

SUMMARY
High fidelity DNA synthesis requires that polymerases display a strong preference for right nucleotide insertion. When the wrong nucleotide is inserted, the polymerase deters extension from the mismatched DNA terminus. Twenty-three crystallographic structures of DNA polymerase β with terminal template-primer mismatches were determined as binary DNA and ternary pre-catalytic substrate complexes. These structures indicate that the mismatched termini adopt various distorted conformations that attempt to satisfy stacking and hydrogen bonding interactions. The binary complex structures indicate an induced strain in the mismatched template nucleotide. Addition of a non-hydrolysable incoming nucleotide stabilizes the templating nucleotide with concomitant strain in the primer terminus. Several dead-end ternary complex structures suggest that DNA synthesis might occur as the enzyme transitions from an open to closed complex. The structures are consistent with an induced fit mechanism where a mismatched terminus is misaligned relative to the correct incoming nucleotide to deter or delay further DNA synthesis.
Graphical abstract

INTRODUCTION
DNA polymerases catalyze a metal-dependent nucleophilic displacement reaction. Metal-assisted deprotonation of the deoxyribose 3′-hydroxyl of the primer terminus generates an oxyanion. An in-line nucleophilic attack on Pα of the incoming nucleoside triphosphate (dNTP) by this oxyanion extends the primer terminus by one nucleotide with PPi elimination. Most DNA polymerases utilize an induced fit mechanism to enhance their fidelity where binding of the right nucleotide aligns reactive atoms (primer O3′ and Pα of the incoming dNTP) for efficient nucleotide insertion, and the wrong nucleotide misaligns these atoms to deter misinsertion. Although induced fit is typically discussed in the context of nucleotide selectivity, it is also relevant to the extension of a terminal DNA mispair subsequent to a misinsertion event. In this context, binding of a correct nucleotide with a matched primer terminus aligns reactive atoms, but would distort a mismatched terminus to discourage further extension. This provides an opportunity for a proofreading enzyme to remove the misinserted nucleotide thereby enhancing overall fidelity and reducing mutagenesis.
DNA polymerase (pol) β is the smallest eukaryotic DNA polymerase and a member of the X-family of DNA polymerases. Crystallographic structures of pol β have revealed the global conformation and detailed active site architecture of binary DNA and ternary substrate pre-catalytic complexes (Batra et al., 2006; Freudenthal et al., 2013; Sawaya et al., 1997). As with several high fidelity DNA polymerases, pol β undergoes a large conformational change when the DNA binary complex binds a dNTP (Figure 1A). The carboxyl-terminal N-subdomain (referred to as fingers of right-handed polymerases) closes around the nascent base pair to ‘trap’ it between protein (e.g., α-helix N) and the DNA duplex terminus. The polymerase active site includes two-metal ions (magnesium), the DNA primer terminus (deoxyribose O3′), the incoming dNTP, and protein side chains that coordinate metals and substrates. The interactions among these components are interdependent such that the positions of the template-primer (TP) terminal base pair and the primer O3′ depend on interactions with enzyme side chains, metals, and nascent base pair. Likewise, the position of the incoming nucleotide (i.e., Pα) also depends on the precise interactions of these components. Accordingly, efficient DNA synthesis requires a geometry and an electrostatic environment that optimizes the reactive atoms. Since the terminal base pair of the growing DNA duplex forms a portion of the binding pocket of the incoming nucleotide, the kinetic consequences of modifying the binding pocket by introducing TP mismatches are of biological and structural interest. Previous kinetic analysis indicated that insertion efficiency is altered by as much as 25-fold by altering the identity of a matched primer terminus (e.g., insertion on G-C > C-G, template-primer). The correct insertion efficiency on mispaired primer termini depends on the identity of the terminal mismatch and is diminished from 30- (T-G) to 67,000-fold (G-G) (Beard et al., 2004).
Figure 1. Conformational Changes Associated with the Transition from Binary DNA Complex to a Ternary Substrate Complex (+dNTP).
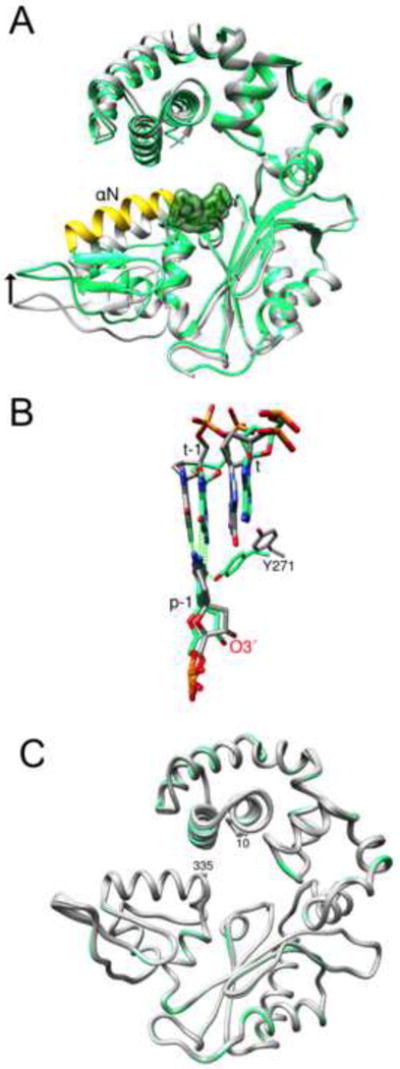
(A) The backbone of the binary complex (PDB: 3ISB) is grey and the ternary complex (PDB: 2FMS) is colored. Helix N (yellow) is repositioned (arrow) upon binding dCTP (green semitransparent surface).
(B) The transition to a pre-catalytic ternary complex repositions the template strand. The coding (t) and upstream (t-1) templating nucleotides move downstream (~2 Å) upon transitioning from a binary (grey carbons) to a ternary complex (green carbons). The shift in the template strand alters the position of O3′ of the primer terminus (p-1) that is stabilized through a minor groove hydrogen bond with Tyr271 (Y271).
(C) Licorice protein backbone representation of the 12 ternary pre-catalytic complexes with primer terminus mismatches (grey) compared to enzyme with a matched (G-C) primer terminus (green). The first (10) and last (335) protein residues observed in the electron density are indicated. The root-mean-square deviation (all Cαs) of the mismatch structures compared to pol β with a matched primer terminus varied from 0.18 to 0.40 Å.
In this study, we analyze the structures of 12 possible mismatched primer termini in the context of the pol β active site. The structure of the binary DNA complex is compared to that observed in the ternary substrate complex. The ternary complex mimics a pre-catalytic state with a correct incoming nucleotide situated adjacent to the terminal mismatch. This comparison provides an opportunity to analyze how nucleotide binding impacts active site architecture subsequent to a misinsertion event. The binary complexes with terminal mismatches exhibit a distorted mismatched template nucleotide and binding of the correct incoming nucleotide to form a ternary pre-catalytic complex relieves this distortion. More importantly, a distortion in the primer terminus occurs upon ternary complex formation that deters further extension thereby supporting an induced fit mechanism that increases base substitution fidelity.
RESULTS
Binary DNA Complex Structures
Pol β binary complex crystal structures with mismatched TP termini were obtained after annealing template, primer and downstream oligonucleotides to form double-stranded DNA with a 1-nucleotide gap. Comparison of structures of binary pol/DNA complexes with the corresponding ternary complexes with a correct incoming dNTP indicates that protein and/or substrate conformational changes must occur to produce the correctly aligned pre-chemistry complex. For pol β, a feature of this conformational change is movement of α-helix N of the carboxyl-terminal N-subdomain into close proximity to the nascent (i.e., template-incoming nucleotide) base pair (Figure 1A). The conformation of the binary pol/DNA complex is referred to as “open,” whereas the enzyme in the ternary substrate complex is “closed.” In addition, this transition results in re-alignment of the templating nucleotide to optimize Watson-Crick (WC) hydrogen bonding (Figure 1B). The primer terminus is stabilized through WC hydrogen bonding with its templating base, stacking interactions with the incoming nucleotide, and a minor groove hydrogen bond with Tyr271.
We were able to obtain 11 of the 12 possible TP terminus mismatch structures. The poor density for the binary complex with the C-C mismatch precluded structure determination. All binary pol β complexes with terminal mismatches were in open conformations. The crystallographic statistics and base pair parameters for these terminal mispairs are summarized in Tables 1 and S1 (available online), respectively. In general, nucleotides of the primer terminus are in anti-conformations and planar (Figures S1A–S4A). The largest distortion is observed for the A-C mismatch where O3′ of the primer terminus sugar is positioned away from the active site. A water molecule bridges N6 of adenine and N4 of cytosine (Figure S1A, middle panel). In addition, the primer terminus adenine in the A-A mismatch has reversed its orientation positioning N6 toward the minor groove (Figure S1A, top panel). Other structural features of the binary complex terminal mismatches will be highlighted during description of the respective ternary complex structures.
Table 1.
Data Collection and Refinement Statistics for Binary DNA Complexes
| Mispair Template-Primer | A-A | A-C | A-G | C-A | C-T | G-A | G-G | G-T | T-C | T-G | T-T |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Data Collection | |||||||||||
|
| |||||||||||
| Space group | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 |
|
| |||||||||||
| Cell dimensions | |||||||||||
|
| |||||||||||
| a, b, c (Å) | 54.1, 79.5, 54.7 | 54.1, 79.6, 54.6 | 54.7, 79.4, 55.0 | 54.6, 79.5, 54.9 | 54.6, 79.5, 54.9 | 54.4, 79.2, 54.7 | 54.5, 79.5, 55.0 | 54.5, 79.4, 54.7 | 54.2, 79.1, 54.7 | 54.4, 79.3, 54.7 | 54.6, 79.1, 54.5 |
| α, β, γ, (°) | 90, 105.3, 90 | 90, 105.6, 90 | 90, 105.9, 90 | 90, 105.6, 90 | 90, 105.7, 90 | 90, 106.1, 90 | 90, 105.7, 90 | 90, 105.8, 90 | 90, 105.9, 90 | 90, 105.5, 90 | 90, 106.1, 90 |
| Resolution (Å) | 50 – 2.00 (2.07 –2.0)a | 50 – 2.20 (2.28 –2.20) | 50 – 2.00 (2.07 –2.0) | 50 – 2.00 (2.07 –2.0) | 50 – 2.00 (2.07 –2.0) | 50 – 2.00 (2.07 –2.0) | 50 – 2.10 (2.18 –2.10) | 50 – 2.00 (2.07 –2.0) | 50 – 2.40 (2.49 –2.40) | 50 – 2.00 (2.07 –2.0) | 50 – 2.00 (2.07 –2.0) |
| Rsym or Rmerge | 0.066 (0.252) | 0.079 (0.562) | 0.090 (0.289) | 0.109 (0.387) | 0.121 (0.409) | 0.112 (0.467) | 0.142 (0.489) | 0.059 (0.181) | 0.097 (0.501) | 0.107 (0.306) | 0.058 (0.215) |
| I/σI | 19.3 (4.05) | 12.3 (2.22) | 13.6 (3.53) | 9.2 (2.76) | 9.6 (3.09) | 8.2 (2.53) | 6.5 (1.96) | 17.5 (6.19) | 13.1 (2.53) | 12.1 (3.84) | 16.7 (5.53) |
| Completeness (%) | 99.0 (95.9) | 96.5 (95.0) | 98.8 (96.8) | 99.2 (97.6) | 99.4 (95.4) | 99.3 (94.4) | 98.0 (91.7) | 98.9 (94.7) | 97.6 (96.9) | 99.7 (97.9) | 98.1 (93.1) |
| Redundancy | 3.6 (3.1) | 3.6 (3.6) | 3.4 (3.1) | 3.2 (2.9) | 3.6 (3.2) | 3.6 (3.3) | 2.8 (2.2) | 3.7 (3.3) | 3.4 (3.4) | 3.6 (3.2) | 3.7 (3.4) |
|
| |||||||||||
| Refinement | |||||||||||
|
| |||||||||||
| Resolution (Å) | 2.00 | 2.20 | 2.00 | 2.00 | 2.00 | 2.00 | 2.10 | 2.00 | 2.40 | 2.00 | 2.00 |
| No. reflections | 107384 | 78534 | 103148 | 95755 | 109418 | 109468 | 73145 | 110817 | 58338 | 107783 | 109528 |
| Rwork/Rfree | 21.0/26.4 | 24.9/32.0 | 20.7/26.1 | 20.8/27.2 | 19.9/24.6 | 20.1/25.8 | 20.5/28.1 | 17.4/22.9 | 23.4/30.6 | 19.3/24.2 | 18.9/24.5 |
|
| |||||||||||
| No. of atoms | |||||||||||
|
| |||||||||||
| Protein | 2615 | 2640 | 2640 | 2619 | 2619 | 2613 | 2598 | 2620 | 2627 | 2620 | 2609 |
| Ligand/ion | 634 | 632 | 635 | 634 | 633 | 637 | 638 | 636 | 631 | 636 | 636 |
| Water | 419 | 261 | 617 | 476 | 602 | 501 | 400 | 591 | 263 | 547 | 560 |
| B-factors | |||||||||||
| Protein | 34.5 | 38.8 | 24.2 | 24.1 | 24.9 | 26.2 | 29.2 | 20.6 | 28.0 | 25.1 | 24.2 |
| Ligand/ion | 27.8 | 46.8 | 20.2 | 23.7 | 22.5 | 22.0 | 26.0 | 18.5 | 26.7 | 23.1 | 22.7 |
| Water | 35.1 | 38.0 | 28.6 | 26.8 | 31.2 | 30.2 | 30.0 | 25.6 | 23.5 | 31.6 | 30.9 |
| Rmsds | |||||||||||
| Bond lengths (Å) | 0.005 | 0.006 | 0.005 | 0.008 | 0.005 | 0.005 | 0.008 | 0.008 | 0.006 | 0.005 | 0.005 |
| Bond angles (°) | 1.05 | 1.12 | 1.05 | 0.966 | 1.07 | 1.06 | 0.965 | 0.915 | 1.10 | 1.06 | 1.03 |
| RCSB ID code | 5J0O | 5J0P | 5J0Q | 5J0R | 5J0S | 5J0T | 5J0U | 5J0V | 5J0W | 5J0X | 5J0Y |
Values in parentheses are for highest-resolution shell.
General Features of Ternary Complex Structures with Primer Terminus Mismatches
Ternary complex structures were obtained by soaking binary complex crystals in cryo-protectant containing the non-hydrolzable dNTP analog dUMPNPP. This nucleotide analog forms a WC base pair with the templating adenine, but is not inserted due to nitrogen substitution for the bridging oxygen between Pα and Pβ. The enzyme is in the closed conformation despite distortions in the mismatched TP terminus for all 12 ternary complex structures. The closed conformation of these complexes is similar to those of the reference matched TP terminus (G-C, PDB: 2FMS); root mean square deviations (rmsd) for 326 Cα 0.2–0.4 Å (Figure 1C). The structure of the terminal mismatches are illustrated relative to reference matched termini in Figures S1B–S4B.
Although binary and ternary complex reference structures of the matched G-C TP terminus base pair reveal WC hydrogen bonding, the distance between acceptor and donor atoms are shorter in the ternary substrate complex. This is probably the result of subtle template slippage that aligns O3′ of the primer terminus with Pα of the incoming dUMPNPP (Figure 1B). This modest movement positions O3′ 3.4 Å from Pα generating a virtual attack angle (O3′–Pα–N) of ~160° (Table 2). The sugar pucker of the primer terminus is also altered (binary complex, C4′-exo) during formation of the ternary complex resulting in a C3′-endo pucker that facilitates catalytic metal coordination and alignment.
Table 2.
Ternary Pre-Catalytic Complexes Primer Terminus Mismatches
| TP | Primer terminus properties
|
||||
|---|---|---|---|---|---|
| B-factor (upstream nucleotide) |
B-factor (primer terminus) |
Differencea |
d (O3′–Pα) |
O3′–Pα–N | |
| (Å2) | (Å2) | (%) | (Å) | (°) | |
| GCb | 27.7 | 25.0 | −9.7 | 3.4 | 159.7 |
| TAc | 25.6 | 18.4 | −28.1 | 3.5 | 167.9 |
| AA | 26.4 | 40.0 | 51.5 | 8.8d | 105.8 |
| AC | 24.8 | 58.0 | 133.9 | 5.6 | 147.3 |
| AG | 28.2 | 41.5 | 47.2 | 8.5d | 98.5 |
| CA | 29.9 | 40.8 | 36.5 | 9.1d | 102.8 |
| CC | 27.3 | 37.2 | 36.3 | 5.2d,e | 145.3 |
| CT | 21.1 | 38.8 | 83.9 | 5.2d,e | 144.2 |
| GA | 27.3 | 37.7 | 38.1 | 3.4 | 165.6 |
| GG | 24.8 | 38.8 | 56.5 | 9.4d | 102.4 |
| GT | 27.7 | 43.8 | 58.1 | 4.6d | 141.7 |
| TC | 42.9 | 72.5 | 69.0 | 4.8 | 72.0 |
| TG | 23.1 | 34.1 | 47.6 | 4.4d | 145.3 |
| TT | 26.0 | 28.5 | 9.6 | 4.6d | 139.9 |
Difference = [(Bf)pt − (Bf)(pt-1)/(Bf)(pt-1)] × 100 where (Bf)pt and (Bf)(pt-1) are the B-factors for the primer terminus and its 5′-upstream neighboring nucleotide, respectively.
PDB ID 2FMS (Batra et al., 2006)
PDB ID 3RJI (Batra et al., 2012)
Water observed in position vacated by primer terminus O3′
Sugar-phosphate backbone not well-resolved
The nucleotide and base pair parameters for matched and mismatched primer termini for both the binary and ternary complex structures are tabulated in Table S1. The crystallographic statistics for the ternary complex structures are summarized in Table 3. The B-factors of the primer terminus in the ternary complexes are 10–134% higher than that of the neighboring upstream nucleotide that forms a WC base pair (Table 2). The description of the terminal mismatch structures will focus on the architecture of the mismatched TP, rather than enzyme side chains and emphasize the spatial relationship between the primer O3′ and Pα of the incoming nucleotide since insertion efficiency should be exquisitely sensitive to their specific positions. The active site distortions are localized around these reacting atoms since the nascent templating adenine, incoming uracil base and two magnesium ions are in identical positions for all ternary complex structures.
Table 3.
Data Collection and Refinement Statistics for Pre-Catalytic Ternary Complexes
| Mispair Template-Primer | A-A | A-C | A-G | C-A | C-C | C-T | G-A | G-G | G-T | T-C | T-G | T-T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Data Collection | ||||||||||||
|
| ||||||||||||
| Space group | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 |
|
| ||||||||||||
| Cell dimensions | ||||||||||||
| a, b, c (Å) | 50.8, 79.8, 55.5 | 50.4, 79.2, 55.5 | 50.7, 79.9, 55.5 | 50.8, 80.3, 55.5 | 50.7, 79.9, 55.5 | 50.9, 79.9, 55.5 | 50.7, 79.9, 55.5 | 50.7, 80.5, 55.4 | 50.8, 79.7, 55.4 | 50.6, 80.2, 55.5 | 50.8, 80.2, 55.5 | 50.8, 80.2, 55.4 |
| α, β, γ, (°) | 90, 107.6, 90 | 90, 107.1, 90 | 90, 107.6, 90 | 90, 107.7, 90 | 90, 107.6, 90 | 90, 107.7, 90 | 90, 107.7, 90 | 90, 108.1, 90 | 90, 107.5, 90 | 90, 107.5, 90 | 90, 107.9, 90 | 90, 107.9, 90 |
| Resolution (Å) | 50 – 2.20 (2.28 –2.20)a | 50 – 2.50 (2.59 –2.50) | 50 – 2.50 (2.59 –2.50) | 50 – 2.00 (2.18 –2.10) | 50 – 2.10 (2.18 –2.10) | 50 – 2.00 (2.18 –2.10) | 50 – 2.00 (2.18 –2.10) | 50 – 2.10 (2.18 –2.10) | 50 – 2.30 (2.38 –2.30) | 50 – 2.40 (2.49 –2.40) | 50 – 2.20 (2.28 –2.20) | 50 – 2.10 (2.18 –2.10) |
| Rsym or Rmerge | 0.110 (0.477) | 0.114 (0.564) | 0.101 (0.455) | 0.095 (0.387) | 0.072 (0.240) | 0.137 (0.461) | 0.106 (0.534) | 0.090 (0.366) | 0.114 (0.564) | 0.072 (0.408) | 0.111 (0.444) | 0.101 (0.414) |
| I/σI | 9.9 (2.4) | 10.0 (2.0) | 13.1 (2.5) | 11.0 (3.3) | 16.6 (3.3) | 8.1 | 8.9 (2.2) | 11.9 | 9.9 (2.3) | 18.9 (1.8) | 9.5 (2.7) | 11.0 |
| Completeness (%) | 99.2 (97.9) | 100 (99.8) | 99.4 (99.3) | 100 (99.8) | 99.0 (93.4) | 97.9 (97.1) | 98.6 (87.5) | 95.7 (93.7) | 99.8 (100) | 94.7 (91.4) | 96.5 (94.4) | 95.2 (94.8) |
| Redundancy | 3.3 (3.1) | 3.2 (3.1) | 3.5 (3.2) | 3.2 (2.9) | 3.3 (2.5) | 3.5 (3.6) | 3.5 (2.9) | 3.8 (3.8) | 3.5 (3.4) | 2.4 (1.9) | 3.7 (3.8) | 3.0 (2.8) |
|
| ||||||||||||
| Refinement | ||||||||||||
|
| ||||||||||||
| Resolution (Å) | 2.20 | 2.50 | 2.50 | 2.10 | 2.10 | 2.10 | 2.10 | 2.10 | 2.30 | 2.40 | 2.20 | 2.10 |
| No. reflections | 70905 | 65503 | 50558 | 88016 | 81520 | 85725 | 85562 | 89233 | 65503 | 38421 | 78185 | 70004 |
| Rwork/Rfree | 21.0/28.3 | 23.4/29.8 | 21.2/28.3 | 21.2/26.6 | 20.1/25.7 | 24.0/28.9 | 20.0/26.0 | 20.8/26.7 | 20.3/26.8 | 22.4/28.7 | 20.0/26.6 | 22.5/28.4 |
| No. atoms | ||||||||||||
| Protein | 2608 | 2608 | 2608 | 2645 | 2608 | 2612 | 2614 | 2612 | 2630 | 2610 | 2631 | 2643 |
| Ligand/ion | 664 | 664 | 667 | 666 | 664 | 666 | 670 | 671 | 669 | 661 | 668 | 666 |
| Water | 349 | 257 | 225 | 328 | 356 | 359 | 365 | 333 | 273 | 234 | 385 | 389 |
| B-factors | ||||||||||||
| Protein | 26.6 | 25.6 | 26.8 | 27.7 | 23.9 | 25.4 | 29.4 | 26.8 | 29.5 | 39.1 | 24.8 | 25.9 |
| Ligand/ion | 33.2 | 33.9 | 34.5 | 36.3 | 31.9 | 33.4 | 37.2 | 33.4 | 36.3 | 38.7 | 47.6 | 34.9 |
| Water | 31.6 | 25.9 | 26.7 | 31.4 | 32.0 | 30.5 | 36.8 | 32.4 | 31.1 | 29.6 | 41.9 | 32.5 |
| Rmsds | ||||||||||||
| Bond lengths (Å) | 0.006 | 0.006 | 0.006 | 0.005 | 0.005 | 0.006 | 0.005 | 0.006 | 0.005 | 0.006 | 0.008 | 0.005 |
| Bond angles (°) | 1.08 | 1.13 | 1.11 | 1.10 | 1.07 | 1.13 | 1.09 | 1.08 | 1.09 | 1.12 | 0.976 | 1.08 |
| RCSB ID code | 5J29 | 5J2A | 5J2B | 5J2C | 5J2D | 5J2E | 5J2F | 5J2G | 5J2H | 5J2I | 5J2J | 5J2K |
Values in parentheses are for highest-resolution shell.
In general, the binary complex structures of the terminal mismatches indicate that the backbone phosphate of the templating nucleotide in the mispair is displaced relative to that observed in the reference structure with a matched primer terminus (Figures 2A and S1A–S4A). In contrast, the closed ternary pre-catalytic complexes with the mispaired termini indicate that the template backbone of the mispair now resembles that of the matched reference structure (Figures 2A and S1B–S4B).
Figure 2. Displacement of the Primer and Template Backbone in Binary and Ternary Complexes with Terminal Primer Mismatches.

Each structure with a terminal primer mismatch was superimposed with a structure with a matched (G-C) terminus (i.e., binary complex, PDB: 3ISB; ternary complex, PDB: 2FMS).
(A) The relative position of the phosphate of the template nucleotide opposite the primer terminus (Pt (n-1)) was determined for the binary (grey bar) and ternary (black bar) complexes as compared to the reference structure. The phosphate backbone of the templating nucleotide of the mismatch is displaced to a much greater extent in the binary complex than the ternary complex.
(B) The relative position of the phosphate of the primer terminus nucleotide (Pp (n-1)) was determined for the binary (grey bar) and ternary (black bar) complexes. In contrast to the template nucleotide phosphate, the phosphate of the primer terminus is displaced to a greater extent in the ternary complex than the binary complex.
Template ‘A’ Primer Terminus Mismatches
In the binary complex structures, the primer terminus with adenine or guanine bases indicate two hydrogen bonding interactions with the templating adenine (Figure S1A). To accommodate the two purines, the C1′-C1′ distance for guanosine at the primer terminus is elevated (>12 Å), while the polarity of the sugar of the terminal adenosine has been altered so that O3′ faces the DNA major groove increasing C1′-C1′ (>13 Å). As noted above, the A-C mismatch positions the cytosine in the active site precluding direct template base interactions. A water-mediated indirect interaction can occur between the bases (Figure S1A, middle panel).
In the ternary complex structures (Figure S1B), the adenosine at the primer terminus retains it reverse orientation and hydrogen bonding pattern. Like adenosine, the sugar of the guanosine has altered its polarity so that O3′ faces the major groove. This results in a loss of one hydrogen bond relative to what is observed with the binary complex structure. The position of O3′ relative to Pα (>8 Å) of the incoming nucleotide removes a coordinating ligand from the catalytic metal site and effectively deters further DNA synthesis.
The ternary complex structure with the A-C terminal mismatch indicates that the cytosine can now interact with the templating adenine (Figure S1B, middle panel). The proximity of N1 of adenine with O2 of cytosine suggest that it may be protonated as observed previously with an A-C mismatch embedded in duplex DNA (Hunter et al., 1986).
Template ‘C’ Primer Terminus Mismatches
The binary complex structures with adenine or thymine primer termini indicate a planar arrangement of the bases with a single direct hydrogen bond between the bases (Figure S2A). For the C-T mismatch, a water molecule intervenes between O2 of these pyrimidines providing an additional stabilizing interaction.
In the ternary complex structure for the C-A terminus, the polarity of the adenosine residue is altered thereby positioning O3′ in the major groove over 9 Å away from Pα of the incoming nucleotide (Figure S2B, top panel); the bases exhibit two weak hydrogen bonds. In the ternary complex structure with the C-C terminus (Figure S2B, middle panel), there is a single hydrogen bond where N4 of the primer terminus interacts with N3 of the template cytosine. Additionally, Tyr271 hydrogen bonds with the minor groove edge of the primer terminus cytosine (O2). This is the only structure with a terminal mismatch where a direct interaction between Tyr271 and the primer terminus is observed. The C1′-C1′ distance for these two pyrimidines is narrowed relative to that expected for a WC base pair (Table S1). The ternary complex structure for the CT mispair exhibits two hydrogen bonds with a narrowed C1′-C1′ distance (Table S1 and Figure S2B, bottom panel).
Template ‘G’ Primer Terminus Mismatches
For the G-A mispair, the binary complex structure reveals both purine bases are in anti-conformations with two hydrogen bonds (Figure S3A, top panel). Compared to the reference G-C terminus, the phosphodiester backbone and sugar of the templating G has shifted 1.2 Å and the base has moved into the minor groove. In the ternary complex (Figure S3B, top panel), the adenosine has transitioned to a syn-conformation with Hoogsteen base pairing. Other key distances (C1′–C1′, O3′–Pα) and O3′–Pα–N angle are similar to the G-C reference structure (Tables 2 and S1). However, the primer terminus sugar and backbone phosphate are displaced modestly (Figures 2B and S3B, top panel).
In the binary complex of the G-G mispair, the two purines are again observed in anti-conformations with a single hydrogen bond (Figure S3A, middle panel), widened C1′-C1′ distance (Table S1), and template strand phosphate displaced by 1.4 Å (Figure 2A). In contrast with G-A in the ternary complex, the polarity of the primer terminus is altered so that O3′ now points into the major groove (Figure S3B, middle panel). In this position, the G-G mispair forms two hydrogen bonds. Thus in this ternary complex, the architecture of the G-G template-primer base pair is profoundly distorted. The C1′–C1′ and O3′–Pα distances are much greater than their reference distances (Tables 2 and S1). In the absence of a sugar near the active site, a water molecule replaces the position occupied by O3′ completing the octahedral coordination of the catalytic Mg2+.
In the G-T binary complex, the bases adopt a “Wobble” conformation with two hydrogen bonds (Figure S3A, bottom panel). As noted above for binary complexes, the template phosphate backbone is modestly displaced with the templating guanine positioned deeper into the major groove (Figure 2A). For the ternary complex, the mispair has opened relative to the binary complex altering its hydrogen-bonding pattern (Table S1 and Figure S3B, bottom panel). Except for the phosphate, the primer terminus is displaced into the DNA major groove. The C1′–C1′ distance is narrowed and other key distance and geometrical features of the active site are altered (Tables 2 and S1). The displaced primer terminus permits a water molecule to occupy the position vacated by O3′.
Template ‘T’ Primer Terminus Mismatches
In the binary complex structure of the T-C terminus, there are three potential hydrogen bonding interactions; N3 of template T can hydrogen bond with N3 and O2 of the primer terminus C (Figure S4A, top panel) and an additional hydrogen bond between the atoms at the 4-position of these pyrimidines. With two pyrimidines, the C1′–C1′ has narrowed to permit the observed hydrogen bonding (Table S1). The template strand backbone phosphate is displaced ~1.9 Å. In the ternary complex structure (Figure S4B, top panel), the thymidine adopts a frayed distorted conformation thereby failing to form inter-base hydrogen bonds. The distance and geometry between O3′ and Pα precludes nucleotidyl transfer.
In the binary complex T-G structure (Figure S4A, middle panel), the bases form a Wobble conformation with two hydrogen bonds. The primer terminus guanine has moved into the minor groove, while the phosphate backbone of the template thymidine is displaced 1.1 Å. In the ternary complex structure (Figure S4B, middle panel), guanosine adopts a syn-conformation forming a weak Hoogsteen hydrogen bond. The sugar has moved toward the major groove thereby moving O3′ of the primer terminus away from Pα of the incoming nucleotide.
The binary complex T-T structure (Figure S4A, bottom panel) shows a Wobble conformation with two hydrogen bonds between the mispaired thymines. Again, the C1′–C1′ distance has been narrowed to permit these pyrimidines to interact. In the ternary complex structure (Figure S4B, bottom panel), the Wobble conformation with two hydrogen bonds is maintained. Likewise, the narrow C1′–C1′ distance aids in displacing O3′ and deterring further DNA synthesis (Tables 2 and S1).
DISCUSSION
DNA polymerase dependent base substitution errors require both misinsertion and mismatch extension. Accordingly, a molecular understanding of mismatch extension is vital to our understanding of mutagenesis. By substituting a matched TP terminus with DNA mismatches, a variety of pol β active site perturbations have been observed in both binary DNA and pre-catalytic ternary complexes trapped with a non-hydrolyzable incoming nucleotide. Since the structures of both the binary DNA and ternary substrate complexes have been determined, the effect of correct nucleotide binding, as well as polymerase subdomain motions, on the conformation of the primer terminus could be assessed providing a molecular understanding of this key fidelity checkpoint.
Induced Distortion in the TP Terminus Transitioning to the Ternary Complex
Comparison of the mismatched binary and ternary complex structures reveals that the TP terminus was dramatically altered in several cases; three of these are illustrated in Figure 3. In the binary complexes, the bases at the TP termini are generally planar and attempt to hydrogen bond with one another. Formation of the ternary complex with a correct incoming nucleotide sometimes results in the primer terminus altering its polarity as the nucleoside retains its anti-conformation. This displaces O3′ into the major groove (Figure 3).
Figure 3. Mismatched Primer Termini with Altered Polarity Transitioning from Binary to Ternary Complex.

The binary complex structure of the terminal mismatch is shown in a wire representation and the ternary complex is shown in a stick representation. Hydrogen bonding in the binary and ternary complexes is shown with black and green dashed lines, respectively. Several structures with primer terminal mismatches exhibited a dramatic change in the orientation of the primer terminal nucleotide upon ternary complex formation. In these cases, the major groove edge of the primer base faces the minor groove (i.e., faces down in the illustrated orientation) of the DNA duplex. This positions the primer terminus in the major groove with the opposite polarity. The carbons of the respective nucleotides are colored: A, light blue; C, yellow; G, pink. The At-Gp (top panel), Ct-Ap (middle panel), and Gt-Gp (lower panel) mispairs are extended the least efficiently (see text).
In the binary DNA complexes, the backbone phosphate of the templating nucleotide opposite the mispaired primer terminus is displaced relative to the position observed in the matched reference state (Figures 2A and S1A–S4A). This reflects molecular adjustments necessary to relieve strain generated by the mispair. Importantly, binding of the correct nucleotide to generate a ternary complex aligns the template backbone phosphates to that observed in the matched reference structure (Figures 2A and S1B–S4B). However, this often results in re-positioning of the primer terminus O3′ or phosphate (Figures 3 and 2B, respectively).
Comparison of various ternary complex structures with active site mismatches or lesions indicates that the incoming nucleotide positions are usually very similar. These structures include active site mismatches (Batra et al., 2008), an abasic site in the templating base position (Beard et al., 2009), or a template lesion (i.e., 8-oxoguanune) opposite the primer terminus (Batra et al., 2012). This suggests that the nucleotide-binding pocket in the closed complex is rigid, and that active site distortions can be accommodated through adjustments in the DNA (e.g., primer terminus) forming the binding pocket. A mismatched primer terminus will be exquisitely susceptible to distortion upon nucleotide binding. Thus, as with an induced fit mechanism to deter incorrect nucleotide insertion, an induced fit mechanism amplifies DNA polymerase fidelity by discouraging mismatch extension through conformational adjustments in the primer terminus deterring chemistry. This can be achieved by altering catalytic metal coordination, stacking interactions between the bases of the primer terminus and incoming nucleotide, position and electrostatic environment of primer terminus O3′ and Pα (dNTP), and/or open-closed subdomain dynamics.
Previous studies of the G-A mismatch in the confines of duplex DNA had indicated that while G is observed in an anti-conformation, A can be in an either an anti- or syn-conformation suggesting that they are energetically similar (Brown et al., 1986). In the binary complex structure with the G-A terminus, the A is in an anti-conformation with a two hydrogen bonds (Figure S3A, top panel). In contrast, the ternary complex structure indicates that A has transitioned to a syn-conformation (Figure S3B, top panel).
Induced Disorder at the Primer Terminus
As noted above (Table 2), the average B-factor for the primer terminus of several mismatches in ternary complexes are higher than that observed in the upstream nucleotide suggesting greater disorder at the 3′-primer terminus. The relative B-factors [B-factor(nucleotide)/B-factor(protein)] for the templating (coding) nucleotide (T(n)), primer terminus (P(n-1)), and its templating base (T(n-1)) in the binary DNA complex are between 1 (i.e., similar to that observed for the open polymerase complex) and 2 for both matched and mismatched primer termini (Figure 4A). For the matched terminus (G-C), the template strand is misaligned (Figure 1B) and the templating nucleotides (T(n) and T(n-1)) exhibit elevated B-factors whereas the primer terminus B-factor is similar to that observed for the polymerase.
Figure 4. Altered Nucleotide B-Factor in Response to Ternary Complex Formation.

The average B-factor for each active site nucleotide was determined: template coding nucleotide (T(n), blue circle) and template nucleotide opposite the primer terminus (T(n-1), blue square); primer terminus (P(n-1), red square) and dUMPNPP (red circle). The relative B-factor is calculated relative to the average B-factor for the polymerase of each structure.
(A) The relative nucleotide B-factors for the matched and mismatched primer termini structures of the binary complexes.
(B) The relative nucleotide B-factors for the matched and mismatched primer termini structures of the ternary complexes.
(C) Comparison of the relative B-factors of the primer terminus (ternary/binary) indicating general increases in relative B-factor upon ternary complex formation.
Addition of dUMPNPP to form the closed polymerase complex generates ternary complex structures that alter the relative B-factors dramatically. In the case of the matched primer terminus, the relative B-factors are similar and lower than that observed in the binary complex (i.e., <1) suggesting a decrease in disorder for these residues (Figure 4B). For the 12 ternary complex structures with terminal mismatches, the coding templating nucleotide (i.e., correctly base paired with the incoming nucleotide) and its upstream neighbor (i.e., mismatched nucleotide opposite the primer terminus) have comparable relative B-factors that are similar to that of the polymerase (i.e., ~1). In contrast, the relative B-factor of the primer terminus of the mismatched termini is generally greater than that of pol β (>1). In three instances where the primer terminus is C (i.e., A-C, C-C, T-C), the relative B-factor is the greatest (≥2). Thus, the greatest disorder appears to reside in the primer terminus of mismatched termini. The incoming dUMPNPP exhibits the least disorder in all 13 structures (i.e., 1 matched and 12 mismatches) independent of the conformation and observed hydrogen bonding of the primer terminus.
If the relative B-factor of the primer terminus of the binary complex is compared to that observed in the ternary complex, then the effect of transitioning from the binary DNA complex to a pre-catalytic ternary complex can be assessed (Figure 4C). In about half of the cases, formation of the ternary complex results in an increase in the relative B-factor of the primer terminus (>1). In five cases, there is little effect of ternary complex formation (~1).
Active Site Distortion Leads to Water Penetration
Subtle active site distortions are expected to result in dramatic changes in catalytic efficiencies. The resultant active site architecture of the G-T primer terminus mismatch ternary complex is shown in Figure 5. In this case, the distance between O3′ (primer terminus) and Pα (dUMPNPP) is 1.2 Å greater than that observed in a structure with a matched terminus. Critically, a water molecule is now situated at the site vacated by O3′. Accordingly, the loss of metal coordination would result in a significant increase in the pKa of O3′ (Batra et al., 2013). The loss of primer O3′ metal coordination with concomitant water penetration completing catalytic metal coordination is observed for most of the mismatched primer termini; only the A-C and T-C ternary complex structures do not indicate that a water molecule has replaced O3′. The primer terminus O3′ in the G-A ternary complex superimposes with O3′ observed of a matched terminus thereby precluding water penetration at this site.
Figure 5. Induced Active Site Distortion in a Pre-catalytic Ternary Complex.

Active site architecture of the ternary complex structure with a terminal G-T mismatch (stick representation). The reference structure with a matched terminus is superimposed (grey wireframe). The small spheres represent waters (red, mismatch structure; grey, matched structure) and the large green spheres are Mg2+ (mismatch structure). The primer terminus of GT structure is displaced from the ideal position observed in the matched structure. This positions O3′ 4.6 Å from Pα (dUMPNPP), in contrast to the 3.4 Å distance observed in the matched reference structure. The position vacated by O3′ is occupied by an additional water molecule that completes the octahedral geometry of the catalytic Mg2+.
The increase in distance between O3′ and Pα (dUMPNPP) and acute angle between the reacting atoms would also deter nucleotidyl transfer (Batra et al., 2008; Freudenthal et al., 2013; Lin et al., 2008). Similarly, the position of O3′ in the ternary complex structures with an A-A and A-G mismatches (Table 2) indicate that the reacting atoms are too far from one another (>8 Å) to permit catalysis. Since catalysis is known to occur with these terminal mismatches, albeit at a much lower rate (Beard et al., 2004), nucleotidyl transfer must occur from a conformation that more closely mimics the reference state. This might be achieved as the enzyme transitions to the closed inactive conformation observed here.
Active Site Geometry Correlates with Catalytic Efficiency
A general spatial alignment during the chemical reaction involving attack of the primer terminus oxyanion on Pα is required during extension of matched, mismatched, or modified primer termini. The probability of an appropriate alignment would decline as distortion in the primer terminus increases. Previous computational analysis of misinsertion with a misaligned primer terminus indicated a higher energy barrier early in the reaction coordinate was due to geometric adjustments of O3′ and Pα to enable nucleotidyl transfer with a feasible energy barrier (Lin et al., 2008). Accordingly, there would be a higher probability of achieving an appropriate alignment when the architecture of the active site is closer to that of the optimum state (i.e., the reference matched geometry). Thus transition intermediates (purine-pyrimidine, pyrimidine-purine) are extended more efficiently than transversion intermediates (purine-purine, pyrimidine-pyrimidine) (Beard et al., 2004) (Figure 6A). Generally, the observed insertion efficiency inversely correlates with the observed distortion in the active site geometry of the reactive atoms.
Figure 6. Correlation Between Ternary Complex Active Site Geometry and Insertion Efficiency on a Terminal Mismatch.

(A) The insertion efficiency (Eff., kcat/Km) of a correct nucleotide on different primer termini (matched and unmatched) are illustrated on a log plot and were taken from Beard et al. (Beard et al., 2004). The identity of the terminal base pair is given (template–primer) and colored by general base pair identity; red, purine-pyrimidine or pyrimidine-purine; blue, purine-purine; orange, pyrimidine-pyrimidine.
(B) The insertion efficiency (Eff., kcat/Km) of a correct nucleotide on different terminal mismatches are plotted against the distance between O3′ and Pα of dUMPNPP as well a the virtual angle of nucleophilic attack (primer O3′ and Pα–N). The points are colored according to general base-pair identity. In general, extension efficiency decreases with increased active site distortion. The greatest distortion and lowest efficiency corresponds to the lower left portion of the plot.
A plot of the distance (d, Å) between the primer terminus O3′ and Pα (dUMPNPP), the angle for nucleophilic attack of O3′ on Pα (i.e., O3′–Pα–N), and observed catalytic efficiency for correct insertion on each mismatch are compared (Figure 6B) (Beard et al., 2004). Correct nucleotide insertion on a matched primer terminus (G-C) displays the highest efficiency (eff. = 104/μM-s), nearest approach of reacting atoms (d = 3.4 Å), and alignment for nucleophilic attack (O3′–Pα–Oαβ = 160°). This places the corresponding data point in the upper right region of this plot. The corresponding points have also been colored according to their respective general base pair properties: red, purine-pyrimidine or pyrimidine-purine; blue, purine-purine; orange, pyrimidine-pyrimidine.
In general, purine-purine mismatches result in the greatest displacement of the primer terminus O3′ from Pα of the incoming nucleotide resulting in an acute angle between reacting atoms (~100°). In addition, the C-A terminal mismatch has similar characteristics. While extension of these mispairs is the most difficult (i.e, lowest efficiencies), extension does occur. Thus, there is a small population of substrate complexes that better mimic the reference state than observed in this pre-catalytic complex trapped with a non-reactive incoming nucleotide. As suggested above, this state could occur transiently as the enzyme transitions from an open to closed state. The structure of the terminal G-A mismatch appears to have good active site geometry, but exhibits weak catalytic efficiency. In this instance, a Cl− atom is observed in the active site near Arg258 and Tyr271 and ~5 Å from O3′. This negatively charged atom could deter synthesis by elevating the pKa of O3′. Since the primer terminus adenosine is in a syn-conformation, Tyr271 does not form a minor groove hydrogen bond with the base thereby destabilizing the primer terminus consistent with its modestly higher relative B-factor (~30%, Figure 3, middle panel). This Cl− atom is also observed in the ternary complexes of the C-C, CT, and G-T terminal mismatches where it interacts directly with O3′ of the primer terminus.
Another cluster of terminal mismatches correspond to transition intermediates (i.e., A-C, T-G, G-T; red). The geometry of the reactive atoms resembles the reference state more closely than the purine-purine mismatches described above. These mismatches are extended with the greatest efficiencies. The pyrimidine-pyrimidine terminal mismatches (i.e., C-C, C-T, T-T; orange) also appear to cluster. In these cases, the geometry of the reacting atoms is modestly perturbed, but the catalytic efficiency for extension is profoundly diminished. This probably reflects the poor stacking interactions between the nascent base pair and the TP terminus that could destabilize a productive ternary complex.
Structural Comparison with other DNA Polymerases
DNA polymerase structures with terminal mismatches from other polymerase families have been reported. Binary complex structures for seven TP mismatches have been reported for the A-family Bacillus DNA Polymerase I fragment (BF) (Johnson and Beese, 2004; Wang et al., 2011) permitting direct comparison with those reported here. Comparison of the respective BF TP mismatches with pol β complex structures shows that the hydrogen bonding patterns and the base pair geometry are similar in most instances (e.g., Wobble hydrogen bonding for G-T and T-G, Figure 7). In contrast, the primer G adopts a syn-conformer when paired with G in the BF structure, but is in an anti-conformation with unusual hydrogen bonding in the pol β binary complex (Figure 7). Additionally, the primer terminus of the binary pol β A-C structure is not planar, in contrast to the planar geometry observed in the BF structure. A binary complex structure of pol λ, X-family) indicates that the templating G, not the primer terminus G, adopts a syn-conformation, and this mispair can be extended more efficiently than other mispairs (Picher et al., 2006).
Figure 7. Comparison of Binary Complex Primer Terminus Mismatches with Bacillus fragment Pol I and Pol β.

The structures and hydrogen-bonding pattern of the primer terminal mismatches reported for Bacillus fragment (Johnson and Beese, 2004; Wang et al., 2011) and pol β are similar except for the G-G mismatch. Simulated annealing omit maps (Fo-Fc) for the pol β primer terminus mismatches are shown contoured at 3σ; the T-C mismatch is contoured at 2σ.
Ternary complex structures with terminal TP mismatches have also been reported. The ternary complex structure of Dpo4, a Y-family enzyme, from Sulfolobus solfataricus with a G-T mismatch at the terminus indicates that the primer thymidine displays a reverse Wobble conformation that positions O3′ of the primer terminus in the DNA major groove distant from the incoming nucleotide (Trincao et al., 2004). Ternary complex structures of a mutant of RB69 DNA polymerase, a B-family member, with G-T or T-G mismatches at the primer terminus indicates that these mispairs display Wobble hydrogen bonding similar to that seen with pol β (Xia and Konigsberg, 2014).
Influence of DNA Sequence
We previously reported binary and ternary complex structures of pol β with an A-A mismatch at the primer terminus (Batra et al., 2005). In those structures the templating cytosine was removed from the coding position in the binary DNA complex so that the upstream template adenosine could stack with the primer terminal adenosine. Thus, the terminal adenosines are in anti-conformations, but not in a planar arrangement. In the structures described here, the templating (coding) adenosine is not removed from the DNA helix so that the adenosines at the terminus are forced to form a planar arrangement. Each nucleotide is in an anti-conformation, but the primer terminus forms a reverse orientation (Figure S1A, top panel). Thus, the energy barrier to displace a pyrimidine at the coding position is significantly lower than displacing a purine. These observations are consistent with metal- and PPi-induced conformational changes that position the primer terminus in the active site more easily with templating pyrimidines than purines (Kirby et al., 2012).
In the previous study (Batra et al., 2005), addition of the complementary incoming nucleotide induced the templating C into the coding position to form WC hydrogen bonds. In addition, realignment of the primer terminus occurred; the primer A moved to a syn-conformation forming a planar mispair with a single hydrogen bond to the adenosine that moved upstream to make room for the templating cytosine. Since the templating adenosine in the current study did not need to reposition itself, the conformation of the primer terminus is unaltered in the ternary complex relative to the binary complex. However as noted above, the relative B-factor of the primer terminal adenosine is elevated in the ternary complex relative to the binary complex (Figure 3, middle panel).
Rare Base Tautomers
It has been proposed that rare base tautomers that mimic WC geometry/shape, but exhibit altered hydrogen bonding patterns, are intermediates of spontaneous mutagenesis during DNA replication (Watson and Crick, 1953). Crystallographic structures of some nascent mispairs in the confines of the polymerase active site are consistent with this suggestion (Bebenek et al., 2011; Koag et al., 2014; Wang et al., 2011). However, structures of mispairs at the primer terminus indicate that base tautomerization does not occur. For example, structures of the BF with an incoming dCTP/Mn2+ opposite A displays WC geometry consistent with rare base tautomers; however, at the post-insertion site (i.e., primer terminus), this mispair exhibits a Wobble conformation consistent with adenine protonated at N1, similar to that observed here (Figure S1B, middle panel).
Finally, it should be noted that the structures of the mismatches at the primer terminus do not mimic the corresponding mismatches at the polymerase active site. Ternary complex structures of pol β with an incoming non-hydrolyzable dAMPCPP opposite either C or G resulted in closed complexes where the template strand has moved upstream to permit binding of the incoming purine (Batra et al., 2008). Thus, the bases are staggered, rather than planar. More recently, Koag et al. (Koag et al., 2014) characterized ternary complex structures of pol β with transition intermediates (G-dTMPNPP and A-dCMPNPP). Whereas the A-C mismatch suggested a staggered alignment of the bases of the nascent mispair, the G-T mismatch in the presence of Mn2+ generated a closed complex similar to that observed with BF where a WC geometry was observed suggesting hydrogen bonding through rare base tautomers.
EXPERIMENTAL PROCEDURES
Protein Expression and Crystallization
Human pol β was overexpressed in Escherichia coli and purified as previously described (Beard and Wilson, 1995). The DNA consisted of a 16-mer template, a 10-mer upstream primer and a 5-mer downstream oligonucleotide. The DNA sequence was similar (except at the primer terminus) to that used in previous studies (Batra et al., 2006) in order to minimize sequence-dependent structural differences. The sequence of the downstream oligonucleotide was 5′-GTCGG-3′ and the 5′-terminus was phosphorylated. The template sequence was 5′-CCGACAXCGCATCAGC-3′ and the upstream primer sequence was 5′-GCTGATGCGX-3′ (X = A, C, G, or T). Proper annealing of these oligonucleotides resulted in one-nucleotide gapped DNA substrates with all 12 possible mismatches at the primer terminus with adenosine serving as the template coding nucleotide. Oligonucleotides were dissolved in 20 mM MgCl2, 100 mM Tris-HCl, pH 7.5. Template, upstream, and downstream oligonucleotides were mixed in a 1:1:1 ratio and annealed using a thermal cycler by heating 10 min at 90 °C and cooling to 4 °C (1 °C/min) resulting in 1 mM of one-nucleotide gapped duplex DNA. This solution was then mixed with an equal volume of pol β at 4 °C. The mixture was warmed to 35 °C and cooled gradually to 4 °C.
Crystallization and Data Collection
The pol β/DNA mixture was crystallized by sitting-drop vapor diffusion. The drops were incubated at 18 °C and streak seeded after 1 day; crystals grew in approximately 2 to 4 days after seeding. We were able to obtain 11 of the 12 possible TP terminus binary DNA mismatch structures. The poor density for the binary complex with the C-C mismatch precluded structure determination. The ternary complexes were obtained by soaking the binary crystals in cryo-protectant solution containing artificial mother liquor with 90 sodium acetate, 100 mM MgCl2, and 2 mM uridine-5′-[(α,β)-imido]triphosphate (dUMPNPP) with 12% ethylene glycol, and then flash-frozen at 100 K in a nitrogen gas stream. This nucleotide analog forms a WC base pair with the templating adenine, but is not inserted due to nitrogen substitution for the bridging oxygen between Pα and Pβ. We were able to obtain the 12 possible mismatched TP terminus DNA ternary pre-catalytic structures. Data were collected at 100 K on a CCD detector system mounted on a Rigaku MicroMax-007HF rotating anode generator. Data were integrated and reduced with HKL2000 software (Otwinowski and Minor, 1997). Structures were determined by molecular replacement with previously determined structures of pol β complexed with a one-nucleotide gapped DNA (binary complex, PDB: 3ISB) (Beard et al., 2009) or a ternary complex with an incoming dUMPNPP (ternary complex, PDB: 2FMS) (Batra et al., 2006). The crystal structures have similar lattices and are sufficiently isomorphous to determine the molecular replacement model using CNS or PHENIX (Adams et al., 2010). Further refinement and model building were carried out using O (Jones et al., 1991). Nucleotide and base pair parameters were analyzed using 3DNA (Lu and Olson, 2003). The figures were prepared in Chimera (Pettersen et al., 2004).
Primer Terminus Distortions
The backbone displacements illustrated in Figure 2 for binary DNA and ternary pre-catalytic complexes were determined by superimposing each terminal TP mismatch structure with a reference structure with a matched TP terminus. The structures were superimposed using all protein αCs. The reference structures for the binary DNA and ternary pre-catalytic complexes were 3ISB (Beard et al., 2009) and 2FMS (Batra et al., 2006), respectively. The difference in the position of the phosphate of the primer terminus nucleotide (Pp (n-1)) and that of the template nucleotide opposite the primer terminus (Pt (n-1)) was determined. In addition, primer distortion was assessed by analyzing the average B-factor for each active site nucleotide: template coding nucleotide, template nucleotide opposite the primer terminus, primer terminus and dUMPNPP (Figure 4). The relative average B-factors are calculated relative to the average B-factor for the polymerase of each structure.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (project numbers: 1ZIAES050158 and 1ZIAES050161, S.H.W; 1ZIAES102645, L.C.P.) and in association with the National Institutes of Health grant 1U19CA105010. Molecular graphics images were produced using the Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 GM-103311).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
The atomic coordinates and structure factors have been deposited in Protein Data Bank (http://www.pdb.org): 5J0O, 5J0P, 5J0Q, 5J0R, 5J0S, 5J0T, 5J0U, 5J0V, 5J0W, 5J0X, 5J0Y, 5J29, 5J2A, 5J2B, 5J2C, 5J2D, 5J2E 5J2F, 5J2G, 5J2H 5J2I, 5J2J, 5J2K.
AUTHOR CONTRIBUTIONS
Conceptualization, V.K.B., W.A.B. and S.H.W.; Methodology, V.K.B., W.A.B. and S.H.W.; Formal Analysis, V.K.B., W.A.B., L.C.P. and S.H.W.; Investigation, V.K.B.; Writing – Original Draft, V.K.B. and S.H.W.; Writing – Review & Editing, V.K.B., W.A.B., L.C.P. and S.H.W.; Supervision, L.C.P. and S.H.W.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Nucleotide-induced DNA polymerase active site motions accomodating a mutagenic DNA intermediate. Structure. 2005;13:1225–1233. doi: 10.1016/j.str.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA polymerase β with active site mismatches suggest a transient abasic site intermediate during misincorporation. Mol Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra VK, Perera L, Lin P, Shock DD, Beard WA, Pedersen LC, Pedersen LG, Wilson SH. Amino acid substitution in the active site of DNA polymerase β explains the energy barrier of the nucleotidyl transfer reaction. J Am Chem Soc. 2013;135:8078–8088. doi: 10.1021/ja403842j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra VK, Shock DD, Beard WA, McKenna CE, Wilson SH. Binary complex crystal structure of DNA polymerase β reveals multiple conformations of the templating 8-oxoguanine lesion. Proc Natl Acad Sci USA. 2012;109:113–118. doi: 10.1073/pnas.1112235108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard WA, Shock DD, Batra VK, Pedersen LC, Wilson SH. DNA polymerase β substrate specificity: Side chain modulation of the “A-rule”. J Biol Chem. 2009;284:31680–31689. doi: 10.1074/jbc.M109.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard WA, Shock DD, Wilson SH. Influence of DNA structure on DNA polymerase β active site function: Extension of mutagenic DNA intermediates. J Biol Chem. 2004;279:31921–31929. doi: 10.1074/jbc.M404016200. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Purification and domain-mapping of mammalian DNA polymerase β. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- Bebenek K, Pedersen LC, Kunkel TA. Replication infidelity via a mismatch with Watson–Crick geometry. Proc Natl Acad Sci USA. 2011;108:1862–1867. doi: 10.1073/pnas.1012825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T, Hunter WN, Kneale G, Kennard O. Molecular structure of the G•A base pair in DNA and its implications for the mechanism of transversion mutations. Proc Natl Acad Sci USA. 1986;83:2402–2406. doi: 10.1073/pnas.83.8.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal BD, Beard WA, Shock DD, Wilson SH. Observing a DNA polymerase choose right from wrong. Cell. 2013;154:157–168. doi: 10.1016/j.cell.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter WN, Brown T, Anand NN, Kennard O. Structure of an adenine-cytosine base pair in DNA and its implications for mismatch repair. Nature. 1986;320:552–555. doi: 10.1038/320552a0. [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kirby TW, DeRose EF, Cavanaugh NA, Beard WA, Shock DD, Mueller GA, Wilson SH, London RE. Metal-induced DNA translocation leads to DNA polymerase conformational activation. Nucleic Acids Res. 2012;40:2974–2983. doi: 10.1093/nar/gkr1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koag MC, Nam K, Lee S. The spontaneous replication error and the mismatch discrimination mechanisms of human DNA polymerase β. Nucleic Acids Res. 2014;42:11233–11245. doi: 10.1093/nar/gku789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Batra VK, Pedersen LC, Beard WA, Wilson SH, Pedersen LG. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase β. Proc Natl Acad Sci USA. 2008;105:5670–5674. doi: 10.1073/pnas.0801257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processsing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Picher AJ, Garciía-Diaz M, Bebenek K, Pedersen LC, Kunkel TA, Blanco L. Promiscuous mismatch extension by human DNA polymerase lambda. Nucleic Acids Res. 2006;34:3259–3266. doi: 10.1093/nar/gkl377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya MR, Prasad P, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- Trincao J, Johnson RE, Wolfle WT, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Dpo4 is hindered in extending a G•T mismatch by a reverse wobble. Nat Struct Mol Biol. 2004;11:457–462. doi: 10.1038/nsmb755. [DOI] [PubMed] [Google Scholar]
- Wang W, Hellinga HW, Beese LS. Structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis. Proc Natl Acad Sci USA. 2011;108:17644–17648. doi: 10.1073/pnas.1114496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD, Crick FHC. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- Xia S, Konigsberg WH. Mispairs with Watson-Crick base-pair geometry observed in ternary complexes of an RB69 DNA polymerase variant. Protein Sci. 2014;23:508–513. doi: 10.1002/pro.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]