

Abstract
Background
The long‐term risk of stroke increases with age, and stroke is a common cause of disability in the community. Spasticity is considered a significantly disabling impairment that develops in people who have had a stroke. The burden of care is higher in stroke survivors who have spasticity when compared with stroke survivors without spasticity with regard to treatment costs, quality of life, and caregiver burden.
Objectives
To assess if pharmacological interventions for spasticity are more effective than no intervention, normal practice, or control at improving function following stroke.
Search methods
We searched the Cochrane Stroke Group Trials Register (May 2016), the Cochrane Central Register of Controlled Trials (CENTRAL, 2016, Issue 5), MEDLINE (1946 to May 2016), Embase (2008 to May 2016), CINAHL (1982 to May 2016), AMED (1985 to May 2016), and eight further databases and trial registers. In an effort to identify further studies, we undertook handsearches of reference lists and contacted study authors and commercial companies.
Selection criteria
We included randomised controlled trials (RCTs) that compared any systemically acting or locally acting drug versus placebo, control, or comparative drug with the aim of treating spasticity.
Data collection and analysis
Two review authors independently assessed the studies for inclusion and extracted the data. We assessed the included studies for both quality and risk of bias. We contacted study authors to request further information when necessary.
Main results
We included seven RCTs with a total 403 participants. We found a high risk of bias in all but one RCT. Two of the seven RCTs assessed a systemic drug versus placebo. We pooled data on an indirect measure of spasticity (160 participants) from these two studies but found no significant effect (odds ratio (OR) 1.66, 95% confidence interval (CI) 0.21 to 13.07; I2 = 85%). We identified a significant risk of adverse events per participant occurring in the treatment group versus placebo group (risk ratio (RR) 1.65, 95% CI 1.12 to 2.42; 160 participants; I2 = 0%). Only one of these studies used a functional outcome measure, and we found no significant difference between groups.
Of the other five studies, two assessed a systemic drug versus another systemic drug, one assessed a systemic drug versus local drug, and the final two assessed a local drug versus another local drug.
Authors' conclusions
The lack of high‐quality RCTs limited our ability to make specific conclusions. Evidence is insufficient to determine if systemic antispasmodics are effective at improving function following stroke.
Plain language summary
Drugs (with the exception of botulinum toxin) to treat spasticity after stroke
Question
Are drugs (with the exception of botulinum toxin) better than a placebo or control in treating spasticity in people with stroke?
Background
We wanted to learn whether there was evidence for using drugs for spasticity in people with stroke, and if so, to identify whether drugs that acted on the whole body (systemic drugs) differed in their effects to drugs that acted in a localised area of the body (local drugs).
Study characteristics
We included seven studies involving a total of 403 participants. The evidence is current to May 2016. There were variations among the included studies. Two of the studies were placebo controlled, while the rest compared one drug to another drug. Two studies compared two systemic drugs against each other; two studies compared two local drugs against each other; and the fifth study compared a systemic drug against a local drug.
Key results
The results for the studies varied, meaning that we could make no clear conclusions. The two studies that used a placebo as a control provided conflicting results; when the results were combined we identified a slight benefit in favour of the treatment group. These two studies provided clear evidence that taking the treatment drug was likely to result in an increased risk of having an adverse event. This review identified a lack of studies and subsequent evidence relating to the use of pharmacological interventions (with the exception of botulinum toxin) to treat spasticity. Future research to identify the best time to start treatment and the optimal dose for treatment is recommended.
Quality of the evidence
The quality of the evidence varied. We judged all but one of the studies to be at high risk of bias in at least one of the six areas considered. The sample sizes in the included studies were relatively low for drug trials; three of the seven studies had 30 or fewer participants.
Summary of findings
Summary of findings for the main comparison. Systemic antispasticity drugs compared with placebo for spasticity after stroke.
| Systemic antispasticity drugs compared with placebo for spasticity after stroke | ||||||
| Patient or population: people with spasticity following stroke Settings: community Intervention: systemic antispasticity drugs Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Systemic antispasticity drugs | |||||
| Indirect measure of spasticity Modified Ashworth Scale Follow‐up: 4 or 6 weeks | Study population | OR 1.66 (0.12 to 15.38) | 160 (2 studies) | ⊕⊝⊝⊝ very low1,2,3,4 | ‐ | |
| 49 per 100 | 57 per 100 (10 to 94) | |||||
| Low | ||||||
| ‐ | ‐ | |||||
| Adverse events Adverse events per participant Follow‐up: 4 or 6 weeks | Study population | RR 1.65 (1.12 to 2.42) | 160 (2 studies) | ⊕⊝⊝⊝ very low1,3 | ‐ | |
| 40 per 100 | 65 per 100 (44 to 96) | |||||
| Low | ||||||
| ‐ | ‐ | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Unclear how random sequence generation was carried out in either study. 2Results very heterogenous (I2 = 90%). 3The studies did not investigate the same drug against a placebo. 4Both studies appeared to selectively report results. The corresponding author of the Stamenova 2005 study was an employee of the company that manufactured the drug.
Background
The long‐term risk of stroke increases with age. The risk of stroke over an expected lifetime has been calculated at between 1 in 5 for females and 1 in 6 for males in a group of middle‐aged adults (Seshadri 2006). A stroke will have a greater impact on a person than any other chronic disease (Adamson 2004). Stroke causes a range of disabilities, one of which is spasticity. The burden of care is higher in stroke patients who have spasticity when compared with stroke patients without spasticity with regard to treatment costs, quality of life, caregiver burden, and the effects of comorbidities (Esquenazi 2011).
Description of the condition
Spasticity occurs as a result of damage to the descending tracts of the upper motor neuron system. This generally involves damage to the corticoreticular pathways (para‐pyramidal pathways), tracts originating at the brain stem or reticular formation (e.g. the reticulospinal and vestibulospinal tracts) (Burke 1988). Such damage to the upper motor neuron system results in a loss of descending control that in turn manifests as abnormal muscle activity to an externally imposed stimuli.
Spasticity is poorly defined in the literature (Malhotra 2009). For the purposes of this review, in order to ensure that no data were missed, we defined spasticity as "disordered sensory‐motor control, resulting from an upper motor neuron lesion, presenting as intermittent or sustained involuntary activation of muscles" (Pandyan 2005).
Spasticity is common, with prevalence ranging from 30% to 80% of stroke survivors (Malhotra 2008; Watkins 2002). Spasticity that is not treated effectively can result in increased pain and stiffness, decreased range of movement, and altered posturing (Ada 2006; Malhotra 2008). It may also interfere with functional recovery (Sorinola 2009). However, the complexity of differentiating between neural and intrinsic stiffness makes the correlation between function and spasticity difficult to identify (Mirbagheri 2011). In spite of this, many studies have used a functional measure to primarily assess the efficacy of the intervention. As the aim of rehabilitation is to facilitate and optimise the recovery of function, this review focused primarily on whether current treatments of spasticity lead to improvements in function, despite there being a lack of evidence of a causal relationship between spasticity and function.
Description of the intervention
The treatment of spasticity can be complex and relies on a multidisciplinary approach (Barnes 1998). Treatment of spasticity has traditionally taken a stepped approach whereby conservative procedures are used first and progressively more invasive procedures are then employed (Bogey 2004; Rekand 2010). Physiotherapeutic approaches are deemed the most conservative and are therefore used initially. This is reinforced by consensus guidelines (Royal College of Physicians 2009; Sheean 2010). However, physiotherapeutic interventions do not have a direct effect on abnormal muscle activity (there are some unproven claims that there may be an indirect effect) resulting from spasticity; this can only be achieved by pharmacological interventions.
Pharmacological interventions for stroke spasticity are generally termed antispasmodics and can be divided into two groups: those that act systemically and those that act locally (Sheean 2006), with the locally acting treatments tending to be more invasive. The stepped approach advocates that systemically acting drugs be used initially (Gormley 1997). Such drugs include baclofen, tizanidine, or dantrolene. If these are not successful in alleviating the problem, then locally acting drugs are employed (Bogey 2004), such as injections of botulinum toxin to the muscles or alcohol or phenol to the peripheral nerves (Kocabas 2010). Should none of these treatments be effective, then surgery is the final treatment option. However, this is rarely used in the stroke population.
The use of physical therapeutic methods to manage spasticity is currently being systematically reviewed (Monaghan 2011), as is the use of botulinum toxin, which is the most widely used locally acting pharmacological therapy (Lyons 2007). However, the use of systemically acting drugs and other locally acting drugs has not been systematically reviewed. This review therefore focused on these systemically and locally acting drugs, excluding those technologies already under review (i.e. botulinum toxin and physiotherapeutic interventions).
How the intervention might work
Drugs act either systemically or peripherally. Systemic drugs act in a number of different ways but generally aim to inhibit neurotransmitter activity at one or more sites within the central nervous system. Sites of action include both pre‐ and post‐synaptic terminals of the spinal interneurons (at varying levels of the upper motor neuron pathway), alpha motor neurons, and primary sensory afferent neurons. This inhibitory effect acts on the neurotransmitters throughout the central nervous system and can result in drowsiness amongst other side effects (Gracies 1997). Administration tends to be oral, so doses need to be high for the active agent in the drug to cross the blood‐brain barrier. In order to lessen these negative effects it is possible to introduce some drugs directly into the cerebrospinal fluid via an intrathecal pump. One particular group of drugs inhibits force production at the level of the muscle; however, this effect can also be systemic.
Other drugs are given peripherally via injection directly to the nerve. This is a far more invasive procedure; however, the systemic side effects are fewer. The aim of such injections is to destroy conduction in the nerves or the release of transmitters at the motor neuron junction or to decrease muscle contraction.
We have provided an outline of the most regularly used drugs. While not systemically acting, we have also included alcohol and phenol in Table 2.
1. Pharmacological agents for treating spasticity after stroke.
| Generic name | Method of administration | Method of action |
| Baclofen | Oral, intrathecal pump | Baclofen is a centrally acting gamma‐aminobutyric acid (GABA) analogue that limits the release of excitatory neurotransmitters in the spinal cord (Mukherjee 2010). It binds to GABA receptors at the presynaptic terminal and inhibits the influx of calcium into the presynaptic terminal, decreasing both mono‐ and polysynaptic reflexes (Davidoff 1985; Krach 2001). It also binds on the postsynaptic terminal of the 1a sensory afferent (Howe 1987) |
| Tizanidine | Oral | An imidazole derivative with agonist action on alpha‐2‐adrenergic receptors in the central nervous system (Mukherjee 2010). It has an effect both pre‐ and postsynaptically and decreases the level of excitatory neurotransmitter between the spinal interneurons to the alpha motor neurons through presynaptic inhibition (Gallichio 2004). It may also inhibit abnormal activity by acting on locus coeruleus and inhibiting activity in the coerulospinal pathway (Palmeri 1990) |
| Dantrolene | Oral | This drug acts at the muscle level rather than the neural level and affects both intra‐ and extrafusal muscles (Gallichio 2004). It interferes with the release of calcium from the sarcoplasmic reticulum of the muscle (Mukherjee 2010). This decrease in available calcium reduces the force produced during a contraction (Gallichio 2004) |
| Tolperisone | Oral | Centrally acting muscle relaxant that acts at the spinal cord level. Primarily acts presynaptically to inhibit both calcium and sodium channels. Decreases reflex activity by inhibiting release from the primary afferent (Vora 2010) |
| Alcohol | Injection | The effect of alcohol varies depending on the concentration. At concentrations below 35%, it acts as a local anaesthetic. In concentrations between 35% to 50% small‐fibre demyelination was observed, but in concentrations above this level, Wallerian degeneration and fibrosis are observed (Kocabas 2010) |
| Phenol | Injection, intrathecal | The effect of phenol also varies depending on concentrations used. It can be injected either into the muscle or directly into the nerve. At concentrations greater than 3%, protein denaturation and axonal degeneration destroy the neurons. 5% phenol injection to the motor points of the muscle lead to demyelination of the neuron (Kocabas 2010). |
Why it is important to do this review
Using antispasmodics that have systemic effects appears to remain pervasive in the clinical setting. There is evidence that global antispasmodics tend to depress the nervous system (Gracies 1997; Simpson 2009). This in turn can be detrimental to motor skill acquisition (Sheean 2009; Willerslev‐Olsen 2011). As these pharmacological therapies are started during rehabilitation, where the main objective is to relearn motor skills, such drugs may not be appropriate. There was therefore a need to review the level of evidence on which these drugs are used.
Phenol and alcohol are locally acting drug treatments, but their effectiveness has not been systematically reviewed with regard to poststroke spasticity, and so we have included these in our review.
The management of spasticity poststroke using physical therapeutic means is being systematically reviewed, having been accepted as a protocol (Monaghan 2011), and a systematic review of botulinum toxin poststroke is currently being reviewed by editors (Lyons 2007); we therefore have not included these in this review.
It is unclear whether one drug is more effective than another in treating both spasticity and the prevention of secondary consequences, and whether any effects translate into functional improvements.
It is also important to assess what common adverse events are identified with the use of such drugs.
Objectives
To assess if pharmacological interventions for spasticity are more effective than no intervention, normal practice, or control at improving function following stroke.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials in this review. Studies had to have investigated any pharmacological therapy; this was not limited to the drugs documented in Table 2. Systemically acting drugs tend to have a long half‐life and will therefore carry a high risk of carry‐over effect. We planned to include studies with a cross‐over design if the results of the first period of the data (i.e. before cross‐over) were available. We contacted the study authors to request this information if it was not included in the published article. Some studies combined complex combinations of pharmacological interventions with physical therapeutic interventions where there was no clear placebo or normal treatment group. We identified in the protocol that we would not include such studies, as it would be difficult to ascertain what part of the treatment provided the main effect.
Types of participants
We included participants who have had a stroke resulting in spasticity. There is evidence that spasticity is poorly defined and measured in the literature (Malhotra 2009). Excluding papers using invalid outcome measures was likely to result in very few papers left to review. We therefore included all trials that explicitly stated that the aim was to treat spasticity (it was possible to ascertain that this was the case by reading the appropriately selected papers). A variety of methods are available to measure aspects of spasticity, some that are direct (neurophysiological methods such as Hoffmann reflex (H‐reflex) and electromyography) and others that are not (biomechanical measures such as stiffness or clinical scales such as the Ashworth Scale or Tone Assessment Scale). Most of the commonly used measures are indirect and confounded (Fleuren 2010). We excluded studies where no outcome measure of spasticity had been used.
We included participants irrespective of gender and older than 18 years of age. Some studies included participants with a variety of diagnoses (e.g. traumatic brain injuries or multiple sclerosis as well as stroke). In such cases we contacted the study authors to request stroke‐specific data. If study authors were not able to provide stroke‐specific data, we still included the studies if the proportion of participants with stroke was greater than 80%. We did not include studies where the proportion of participants with stroke was less than 80%.
Types of interventions
We included any pharmacological intervention that aimed to reduce spasticity regardless of dose or mode of delivery.
We included any pharmacological intervention that was compared with a placebo, normal practice, or no intervention. We also planned to include trials that compared two forms of antispasticity intervention, as long as at least one intervention was pharmacological; however, we did not identify any such trials.
Types of outcome measures
There are wide variations in the types of outcome measures used to assess the efficacy of antispasticity treatments. Many studies use a variety of scales that attempt to assess disability at the level of impairment, activity, and participation. As elucidated already, the main aim of rehabilitation is to achieve functionally relevant activity that transfers into usual activities of daily living. Such scales tend to be general and not dependent on specific areas of the body. However, other activity scales are more localised. We decided that such specific outcome measures should be divided into those assessing upper limb and those assessing the lower limb.
Primary outcomes
Functional ability during activities of daily living.
Secondary outcomes
We identified functional movement scales that measure activity at the upper limb separately to those that measure activity at the lower limb with a view to analysing them separately; however, this was not possible due to the methods used in the included studies.
Secondary consequences of spasticity, assessed by specific objective measures such as pain and range of movement.
Spasticity, either measured through neurophysiological methods, such as electromyographic (EMG) activity or H‐reflex activity, or using indirect measures of stiffness such as the Modified Ashworth Scale or Tone Assessment Scale. While the Modified Ashworth Scale and Tone Assessment Scale are not necessarily measures of spasticity, such measures are commonly used so we also included them.
Adverse events.
Search methods for identification of studies
See the 'Specialized register' section in the Cochrane Stroke Group module. We searched for trials in any language and attempted to arrange translation of relevant papers published in languages other than English.
Electronic searches
We searched the Cochrane Stroke Group Trials Register (last searched 19 May 2016) and the following electronic bibliographic databases and trials registers:
The Cochrane Central Register of Controlled Trials (CENTRAL 2016, Issue 5) (Appendix 1);
MEDLINE (Ovid) (1946 to 17 May 2016) (Appendix 2);
Embase (Ovid) (2008 to 17 May 2016) (Appendix 3);
CINAHL (EBSCO) (1982 to 17 May 2016) (Appendix 4);
AMED (Ovid) (1985 to 17 May 2016) (Appendix 5);
Physiotherapy Evidence Database (PEDro) (www.pedro.org.au/) (13 May 2016);
REHABDATA (www.naric.com/?q=en/REHABDATA) (13 May 2016);
Center for International Rehabilitation Research Information and Exchange (CIRRIE) (cirrie.buffalo.edu/) (13 May 2016);
ClinicalTrials.gov (www.clinicaltrials.gov/) (13 May 2016);
EU Clinical Trials Register (www.clinicaltrialsregister.eu) (13 May 2016);
Stroke Trials Registry (www.strokecenter.org/trials/) (13 May 2016);
ISRCTN Registry (www.isrctn.com/) (previously Current Controlled Trials: www.controlled‐trials.com) (13 May 2016);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch/) (13 May 2016).
We developed the MEDLINE search strategy with the help of the Cochrane Stroke Group Information Specialist and adapted it for the other databases.
Using a comprehensive search strategy, the Cochrane Stroke Group Information Specialist has already completed a retrospective search of Embase for all stroke trials to January 2008 and added all relevant trials to the Cochrane Stroke Group Trials Register. In order to avoid duplication of efforts, we have limited the search of this database from January 2008 onwards.
Searching other resources
In order to identify further published, unpublished, and ongoing trials, we:
searched the reference lists of published reviews and trials identified by the above methods;
contacted the following pharmaceutical companies: Novartis (baclofen) April 2014; Strathmann (tolperisone) April 2014; Sanochemia (tolperisone) April 2014; Acorda (tizanidine) April 2014;
contacted authors and researchers in the field;
used Science Citation Index Cited Reference Search for forward tracking of important articles.
Data collection and analysis
Two review authors (CL and AK) independently screened the titles and abstracts of all the records obtained from the electronic searches and excluded obviously irrelevant studies. We then obtained the full text of the remaining studies, and the same two review authors independently selected studies that met the review inclusion criteria (types of studies, types of participants, aims of interventions, and outcome measures).
Selection of studies
Two review authors (CL and AK) independently considered all intervention studies and identified those that were randomised controlled trials. If there was disagreement between review authors following the independent review process, we reached consensus through discussion.
As recommended in the PRISMA statement (Moher 2009), we have included a flow diagram outlining the phases for selection of studies included in the review using the PRISMA template available in Review Manager 5 (RevMan 2014).
Data extraction and management
Having identified all randomised controlled trials, two review authors (CL and CS) independently assessed each study and extracted the relevant details using a data extraction form (Appendix 6) based on a modified version of the van Tulder pro‐forma previously advocated for assessment of risk of bias by the Cochrane Back Review Group (van Tulder 1997). This pro‐forma is designed to assess risk of bias, but also contains items that relate to quality and precision of reporting rather than internal validity.
Assessment of risk of bias in included studies
Two review authors (CL and CS) independently assessed each study for risk of bias using Cochrane's tool for assessing risk of bias in included studies (Higgins 2011). In cases where there was not enough information to make a clear decision, we contacted the study authors and requested clarification or further information. We then made a decision on whether there was a low risk, high risk, or unclear risk of bias for all included studies in each of the five domains of the Cochrane 'Risk of bias' tool (Higgins 2011). These domains are presented as five questions.
Was random sequence generation adequate?
We deemed studies to have a low risk of bias if the study described an adequate random component in the sequence generation process. Methods might have included computer‐generated random number, coin tossing, throwing dice, or drawing lots. We considered studies to be at high risk of bias if a non‐random method for sequence generation was described. Such methods might have included birthdays, hospital numbers, or non‐random categorisation. In studies where there was not enough information to make a judgement of 'low risk' or 'high risk', we classified the study as 'unclear'.
Was allocation adequately concealed?
If a centralised randomisation, computerised allocation, or the picking of opaque envelopes (which all required participant details to be provided prior to allocation) was used, then we considered concealment to be adequate. We decided to deem a study to have inadequate concealment if it included using an open list containing random numbers or contained on an open computer system. If there was no or inadequate information described in the report, then we classified the study as 'unclear'.
Was the allocated intervention adequately concealed from the outcome assessor?
In studies where the outcome assessor was masked or blinded and the report did not identify any unmasking, we deemed the study to have adequate concealment. Inadequately concealed studies might have described an unblinded assessor or assessor who became unblinded. In studies where no description of the assessors masked status was described, we considered the study to be 'unclear'.
Were incomplete outcome data adequately addressed?
We considered studies that reported all outcome data to have a low risk of attrition bias. In cases where there was missing data, but we deemed this data to be balanced across groups or unlikely to relate to the outcome, we considered the study to be at low risk of attrition bias. We deemed studies that inadequately addressed missing outcome data as at high risk of attrition bias. In studies where there was insufficient information to make a judgement, we deemed attrition bias to be 'unclear'.
Were any other issues identified that increased the high risk of bias?
We added any additional potential sources of bias identified during the review in this section. Only those studies that were identified as having a high risk of bias were marked. If there was no clear indication of a high risk of bias, then we left the field blank. This was because a lack of reporting made it difficult to distinguish between a study that was unclear or had a low risk of bias.
Measures of treatment effect
By entering details from each study into Review Manager 5 (RevMan 2014), we were able to statistically analyse the treatment effect. The outcomes studied used a variety of data. We analysed dichotomous data as odds ratios (ORs) with 95% confidence intervals (CIs). We presented pain and the presence of side effects as dichotomous data. We also re‐coded measures of stiffness such as the Ashworth Scale into dichotomous data, identifying if there was an improvement or not. This was necessary because (as expected) there were many variations on the scale system, and it was not possible to compare such poorly defined ordinal scales as continuous data. There is also no clinically significant cutoff point available to use for these scales.
We analysed continuous data in two ways, depending on the measures used in studies. Examples of continuous data that we analysed in this way were the H‐reflex or Barthel Index.
For separate studies where the same measure was used, we used the mean difference (MD) with 95% CI to calculate treatment effect. We intended to use standardised mean difference (SMD) with 95% CI in cases where separate studies used different scales to measure the same outcome; however, this was not necessary due to the lack of studies.
As some participants could have had more than one adverse event and some participants may have had none, we analysed adverse events as risk ratio (RR) with 95% CI.
Where an important outcome could not be summarised in the way described above, we tabulated the results.
Unit of analysis issues
Due to the high risk of a carry‐over effect in cross‐over trials of systemic medications, we planned to include only studies where the the first‐period data were available (i.e. a baseline measure and then a further measure just before cross‐over). We contacted the study authors to request these data if they were not available in the published article.
Dealing with missing data
In the case of missing data we contacted the original investigators to request the information. As planned, it was possible to calculate some missing data using calculations of provided data (as might be the case in missing standard deviations). Where a study did not report a designated outcome, we excluded the study from the analyses of the outcome.
Assessment of heterogeneity
We expected that the studies would be clinically heterogeneous, with variation in interventions and outcomes observed. In all instances we could observe such variation visually using the forest plot. We also used statistical methods to assess heterogeneity. We deemed a Chi2 test with an χ2 greater than the number of studies included minus 1 as significant heterogeneity.
We were also able to quantify the inconsistency and impact on the meta‐analysis using the I2 statistic (Higgins 2002). In this meta‐analysis we set an I2 statistic of up to 40% as an acceptable level of heterogeneity.
Assessment of reporting biases
By using the search strategy outlined above to include and analyse all positive or negative available data in any language, reporting bias was kept to a minimum. We planned to subject any unpublished trials to a detailed review prior to inclusion.
Data synthesis
The aim of this review was to establish whether there is evidence of an effect. This type of review makes conducting a meta‐analysis appropriate. We expected that this review would identify studies that used a variety of drugs to achieve the same outcome, though measured in various ways. Should the pooled data from these studies demonstrate statistical homogeneity, then we intended to use a fixed‐effect meta‐analysis. We considered it more likely that we would need to use a random‐effects model. We planned to report the most conservative outcome in cases where there was divergence (Deeks 2011).
In order to investigate the primary objective, we pooled studies using continuous data. As described in the Measures of treatment effect section, for separate studies where the same measure was used, we used MD with 95% CI to calculate treatment effect; for separate studies that used different scales to measure the same outcome, we planned to employ SMD with 95% CI.
We intended to pool the timed functional tests using SMD with 95% CI.
In order to investigate the secondary outcomes of spasticity (and pain and secondary consequences of spasticity), we pooled studies using dichotomous outcome measures. We calculated an OR using the Mantel‐Haenszel statistical method with 95% CI to assess treatment effect.
We intended to pool studies that used EMG or H‐reflex to assess spasticity and use MD with 95% CI to calculate treatment effect.
In order to determine if global antispasmodic interventions are more effective than local treatments at improving function after stroke, we intended to pool studies using the same drug to study the effect of particular drugs. We intended to then pool the results of locally or globally acting drugs.
In order to determine the rate of side effects, we planned to pool the RRs of each trial using the Mantel‐Haenszel method with 95% CI.
GRADE and 'Summary of findings' table
We used the GRADE approach to assess the quality of the evidence in the included studies. This system required two review authors (CL and AK) to assess the quality of the evidence for each individual outcome and rate it on a four‐point scale from high to very low. Randomised controlled trials began as high evidence and were sequentially downgraded from moderate to low to very low depending on the presence or lack of five factors described by Higgins 2011 (see Table 3). We have presented our judgement of the quality of the evidence with reasons in Table 1.
2. Factors that may decrease the quality of the evidence.
| 1. Limitations in the design and implementation of available studies suggesting high likelihood of bias |
| 2. Indirectness of evidence (indirect population, intervention, control, outcomes) |
| 3. Unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses) |
| 4. Imprecision of results (wide confidence intervals) |
| 5. High probability of publication bias |
Subgroup analysis and investigation of heterogeneity
If deemed appropriate, we intended to conduct subgroup analysis to investigate if the treatment effects on the primary outcome (functional recovery) varied in subpopulations. We planned the following subgroup analyses.
Time since stroke onset: to explore the effect of beginning the intervention at varying times since stroke. The subgroups were those where the intervention began within one month poststroke, within one to three months poststroke, and between three and six months poststroke. We used the mean time from stroke to intervention to identify participants that qualified for inclusion.
Treatments focused specifically on arm function versus specifically on leg function.
To determine if there was a difference between the side effects in drugs that were globally acting or locally acting.
Sensitivity analysis
We planned a sensitivity analysis to assess the difference between using RR and OR for the dichotomised outcomes.
Results
Description of studies
See Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification.
Results of the search
Electronic and manual searches recovered 4064 records, of which we identified 507 as being duplicates. Following review by two review authors (CL and AK) of the remaining 3557 titles and abstracts, we selected 59 for closer review, including eight where there was no abstract available; see Figure 1. After reviewing the full articles, we immediately excluded 20 articles, including all eight without an abstract, as they did not meet the initial criteria. At this point, we emailed the authors of 19 studies in which less than 80% of included participants had a diagnosis of stroke or which were cross‐over studies to request further information. As none of the authors responded, it was not possible to include these studies. We finally accepted 20 full‐text articles, of which 12 are awaiting translation and so have been placed in Studies awaiting classification. We therefore included eight articles. We did not identify any ongoing studies.
1.

Study flow diagram.
Included studies
We included seven randomised controlled trials involving 403 participants (Bes 1988; Kirazli 1998; Kocabas 2010; Medici 1989; Simpson 2009; Stamenova 2005; Yazdchi 2013); two articles reported on the same study (Kirazli 1998).
Design
Only two studies were placebo controlled (Simpson 2009; Stamenova 2005). Stamenova 2005 assessed a maximum dose of 900 mg tolperisone, while Simpson 2009 investigated both a maximum dose of 36 mg tizanidine and onabotulinumtoxinA against placebo. As the stated objective of this review was to exclude botulinum toxin as a drug, we have not presented the 20 participants in this trial who received onabotulinumtoxinA. We analysed these two studies together as 'systemic drug versus placebo'.
The other five studies were all comparative studies investigating one drug against another drug. Two of these studies compared a systemic drug versus another systemic drug. Both of these studies used tizanidine as the interventional drug but used different comparator drugs: Bes 1988 compared a maximum 12 mg tizanidine with 15 mg diazepam, and Medici 1989 compared a maximum 20 mg tizanidine against a maximum dose of 50 mg baclofen.
One study investigated a systemic drug versus a locally acting drug (Yazdchi 2013). This study assessed a maximum dose of 24 mg tizanidine against a maximum dose of 1000 units abobotulinumtoxinA.
The other two studies investigated a locally acting drug versus another locally acting drug (Kirazli 1998; Kocabas 2010). Kirazli 1998 compared 3 ml of 5% phenol to 400 units onabotulinumtoxinA, while Kocabas 2010 compared 5 ml of 5% phenol to 5 ml of 50% ethyl alcohol.
We have presented the results of these four distinct study groups separately.
Sample sizes
The seven studies included 423 randomised participants, of which 403 participants relevant to this review were included (the remaining 20 participants were randomised to botulinum toxin in the Simpson 2009 study). Two of the studies had over 100 participants each, meaning these two studies accounted for 56% of the entire sample: Bes 1988 (105 participants) and Stamenova 2005 (120 participants). The two studies that assessed a systemic drug versus a placebo had a combined total of 160 participants. The two studies that compared a systemic drug versus another systemic drug had a combined number of 135 participants (Bes 1988; Medici 1989). The one study investigating a systemic drug versus a locally acting drug had a sample size of 68 (Yazdchi 2013). The two studies that compared a locally acting drug versus another locally acting drug had notably small samples of just 10 participants in each group, therefore the combined number of participants totaled 40 (Kirazli 1998; Kocabas 2010).
Interventions
Despite the variety of drugs that are currently used clinically to treat spasticity, only three different drugs (tolperisone, tizanidine, and phenol) were investigated against four different control drugs (diazepam, baclofen, botulinum toxin, and alcohol). Of the three trial drugs investigated, one was a locally acting drug (phenol), and the other two were systemically acting drugs (tizanidine and tolperisone).
Of the four control drugs used, two were locally acting (botulinum toxin or alcohol), and two were systemically acting (baclofen and diazepam). None of the studies investigated exactly the same treatments. Simpson 2009 and Yazdchi 2013 both compared tizanidine with botulinum toxin; however, as previously described, Simpson 2009 used a placebo arm as well. This review included only data from the placebo and tizanidine arms of Simpson 2009; we will include the botulinum toxin arm when we update the review to include botulinum toxin.
Outcomes
In order to assess the primary research objective, we identified outcome measures of general functional ability. Only two studies included a general functional measure (Medici 1989; Stamenova 2005), while the other studies used measures of activity or impairment. These two studies did not fall into the same category of study design. Medici 1989 ('systemic drug versus systemic drug' design) used a functional assessment called the Pedersen scale, which is very rarely used, and dichotomised the results to improved or deteriorated. They only reported results at 12 months, despite assessments being carried out at monthly intervals up to six months and then every other month to the 12‐month assessment. We did not attempt to contact these authors, as the publication was more than 25 years old. These results are presented as odds ratio. Stamenova 2005 (placebo design) assessed the Barthel Index; however, they only presented the mean difference in Barthel. As secondary anonymised data, such as standard deviation or 95% confidence interval, were not available to review, this data could not be pooled.
We intended to investigate activity measures of the arm separately to the leg; however, given the lack of studies, this was not possible.
Systemic drug versus a placebo
The two studies that investigated a systemically acting drug versus a placebo both used functional measures, which were measured at 12 weeks. Simpson 2009 used the disability assessment scale, and this data could be plotted. Stamenova 2005 used two measures of functional ability; however, we were unable to analyse either. They used the two‐minute walk test, but only presented the results in graph form, and a five‐point ordinal scale of ability to perform routine activities. This was deficient in that the table only gave numbers in each group at pre‐ and end of study so that the number who improved or deteriorated was not provided.
Systemic drug versus another systemic drug
The two studies that compared a systemic drug against another systemic drug both measured a functional outcome. Medici 1989 used the Pedersen scale but also used a patient self assessment of disability. The only stated result for the latter measure was an improvement from baseline to 12 months (P < 0.001) in both groups, but there was no significant difference between groups. Bes 1988 measured walking distance over flat ground and rough ground at eight weeks from baseline. The study authors did not provide information of how the distance was achieved, as they did not detail whether it was a timed walk or an endurance‐limited distance where the participant stopped when they could do no more. As fewer participants were able to complete the walk on rough ground, only the results of the flat ground were used. The mean distance and standard deviation at baseline and eight weeks were provided. In order to assess efficacy, we subtracted the mean outcome measures at eight weeks from baseline. We calculated the corresponding standard deviation (with an assumption for equal variance) using methodology described in Daniel 1999.
Systemic drug versus a locally acting drug
In the one study that investigated a systemic drug against a locally acting drug, the baseline Action Research Arm Test (ARAT) scores showed a significant difference between groups (P < 0.001) (Yazdchi 2013). We used the same methodology as that of Daniel 1999 to estimate the corresponding standard deviation, having subtracted the mean outcome measures at 12 weeks from baseline.
Locally acting drug versus another locally acting drug
Of the two studies that compared a locally acting drug against another locally acting drug, Kocabas 2010 reported that visual analysis of gait showed that all participants in both groups improved from baseline; however, the time at which this gait assessment was completed was not described. This was the only outcome in this study that related to function. Kirazli 1998 used a 25‐foot timed walk to measure function, which was completed at eight and 12 weeks postintervention. The report provided the mean difference between baseline and 12 weeks with standard deviation.
As expected, the scales used to assess spasticity were diverse. Three studies used the Ashworth scale (Kirazli 1998; Medici 1989; Stamenova 2005), three used a Modified Ashworth Scale (Kocabas 2010; Simpson 2009; Yazdchi 2013), and Bes 1988 used a novel ordinal scale with similarities to the Ashworth Scale. As described in the Methods, we planned to dichotomise the data, and we performed this for five of the studies. We contacted the authors of the two studies in which only the mean and standard deviation data were provided, but they did not respond (Kirazli 1998; Yazdchi 2013).
Only Kirazli 1998 used an outcome measure that directly assessed the abnormal muscle activity that is specific to spasticity. This study presented a number of neurophysiological measures (see Characteristics of included studies); however, only the H‐reflex and H(max):M(max) ratio were analysed.
Other measures of abnormal muscle activity included muscle spasms and clonus as reported by Kocabas 2010 and Kirazli 1998. These two studies also provided results of change in range of movement at a joint; however, Kirazli 1998 only provided a mean change with no other data.
Adverse events were universally reported. They fell into three defined categories (adverse events per participant, total adverse events reported, and number of participants withdrawn due to side effects), with some studies reporting just one of these categories, and others reporting two of the categories. We were able to pool all the data from each category in the form of risk ratios.
Excluded studies
We excluded 19 studies (see Excluded studies). We excluded them on the grounds that less than 80% of the participants had a diagnosis of stroke (11 studies: Burke 1975; Caruso 1977; Chipman 1974; Glass 1974; Harvey 1974; Knutsson 1982; Manca 2010; Meythaler 2001a; Monster 1973; Monster 1974; Thompson 1966), or that a cross‐over trial method had been used and the pre cross‐over data were not available (8 studies; Basmajian 1984; Bovier 1985; Burt 1978; Civardi 1994; Katrak 1992; Maupas 2004; Medaer 1991; Meythaler 2001). We specified these two criteria in the protocol and attempted to contact the lead author, corresponding author, or both to obtain the data needed to include the study.
The eight cross‐over studies had a mean (and median) date of publication of 1991 (range 1978 to 2004). This is compared to the included studies, where date of publication was mean 2002 and median 2005 (range 1988 to 2013).
Interventions of excluded studies
A total of 10 drugs were investigated in the 19 excluded trials, reflecting the diversity of potential drugs available. Five of these 19 studies investigated dantrolene. None of the included studies investigated this drug, despite its widespread clinical use.
Studies awaiting classification
Twelve studies are awaiting classification (see Characteristics of studies awaiting classification). All of these studies require translation into English: four studies are Russian (Gekht 1998; Kovalchuk 2008; Kovalchuk 2013; Stamenova 2006), three are in Chinese (Cui 2009; Gan 2004; Yao 2004), and the rest are in Dutch (Weynants 1990), Polish (Musiol 1977), Italian (Prati 1974), Spanish (Arbizu 1988), and Korean (Kim 2003).
These studies appear to have much larger sample sizes; for 10 studies where we could identify the numbers recruited, there were a total of 964 participants.
Risk of bias in included studies
A summary of our judgement for each potential form of bias across the included studies is provided in the 'Risk of bias' graph in Figure 2. Our assessment of risk of bias for each individual study can be found in the Characteristics of included studies table. The risk of bias for each individual study is presented in a figurative form in the 'Risk of bias' summary in Figure 3. This helps identify that only one study did not have a high risk of bias in any area.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
A poor response from study authors for further information necessitated our assigning an unclear risk of bias as a large proportion of our judgements. With the exception of Kocabas 2010, no other study authors responded to our requests for further information.
Allocation
Random sequence generation and allocation concealment were poorly described in all studies. As study authors did not respond to our requests for further information, we had to assess all of these studies as unclear risk of bias. Only Kocabas 2010 described the sequence generation, which was by pitch and toss.
Blinding
Performance bias (blinding of participants and physician) tended to be well reported, and we were able to make a clear decision from the study reports in many cases; however, this was not the case with detection bias (blinding of outcome assessors), which was poorly reported. Due to this we classified four of the seven studies as at unclear risk of bias (Bes 1988; Kocabas 2010; Medici 1989; Stamenova 2005).
Incomplete outcome data
There was a good level of reporting on participants lost to follow‐up in many studies, and in some cases no one was lost to follow‐up, allowing us to make a clear decision on whether there was attrition bias.
Selective reporting
We found the highest risk of bias to be in the selective reporting of data. Four of the seven studies did not adequately report all the outcome measures described in the methodology. We reviewed ClinicalTrials.gov and other potential resources to identify whether results had been registered in open access public forums, but this was not the case.
Other potential sources of bias
We identified where commercial involvement appeared to have occurred in Stamenova 2005, with the corresponding author an employee of the drug company. Based on the available information, it was not possible to identify any other sources of bias in the studies.
Effects of interventions
See: Table 1
See Table 1.
As described, there were four distinct groups of studies, and we made comparisons between studies that for each of these four categories.
Systemically acting drug versus a placebo
Functional ability during activities of daily living
Of the two studies that assessed a systemic drug against a placebo (Simpson 2009; Stamenova 2005), only Simpson 2009 employed any usable measure of function (Disability Assessment Scale). The mean difference (MD) between the group receiving tizanidine (n = 21) and the group receiving placebo (n = 19) was not significant (MD ‐0.2, 95% confidence interval (CI) ‐0.9 to 0.5; Analysis 1.1).
1.1. Analysis.
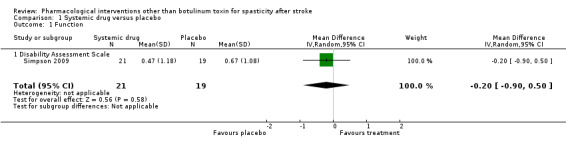
Comparison 1 Systemic drug versus placebo, Outcome 1 Function.
Spasticity: indirect measure
Both of these studies had the measures of spasticity dichotomised; however, this produced very heterogenous results (I2 = 85%). While the results of Simpson 2009 were non‐significant, Stamenova 2005 showed a significant benefit for the treatment group. When we pooled the data (160 participants), we did not identify any significant effect (odds ratio (OR) 1.66, 95% CI 0.21 to 13.07; Analysis 1.2).
1.2. Analysis.

Comparison 1 Systemic drug versus placebo, Outcome 2 Spasticity ‐ indirect measure.
Adverse events
Stamenova 2005 reported two of the three defined categories of adverse events (adverse events per participant and withdrawals due to side effects), while Simpson 2009 reported all three categories, including the total number of adverse events reported. The combined data from both studies (160 participants) on adverse events with a possible causal relationship to drug per participant demonstrated a statistically significant (P = 0.01) risk to participants receiving the treatment drug compared with placebo (risk ratio (RR) 1.65, 95% CI 1.12 to 2.42). There was very little heterogeneity in these results (I2 = 0%) (Analysis 1.3). Simpson 2009 also found the total number of adverse events reported in each group to be different. Ten adverse events were reported in the 19 participants receiving placebo, whereas 24 adverse events were reported in 21 participants receiving treatment.
1.3. Analysis.

Comparison 1 Systemic drug versus placebo, Outcome 3 Adverse events.
Despite the homogeneity of results for adverse events per participant, the two studies reported very heterogenous results on numbers withdrawn due to side effects (I2 = 62%). We therefore did not identify any significant results (RR 1.16, 95% CI 0.04 to 34.40; 160 participants).
Systemic drug versus another systemic drug
Functional ability during activities of daily living
The two studies that compared one systemic drug against another systemic drug measured functional change in different ways (Bes 1988; Medici 1989).
Bes 1988 compared tizanidine against diazepam and assessed benefit using a distance‐walked test over flat ground (metres) as a functional measure (79 participants, as not all participants could walk). The study report stated that "only the patients on tizanidine showed a statistically significant improvement in walking distance on flat ground"; however, both groups achieved a statistically significant improvement in distance walked. Indeed, there was a statistically significant difference (P = 0.01) between groups in favour of diazepam when we calculated mean change from baseline to eight weeks (MD ‐183.30, 95% CI ‐330.40 to ‐36.20; Analysis 2.1).
2.1. Analysis.

Comparison 2 Systemic drug versus another systemic drug, Outcome 1 Function.
Medici 1989 compared tizanidine against baclofen and assessed benefit using dichotomised data from the Pedersen scale, to measure function, and presented the arm and leg scores separately (30 participants). Both groups made equal improvements in the arm and leg function, so there was no difference between groups for the arm measure (OR 1.0, 95% CI 0.23 to 4.31) and the leg measure (OR 1.0, 95% CI 0.17 to 5.98; Analysis 2.2).
2.2. Analysis.

Comparison 2 Systemic drug versus another systemic drug, Outcome 2 Function dichotomous.
Spasticity: indirect measure
Although the two studies used different scales to measure spasticity, the response to treatment was dichotomised to identify responders. However, we did not pool the results, as the compared drugs in each study were different. No significant differences between drugs were identified in either study: Medici 1989 compared tizanidine with baclofen (30 participants) and identified no significant difference between groups (OR 2.36, 95% CI 0.36 to 15.45), and Bes 1988 compared tizanidine and diazepam (78 participants) and identified no significant difference between groups (OR 0.90, 95% CI 0.37 to 2.20; Analysis 2.3).
2.3. Analysis.

Comparison 2 Systemic drug versus another systemic drug, Outcome 3 Spasticity ‐ indirect measure.
Clonus and muscle spasms
Both studies also dichotomised signs of abnormal muscle activity (e.g. clonus or muscle spasms) using customised scales. Not all participants in the group had these signs, so the sample sizes in the analyses were smaller than the study sample sizes. Both studies presented the improvement in clonus. In Medici 1989 five out of seven participants improved on tizanidine, compared with four out of five on baclofen (0.63, 95% CI 0.04 to 9.65; 12 participants), whereas in Bes 1988 14 out of 29 participants in the tizanidine group improved, compared with eight out of 20 on diazepam (1.40, 95% CI 0.44 to 4.44; 49 participants). Both studies showed no significant differences between groups (Analysis 2.4). In addition to clonus, Medici 1989 also assessed muscle spasms, which were improved in five out of eight participants receiving tizanidine and five out of six participants receiving baclofen (0.33, 95% CI 0.03 to 4.40; 14 participants; Analysis 2.5).
2.4. Analysis.

Comparison 2 Systemic drug versus another systemic drug, Outcome 4 Clonus.
2.5. Analysis.

Comparison 2 Systemic drug versus another systemic drug, Outcome 5 Muscle spasms.
Adverse events
Bes 1988 reported two of the three defined categories adverse events (total adverse events and withdrawals due to side effects), while Medici 1989 reported all three categories, including the number of adverse events per participant. In Medici 1989 there was no significant difference between groups in the number of adverse events per participant (RR 2.10, 95% CI 0.83 to 5.30; 29 participants). In both studies there were a greater number of total adverse events in the tizanidine groups when compared with either of the comparative drugs. While this difference was not significant in Medici 1989 (RR 1.26, 95% CI 0.94 to 1.69; 29 participants), there was a significant difference (P = 0.04) in favour of diazepam in Bes 1988 (RR 1.22, 95% CI 1.01 to 1.48; 105 participants).
In both studies more participants were withdrawn from the control group when compared with the tizanidine group; however, this was not significant. Medici 1989 identified a non‐significant RR of 0.13 (95% CI 0.01 to 2.38), while Bes 1988 found an RR of 0.42 (95% CI 0.18 to 1.01) (Analysis 2.6).
2.6. Analysis.

Comparison 2 Systemic drug versus another systemic drug, Outcome 6 Adverse events.
Systemic drug versus a locally acting drug
Functional ability during activities of daily living
Yazdchi 2013 (68 participants) compared tizanidine against abobotulinumtoxinA and used the Action Research Arm Test (ARAT) as the outcome measure. We calculated the mean difference from baseline to week 12 and identified a significant difference (P = 0.02) in favour of botulinum toxin (MD 3.8, 95% CI ‐6.93 to ‐0.65; Analysis 3.1).
3.1. Analysis.

Comparison 3 Systemically acting drug versus a locally acting drug, Outcome 1 Function.
Spasticity
This study did not present the results in a way that allowed any measure of spasticity to be dichotomised. We contacted the corresponding author using the email address provided but did not receive a response.
Adverse events
Adverse events were only presented as number of participants withdrawn due to side effects. This showed a significant risk ratio in favour of botulinum toxin (RR 41.00, 95% CI 2.6 to 651.7; P = 0.009; 68 participants; Analysis 3.2).
3.2. Analysis.

Comparison 3 Systemically acting drug versus a locally acting drug, Outcome 2 Adverse events.
Locally acting drug against another locally acting drug
Functional ability during activities of daily living
Kocabas 2010 and Kirazli 1998 both compared phenol injections against another locally acting drug (alcohol and onabotulinumtoxinA, respectively). Kocabas 2010 only measured change at the level of impairment, whereas Kirazli 1998 (20 participants) identified no significant mean difference between groups at 12 weeks in a 25‐foot timed walk test (MD 2.70, 95% CI ‐2.83 to 8.23; Analysis 4.1).
4.1. Analysis.

Comparison 4 Locally acting drug versus another locally acting drug, Outcome 1 Function.
Passive range of movement
Both studies provided information on range of movement at the ankle; however, Kirazli 1998 only presented the mean change and no other information with which to analyse the data. Kocabas 2010 presented this data, which once again showed no significant difference between the two comparison drugs (P = 0.84; Analysis 4.2).
4.2. Analysis.

Comparison 4 Locally acting drug versus another locally acting drug, Outcome 2 Passive range of movement.
Spasticity: neurophysiological
Neither of the neurophysiological outcome measures used in Kirazli 1998 (20 participants) demonstrated a significant difference between the groups. The mean difference between the H(max):M(max) ratio was ‐0.01 (95% CI ‐0.48 to 0.46; P = 0.97), and the mean difference in H‐reflex (amplitude) was 1.24 (95% CI ‐1.44 to 3.92; P = 0.36; Analysis 4.3).
4.3. Analysis.

Comparison 4 Locally acting drug versus another locally acting drug, Outcome 3 Spasticity ‐ neurophysiological.
Spasticity: indirect measure
Kocabas 2010 (20 participants) provided dichotomised data for the modified Ashworth scale only at the six‐month assessment but reported that all 20 participants had a decrease in the scale immediately after the injection. This benefit was still present at six‐month follow‐up in nine of the 10 participants treated with alcohol and seven of the 10 in the phenol group, indicating no significant difference between groups (OR 0.26, 95% CI 0.02 to 3.06; Analysis 4.4).
4.4. Analysis.

Comparison 4 Locally acting drug versus another locally acting drug, Outcome 4 Spasticity ‐ indirect measure.
Clonus
In Kocabas 2010 the dichotomised results of clonus improvement showed that all five participants treated with phenol and four of seven participants treated with alcohol improved in clonus score. There was no significant difference between the groups (OR 8.56, 95% CI 0.34 to 212.94; Analysis 4.5).
4.5. Analysis.

Comparison 4 Locally acting drug versus another locally acting drug, Outcome 5 Clonus.
Adverse events
Kocabas 2010 and Kirazli 1998 both reported the total adverse events occurring in the study. In both studies three of the 10 participants in the phenol groups reported adverse events while none of the 10 participants in the control groups reported any adverse events. There was no significant difference between groups (RR 7.0, 95% CI 0.41 to 120.16; P = 0.18 ; I2 = 0%).
Discussion
Summary of main results
We identified seven randomised controlled trials with a combined sample size of 403 participants that investigated a pharmacological intervention for spasticity after stroke.
Just two of these studies were placebo‐controlled trials, whereas in the other five trials one drug was compared against another drug. Only three different drugs (tolperisone, tizanidine, and phenol) were investigated against four different control drugs (diazepam, baclofen, botulinum toxin, and alcohol), and none of the studies compared the same two drugs. We were able to pool data from the two placebo trials with a combined sample size of 160 participants (Simpson 2009; Stamenova 2005).
In addition to the two placebo studies, the studies could be divided into three further categories. Two compared two systemic drugs (Bes 1988; Medici 1989); one compared a systemic drug with a local drug (Yazdchi 2013); and two compared two local drugs (Kirazli 1998; Kocabas 2010).
Six studies used at least one functional outcome measure; however, the results were not always reported in such a way as to allow for data extraction and analyses (Bes 1988; Kirazli 1998; Medici 1989; Simpson 2009; Stamenova 2005; Yazdchi 2013). Kocabas 2010 measured all outcomes at an impairment level and did not attempt to assess functional change.
Our primary objective was to determine if pharmacological interventions for spasticity are more effective than no intervention or control at improving function following stroke. Clear evidence for this was not available due to the lack of placebo‐controlled trials and the large variation in comparator trials. In the only placebo‐controlled study that employed usable outcome measures, results were inconclusive (Simpson 2009). This trial was also the least likely to be at risk of bias.
The results between the two placebo trials were divergent for treating spasticity, and meta‐analysis of the data produced an I2 of 85%. Despite this heterogeneity in treating spasticity, there was a clear increased risk of having an adverse event associated with receiving tolperisone when compared with placebo.
Due to the methodology employed in comparing two drugs against each other, further results were limited. Treatment in current clinical practice varies among a number of drugs, so comparative methods are not useful in this area of investigation.
The methodology used to investigate whether the drug had indeed treated the specific impairment of spasticity varied. All of the studies used a method based on the (modified) Ashworth Scale, which has been shown to lack validity (Fleuren 2010; Pandyan 1999). Of the 79 participants who received a placebo drug, 39 (49.4%) of these improved according to the (modified) Ashworth Scale. The one study using a direct measure of one aspect of spasticity suggested that there was no difference between phenol and botulinum toxin, both of which are locally acting. This suggests a possibly large placebo effect or the effects of confounded measurement (Fleuren 2010; Pandyan 1999).
In summary, this review has identified a lack of studies and subsequent evidence relating to the use of pharmacological interventions (with the exception of botulinum toxin) to treat spasticity. The quality of the evidence is low to very low.
Overall completeness and applicability of evidence
The completeness of evidence is limited by the methodology used in many studies. Just two out of the seven studies used a placebo control. We identified some studies using a cross‐over placebo‐controlled methodology. We contacted the authors of these cross‐over studies, but none of them responded, which meant that we could not include any of these studies in the analysis.
The majority of outcome measures that measured function were at the activity level rather than the participation level. The measures employed differed for each study, and appropriate data were not always reported to allow analysis.
In the two placebo‐controlled trials, there was marked heterogeneity in the results for whether the intervention had treated spasticity, which means that it is difficult to apply the evidence. The only neurophysiological measure of spasticity did not show a significant difference between phenol and botulinum toxin at 12 weeks.
Unfortunately, despite attempts to do so, it was not possible to translate any of the full texts published in different languages into English. Until this is achieved the evidence is not fully complete. We expect to translate all of these studies into English by the time that the review is updated.
Furthermore, due to initial editorial restrictions, we were not in a position to include botulinum toxin as a pharmacological agent. In the recent past the Cochrane Stroke Group Editorial Board has approved that this review is updated with the inclusion of botulinum neurotoxin type A. This process is ongoing, and the results will be updated once we have collated the evidence and carried out meta‐analyses.
Quality of the evidence
The quality of the evidence varied. We judged six out of seven studies to be at high risk of bias in at least one of the six methodological criteria assessed. Three of these studies had two components that we judged to be at high risk of bias. The one study we judged to have no evidence of high risk of bias did not have a judgement made in three of the six components. We judged only one study to be at low risk of bias in more than three components (Simpson 2009). This study was one of the two placebo‐controlled studies; however, we judged the quality of the other study using this methodology to have two components at high risk of bias.
There is a risk of reduced internal validity associated with the use of confounded measures in both the inclusion criteria for studies and outcome measures. There was also inconsistency in reporting of study results. In addition, the sample sizes in the included studies were relatively low for drug trials; three studies had 30 or fewer participants, and only two studies had a sample size of more than 100 participants.
Potential biases in the review process
Despite our extensive literature searching, selection bias is possible, and in particular publication bias. Furthermore, the inability to gain access to the secondary anonymised data from the source publications is also likely to have increased the chance of publication bias. Given the large numbers of participants in the studies awaiting classification and translation, it is possible that the current conclusions may be biased towards studies that have been published in English. We expect to translate all of these studies into English by the time that the review is updated.
The inclusion of participants who had not had a stroke is a possible concern. However, the two studies that included participants who had survived something other than a stroke only included participants who had sustained a traumatic brain injury, and these 24 participants are unlikely to have had a major impact on any conclusions made.
Agreements and disagreements with other studies or reviews
Two previous systematic reviews have investigated this general area. A review by Montane 2004 investigated oral antispastic drugs in non‐progressive neurological diseases, meaning that studies investigating traumatic brain injury, spinal cord injury, and cerebral palsy were analysed as well as stroke. This review also included the end data from cross‐over trials, which we had stated in our original protocol we would not include in our review. The authors only identified 12 studies, two of which we have included in our current review (Bes 1988; Medici 1989). They also included three studies that we excluded from our review (Katrak 1992; Medaer 1991; Meythaler 2001a). We excluded Meythaler 2001a because less than 80% of the participants had a diagnosis of stroke, and the other two studies because they were studies with a cross‐over design. Despite the methodological differences between the Montane 2004 review and our review, the conclusions were similar.
A more recent review by Olvey 2010 investigated only people with a history of stroke but also specifically arm spasticity. This review used just nine search terms and included only drugs available commercially in the United States. The methodology also included studies that were not randomised. Importantly, this review included studies which investigated botulinum toxin. Of the 54 studies that met the inclusion criteria, 51 of them involved botulinum toxin. The other three studies were uncontrolled trials of tizanidine and intrathecal baclofen, and one cross‐over trial investigating intrathecal baclofen that we excluded from our review (Meythaler 2001). The one trial that was included in both the review by Olvey 2010 and our review was Simpson 2009. Due to methodological differences, it is not appropriate to compare our review with Olvey 2010.
Authors' conclusions
Implications for practice.
There is currently insufficient high‐quality evidence to make generalisable conclusions about the effect of pharmacological interventions on spasticity in people with stroke. Furthermore, we found some very low‐quality evidence that suggests there is an increased risk of adverse effects in people who take antispasmodics when compared with placebo. We therefore cannot recommend pharmacological interventions as first‐line treatment for people with spasticity after stroke. If a clinical decision is made to administer any of these interventions, particular care should be taken to monitor for adverse effects.
Implications for research.
This review has highlighted the lack of high‐quality research into the use of pharmacological interventions to treat spasticity following stroke (with the exception of botulinum toxin).
Future research should implement high‐quality randomised trial methodology, which will help in reducing potential bias in the results. Future research should also employ placebo controls to assess the efficacy of an intervention. The use of comparative drug trials has meant we have been unable to meta‐analyse the studies, with the exception of the two studies that did use a placebo control. If a clear gold standard first‐line drug is identified, then comparative drug trials may be worthwhile in identifying novel drugs; however, this does not appear to currently be the case. If a comparison study is employed, a clear rationale for use of the comparative drug should be given.
Future investigations should also use an assessment tool that is capable of identifying whether the drug being investigated has actually stopped the abnormal muscle activity related to spasticity from occurring. Current approaches that use indirect measures of stiffness are confounded by coexisting contractures. Only when it is evident that spasticity has been treated should methods that investigate functional changes also be measured. Even if the current review had identified a number of high‐quality placebo‐controlled trials with large sample sizes, it might not have been possible to conclude that antispasmodics were effective. If the primary impairment being measured cannot be shown to have been improved, then any functional recovery seen between groups cannot be causatively associated with the drug.
Once these questions have been answered it will be important for future research to then focus on the optimal time at which to initiate treatments and the ideal dose associated with each treatment.
Notes
Simpson 2009 was a three‐arm trial comparing tizanidine versus botulinum toxin versus placebo. On initial data extraction we considered that only the comparison of tizanidine versus placebo was relevant to this review, and we have included only data for these arms. However, we identified and have included other trials that had botulinum toxin as the comparator (Kirazli 1998 compared phenol to botulinum toxin, and Yazdchi 2013 compared tizanidine to botulinum toxin). On reflection, our decision to exclude the botulinum toxin arm of the Simpson 2009 trial was incorrect, and the comparison of tizanidine versus botulinum toxin should have been included in the comparison 'Systemic drug versus a locally acting drug'. This inconsistency was noted at the final editorial stages prior to publication of this review, and we have made no correction to this version of the review. We will broaden future updates of this review to include all pharmacological interventions for spasticity (i.e. we will include botulinum toxin as an active intervention). We will therefore include the botulinum toxin arm of the Simpson 2009 trial in future updates.
Acknowledgements
The review authors acknowledge the staff at the UK Cochrane Centre in Oxford for providing training and advice during the writing of this protocol. The review authors also thank Brenda Thomas and Hazel Fraser from the Cochrane Stroke Group for their guidance and advice, and acknowledge the time and assistance of Julie Gildie in providing consumer peer review of the completed review.
Appendices
Appendix 1. Cochrane Central Register of Controlled Trials (CENTRAL)
MeSH descriptor: [Cerebrovascular Disorders] explode all trees
stroke or poststroke or post‐stroke or cerebrovasc* or brain vasc* or cerebral vasc* or cva* or apoplex* or SAH:ti,ab,kw (Word variations have been searched)
(brain* or cerebr* or cerebell* or intracran* or intracerebral) near/5 (isch?emi* or infarct* or thrombo* or emboli* or occlus*):ti,ab,kw (Word variations have been searched)
(brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid) near/5 (haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed*):ti,ab,kw (Word variations have been searched)
MeSH descriptor: [Hemiplegia] this term only
MeSH descriptor: [Paresis] explode all trees
MeSH descriptor: [Brain Injuries] this term only
MeSH descriptor: [Brain Injury, Chronic] this term only
MeSH descriptor: [Brain Damage, Chronic] this term only
#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9
MeSH descriptor: [Muscle Spasticity] this term only
MeSH descriptor: [Muscle Hypertonia] this term only
MeSH descriptor: [Muscle Rigidity] this term only
MeSH descriptor: [Muscle Tonus] this term only
MeSH descriptor: [Spasm] this term only
MeSH descriptor: [Dystonia] this term only
MeSH descriptor: [Paraparesis, Spastic] this term only
spastic* or high tone:ti,ab,kw (Word variations have been searched)
muscle* near/5 (spasm or spasms or rigid* or tone or tonus or hyperton* or hypermyoton* or dyston*):ti,ab,kw (Word variations have been searched)
#11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19
MeSH descriptor: [Muscle Relaxants, Central] explode all trees
MeSH descriptor: [Cannabis] this term only
MeSH descriptor: [Cannabinoids] explode all trees
MeSH descriptor: [Phenols] explode all trees
MeSH descriptor: [Ethanol] this term only
MeSH descriptor: [Injections, Intramuscular] this term only
MeSH descriptor: [Nerve Block] this term only
MeSH descriptor: [Neuromuscular Blockade] this term only
MeSH descriptor: [Injections, Spinal] this term only
intrathecal:ti,ab,kw (Word variations have been searched)
baclofen or carisoprodol or chlormezanone or chlorphenesin or chlorzoxazone or cyclobenzaprine or dantrolene or dimethothiazine or diazepam or eperisone or gabapentin or idrocilamide or ketazolam or medazepam or mephenesin or meprobamate or methocarbamol or nefopam or orphenadrine or quinine or tetrazepam or tizanidine or tolperisone or zoxazolamine or cannabis or cannabinoid* or sativex or "GW‐1000" or phenol or alcohol or ethanol:ti,ab,kw (Word variations have been searched)
antispastic* near/5 (agent* or drug* or medication):ti,ab,kw (Word variations have been searched)
(nerve or neuromuscular or motor point) near/5 block*:ti,ab,kw (Word variations have been searched)
intramuscular near/3 (drug* or injection* or treatment* or medication* or neurolysis):ti,ab,kw (Word variations have been searched)
#21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34
#10 and #20 and #35
MeSH descriptor: [Cerebral Palsy] this term only
"cerebral palsy":ti,ab,kw (Word variations have been searched)
#37 or #38
#36 not #39
Appendix 2. MEDLINE search strategy
cerebrovascular disorders/ or exp basal ganglia cerebrovascular disease/ or exp brain ischemia/ or exp carotid artery diseases/ or exp intracranial arterial diseases/ or exp "intracranial embolism and thrombosis"/ or exp intracranial hemorrhages/ or stroke/ or exp brain infarction/ or vertebral artery dissection/
(stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
hemiplegia/ or exp paresis/ or brain injuries/ or brain injury, chronic/ or brain damage, chronic/
(hemipleg$ or hemipar$ or paresis or paretic or acquired brain injur$).tw.
1 or 2 or 3 or 4 or 5 or 6
muscle spasticity/ or muscle hypertonia/ or muscle rigidity/ or muscle tonus/
spasm/ or dystonia/ or paraparesis, spastic/
(spastic$ or high tone).tw.
(muscle$ adj5 (spasm or spasms or rigid$ or tone or tonus or hyperton$ or hypermyoton$ or dyston$)).tw.
8 or 9 or 10 or 11
exp muscle relaxants, central/
cannabis/ or exp cannabinoids/
exp phenols/ or ethanol/ or injections, intramuscular/ or nerve block/ or neuromuscular blockade/ or injections, spinal/ or intrathecal.tw
(baclofen or carisoprodol or chlormezanone or chlorphenesin or chlorzoxazone or cyclobenzaprine or dantrolene or dimethothiazine or diazepam or eperisone or gabapentin or idrocilamide or ketazolam or medazepam or mephenesin or meprobamate or methocarbamol or nefopam or orphenadrine or quinine or tetrazepam or tizanidine or tolperisone or zoxazolamine or cannabis or cannabinoid* or sativex or GW‐1000 or phenol or alcohol or ethanol).tw,nm.
(antispastic$ adj5 (agent$ or drug$ or medication)).tw.
((nerve or neuromuscular or motor point) adj5 block$).tw.
(intramuscular adj3 (drug$ or injection$ or treatment$ or medication$ or neurolysis)).tw.
(drug therapy or drug effects).fs.
or/13‐20
7 and 12 and 21
cerebral palsy/ or cerebral palsy.tw.
22 not 23
exp animals/ not humans.sh.
24 not 25
Randomized Controlled Trials as Topic/
random allocation/
Controlled Clinical Trials as Topic/
control groups/
clinical trials as topic/ or clinical trials, phase i as topic/ or clinical trials, phase ii as topic/ or clinical trials, phase iii as topic/ or clinical trials, phase iv as topic/
double‐blind method/
single‐blind method/
Placebos/
placebo effect/
Therapies, Investigational/
Drug Evaluation/
Research Design/
randomized controlled trial.pt.
controlled clinical trial.pt.
(clinical trial or clinical trial phase i or clinical trial phase ii or clinical trial phase iii or clinical trial phase iv).pt.
(random$ or RCT$).tw.
(controlled adj5 (trial$ or stud$)).tw.
(clinical$ adj5 trial$).tw.
((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
(quasi‐random$ or quasi random$ or pseudo‐random$ or pseudo random$).tw.
((control or experiment$ or conservative) adj5 (treatment or therapy or procedure or manage$)).tw.
((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
placebo$.tw.
(assign$ or allocat$).tw.
controls.tw.
or/27‐51
26 and 52
Appendix 3. Embase search strategy
cerebrovascular disease/ or basal ganglion hemorrhage/ or cerebral artery disease/ or cerebrovascular accident/ or stroke/ or exp carotid artery disease/ or exp brain hematoma/ or exp brain hemorrhage/ or exp brain infarction/ or exp brain ischemia/ or exp intracranial aneurysm/ or exp occlusive cerebrovascular disease/
stroke unit/ or stroke patient/
(stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
hemiplegia/ or paresis/ or brain injury/ or acquired brain injury/
(hemipleg$ or hemipar$ or paresis or paretic or acquired brain injur$).tw.
1 or 2 or 3 or 4 or 5 or 6 or 7
muscle hypertonia/ or muscle rigidity/ or spastic paresis/ or spasticity/
muscle spasm/ or muscle tone/ or dystonia/ or paraplegia/
(spastic$ or high tone).tw.
((muscle$ or muscular) adj5 (spasm or spasms or rigid$ or tone or tonus or hyperton$ or hypermyoton$ or dyston$)).tw.
9 or 10 or 11 or 12
muscle relaxant agent/ or exp central muscle relaxant/ or cyclobenzaprine/ or exp directly acting muscle relaxant/ or exp neuromuscular blocking agent/ or exp neuromuscular depolarizing agent/
exp cannabinoid/ or cannabis/
exp phenol derivative/ or alcohol/ or intramuscular drug administration/ or exp nerve block/ or exp neuromuscular blocking/ or exp intraspinal drug administration/ or intrathecal.tw.
(baclofen or carisoprodol or chlormezanone or chlorphenesin or chlorzoxazone or cyclobenzaprine or dantrolene or dimethothiazine or diazepam or eperisone or gabapentin or idrocilamide or ketazolam or medazepam or mephenesin or meprobamate or methocarbamol or nefopam or orphenadrine or quinine or tetrazepam or tizanidine or tolperisone or zoxazolamine or cannabis or cannabinoid* or sativex or GW‐1000 or phenol or alcohol or ethanol).tw.
(antispastic$ adj5 (agent$ or drug$ or medication)).tw.
((nerve or neuromuscular or motor point) adj5 block$).tw.
(intramuscular adj3 (drug$ or injection$ or treatment$ or medication$ or neurolysis)).tw.
dt.fs.
or/9‐21
Randomized Controlled Trial/
Randomization/
Controlled Study/
control group/
clinical trial/ or phase 1 clinical trial/ or phase 2 clinical trial/ or phase 3 clinical trial/ or phase 4 clinical trial/ or controlled clinical trial/
Double Blind Procedure/
Single Blind Procedure/ or triple blind procedure/
placebo/
"types of study"/
random$.tw.
(controlled adj5 (trial$ or stud$)).tw.
(clinical$ adj5 trial$).tw.
((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
(quasi‐random$ or quasi random$ or pseudo‐random$ or pseudo random$).tw.
((control or experiment$ or conservative) adj5 (treatment or therapy or procedure or manage$)).tw.
((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
placebo$.tw.
(assign$ or allocat$).tw.
controls.tw.
drug research/
(RCT or RCTS).tw. or trial.ti.
or/23‐43
8 and 13 and 22 and 44
cerebral palsy/ or cerebral palsy.tw.
45 not 46
(exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.)
47 not 48
(2008$ or 2009$ or 201$).em.
49 and 50
Appendix 4. CINAHL search strategy
(MH "Cerebrovascular Disorders+") or (MH "stroke patients") or (MH "stroke units")
TI ( stroke or poststroke or post‐stroke or cerebrovasc* or brain vasc* or cerebral vasc or cva or apoplex or SAH ) or AB ( stroke or poststroke or post‐stroke or cerebrovasc* or brain vasc* or cerebral vasc or cva or apoplex or SAH )
TI ( brain* or cerebr* or cerebell* or intracran* or intracerebral ) or AB ( brain* or cerebr* or cerebell* or intracran* or intracerebral )
TI ( ischemi* or ischaemi* or infarct* or thrombo* or emboli* or occlus* ) or AB ( ischemi* or ischaemi* or infarct* or thrombo* or emboli* or occlus* )
S3 and S4
TI ( brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid ) or AB ( brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid )
TI ( haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed* ) or AB ( haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed* )
S6 and S7
(MH "Hemiplegia")
TI ( hemipleg* or hemipar* or paresis or paretic or acquired brain injur* ) or AB ( hemipleg* or hemipar* or paresis or paretic or acquired brain injur* )
(MH "Brain Injuries")
S1 or S2 or S5 or S8 or S9 or S10 or S11
(MH "Muscle Spasticity") OR (MH "Muscle Hypertonia") OR (MH "Muscle Tonus")
(MH "spasm")
(MH "dystonia")
(MH "gait disorders, neurologic")
TI ( spastic* or high tone ) or AB ( spastic* or high tone )
TI ( muscle N5 (spasm or spasms or rigid* or tone or tonus or hyperton* or hypermyoton* or dyston*) ) OR AB ( muscle N5 (spasm or spasms or rigid* or tone or tonus or hyperton* or hypermyoton* or dyston*) )
S13 OR S14 OR S15 OR S16 OR S17 OR S18
(MH "Neuromuscular Agents") OR (MH "Muscle Relaxants, Central+") OR (MH "Neuromuscular Blocking Agents+")
(MH "Cannabis")
(MH "Phenols+")
(MH "Ethanol+")
(MH "Injections, Intramuscular+")
(MH "Nerve Block")
(MH "Neuromuscular Blockade")
(MH "Injections, Intraspinal+")
TI intrathecal OR AB intrathecal
TI ( baclofen or carisoprodol or chlormezanone or chlorphenesin or chlorzoxazone or cyclobenzaprine or dantrolene or dimethothiazine or diazepam or eperisone or gabapentin or idrocilamide or ketazolam or medazepam or mephenesin or meprobamate or methocarbamol or nefopam or orphenadrine or quinine or tetrazepam or tizanidine or tolperisone or zoxazolamine or cannabis or cannabinoid* or sativex or GW‐1000 or phenol or alcohol or ethanol ) OR AB ( baclofen or carisoprodol or chlormezanone or chlorphenesin or chlorzoxazone or cyclobenzaprine or dantrolene or dimethothiazine or diazepam or eperisone or gabapentin or idrocilamide or ketazolam or medazepam or mephenesin or meprobamate or methocarbamol or nefopam or orphenadrine or quinine or tetrazepam or tizanidine or tolperisone or zoxazolamine or cannabis or cannabinoid* or sativex or GW‐1000 or phenol or alcohol or ethanol )
TI ( antispastic* N5 (agent* or drug* or medication) ) OR AB ( antispastic* N5 (agent* or drug* or medication) )
TI ( (nerve or neuromuscular or motor point) N5 block* ) OR AB ( (nerve or neuromuscular or motor point) N5 block* )
TI ( intramuscular N3 (drug* or injection* or treatment* or medication* or neurolysis) ) OR AB ( intramuscular N3 (drug* or injection* or treatment* or medication* or neurolysis) )
S20 OR S21 OR S22 OR S23 OR S24 OR S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 OR S32
S12 AND S19 AND S33
Appendix 5. AMED search strategy
cerebrovascular disorders/ or cerebral hemorrhage/ or cerebral infarction/ or cerebral ischemia/ or cerebrovascular accident/ or stroke/
(stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
hemiplegia/ or brain injuries/
(hemipleg$ or hemipar$ or paresis or paretic or acquired brain injur$).tw.
1 or 2 or 3 or 4 or 5 or 6
muscle hypertonia/ or muscle spasticity/ or muscle tonus/
spasm/ or dystonia/
(spastic$ or high tone).tw.
(muscle$ adj5 (spasm or spasms or rigid$ or tone or tonus or hyperton$ or hypermyoton$ or dyston$)).tw.
8 or 9 or 10 or 11
muscle relaxants central/
cannabis/
neuromuscular blocking agents/ or nerve block/ or injections/ or intrathecal.tw.
(baclofen or carisoprodol or chlormezanone or chlorphenesin or chlorzoxazone or cyclobenzaprine or dantrolene or dimethothiazine or diazepam or eperisone or gabapentin or idrocilamide or ketazolam or medazepam or mephenesin or meprobamate or methocarbamol or nefopam or orphenadrine or quinine or tetrazepam or tizanidine or tolperisone or zoxazolamine or cannabis or cannabinoid* or sativex or GW‐1000 or phenol or alcohol or ethanol).tw.
(antispastic$ adj5 (agent$ or drug$ or medication)).tw.
((nerve or neuromuscular or motor point) adj5 block$).tw.
(intramuscular adj3 (drug$ or injection$ or treatment$ or medication$ or neurolysis)).tw.
drug effects/ or drug therapy/
13 or 14 or 15 or 16 or 17 or 18 or 19 or 20
7 and 12 and 21
cerebral palsy/ or cerebral palsy.tw.
22 not 23
animals/
24 not 25
Appendix 6. Data extraction form for included trials
Study details
Study ID
Citation
Contact details
Language
Title of study
Authors of study
Country of study
Objective stated
Criteria
Inclusion criteria
Exclusion criteria
Stroke/spasticity
Definition of stroke
Type of stroke
Time from stroke to inclusion as criterion
Definition of spasticity
Participants
Age
Gender
Time post stroke
Laterality of stroke
Randomisation
Type of study
Treatment allocation/method of randomisation
Blinding–patient
Blinding–drug
Blinding–assessor
Interventions
Number of interventions
Pharmaceutical treatment
Placebo treatment
Dosage of trial drug
Co‐interventions (comparable/avoided)
Durations of trial
Outcome measures
Outcome definition
Primary outcome measure
Secondary Outcome measures
If measure is a scale include upper and lower limits and whether high or low score is positive
Outcomes and time points collected
Intervention effect on the outcome measures chosen for the study
Short term assessment
Long term assessment
Results
Sample size
Withdrawal/drop‐out rate
Missing participants
Compliance to treatment reported
Summary data for each intervention group. 2 × 2 table for dichotomous data; means and SDs for continuous data
Estimate of effect with confidence interval and P value
Subgroup analyses
Intention to treat
Safety
Data and analyses
Comparison 1. Systemic drug versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Function | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.90, 0.50] |
| 1.1 Disability Assessment Scale | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.90, 0.50] |
| 2 Spasticity ‐ indirect measure | 2 | 160 | Odds Ratio (M‐H, Random, 95% CI) | 1.66 [0.21, 13.07] |
| 3 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Adverse events per participant | 2 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.12, 2.42] |
| 3.2 Participants withdrawn due to side effects | 2 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.04, 34.40] |
Comparison 2. Systemic drug versus another systemic drug.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Function | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Gait | 1 | 79 | Mean Difference (IV, Random, 95% CI) | ‐183.3 [‐330.40, ‐36.20] |
| 2 Function dichotomous | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Pedersen Scale ‐ upper limb | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.23, 4.31] |
| 2.2 Pedersen Scale ‐ lower limb | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.17, 5.98] |
| 3 Spasticity ‐ indirect measure | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Tizanidine versus baclofen | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 2.36 [0.36, 15.45] |
| 3.2 Tizanidine versus diazepam | 1 | 78 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.37, 2.20] |
| 4 Clonus | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Tizanidine versus baclofen | 1 | 12 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.04, 9.65] |
| 4.2 Tizanidine versus diazepam | 1 | 49 | Odds Ratio (M‐H, Random, 95% CI) | 1.4 [0.44, 4.44] |
| 5 Muscle spasms | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Tizanidine versus baclofen | 1 | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.03, 4.40] |
| 6 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Per participant ‐ tizanidine versus baclofen | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.1 [0.83, 5.30] |
| 6.2 Total‐ tizanidine versus baclofen | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.94, 1.69] |
| 6.3 Total ‐ tizanidine versus diazepam | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [1.01, 1.48] |
| 6.4 Withdrew ‐ tizanidine versus baclofen | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 2.38] |
| 6.5 Withdrew ‐ tizanidine versus diazepam | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.18, 1.01] |
Comparison 3. Systemically acting drug versus a locally acting drug.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Function | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Action Research Arm Test | 1 | 68 | Mean Difference (IV, Random, 95% CI) | ‐3.79 [‐6.93, ‐0.65] |
| 2 Adverse events | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 41.00 [2.58, 651.70] |
Comparison 4. Locally acting drug versus another locally acting drug.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Function | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Gait (25 feet timed walk) | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 2.70 [‐2.83, 8.23] |
| 2 Passive range of movement | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Phenol versus alcohol | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 1.0 [‐7.00, 11.00] |
| 3 Spasticity ‐ neurophysiological | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 H:M Ratio | 1 | 20 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.48, 0.46] |
| 3.2 H‐reflex | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 1.24 [‐1.44, 3.92] |
| 4 Spasticity ‐ indirect measure | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Phenol versus alcohol | 1 | 20 | Odds Ratio (M‐H, Random, 95% CI) | 0.26 [0.02, 3.06] |
| 5 Clonus | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Phenol versus alcohol | 1 | 12 | Odds Ratio (M‐H, Random, 95% CI) | 8.56 [0.34, 212.94] |
| 6 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Total ‐ phenol versus alcohol | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.41, 120.16] |
| 6.2 Total ‐ phenol versus botulinum toxin | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.41, 120.16] |
4.6. Analysis.

Comparison 4 Locally acting drug versus another locally acting drug, Outcome 6 Adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bes 1988.
| Methods | Randomised, double‐blind, comparative study Objective: to compare the efficacy and tolerability of tizanidine with that of diazepam |
|
| Participants | France 105 recruited adults (28 females, 77 males), of which 89 had a diagnosis of stroke, were randomly divided in 2 groups (the other 16 had TBI) Tizanidine group had a mean age of 51.4 (SD 2.2) Diazepam group had a mean age of 51.8 (SD 2.4) |
|
| Interventions | A starting dose of one 2 mg tizanidine (Sirdalud) capsule was titrated up by 2 mg every 2 days to a maximum of 24 mg/day A starting dose of one 2.5 mg diazepam capsule was titrated up by 2.5 mg every 2 days to a maximum of 30 mg/day |
|
| Outcomes | Dichotomised data for clonus, spasticity, muscle strength. The stretch reflex was analysed during extension at a constant rate (defined as the rate of fall of the limb due to gravity) of four groups of muscles ‐ forearm flexors, quadriceps, knee flexors and triceps surae. The duration of any resulting contracture and the angle at which it appeared were noted Walking distance was also measured. Measures at 2 and 8 weeks |
|
| Spasticity and inclusion | No definition of spasticity provided. but inclusion criteria specified that spasticity "was severe enough to interfere with daily activities". Spasticity was measured using a 1 to 5 rating scale | |
| Notes | Not able to make contact with authors (26 years since study) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Allocation concealment (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Appears that capsules for both drugs were the same so that investigator and participant were blinded to treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is unclear whether the evaluating physician was blinded at all outcomes |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 28% of 1 group was lost to follow‐up at 8‐week assessment |
| Selective reporting (reporting bias) | High risk | All assessment measures described in the methodology were subsequently reported. The authors have deemed reporting bias to be high risk because a statistically significant improvement in walking distance on flat ground was reported in the tizanidine group only. On calculation, both the tizanidine and diazepam group had made statistically significant improvements |
Kirazli 1998.
| Methods | Randomised, double‐blind comparison study Objective: "Does botulinus toxin Type A and phenol relieve the signs and symptoms of ankle plantar flexor and foot invertor spasticity after stroke and do either of these methods offer any advantages and disadvantages over the other?" |
|
| Participants | Turkey 20 adults (6 female, 14 male) Mean age of 50.3 years (SD 17.8) with a diagnosis of stroke (10 thrombotic, 6 embolic, 4 haemorrhagic) at least 6 months ago were randomly divided into 2 groups. |
|
| Interventions | 3 ml of 5% phenol injected into tibial nerve approximately 7 cm above popliteal crease and localised by percutaneous stimulation Total of 400 units onabotulinumtoxinA (50 units/1 ml injected with electrical stimulation guidance) 100 units into soleus, 100 units into tibialis posterior, 100 units into lateral head of gastrocnemius, and 100 units into medial head of gastrocnemius |
|
| Outcomes | Electrophysiological measures (Achilles Tendon Reflex (ATR) (milliVolts (mV)), M‐wave (mV), H‐reflex (mV), H:M ratio, ATR:H ratio) Ashworth Scale: mean and standard deviations at ankle dorsiflexion and eversion Timed walk (25 feet) (seconds) Clonus duration (seconds) Global assessment of spasticity: participant subjective report Dichotomous improvement in active and passive range of movement at ankle and in wearing of ankle brace Adverse events |
|
| Spasticity and inclusion | Spasticity was defined as "typically includes hyperactive phasic stretch reflexes with velocity dependent hypertonus" To be included spasticity had to be "severe" (Ashworth Scale = 3 or 4) and stable for at least 2 months (0 = no increase in muscle tone, 4 = limb rigid in flexion or extension) |
|
| Notes | We contacted the 2 lead authors by email but received no response. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No explanation provided in either report |
| Allocation concealment (selection bias) | Unclear risk | No explanation provided in either report |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | 2 different procedures carried out by the injector means they were not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Clear description of blinding by assessor |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants were accounted for |
| Selective reporting (reporting bias) | Low risk | All assessment measures described in the methodology of both reports were subsequently reported |
Kocabas 2010.
| Methods | Randomised, double‐blind comparison study Objective: "To determine whether the injection of alcohol or phenol into the tibialis posterior nerve relieves symptoms and signs of ankle plantar flexor spasticity." |
|
| Participants | Turkey 20 adults (6 female, 14 male) with a diagnosis of stroke (14 infarct and 6 haemorrhagic) were randomly divided into 2 groups Alcohol group had mean age of 55.6 (SD 13.7) and 21.4 (SD 16.0) months poststroke Phenol group had mean age of 51.9 (SD 27.7) and 22.8 (SD 27.8) months poststroke |
|
| Interventions | 5 ml of 5% phenol – motor branches of tibial nerve 5 ml of 50% ethyl alcohol – motor branches of tibial nerve |
|
| Outcomes | Modified Ashworth Scale Clonus Score: 0 (absent), 1 (unsustained), 2 (sustained), 3 (spontaneous/light‐touch provoked) Dichotomous changes in clonus and strength Passive range of movement in degrees |
|
| Spasticity and inclusion | Spasticity was not defined. To be included the person had to have "severe ankle plantar flexor spasticity" (Modified Ashworth Scale ≥ 2) (0 = no increase in muscle tone, 4 = limb rigid in flexion or extension) | |
| Notes | Grateful thanks to Dr Hilal Kocabas who responded to our request for further details | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Pitch and toss method used for randomisation |
| Allocation concealment (selection bias) | Unclear risk | Given method of randomisation, allocation concealment was potentially at risk; as it was not possible to be sure, we judged it as unclear risk |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | It is unclear whether the injecting physician was blinded to which drug the syringe contained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of outcome assessor is not explicitly stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants were accounted for |
| Selective reporting (reporting bias) | High risk | Information on strength was not reported adequately to identify changes. While complications occurred in gait, it was unclear in which groups this occurred |
Medici 1989.
| Methods | Randomised, double‐blind, comparative study Objective: To compare the long‐term efficacy and tolerability of tizanidine with that of baclofen in the treatment of spasticity due to cerebrovascular lesions |
|
| Participants | Uruguay 30 adults (6 female, 24 male) with a diagnosis of stroke (a mean 3.5 (0.1 to 14) years before) were randomly divided into 2 groups Tizanidine (Sirdalud) group had mean age 50 years (22 to 73) Baclofen group had mean age of 49 years (24 to 68) |
|
| Interventions | A starting dose of 2 x 4 mg tizanidine capsules was titrated up by 4 mg every 3 days to a maximum of 20 mg/day A starting dose of 2 x 10 mg baclofen capsules was titrated up by 10 mg every 3 days to a maximum of 50 mg/day |
|
| Outcomes | Dichotomised results of muscle tone (measured using the Ashworth Scale). Ordinal scales for muscle spasms, clonus, and strength were studied. Functional assessment used the Pedersen scale of the arm or leg. A participant self assessment of disability ordinal 4‐point scale and participant and assessor assessment of antispastic effect using an ordinal 4‐point scale Endpoint analysis not explicitly stated but appears to be 12 months |
|
| Spasticity and inclusion | Spasticity was not defined, and the level of spasticity required to participate in the trial was not reported | |
| Notes | Not able to make contact with authors (25 years since study) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Allocation concealment (selection bias) | Unclear risk | There is no description of how allocation concealment was achieved |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | It appears that capsules for both drugs were the same so that investigator and participant were blinded to treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Details on blinding of the assessor are not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT and per‐protocol data provided |
| Selective reporting (reporting bias) | Low risk | All assessment measures described in the methodology were subsequently reported |
Simpson 2009.
| Methods | Randomised, double‐blind, 3‐arm, placebo‐controlled study Due to mandate of review, only the tizanidine and placebo groups are reported ‐ the botulinum toxin arm is not reported |
|
| Participants | USA 40 adults (19 female, 21 male) of which 32 had a diagnosis of stroke were randomly divided into 2 groups (the other 8 had TBI) Tizanidine group had mean age of 51.9 years (SD 17.3) Placebo group had mean age of 51.3 years (SD 14.7) |
|
| Interventions | Tizanidine starting dose of 2 mg/day titrated up by 4 mg every 3 to 4 days to a maximum of 36 mg/day; also received at least 2 placebo injections to forearm muscles Placebo group provided with placebo oral medication; also received at least 2 placebo injections to forearm muscles |
|
| Outcomes | Modified Ashworth Scale at the wrist and finger flexors Modified Ashworth dichotomised Disability Assessment Scale: 1 domain was identified by the participant and assessor as the primary therapeutic target Modified Frenchay Scale, 10‐Metre Walk Test, contralateral grip strength, and Finger Tap Test Other measures included Epworth Sleepiness Scale, Geriatric Depression Scale, and Letter‐Number Sequencing |
|
| Spasticity and inclusion | Spasticity was not defined. Evidence of spasticity was a score of ≥ 3 on the Modified Ashworth Scale (0 normal tone and 5 rigid flexion) | |
| Notes | We contacted 2 of the authors by email but received no response | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Allocation concealment (selection bias) | Low risk | The review authors assume that allocation concealment was maintained |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both participants and injectors were blinded to treatment. Clear explanation provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The outcome assessor was blinded to the treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition was documented for all groups and explanations provided. At primary endpoint 93% of participants were assessed. We noted that there was a high attrition rate between week 6 and week 22 |
| Selective reporting (reporting bias) | High risk | Planned data for report were presented at primary endpoint in the report; however, there has been no follow‐up report of Modified Frenchay Scale and 10‐Metre Walk Test results from the study |
Stamenova 2005.
| Methods | Randomised, double‐blind, placebo‐controlled study Objective: To study the efficacy and safety of tolperisone in the treatment of stroke‐related spasticity |
|
| Participants | Bulgaria and Germany 120 adults (43 female, 77 male) with a diagnosis of stroke (a mean 3.3 (SD 4.4) years before) were randomly divided into 2 groups Tolperisone group had mean age of 64.2 years (SD 10.9) Placebo group had mean age of 62.3 years (SD 10.2) |
|
| Interventions | The starting dose was 2 x 50 mg tablets of tolperisone hydrochloride (Mydocalm) 3 times a day The dose was titrated up to 3, 4, or 6 tablets 3 times a day at 5‐day intervals so that a maximum dose of 900 mg tolperisone was achieved Placebo tablets were given in the same dose and titrated up as per the drug protocol |
|
| Outcomes | The mean change to baseline in the Barthel Index (0 to 100) and Modified Ashworth Scale was reported The Modified Ashworth was dichotomised, and overall efficacy was dichotomised in graphical form 2‐Minute Walk Test was carried out Elbow stiffness was measured using a spring balance Capacity to perform routine activities on a 5‐point ordinal scale Overall efficacy was measured by participant and assessor using a 4‐point ordinal scale |
|
| Spasticity and inclusion | Spasticity was defined as a velocity‐dependent increase in the resistance of muscles to passive stretch associated with exaggerated tendon jerks To be included the person had to have an Ashworth Scale ≥ 2 in at least 1 joint |
|
| Notes | We contacted the corresponding author by email but received no response | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Allocation concealment (selection bias) | Unclear risk | There is no description of how allocation concealment was achieved |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participant blinding appears to have been well maintained. Tablets reported as having same appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is unclear whether the evaluating physician was blinded at all outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT and per‐protocol data provided |
| Selective reporting (reporting bias) | High risk | The results of the force required to extend the elbow are not reported |
| Other bias | High risk | This study appears to have had pharmaceutical industry involvement; however, there is no declaration of interests. The corresponding author was an employee of the company that produced the drug under investigation |
Yazdchi 2013.
| Methods | Randomised, double‐blind, comparative study Objective: "To evaluate the efficacy of focal intramuscular injection of botulinum toxin type A (BoNT) in comparison with oral tizanidine in treatment of post‐stroke upper extremity’s spasticity" |
|
| Participants | Iran 68 adults (29 females, 39 males) Mean age of 66.1 years (35 to 70) with a diagnosis of stroke at least 3 months ago were randomly divided into 2 groups |
|
| Interventions | A starting dose of one 2 mg tizanidine (Sirdalud) (Novartis) capsule was titrated up by 2 mg every week to a maximum of 24 mg/day at week 12 Repeated injections at baseline and 12 weeks; maximum dose of 1000 units of abobotulinumtoxinA into arm muscles deemed appropriate by clinician |
|
| Outcomes | Elbow and wrist Modified Ashworth scale and Action Research Arm Test | |
| Spasticity and inclusion | "Spasticity is an increased resistance impassive movement at a joint that depends upon velocity of an action." Adults with a minimum score of 2 on the Modified Ashworth scale were included | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Allocation concealment (selection bias) | Unclear risk | There is no description of how sequence generation was achieved |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants were either injected or not, indicating that they were not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessor was blinded to treatment at all points |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 20 participants in 1 group withdrew due to side effects, whereas no participants in the other group withdrew. Other eligible participants replaced those who withdrew |
| Selective reporting (reporting bias) | Unclear risk | All assessment measures described in the methodology were subsequently reported. |
H‐reflex: Hoffmann reflex ITT: intention‐to‐treat SD: standard deviation TBI: traumatic brain injury
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Basmajian 1984 | Cross‐over study 1984; unable to contact |
| Bovier 1985 | Cross‐over study 1985; unable to contact |
| Burke 1975 | < 80% diagnosed with stroke (21% stroke) |
| Burt 1978 | Cross‐over study 1978; unable to contact |
| Caruso 1977 | < 80% diagnosed with stroke (61.5% stroke) |
| Chipman 1974 | < 80% diagnosed with stroke (23% stroke) |
| Civardi 1994 | Cross‐over study 1994; unable to contact |
| Glass 1974 | Unclear diagnosis of participants who were in RCT |
| Harvey 1974 | < 80% diagnosed with stroke (21% stroke) |
| Katrak 1992 | Cross‐over study; emailed first author but received no response |
| Knutsson 1982 | < 80% diagnosed with stroke (6% stroke) |
| Manca 2010 | < 80% diagnosed with stroke (77% stroke) Emailed first author but received no response |
| Maupas 2004 | Cross‐over study; emailed first author but received no response |
| Medaer 1991 | Cross‐over study 1991; unable to contact |
| Meythaler 2001 | Cross‐over study; emailed first author but received no response |
| Meythaler 2001a | < 80% diagnosed with stroke (53% stroke) Emailed first author but received no response |
| Monster 1973 | < 80% diagnosed with stroke (34% stroke) |
| Monster 1974 | < 80% diagnosed with stroke (35% stroke) |
| Thompson 1966 | < 80% diagnosed with stroke (39% stroke) |
RCT: randomised controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
Arbizu 1988.
| Methods | Randomised, double‐blind, comparative study |
| Participants | 27 adults (8 females, 19 males) |
| Interventions | Tizanidine (Sirdalud) and baclofen |
| Outcomes | Ashworth Scale |
| Language | Spanish |
| Notes | Information from abstract |
Cui 2009.
| Methods | Randomised, 4‐arm, placebo‐controlled study |
| Participants | 103 adults diagnosed with stroke |
| Interventions | Tizanidine (n = 30), baclofen (n = 25), eperisone (n = 22), and control (n = 26) |
| Outcomes | Modified Ashworth Scale, Fugl‐Meyer Assessment, and Modified Barthel Index 4 weeks prior to trial and 12 weeks after the medication |
| Language | Chinese |
| Notes | Information from abstract |
Gan 2004.
| Methods | RCT |
| Participants | 60 adults diagnosed with stroke |
| Interventions | 50 mg eperisone 3 times/day for 1 month; control group unclear |
| Outcomes | Ashworth Scale, Brunnstrom at weeks 2 and 4 |
| Language | Chinese |
| Notes | Information from abstract |
Gekht 1998.
| Methods | Unclear |
| Participants | 94 adults diagnosed with stroke (aged 42 to 70 years) |
| Interventions | Tizanidine (Sirdalud) |
| Outcomes | Not reported |
| Language | Russian |
| Notes | Information from abstract |
Kim 2003.
| Methods | RCT. Objective: "To evaluate quantitatively the effect of tizanidine on spasticity reduction and to evaluate the effective and tolerable dosage of it in stroke patients" |
| Participants | 24 adults diagnosed with stroke divided into 2 groups |
| Interventions | Tizanidine and control group |
| Outcomes | Biomechanical‐neurophysiological assessment |
| Language | Korean |
| Notes | Information from abstract |
Kovalchuk 2008.
| Methods | Unclear |
| Participants | 360 adults diagnosed with stroke |
| Interventions | Tolperisone, tizanidine, and others |
| Outcomes | Ashworth, Barthel, Lindmark, Scandinavian, and Merton & Sutton scales, 2‐Minute Walk Test |
| Language | Russian |
| Notes | Information from abstract |
Kovalchuk 2013.
| Methods | Not clear what methodology was used |
| Participants | 1700 adults diagnosed with stroke divided into 2 groups (850 in each group) |
| Interventions | Tolperisone and control group |
| Outcomes | Ashworth, Barthel, Lindmark, Scandinavian, and Merton & Sutton scales |
| Language | Russian |
| Notes | Information from abstract |
Musiol 1977.
| Methods | Unclear |
| Participants | 12 adults |
| Interventions | Baclofen (Lioresal) |
| Outcomes | Novel grade system (1 = normal tonus; 5 = high‐grade hypertonus) |
| Language | Polish |
| Notes | Abstract in Polish |
Prati 1974.
| Methods | Unclear |
| Participants | ‐ |
| Interventions | Baclofen |
| Outcomes | ‐ |
| Language | Italian |
| Notes | Abstract in Italian |
Stamenova 2006.
| Methods | Randomised, double‐blind, placebo‐controlled trial |
| Participants | 120 adults diagnosed with stroke |
| Interventions | Tolperisone |
| Outcomes | ‐ |
| Language | Russian |
| Notes | Appears to be the same study as Stamenova 2005 |
Weynants 1990.
| Methods | Unclear |
| Participants | 20 adults (17 diagnosed with stroke), mean age 50 years (range 15 to 69) |
| Interventions | Tizanidine |
| Outcomes | ‐ |
| Language | Dutch |
| Notes | Abstract in Dutch |
Yao 2004.
| Methods | RCT |
| Participants | 144 adults with stroke |
| Interventions | Baclofen (n = 84), control (n = 60) |
| Outcomes | Ashworth, Fugl‐Meyer Assessment, and Modified Barthel Index |
| Language | Chinese |
| Notes | Information from abstract |
RCT: randomised controlled trial
Differences between protocol and review
Outcome measures
We expected to identify studies that investigated varying doses of the same drug. In the protocol we explained how we would deal with such studies. However, we did not identify any such trials, so we have not included this section in the methodology.
We provided examples in our protocol of functional activity scales of the upper limb and lower limb that might be included, such as Action Research Arm Test (ARAT), appropriate subsections of motor assessment scale or Wolf Motor Function Test or Functional Ambulatory Classification or Functional Gait Assessment. It was not possible to analyse the data due to a lack of such objective measures in the included studies.
This was also the case for timed measures of specific activities. Again, such temporal measures can be divided into upper limb and lower limb measures. Upper limb temporal measures might have included the 9‐Hole Peg Test or Jebsen Hand Function Test. Lower limb temporal measures might have included the Timed 10‐Metre Walk Test or Get‐up and Go Test.
Risk of bias
The protocol for this review explained that we would investigate risk of bias in two specific domains first: 'selection bias' and 'performance and detection bias'. Only in studies deemed to have a low risk of bias in these two domains would we carry out further analysis. We suggested in the protocol that we would exclude studies deemed to have a high risk of bias in these two domains. We investigated all included studies for risk of bias in all the domains at the same time point because this was easier for the review authors, but also because we were not going to exclude papers deemed at high risk in these two domains.
Reporting bias
We planned to use a funnel plot of the standard error of the intervention effect to identify asymmetry. This was not completed due to a lack of studies.
Results
The results were presented differently in different studies. Some studies only presented the response at a single joint, other studies presented the results of the joints where most effect was observed, and other studies presented the results of a number of joints. In order to present the results uniformly, we have provided the most conservative results. An example of this are the results from Simpson 2009. The results at the wrist showed 12 people in the placebo group improved versus five in the treatment group, and at the fingers seven in the placebo group and seven in the treatment group improved. In this case we used the five from the wrist to represent the treatment group and the seven from the finger to represent the placebo group. In Bes 1988, arm and leg joints were assessed, but not all participants had spasticity at every joint. We analysed the results of the wrist flexors, as the most participants had spasticity at this joint.
Subgroup analysis and investigation of heterogeneity
We could not conduct any of the intended subgroup analyses because of the lack of data.
Contributions of authors
Cameron Lindsay led the review. Conception and design of the protocol for the review was done in conjunction with Anand Pandyan. Cameron Lindsay ran the searches, excluded the irrelevant references, and applied inclusion criteria during searching. He carried out trial detail, methodology and data extraction. He carried out data analysis and wrote the initial draft of the review.
Anand Pandyan acted as the second review author. Conception and design of the protocol for the review was done in conjunction with Cameron Lindsay. Anand Pandyan carried out data analysis and commented on drafts of the review.
Aphrodite Kouzouna ran the searches, excluded irrelevant references, and applied inclusion criteria during searching. She commented on drafts of the review.
Christopher Simcox carried out trial detail, methodology and data extraction. He commented on drafts of the review.
Sources of support
Internal sources
-
Cameron Lindsay, UK.
Keele University Studentship
External sources
No sources of support supplied
Declarations of interest
CL, CS, and ADP have previously received unconditional funding from Allergan and Ipsen drug companies. ADP has also received unconditional funding from Merz drug company.
AK has no known declarations of interest. In accordance with Cochrane's policy on commercial sponsorship, no money was received from a commercial source to fund this review.
New
References
References to studies included in this review
Bes 1988 {published data only}
- Bes A, Eyssette M, Pierrot‐Deseilligny E, Rohmer F, Warter JM. A multi‐centre, double‐blind trial of tizanidine, a new antispastic agent, in spasticity associated with hemiplegia. Current Medical Research and Opinion 1988;10(10):709‐18. [DOI] [PubMed] [Google Scholar]
Kirazli 1998 {published data only (unpublished sought but not used)}
- Kirazli Y, On AY, Kismali B, Aksit R. Comparison of phenol block and botulinus toxin type A in the treatment of spastic foot after stroke: a randomized, double‐blind trial. American Journal of Physical Medicine & Rehabilitation 1998; Vol. 77, issue 6:510‐5. [DOI] [PubMed]
- On AY, Kirazli Y, Kismali B, Aksit R. Mechanisms of action of phenol block and botulinus toxin type A in relieving spasticity: electrophysiologic investigation and follow‐up. American Journal of Physical Medicine & Rehabilitation 1999;78(4):344‐9. [DOI] [PubMed] [Google Scholar]
Kocabas 2010 {published data only (unpublished sought but not used)}
- Kocabas H, Salli A, Demir AH, Ozerbil OM. Comparison of phenol and alcohol neurolysis of tibial nerve motor branches to the gastrocnemius muscle for treatment of spastic foot after stroke: a randomized controlled pilot study. European Journal of Physical & Rehabilitation Medicine 2010;46(1):5‐10. [PubMed] [Google Scholar]
Medici 1989 {published data only}
- Medici M, Pebet M, Ciblis D. A double‐blind, long‐term study of tizanidine ('Sirdalud') in spasticity due to cerebrovascular lesions. Current Medical Research and Opinion 1989;11(6):398‐407. [DOI] [PubMed] [Google Scholar]
Simpson 2009 {published data only (unpublished sought but not used)}
- Simpson DM, Gracies JM, Yablon SA, Barbano R, Brashear A, Team TBS. Botulinum neurotoxin versus tizanidine in upper limb spasticity: a placebo‐controlled study. Journal of Neurology, Neurosurgery & Psychiatry 2009;80(4):380‐5. [DOI] [PubMed] [Google Scholar]
Stamenova 2005 {published data only (unpublished sought but not used)}
- Stamenova P, Koytchev R, Kuhn K, Hansen C, Horvath F, Ramm S, et al. A randomized, double‐blind, placebo‐controlled study of the efficacy and safety of tolperisone in spasticity following cerebral stroke. European Journal of Neurology 2005;12(6):453‐61. [DOI] [PubMed] [Google Scholar]
Yazdchi 2013 {published data only (unpublished sought but not used)}
- Yazdchi M, Ghasemi Z, Moshayedi H, Rikhtegar R, Mostafayi S, Mikailee H, et al. Comparing the efficacy of botulinum toxin with tizanidine in upper limb post stroke spasticity. Iranian Journal of Neurology 2013;12(2):47‐50. [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Basmajian 1984 {published data only}
- Basmajian JV, Shankardass K, Russell D. Ketazolam treatment for spasticity: double‐blind study of a new drug. Archives of Physical Medicine and Rehabilitation 1984;65(11):698‐701. [PubMed] [Google Scholar]
Bovier 1985 {published data only}
- Bovier P, Cambier J, Morselli PL. Double‐blind trial of progabide in the treatment of spastic syndrome. Therapie 1985;40(3):181‐5. [PubMed] [Google Scholar]
Burke 1975 {published data only}
- Burke D, Hammond C, Skuse N, Jones RF. A phenothiazine derivative in the treatment of spasticity. Journal of Neurology, Neurosurgery & Psychiatry 1975; Vol. 38, issue 5:469‐74. [DOI] [PMC free article] [PubMed]
Burt 1978 {published data only}
- Burt AA, Currie S. A double blind controlled trial of baclofen and diazepam in spasticity due to cerebrovascular lesions. Baclofen: Spasticity and Cerebral Pathology. 1st Edition. Cambridge: Cambridge Medical, 1978:77‐9. [Google Scholar]
Caruso 1977 {published data only}
- Caruso G, Fels A, Santoro L, Perretti A. Effects of dantrolene sodium on pyramidal hypertonia. Clinico‐electrophysiological study. Acta Neurologica 1977; Vol. 32, issue 6:872‐83. [PubMed]
Chipman 1974 {published data only}
- Chipman M, Kaul S, Lambie M. Efficacy of dantrolene sodium in the treatment of spasticity. Diseases of the Nervous System 1974;35(9):427‐31. [PubMed] [Google Scholar]
Civardi 1994 {published data only}
- Civardi C, Naldi P, Cantello R, Gianelli M, Mutani R. Protirelin tartrate (TRH‐T) in upper motoneuron syndrome: a controlled neurophysiological and clinical study. Italian Journal of Neurological Sciences 1994;15(8):395‐406. [DOI] [PubMed] [Google Scholar]
Glass 1974 {published data only}
Harvey 1974 {published data only}
- Harvey MS. A comparative trial of dimethothiazine in spastic conditions. A report from the General Practitioner Research Group. Practitioner 1974;213:101‐5. [PubMed] [Google Scholar]
Katrak 1992 {published data only}
- Katrak PH, Cole AM, Poulos CJ, McCauley JC. Objective assessment of spasticity, strength, and function with early exhibition of Dantrolene sodium after cerebrovascular accident: a randomized double‐blind study. Archives of Physical Medicine and Rehabilitation 1992; Vol. 73, issue 1:4‐9. [PubMed]
Knutsson 1982 {published data only}
- Knutsson E, Martensson A, Gransberg L. Antiparetic and antispastic effects induced by tizanidine in patients with spastic paresis. Journal of the Neurological Sciences 1982;53(2):187‐204. [DOI] [PubMed] [Google Scholar]
Manca 2010 {published data only}
- Manca M, Merlo A, Ferraresi G, Cavazza S, Marchi P. Botulinum toxin type A versus phenol. A clinical and neurophysiological study in the treatment of ankle clonus. European Journal of Physical and Rehabilitation Medicine 2010;46(1):11‐8. [PubMed] [Google Scholar]
Maupas 2004 {published data only}
- Maupas E, Marque P, Roques CF, Simonetta‐Moreau M. Modulation of the transmission in group II heteronymous pathways by tizanidine in spastic hemiplegic patients. Journal of Neurology, Neurosurgery & Psychiatry 2004;75(1):130‐5. [PMC free article] [PubMed] [Google Scholar]
Medaer 1991 {published data only}
- Medaer R, Hellebuyk H, Brande E, Saxena V, Thijs M, Kovacs L, et al. Treatment of spasticity due to stroke. A double‐blind, cross‐over trial comparing baclofen with placebo. Acta Therapeutica 1991;17(4):323‐31. [Google Scholar]
Meythaler 2001 {published data only}
- Meythaler JM, Guin‐Renfroe S, Brunner RC, Hadley MN. Intrathecal baclofen for spastic hypertonia from stroke. Stroke. Department of Physical Medicine and Rehabilitation, Division of Neurological Surgery, University of Alabama at Birmingham School of Medicine, Birmingham, USA. JMeythal@uab.edu, 2001; Vol. 32, issue 9:2099‐109. [DOI] [PubMed]
Meythaler 2001a {published data only}
Monster 1973 {published data only}
- Monster AW, Herman R, Meeks S, McHenry J. Cooperative study for assessing the effects of a pharmacological agent on spasticity. American Journal of Physical Medicine 1973; Vol. 52, issue 4:163‐88. [PubMed]
Monster 1974 {published data only}
- Monster AW. Spasticity and the effect of dantrolene sodium. Archives of Physical Medicine and Rehabilitation 1974; Vol. 55, issue 8:373‐83. [PubMed]
Thompson 1966 {published data only}
- Thompson J. Carisoprodol in the treatment of muscle spasticity in the elderly. Gerontologia Clinica 1966;8(2):100‐5. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Arbizu 1988 {published data only}
- Arbizu Tx, Martinez JA, Rubio F, Aguilar M, Perez J. Clinical evaluation and tolerance of tizanidine (DS 103‐282) and baclofen in patients with chronic spasticity due to cerebrovascular accidents. Revista Espanola de Neurologia 1988;3:291‐6. [Google Scholar]
Cui 2009 {published data only}
- Cui L‐H, Zhang T, Yang L‐Y. Efficacy of three antispasmodics on limb spasticity in patients after stroke: a comparative analysis. Chinese Journal of Cerebrovascular Diseases 2009;6(9):466‐70. [Google Scholar]
Gan 2004 {published data only (unpublished sought but not used)}
- Gan Z, Xiao W‐M, Chen Y‐K. Eperisone in the treatment of hypermyotonia following stroke: a randomised controlled observation. Chinese Journal of Clinical Rehabilitation 2004;8(13):2412‐3. [Google Scholar]
Gekht 1998 {published data only}
- Gekht AB, Burd GS, Selikhova MV, Iaish F, Beliakov VV. Disorders of muscle tonus and their treatment with sirdalud in patients in the early recovery period of ischemic stroke. Zhurnal Nevrologii I Psikhiatrii Imeni SS Korsakova 1998;98(10):22‐9. [PubMed] [Google Scholar]
Kim 2003 {published data only (unpublished sought but not used)}
- Kim DY, Park C, Na S, Park YS. Quantitative assessment of the effect of tizanidine on spasticity in stroke patients. Journal of the Korean Academy of Rehabilitation Medicine 2003;27:471‐9. [Google Scholar]
Kovalchuk 2008 {published data only (unpublished sought but not used)}
- Kovalchuk VV, Skoromets AA, Vasil'eva IV. Comparative efficacy of different muscle relaxants in the rehabilitation of post‐stroke patients with spasticity. Zhurnal Nevrologii I Psikhiatrii Imeni SS Korsakova 2008;108(8):14‐9. [PubMed] [Google Scholar]
Kovalchuk 2013 {published data only}
- Kovalchuk VV. Evaluation of efficacy and safety of Mydocalm in the early rehabilitation of stroke. Zhurnal Nevrologii I Psihiatrii Imeni SS Korsakova 2013; Vol. 113, issue 4:35‐40. [PubMed]
Musiol 1977 {published data only}
- Musiol A, Kazibutowska Z, Kurzbauer H. Lioresal in the treatment of hemiplegia. Clinical and electromyographic evaluation. Preliminary report. Neurologia i Neurochirurgia Polska 1977;11(6):661‐7. [PubMed] [Google Scholar]
Prati 1974 {published data only}
- Prati R, Soriani S. Use of beta‐(p‐clorophenil)‐gamma‐aminobutirric acid in the treatment of hemiplegic patients with hypertonius. Rivista di Patologia Nervosa e Mentale 1974;95(5):651‐2. [PubMed] [Google Scholar]
Stamenova 2006 {published data only}
- Stamenova P, Koytchev R, Kuhn K, Hanasen C, Horvath F, Ramm S, et al. A randomized, double blind, placebo‐controlled study of the efficacy and safety of tolperisone in spasticity following cerebral stroke. Zhurnal Nevrologii I Psikhiatrii Imeni SS Korsakova 2006; Vol. 106, issue 1:34‐42. [PubMed]
Weynants 1990 {published data only}
- Weynants L, Laere M. Tizanidine in the treatment of spasticity in patients with hemiplegia caused by CVA or brain trauma. Acta Belgica Medica Physica 1990;13(1):19‐22. [PubMed] [Google Scholar]
Yao 2004 {published data only (unpublished sought but not used)}
- Yao J‐R, Wang D‐S, Ni X‐B, Rong D‐M, Zheng H‐M. Efficacy of baclofen combined with rehabilitation training in stroke patients with spastic hemiplegia. Chinese Journal of Clinical Rehabilitation 2004;8(10):1814‐5. [Google Scholar]
Additional references
Ada 2006
- Ada L, O'Dwyer N, O'Neill E. Relation between spasticity, weakness and contracture of the elbow flexors and upper limb activity after stroke: an observational study. Disability & Rehabilitation 2006;28(13):891‐7. [DOI] [PubMed] [Google Scholar]
Adamson 2004
Barnes 1998
- Barnes MP. Management of spasticity. Age and Ageing 1998;27(2):239‐45. [DOI] [PubMed] [Google Scholar]
Bogey 2004
- Bogey RA, Geis CC, Bryant PR, Moroz A, O'Neill BJ. Stroke and neurodegenerative disorders. 3. Stroke: rehabilitation management. Archives of Physical Medicine and Rehabilitation 2004;85(3):S15‐20, S34‐40. [DOI] [PubMed] [Google Scholar]
Burke 1988
- Burke D. Spasticity as an adaptation to pyramidal tract injury. Advances in Neurology 1988;47:401. [PubMed] [Google Scholar]
Daniel 1999
- Daniel WW. Biostatistics: A Foundation for Analysis in the Health Sciences. 7th Edition. London: Wiley and Sons, 1999. [Google Scholar]
Davidoff 1985
- Davidoff RA. Antispasticity drugs: mechanisms of action. Annals of Neurology 1985;17(2):107‐16. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Esquenazi 2011
- Esquenazi A. The human and economic burden of poststroke spasticity and muscle overactivity. Journal of Clinical Outcomes Management 2011;18(1):34‐44. [Google Scholar]
Fleuren 2010
- Fleuren JF, Voerman GE, Erren‐Wolters CV, Snoek GJ, Rietman JS, Hermens HJ, et al. Stop using the Ashworth scale for the assessment of spasticity. Journal of Neurology, Neurosurgery & Psychiatry 2010;81(1):46‐52. [DOI] [PubMed] [Google Scholar]
Gallichio 2004
- Gallichio JE. Pharmacologic management of spasticity following stroke. Physical Therapy 2004;84(10):973‐81. [PubMed] [Google Scholar]
Gormley 1997
- Gormley ME, O'Brien CF, Yablon SA. A clinical overview of treatment decisions in the management of spasticity. Muscle and Nerve Supplement 1997;20(S6):S14‐20. [PubMed] [Google Scholar]
Gracies 1997
- Gracies JM, Nance P, Elovic E, McGuire J, Simpson DM. Traditional pharmacological treatments for spasticity. Part II: General and regional treatments. Muscle & Nerve Supplement. Wiley, 1997; Vol. 20, issue S6:S92‐120. [PubMed]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Howe 1987
- Howe JR, Sutor B, Zieglgänsberger W. Baclofen reduces post‐synaptic potentials of rat cortical neurones by an action other than its hyperpolarizing action. Journal of Physiology 1987;384:539‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Krach 2001
- Krach LE. Pharmacotherapy of spasticity: oral medications and intrathecal baclofen. Journal of Child Neurology 2001;16(1):31‐6. [DOI] [PubMed] [Google Scholar]
Lyons 2007
- Lyons BE, Moore P, Bhakta BB, Bamford JM, Cardwell C, Logan ID. Botulinum toxin for adult spasticity after stroke or non‐progressive brain lesion [Protocol withdrawn 2013]. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD002926.pub2] [DOI] [Google Scholar]
Malhotra 2008
- Malhotra S, Cousins E, Ward A, Day C, Jones P, Roffe C, et al. An investigation into the agreement between clinical, biomechanical and neurophysiological measures of spasticity. Clinical Rehabilitation 2008;22(12):1105‐15. [DOI] [PubMed] [Google Scholar]
Malhotra 2009
- Malhotra S, Pandyan AD, Day CR, Jones PW, Hermens H. Spasticity, an impairment that is poorly defined and poorly measured. Clinical Rehabilitation 2009;23(7):651‐8. [DOI] [PubMed] [Google Scholar]
Mirbagheri 2011
- Mirbagheri MM, Lilaonitkul T, Rymer WZ. Prediction of natural history of neuromuscular properties after stroke using Fugl‐Meyer scores at 1 month. Neurorehabilitation and Neural Repair 2011;25(5):458‐68. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. Annals of Internal Medicine 2009;151(4):264. [DOI] [PubMed] [Google Scholar]
Monaghan 2011
- Monaghan K, Horgan F, Blake C, Cornall C, Hickey PPM, Lyons BE, et al. Physical treatment interventions for managing spasticity after stroke. Cochrane Database of Systematic Reviews 2011, Issue 7. [DOI: 10.1002/14651858.CD009188] [DOI] [Google Scholar]
Montane 2004
- Montane E, Vallano A, Laporte JR. Oral antispastic drugs in nonprogressive neurologic diseases: a systematic review. Neurology 2004;63:1357‐63. [DOI] [PubMed] [Google Scholar]
Mukherjee 2010
- Mukherjee A, Chakravarty A. Spasticity mechanisms ‐ for the clinician. Frontiers in Neurology 2010;1:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Olvey 2010
- Olvey EL, Armstrong EP, Grizzle AJ. Contemporary pharmacologic treatments for spasticity of the upper limb after stroke: a systematic review. Clinical Therapeutics 2010;32(14):2282‐303. [DOI] [PubMed] [Google Scholar]
Palmeri 1990
- Palmeri A, Wiesendanger M. Concomitant depression of locus coeruleus neurons and of flexor reflexes by an alpha 2‐adrenergic agonist in rats: a possible mechanism for an alpha 2‐mediated muscle relaxation. Neuroscience 1990;34(1):177‐87. [DOI] [PubMed] [Google Scholar]
Pandyan 1999
- Pandyan AD, Johnson GR, Price CI, Curless RH, Barnes MP, Rodgers H. A review of the properties and limitations of the Ashworth and modified Ashworth scales as measures of spasticity. Clinical Rehabilitation 1999;13(5):373‐83. [DOI] [PubMed] [Google Scholar]
Pandyan 2005
- Pandyan AD, Gregoric M, Barnes MP, Wood D, Wijck F, Burridge J, et al. Spasticity: clinical perceptions, neurological realities and meaningful measurement. Disability & Rehabilitation 2005;27(1):2‐6. [DOI] [PubMed] [Google Scholar]
Rekand 2010
- Rekand T. Clinical assessment and management of spasticity: a review. Acta Neurologica Scandinavica 2010;122 Suppl 190:62‐6. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Royal College of Physicians 2009
- Royal College of Physicians, British Society of Rehabilitation Medicine, Chartered Society of Physiotherapy, Association of Chartered Physiotherapists Interested in Neurology. Spasticity in Adults: Management Using Botulinum Toxin. National Guidelines. London: RCP, 2009. [Google Scholar]
Seshadri 2006
- Seshadri S, Beiser A, Kelly‐Hayes M, Kase CS, Au R, Kannel WB, et al. The lifetime risk of stroke. Stroke 2006;37(2):345‐50. [DOI] [PubMed] [Google Scholar]
Sheean 2006
- Sheean G. Botulinum toxin treatment of adult spasticity: a benefit‐risk assessment. Drug Safety 2006;29(1):31‐48. [DOI] [PubMed] [Google Scholar]
Sheean 2009
- Sheean G. Botulinum toxin should be first‐line treatment for poststroke spasticity. Journal of Neurology, Neurosurgery & Psychiatry 2009;80(4):359. [DOI] [PubMed] [Google Scholar]
Sheean 2010
- Sheean G, Lannin NA, Turner‐Stokes L, Rawicki B, Snow BJ. Botulinum toxin assessment, intervention and after‐care for upper limb hypertonicity in adults: international consensus statement. European Journal of Neurology 2010;17:74‐93. [DOI] [PubMed] [Google Scholar]
Sorinola 2009
- Sorinola IO, White CM, Rushton DN, Newham DJ. Electromyographic response to manual passive stretch of the hemiplegic wrist: accuracy, reliability, and correlation with clinical spasticity assessment and function. Neurorehabilitation and Neural Repair 2009;23(3):287‐94. [DOI] [PubMed] [Google Scholar]
van Tulder 1997
- Tulder MW, Assendelft WJJ, Koes BW, Bouter LM, the Editorial Board of The Cochrane Collaboration Back Review Group. Method guidelines for systematic reviews in The Cochrane Collaboration Back Review Group for Spinal Disorders. Spine 1997;22(20):2323‐30. [DOI] [PubMed] [Google Scholar]
Vora 2010
- Vora A. Tolperisone. Journal of the Association of Physicians of India 2010;58:127‐8. [PubMed] [Google Scholar]
Watkins 2002
- Watkins CL, Leathley MJ, Gregson JM, Moore AP, Smith TL, Sharma AK. Prevalence of spasticity post stroke. Clinical Rehabilitation 2002;16(5):515‐22. [DOI] [PubMed] [Google Scholar]