

Abstract
Electronic structure calculations and nonadiabatic dynamics simulations (more than 2000 trajectories) are used to explore the Z–E photoisomerization mechanism and excited‐state decay dynamics of two arylazopyrazole photoswitches. Two chiral S1/S0 conical intersections with associated enantiomeric S1 relaxation paths that are barrierless and efficient (timescale of ca. 50 fs) were found. For the parent arylazopyrazole (Z8) both paths contribute evenly to the S1 excited‐state decay, whereas for the dimethyl derivative (Z11) each of the two chiral cis minima decays almost exclusively through one specific enantiomeric S1 relaxation path. To our knowledge, the Z11 arylazopyrazole is thus the first example for nearly stereospecific unidirectional excited‐state relaxation.
Keywords: arylazopyrazoles, conical intersections, nonadiabatic dynamics, photoisomerization, photoswitches
Photoswitchable compounds have many potential applications ranging from photopharmacology through optochemical genetics to data storage.1, 2 Azobenzenes are among the most studied photoswitchable compounds, since they are easily synthesized and highly stable.3, 4 Their high extinction coefficients and isomerization quantum yields enable repeated photoswitching with low‐intensity light. Photoinduced isomerization leads to a remarkable change of the shape and length of these azobenzenes, which has been exploited, for example, in tuning the folding and unfolding of peptides and proteins.5, 6, 7, 8
Many azobenzenes have been explored experimentally and computationally in the past decade,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22 but there remain a few drawbacks that limit their broad application. These include incomplete photoswitching because of overlapping absorbance of the two isomers and fast thermal rearrangement of the cis isomer (Z) back to the trans isomer (E). To improve overall performance, many groups have explored the properties of azobenzene variants by altering the substituents on the aromatic rings, for example, in bridged and ortho‐hydroxy azobenzenes.23, 24, 25, 26, 27, 28, 29, 30
Recently, novel classes of azoheterocycle photoswitches were reported,31, 32, 33, 34, 35, 36 especially arylazopyrazoles, which provide quantitative photoswitching and high thermal stability (ca. 1000 days).35, 36 The absorption band maxima of the E‐ and Z‐isomers of arylazopyrazoles are found to be well separated, which enables quantitative two‐way photoswitching.35 Furthermore, the substitution on the heterocyclic ring offers photophysical and photochemical properties that cannot be accessed with azobenzenes. However, the photoinduced isomerization mechanism and the dynamical behavior of these arylazopyrazoles are still unexplored at the atomistic level. Obvious questions that remain are: 1) Whether azobenzenes and arylazopyrazoles share similar photophysical and photochemical mechanisms and 2) whether they show analogous dynamical behavior (lifetimes, conical intersections, path selectivities). A detailed mechanistic understanding will be helpful to further improve their performance through rational design. Motivated by these considerations, we have combined static electronic structure calculations (TD‐DFT, CASSCF, and MS‐CASPT2) and OM2/MRCI nonadiabatic dynamics simulations to comprehensively explore the Z–E photoisomerization dynamics of arylazopyrazoles in the lowest 1nπ* excited singlet state (see Supporting Information for computational details).
We first studied the Z11 arylazopyrazole (R1=R2=CH3 in Figure 1). At the CASSCF level, we find two conformational enantiomers with different helicity for the cis S0 minimum (M‐Z11(S0) and P‐Z11(S0); see Figure 1), which obviously have the same energy (see the Supporting Information), as in the case of cis azobenzene.17 At the Franck–Condon point (for example, M‐Z11(S0)), the lowest gas‐phase vertical excitation energy is computed to be 60.3, 62.5, and 61.7 kcal mol−1 at the TD‐B3LYP, TD‐CAM‐B3LYP, and MS‐CASPT2 levels (6‐31G* basis), respectively, slightly above that of cis azobenzene (58.3 kcal mol−1, CASPT2).15 The corresponding values calculated in acetonitrile solution are 61.5, 63.8, and 62.7 kcal mol−1, close to the experimentally measured absorption maximum of 441 nm (64.8 kcal mol−1).35 The lowest excited singlet state of Z11 stems from an n→π* electronic excitation, which is not spectroscopically dark but carries some oscillator strength (0.05 at the TD‐B3LYP/6‐31G* level).
Figure 1.

(top) Arylazopyrazoles studied in this work (R1=R2=H and R1=R2=CH3). Upon irradiation, both Z–E and E–Z photoisomerizations can occur. (bottom) Enantiomeric ground‐state cis minimum (left) and S1/S0 conical intersection (right) of Z11 with M helicity optimized at the CASSCF level (N=blue, C=gray, and H=white). Also shown is the atomic numbering. See the Supporting Information for plots of the enantiomeric species P‐Z11(S0) and P‐CI(S1S0) with P helicity and for Cartesian coordinates and relative energies.
This nπ* state is primarily composed of the HOMO–LUMO single excitation (see the Supporting Information, Table S1). The HOMO represents an out‐of‐phase combination of the lone pairs of the two azo nitrogen atoms (n−), with some π contributions from both rings. The LUMO is an antibonding π*N=N orbital of the azo group, with π contributions mainly from the phenyl group. Owing to the π admixture from the rings in both orbitals, the n→π* electronic excitation gains some oscillator strength. In the nπ* state, we could not find any cis S1 minimum. Trial optimizations starting from the Franck–Condon points (M‐Z11(S0) and P‐Z11(S0)) always converged toward S1/S0 conical intersections. At the CASSCF level, we located two minimum‐energy conical intersection structures (M‐CI(S1S0) and P‐CI(S1S0); see Figure 1). They are enantiomers and thus have the same CASSCF energy, with single‐point MS‐CASPT2 energies of 29.4 (S0) and 35.8 (S1) kcal mol−1 (see Supporting Information). We also computed the linearly interpolated internal coordinate (LIIC) paths connecting the S0 minimum geometry (M‐Z11(S0)) and the S1/S0 conical intersection (M‐CI(S1S0)). As shown in the Supporting Information Figure S4, the S1 path from M‐Z11(S0) to M‐CI(S1S0) is barrierless, implying that the S1 decay should be efficient and ultrafast. The potential energy surfaces thus have the same topology as in the case of cis azobenzene;11, 17, 22 however, the dynamical roles of the two enantiomeric S1/S0 conical intersections in the 1nπ* excited‐state decay are distinct in azobenzenes and arylazopyrazoles (see below).
To further explore the S1 excited‐state properties of the Z11 arylazopyrazole (such as lifetimes and decay channels), we performed 700 OM2/MRCI nonadiabatic dynamics runs for each cis S0 conformer (M‐Z11(S0) and P‐Z11(S0)) and analyzed the successful respective 688 and 685 trajectories (some failed due to convergence issues). During the 300 fs simulation time, 91 % and 90 % of the trajectories hop to the S0 state from the initially populated S1(1nπ*) state of M‐Z11(S0) and P‐Z11(S0), respectively (Table 1). Counting the number of trajectories that arrive at the cis and trans ground‐state conformers, we estimate the quantum yield of the Z→E photoisomerization to be 0.52 and 0.51 for M‐Z11(S0) and P‐Z11(S0), respectively, which is consistent with the experimentally measured value of 0.56±0.04 for the Z11 arylazopyrazole in acetonitrile.35
Table 1.
Numbers of started trajectories, successful trajectories, hops, arrivals at Z and E ground‐state isomers, and quantum yield of Z–E photoisomerization from the OM2/MRCI dynamics simulations.
| from M‐Z11(S0) | from P‐Z11(S0) | |
|---|---|---|
| Started trajectories | 700 | 695 |
| Successful trajectories | 688 | 685 |
| Hops | 623 | 616 |
| Z ground‐state isomers | 296 | 305 |
| E ground‐state isomers | 327 | 311 |
| Quantum yield (φ Z‐E) | 0.52 | 0.51 |
We have analyzed the distribution of the C2N3N4C5 dihedral angle at all S1→S0 hopping points (see top panel of Figure 2). It is clear that each conformer adopts a stereospecific excited‐state decay channel. For M‐Z11(S0), 97 % of the trajectories hop to the S0 state in the vicinity of the M‐CI(S1S0) conical intersection (only ca. 3 % at P‐CI(S1S0)); whereas, for P‐Z11(S0), 97 % of the trajectories jump to the S0 state near the P‐CI(S1S0) conical intersection. This S1 excited‐state behavior is quite different from the situation in cis azobenzene, which also has two enantiomeric S1 excited‐state decay routes; however, its excited‐state decay does not exhibit stereospecific excited‐state motion. In our previous OM2/MRCI dynamics simulations of cis azobenzene, the two enantiomeric excited‐state decay channels for each S0 conformer were both found to play a significant role (ca. 77 % vs. 22 %).17 To our best knowledge, the Z11 arylazopyrazole is thus the first example of nearly stereospecific excited‐state decay. The bottom panel of Figure 2 illustrates the distribution of the S1→S0 hopping times from either M‐Z11(S0) (left) or P‐Z11(S0) (right). In the trajectories starting from M‐Z11(S0), the hops to the S0 state through M‐CI(S1S0) are overwhelmingly dominant (blue in the left panel of Figure 2); in those starting from P‐Z11(S0), the hops through P‐CI(S1S0) dominate analogously (red in the right panel of Figure 2). In both cases, the distribution of the hopping times shows an oscillatory pattern (bottom panel of Figure 2), which indicates that the approach to the conical intersection seam is modulated by a periodic structural change. The average S1→S0 hopping time is estimated to be around 50 fs. A periodic excited‐state decay has also been seen in cis azobenzene and a retinal chromophore model17, 37 but the stereospecific excited state decay is unique to Z11.
Figure 2.
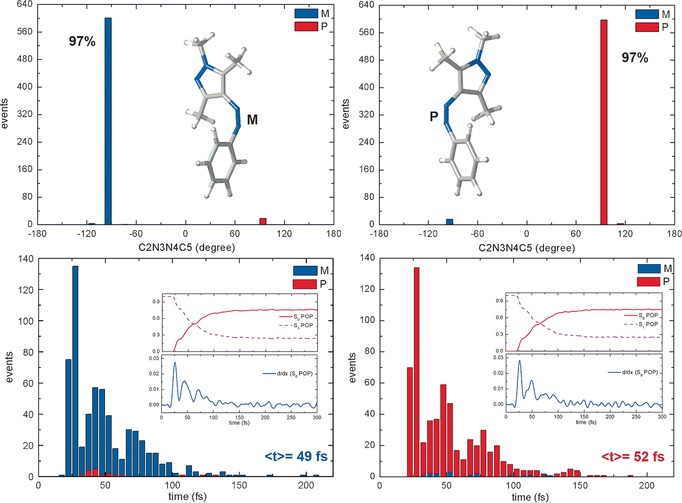
Distribution of (top) the C2N3N4C5 dihedral angle and (bottom) the hopping times at all S1→S0 hopping points, with time‐dependent state populations and their time derivatives (insets). Left panels, trajectories starting from M‐Z11(S0); right panels, trajectories starting from P‐Z11(S0). Hops through M‐CI(S1S0) in blue, hops through P‐CI(S1S0) in red. See text for discussion.
The left panel of Figure 3 shows the evolution of the distribution of the C1C2N3N4 and C2N3N4C5 dihedral angles from the initial points to the S1→S0 hopping points, for trajectories starting from M‐Z11(S0) (see Supporting Information for those from P‐Z11(S0)). In the beginning, all trajectories are in the Franck–Condon region, and hence, the C2N3N4C5 dihedral angle is around 0°. In the M‐CI(S1S0) and P‐CI(S1S0) hopping regions, the C2N3N4C5 dihedral angle has reached ca. −90° and 90°, respectively, whereas the C1C2N3N4 and N3N4C5C6 dihedral angles have merely changed by about 30° (for 97 % of the trajectories). Hence, the main reaction coordinate for the Z–E photoisomerization of the Z11 arylazopyrazole is the rotational motion of the central N=N group. The rotations of the nearby phenyl and pyrazole rings only play a minor role, as also found in previous dynamics simulations for azobenzenes.16, 17, 20, 22
Figure 3.

Distribution of the C1C2N3N4 and C2N3N4C5 dihedral angles at the initial points (blue squares) and hopping points (red circles) of (left) Z11 and (right) Z8. Also shown are the percentages of hops through different S1/S0 conical intersection regions. See the Supporting Information for the distributions of other dihedral angles.
For comparison, we performed electronic structure calculations and dynamics simulations for the Z8 arylazopyrazole (Figure 1; R1=R2=H). This compound has only one stable ground‐state minimum, in which the pyrazole and phenyl rings are perpendicular to each other (see the Supporting Information, Figure S1). However, it still has two enantiomeric S1/S0 conical intersections (M‐CI(S1S0) and P‐CI(S1S0); see the Supporting Information, Figure S1). Their potential energies relative to the S0 minimum are the same at the CASSCF level, with single‐point MS‐CASPT2 energies of 32.6 (S0) and 39.9 (S1) kcal mol−1, that is, about 3–4 kcal mol−1 higher than in the case of Z11 (see above). The S1 relaxation processes from the Franck–Condon point to these two enantiomeric S1/S0 conical intersections are again barrierless, as in Z11. In spite of this similarity, the excited‐state dynamics is qualitatively different in Z8 and Z11. While the S1 decay path is enantiomer‐specific in Z11 (with each conformer following almost exclusively one of the clockwise and counterclockwise isomerization decay channels, 97 % versus 3 % in the left panel of Figure 3), both clockwise and counterclockwise S1 decay paths play an evenly important role in Z8. The C2N3N4C5 dihedral angle distribution at the S1→S0 hopping points of Z8 (Figure 3) shows that the hops take place near both S1/S0 conical intersections in similar number (48 % vs. 52 %). This feature is also reflected in the time distribution of the S1→S0 hops (Supporting Information, Figure S6). These findings are not surprising since, after vertical excitation, the S1 dynamics starts from geometries with a phenyl ring that is perpendicular to the azopyrazole moiety (see above) and can thus twist in either direction with essentially equal probability.
The enantiomer‐specific relaxation selectivity of Z11 can be understood by considering the relevant LIIC paths. As shown in Figure 4, the paths from M‐Z11(S0) to M‐CI(S1S0) are barrierless, proceeding steeply downhill both for Z11 and cis azobenzene (solid lines), and are thus the dominant S1 relaxation channels. By contrast, the paths from M‐Z11(S0) to P‐CI(S1S0) are less steep (dashed lines) and even show small barriers of 0.03 kcal mol−1 for cis azobenzene and 0.51 kcal mol−1 for Z11. Although these barriers are very low, they appear to be dynamically important in increasing the path selectivity of the ultrafast S1 relaxation from 77 %:22 % in cis azobenzene to 97 %:3 % in Z11. For comparison, both pathways are equivalent in Z8, which thus lacks any path selectivity (see the Supporting Information, Figure S5).
Figure 4.

OM2/MRCI‐computed linearly interpolated internal coordinate (LIIC) paths (M‐M and M‐P) connecting the Franck–Condon point (M‐Z11(S0)) and the two S1/S0 conical intersections (M‐CI(S1S0) and P‐CI(S1S0)). Total OM2/MRCI energies are given for Z11 (left) and azobenzene (right). The LIIC paths are expected to approximate the intrinsic reaction coordinate (IRC) paths.
To summarize, we have combined static electronic structure calculations and nonadiabatic dynamic simulations to explore the Z–E photoisomerization mechanism of two novel azo photoswitches, namely, arylazopyrazoles.35 We find that arylazopyrazoles and azobenzenes share similar vertical excitation energies, lifetimes, and conical intersections, whereas their excited‐state decay dynamics are qualitatively different. For the Z11 arylazopyrazole, each conformer almost exclusively adopts a stereospecific excited‐state decay pathway for the Z–E photoisomerization (97 % vs. 3 %), while the Z8 arylazopyrazole utilizes both available excited‐state decay channels nearly evenly (48 % vs. 52 %). This is different from the situation in cis azobenzene17 and in a recently studied model for a chiral rotary motor,38 in which the two enantiomeric decay routes play major and minor roles (77 % vs. 22 % and 76 % vs. 24 %, respectively). The distinct excited‐state relaxation behavior of these photoswitches can be attributed to subtle differences in the underlying S1 potential energy surfaces. Full‐dimensional nonadiabatic dynamics simulations are capable of capturing such subtle effects and thus provide detailed insight into the factors that govern excited‐state dynamics.
Finally, we reiterate that enantiomeric excited‐state decay pathways have already been observed in the photodynamics of cis‐azobenzene17 and a rotary motor model.38 However, in these cases, they both play a significant role in the dynamics, whereas to our knowledge the present work on the Z11 arylazopyrazole is the first example of stereospecific unidirectional excited‐state relaxation.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work has been supported by grants NSFC 21522302 (G.C.) and NSFC 21520102005 (G.C. and W.F.) and by an ERC Advanced grant (OMSQC, W.T.).
Y.-T. Wang, X.-Y. Liu, G. Cui, W.-H. Fang, W. Thiel, Angew. Chem. Int. Ed. 2016, 55, 14009.
Contributor Information
Prof. Dr. Ganglong Cui, Email: ganglong.cui@bnu.edu.cn
Prof. Dr. Walter Thiel, Email: thiel@mpg.kofo.de
References
- 1. Szymański W., Beierle J. M., Kistemaker H. A. V., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178. [DOI] [PubMed] [Google Scholar]
- 2. Velema W. A., Szymanski W., Feringa B. L., J. Am. Chem. Soc. 2014, 136, 2178–2191. [DOI] [PubMed] [Google Scholar]
- 3. Beharry A. A., Woolley G. A., Chem. Soc. Rev. 2011, 40, 4422–4437. [DOI] [PubMed] [Google Scholar]
- 4. Bandara H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825. [DOI] [PubMed] [Google Scholar]
- 5. Woolley G. A., Acc. Chem. Res. 2005, 38, 486–493. [DOI] [PubMed] [Google Scholar]
- 6. Zhang F., Sadovski O., Xin S. J., Woolley G. A., J. Am. Chem. Soc. 2007, 129, 14154–14155. [DOI] [PubMed] [Google Scholar]
- 7. Zhang F., Zarrine-Afsar A., Al-Abdul-Wahid M. S., Prosser R. S., Davidson A. R., Woolley G. A., J. Am. Chem. Soc. 2009, 131, 2283–2289. [DOI] [PubMed] [Google Scholar]
- 8. Zhang F., Timm K. A., Arndt K. M., Woolley G. A., Angew. Chem. Int. Ed. 2010, 49, 3943–3946; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4035–4038. [Google Scholar]
- 9. Rau H., Lueddecke E., J. Am. Chem. Soc. 1982, 104, 1616–1620. [Google Scholar]
- 10. Schultz T., Quenneville J., Levine B., Toniolo A., Martínez T. J., Lochbrunner S., Schmitt M., Shaffer J. P., Zgierski M. Z., Stolow A., J. Am. Chem. Soc. 2003, 125, 8098–8099. [DOI] [PubMed] [Google Scholar]
- 11. Cembran A., Bernardi F., Garavelli M., Gagliardi L., Orlandi G., J. Am. Chem. Soc. 2004, 126, 3234–3243. [DOI] [PubMed] [Google Scholar]
- 12. Ciminelli C., Granucci G., Persico M., Chem. Eur. J. 2004, 10, 2327–2341. [DOI] [PubMed] [Google Scholar]
- 13. Pancur T., Renth F., Temps F., Harbaum B., Krüger A., Herges R., Näther C., Phys. Chem. Chem. Phys. 2005, 7, 1985–1989. [DOI] [PubMed] [Google Scholar]
- 14. Toniolo A., Ciminelli C., Persico M., Martinez T. J., J. Chem. Phys. 2005, 123, 234308. [DOI] [PubMed] [Google Scholar]
- 15. Conti I., Garavelli M., Orlandi G., J. Am. Chem. Soc. 2008, 130, 5216–5230. [DOI] [PubMed] [Google Scholar]
- 16. Böckmann M., Doltsinis N., Marx D., J. Phys. Chem. A 2009, 114, 745–754. [DOI] [PubMed] [Google Scholar]
- 17. Weingart O., Lan Z., Koslowski A., Thiel W., J. Phys. Chem. Lett. 2011, 2, 1506–1509. [Google Scholar]
- 18. Pederzoli M., Pittner J., Barbatti M., Lischka H., J. Phys. Chem. A 2011, 115, 11136–11143. [DOI] [PubMed] [Google Scholar]
- 19. Cusati T., Granucci G., Persico M., J. Am. Chem. Soc. 2011, 133, 5109–5123. [DOI] [PubMed] [Google Scholar]
- 20. Gámez J., Weingart O., Koslowski A., Thiel W., J. Chem. Theory Comput. 2012, 8, 2352–2358. [DOI] [PubMed] [Google Scholar]
- 21. Böckmann M., Braun S., Doltsinis N., Marx D., J. Chem. Phys. 2013, 139, 084108. [DOI] [PubMed] [Google Scholar]
- 22. Xia S.-H., Cui G. L., Fang W.-H., Thiel W., Angew. Chem. Int. Ed. 2016, 55, 2067–2072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2107–2112. [Google Scholar]
- 23. Ron S., Hendrikje N., Bengt B. S., Rainer H., Christian N., Falk R., Friedrich T., J. Am. Chem. Soc. 2009, 131, 15594–15595.19827776 [Google Scholar]
- 24. Haberhauer G., Kallweit C., Angew. Chem. Int. Ed. 2010, 49, 2418–2421; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2468–2471. [Google Scholar]
- 25. Böckmann M., Doltsinis N. L., Marx D., Angew. Chem. Int. Ed. 2010, 49, 3382–3384; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3454–3456. [Google Scholar]
- 26. Liu L.-H., Yuan S., Fang W.-H., Zhang Y., J. Phys. Chem. A 2011, 115, 10027–10034. [DOI] [PubMed] [Google Scholar]
- 27. Samanta S., Qin C. G., Lough A. J., Woolley G. A., Angew. Chem. Int. Ed. 2012, 51, 6452–6455; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6558–6561. [Google Scholar]
- 28. Carstensen N. O., Phys. Chem. Chem. Phys. 2013, 15, 15017–15026. [DOI] [PubMed] [Google Scholar]
- 29. Cui G. L., Guan P. J., Fang W.-H., J. Phys. Chem. A 2014, 118, 4732–4739. [DOI] [PubMed] [Google Scholar]
- 30. Guan P. J., Cui G. L., Fang Q., ChemPhysChem 2015, 16, 805–811. [DOI] [PubMed] [Google Scholar]
- 31. Otsuki J., Suwa K., Narutaki K., Sinha C., Yoshikawa I., Araki K., J. Phys. Chem. A 2005, 109, 8064–8069. [DOI] [PubMed] [Google Scholar]
- 32. Otsuki J., Suwa K., Sarker K. K., Sinha C., J. Phys. Chem. A 2007, 111, 1403–1409. [DOI] [PubMed] [Google Scholar]
- 33. Wendler T., Schütt C., Näther C., Herges R., J. Org. Chem. 2012, 77, 3284–3287. [DOI] [PubMed] [Google Scholar]
- 34. Garcia-Amorós J., Castro M. C. R., Coelho P., Raposo M. M. M., Velasco D., Chem. Commun. 2013, 49, 11427–11429. [DOI] [PubMed] [Google Scholar]
- 35. Weston C. E., Richardson R. D., Haycock P. R., White A. J. P., Fuchter M. J., J. Am. Chem. Soc. 2014, 136, 11878–11881. [DOI] [PubMed] [Google Scholar]
- 36. Stricker L., Fritz E.-C., Peterlechner M., Doltsinis N. L., Ravoo B. J., J. Am. Chem. Soc. 2016, 138, 4547–4554. [DOI] [PubMed] [Google Scholar]
- 37. Manathunga M., Yang X., Luk H. L., Gozem S., Frutos L. M., Valentini A., Ferrè N., Olivucci M., J. Chem. Theory Comput. 2016, 12, 839–850. [DOI] [PubMed] [Google Scholar]
- 38. Marchand G., Eng J., Schapiro I., Valentini A., Frutos L. M., Pieri E., Olivucci M., Léonard J., Gindensperger E., J. Phys. Chem. Lett. 2015, 6, 599–604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary