 68, 3830–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jeeninga R. E., Hoogenkamp M., Armand-Ugon M., de Baar M., Verhoef K., and Berkhout B. (2000) Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 74, 3740–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fan Y., and He J. J. (2016) HIV-1 Tat induces unfolded protein response and endoplasmic reticulum stress in astrocytes and causes neurotoxicity through glial fibrillary acidic protein (GFAP) activation and aggregation. J. Biol. Chem. 291, 22819–22829 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.