

Abstract
Death receptor 5 (DR5) is an apoptosis-inducing member of the tumor necrosis factor receptor superfamily, whose activity has been linked to membrane cholesterol content. Upon ligand binding, DR5 forms large clusters within the plasma membrane that have often been assumed to be manifestations of receptor co-localization in cholesterol-rich membrane domains. However, we have recently shown that DR5 clusters are more than just randomly aggregated receptors. Instead, these are highly structured networks held together by receptor dimers. These dimers are stabilized by specific transmembrane helix–helix interactions, including a disulfide bond in the long isoform of the receptor. The complex relationships among DR5 network formation, transmembrane helix dimerization, membrane cholesterol, and receptor activity has not been established. It is unknown whether the membrane itself plays an active role in driving DR5 transmembrane helix interactions or in the formation of the networks. We show that cholesterol depletion in cells does not inhibit the formation of DR5 networks. However, the networks that form in cholesterol-depleted cells fail to induce caspase cleavage. These results suggest a potential structural difference between active and inactive networks. As evidence, we show that cholesterol is necessary for the covalent dimerization of DR5 transmembrane domains. Molecular simulations and experiments in synthetic vesicles on the DR5 transmembrane dimer suggest that dimerization is facilitated by increased helicity in a thicker bilayer.
Keywords: transmembrane domain, ligand/receptor clustering, replica exchange molecular dynamics, disulfide bond, cholesterol-rich membrane domains
Introduction
Death receptor 5 (DR5) is an apoptosis-inducing member of the tumor necrosis factor (TNF) receptor (TNFR) superfamily that is activated by TNF-related apoptosis-inducing ligand (TRAIL) or other agonists [1–5]. The TRAIL–DR5 signaling pathway is of great therapeutic interest, as exogenous TRAIL prevents tumor growth and, in contrast to other apoptosis-inducing ligands such as FasL, does not exhibit systemic cytotoxicity [6]. Thus, DR5 is widely regarded as a potential target in the treatment of cancer [7–10], with a number of its ligands, including recombinant human TRAIL and antibody agonists (as we will study here), currently in clinical trials [11–16] (see also clinicaltrials.gov). However, TRAIL resistance has proven to be a challenging barrier to the success of DR5-based cancer therapies [17,18]. Thus, continued efforts to understand the complex events involved in DR5 signal propagation, including the initiating events at the membrane, will be useful for understanding the therapeutics currently in clinical trials and for discovering novel DR5-targeted therapeutics.
TRAIL- and agonist-induced apoptosis are initiated by the formation of large (~300–500 nm diameter) DR5 receptor networks in the plasma membrane [19–22]. We and others have shown that these networks are held together by dimeric receptor junctions [21,23–26]. Ligand-induced receptor dimerization is consistent with crystallographic and functional evidence, showing that both intracellular protein domains and downstream proteins form stable and functional dimeric complexes [25,27–31]. We also showed that the dimeric junctions of DR5 networks are stabilized through interactions between amino acids in the transmembrane (TM) helices, which form tight dimeric bundles in the membrane [21,32]. In the long isoform of DR5, a disulfide bond forms between TM helices upon activation. Although the short isoform lacks the disulfide bond, both the long and short isoforms contain the well-established GxxxG TM helix interaction motif, which allows for tight packing at the interface of the two monomeric helices [33,34].
We have also recently suggested that the receptor dimer of TNFR1 (a structurally homologous member of the same superfamily as DR5) undergoes a conformational transition upon activation [26]. This is a very different model than what has previously been proposed for TNFR activation. In earlier models, activation was thought to involve receptor trimerization and a stoichiometric change in the cytosolic domain that required no conformational changes (i.e., receptor backbone rearrangements) [35,36]. Our molecular calculations and electron paramagnetic resonance experiments on the TM domains, on the other hand, support a model in which signal initiation is a consequence of the mechanical strain placed on the receptors as the network grows. More specifically, we have suggested that the extracellular domain of the receptor dimer pivots outwards upon network formation and that as a consequence, the TM dimer opens like scissors (closed in the inactive state, open in the active state) [26,32]. This model of activation has been supported by several recent studies on TNFR1 [37–39].
Given that ligand-induced dimeric interactions of DR5 occur in part via TM residues, it stands to reason that the membrane itself may play an active role in driving the dimeric interactions that promote network formation. Cholesterol-rich microdomains in the plasma membrane play an active role in signal transduction in a number of pathways [40–42] including several that involve members of the TNFR superfamily. Ligand-bound TNFR1 localizes to cholesterol-rich domains and recruits downstream signaling proteins [43,44]. The death receptor Fas has been shown to localize to cholesterol-rich domains in a ligand-dependent manner [45], although others have observed ligand-independent cholesterol co-localization [46]. In DR5, Song et al. showed that caspase-8 cleavage in TRAIL-sensitive cells could be inhibited by treatment with methyl-β-cyclodextrin (MβCD), which strips membrane cholesterol [47]. Multiple other studies have shown that DR5 function correlates with its migration into cholesterol-rich domains [47–51]. However, it is unknown whether this localization is associated with receptor oligomeric structure, that is, DR5 dimerization and network formation.
Here, we find that the extent of agonist-induced DR5 clustering is not diminished by cholesterol depletion. However, cholesterol depletion greatly reduced the ability of DR5 to initiate caspase-dependent apoptosis. We attribute this to the formation of non-functional ligand–receptor networks that differ from functional networks by a reduction in the population of constituent disulfide-linked DR5 dimers. Collectively, these results offer the first evidence that membrane heterogeneity plays a central role in dictating the structural details and functional activity of DR5 networks. These results further support a model in which DR5 networks have a specific structure and that cholesterol-rich membrane domains do not simply corral high local concentrations of receptors but play an essential role in driving ligand–receptor network architecture.
Results
DR5 signal transduction in response to agonistic antibody is cholesterol dependent
It was previously shown that the TRAIL activation of DR5 induces cholesterol-dependent caspase-8 activation and concomitant migration of DR5 to cholesterol-rich, detergent-resistant membrane (DRM) fractions [47]. Here, we use the agonistic antibody, mAb631, in lieu of TRAIL to trigger DR5 signaling; thus, we first determined that DR5 activated by mAB631 recapitulates the same cholesterol-dependent behavior as when it is activated by TRAIL. We first depleted the membrane cholesterol with MβCD, then treated the cells with agonistic antibody, and measured caspase-8 activation. Jurkat cells with membrane cholesterol (i.e., not treated with MβCD) efficiently activate caspase-8 upon the addition of DR5 agonist (Fig. 1a, compare gray and black distributions). Pretreatment with MβCD results in a reduced ability of these cells to activate caspase-8 (Fig. 1b, compare gray and black distributions). We then showed that activation by agonistic antibody causes DR5 to relocate from high-density fractions to cholesterol-rich fractions with lower density (Supplementary Fig. 1). These results show that cholesterol-dependent DR5 behavior is consistent when activated by either mAB631 or TRAIL.
Fig. 1.

Membrane cholesterol is required for the efficient activation of caspase-8 by DR5. Jurkat cells were pretreated with either (a) control or (b) MβCD, followed by no treatment or anti-DR5 agonist antibody (α-DR5; gray distribution and black line, respectively), and caspase-8 activity was measured using Red-IETD-FMK and flow cytometry. Plotted is a histogram of caspase-8 activity level showing the ligand-dependent activation of caspase-8 and the reduced activity in cells pretreated with MβCD. (c) The activation of caspase-8 is quantified using identical gating schemes on each population, and the results are plotted as the fold activation of caspase-8.
Ligand–receptor networks form in the absence and presence of membrane cholesterol
DR5 agonistic antibody is known to induce DR5 network formation [21], and we have confirmed above that it drives the co-localization of the receptor to cholesterol-rich membrane fractions. However, there is no clear evidence that cholesterol-rich domains induce DR5 network formation or vice versa. Ligand–receptor networks are routinely identified using confocal fluorescence microscopy, as in this study, in order to quantify cluster size and number [19–21]. Jurkat cells were pretreated with MβCD (or control) to extract membrane cholesterol, and cells were subsequently treated with DR5 antibody agonist. Receptor-bound agonist was labeled with fluorescent secondary anti-mouse antibody and imaged using confocal microscopy to quantitatively determine the extent of network formation in the membrane with and without MβCD treatment (Fig. 2). This labeling scheme prevents potential interference of ligand binding by a non-agonist primary antibody but does not permit live-cell imaging of agonist-free receptor. However, in our previous study (and again below), we showed less receptor clustering in untreated cells [21] by crosslinking Western blot analysis. Treatment of Jurkat cells with MβCD has been shown to deplete cholesterol within 30 min in a dose-dependent manner and extracts cholesterol preferentially from the plasma membrane, since it is not believed to enter live cells [52]. Cell cholesterol content was measured using the Amplex Red assay to confirm ~50% cholesterol depletion in MβCD-treated cells. We measured 0.45 μg cholesterol per μg lysate in untreated versus 0.19 μg cholesterol per μg lysate in MβCD-treated cells, showing a reduction of 58%. Removal of cholesterol much beyond this has been shown to cause substantial cell death in Jurkat cells [52]. Images were taken in three separate experiments, and the mean and standard error of each measurement were calculated.
Fig. 2.
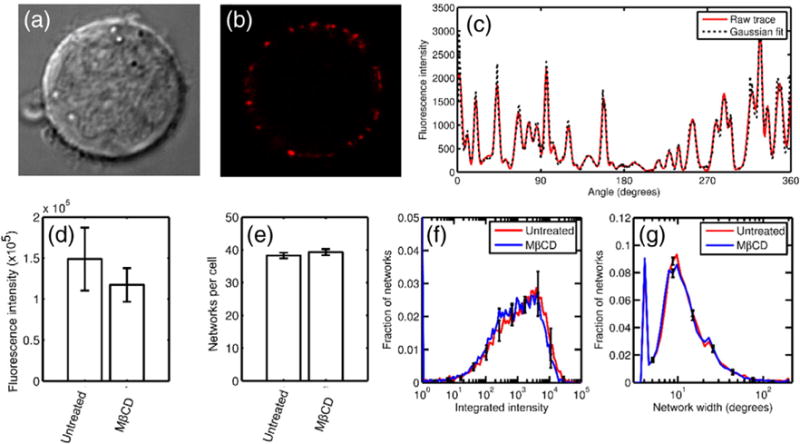
Ligand–receptor cluster formation does not require membrane cholesterol. Jurkat cells were treated with mAb631 and labeled with fluorescent secondary antibody. Shown are (a) Jurkat cells under transmitted luminescence, (b) ligand–receptor clusters illuminated by fluorescently labeled agonist antibody, and (c) the angular fluorescence intensity trace for the cell shown. The raw trace is shown in black and the Gaussian fit in blue. (d) The total ligand bound, (e) number of clusters per cell, and (f, g) distribution of cluster sizes are invariant between untreated and MβCD-treated cells.
In both untreated and MβCD-treated cells, stimulation with labeled mAb631 resulted in punctate staining along the cell membrane, indicating ligand–receptor clustering (Fig. 2a and b) as has been shown previously in DR5 and other members of the TNFR superfamily [21,22,53–57]. To determine the number and size distribution of these DR5 networks, we fit the angular intensity trace to a 1D mixed Gaussian distribution (Fig. 2c). The total integrated label intensity reports the total ligand bound per cell, while the total number of peaks in each cell reports the number of independent networks, and the heights and widths of the individual Gaussians were used to determine network size. Quantitative analysis revealed no significant differences in the extent of network formation in untreated and MβCD-treated cells. Total fluorescence per cell was determined by taking the area under the curve of the raw angular intensity trace. The area under the curve in the untreated and MβCD-treated cells were 1.49 × 105 ± 3.8 × 104 and 1.17 × 105 ± 2.06 × 104, respectively (Fig. 2d), indicating that cholesterol depletion did not inhibit mAb631 binding. The average number of clusters per cell was 38.2 ± 0.85 and 39.3 ± 0.95 in the control and MβCD-treated cells, respectively (Fig. 2e). Peaks were counted as any bin along the angular integration with higher intensity than both neighboring bins, and therefore, any fluorescence above background is counted. The Gaussian integral of each of the networks, calculated as a|c| , where a is the height of the curve peak and c is the standard deviation, reveals the distribution of cluster sizes (Fig. 2f). DR5 in both the control and MβCD-treated cells formed networks with integrated intensity varying widely between ~ 10 and 2 × 104 (intensity units) with equivalent distributions. Error bars are calculated from triplicate experiments and reported for every eighth bin for clarity. Lastly, we plotted the distribution of network widths (calculated as 4 × c to represent the arc length in degrees containing 95.4% of the labeled receptor; Fig. 2g). Again, both control and MβCD cells behaved the same, with network sizes centered at 10°. Assuming a cell with a radius of 3 μm, a 10° arc spans 524 nm, which is on the same scale as the Fas networks measured by Siegel et al. [20]. To conclude, treatment with MβCD does not prevent agonist binding, and no difference in average network size or number is discernible in cholesterol-depleted versus control cells, strongly suggesting that mAb631–DR5 clustering is not cholesterol dependent.
Membrane cholesterol is required for the dimerization of DR5 in response to ligand
We have shown previously that upon the addition of antibody agonist, DR5 forms disulfide-linked dimers that are subsumed within ligand–receptor networks [21]. Figure 3 repeats this result and is expanded to assess whether the extraction of membrane cholesterol has any effect on ligand-induced DR5 dimerization. Jurkat cells were pretreated with MβCD (5 mM) or left untreated, and cells were subsequently washed and treated with DR5 antibody agonist, crosslinked, and lysed. Equal amounts of total protein were run on non-reducing SDS-PAGE, and DR5 dimerization and oligomerization were analyzed by Western blot (Fig. 3a). As can be clearly seen in the control cells, the ligand-induced DR5 dimer band disappears upon crosslinking (compare lane 2 to 4) because the dimer becomes part of the larger crosslinked network. The dimer band is present in the uncrosslinked sample (lane 2) because it is held together by a disulfide bond between TM helices via Cys209, as previously shown [21]. The networks can be seen clearly as high-molecular-weight species larger than 260 kDa and are induced by the addition of ligand in both untreated and MβCD-treated cells (lanes 4 and 8).
Fig. 3.
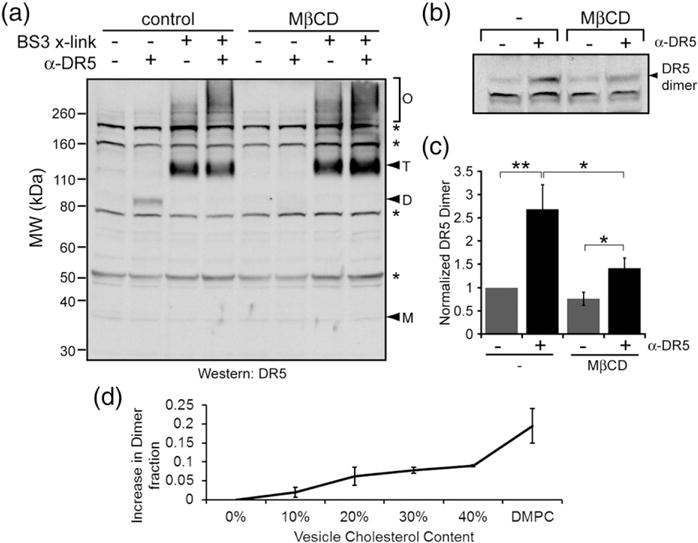
Extraction of membrane cholesterol inhibits ligand-induced DR5 dimer formation. Jurkat cells were pretreated with either control or MβCD, as indicated, to extract membrane cholesterol. Cells were subsequently washed and treated with α-DR5 agonist antibody and lysed. Lysates were analyzed via non-reducing SDS-PAGE and Western blot using an antibody against DR5. (a) In lanes 4 and 8, agonist-bound DR5 formed oligomeric networks in both untreated and MβCD-treated cells. Asterisks indicate non-specific bands, which do not appear at the known molecular weight of DR5 or any multiple of it and are invariant with ligand treatment. (a) In lane 2, disulfide-linked dimerization of DR5 via Cys209 occurred only in untreated cells. (b) Shown is the dimeric form of DR5 at ~85 kDa from a separate experiment. (c) Quantification of DR5 dimer bands shows a significant increase in DR5 dimerization upon treatment with ligand, and pretreatment with MβCD diminishes DR5 dimerization. (d) The dimer fraction of DR5-L TM peptide increases with increasing cholesterol in DLPC vesicles. DMPC vesicles also favor a larger dimer fraction than DLPC vesicles in the absence of cholesterol.
Cells pretreated with MβCD have a markedly diminished ability to form DR5 dimers (Fig. 3a and b). To better evaluate the extent to which MβCD inhibits disulfide-linked dimerization of DR5, we ran the agonist-treated and untreated samples with and without MβCD and then densitometrically quantified the amount of dimer using ImageJ. The Western blot in Fig. 3b shows a smaller reduction in dimerization than that in Fig. 3a; however, quantification reveals significant differences in dimerization between untreated versus MβCD-treated cells (from seven independent experiments). In cells containing cholesterol, the addition of ligand causes a 2.7-fold increase in the dimeric population of DR5. After pretreatment of Jurkat cells with MβCD, ligand causes only a 1.8-fold increase in DR5 dimerization (Fig. 3c). Therefore, while the depletion of membrane cholesterol does not alter the formation of large DR5 networks, it does influence the covalent structure of DR5 dimers subsumed within these oligomers.
Bilayer thickness modulates dimer formation of the synthetic DR5-L TM domain in lipid vesicles
To further test whether DR5-L dimerization is increased by membrane cholesterol (and concomitant bilayer thickening), we synthesized the DR5-L TM peptide and incorporated it into synthetic vesicles. Our approach follows Cristian et al. [58], in which they have shown that the dimerization of the influenza M2 TM helix (also by disulfide linkage) is sensitive to bilayer thickness: dimerization increased in lipid vesicles with increased cholesterol concentration and/or lipid acyl chain length. These experiments are carefully controlled to ensure equilibrium distributions of monomeric/dimeric states by freeze/thaw cycles and inclusion of glutathione to mediate disulfide exchange. We have recapitulated the test of these two potential drivers, cholesterol content and thickness, of peptide dimerization in the case of DR5-L. First, in a short-chain 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) (di12:0) bilayer, we observed a dose-dependent increase in the DR5-L peptide dimer fraction with increasing mole fraction of cholesterol (Fig. 3d and Supplementary Fig. 2). At the highest cholesterol content (40 mol%), we see an increase of ~10% in dimer content. The magnitude of this effect is consistent with the sensitivity of the M2 TM helix to cholesterol [58].
Second, and equally compelling, we compared the dimerization propensity of the DR5-L TM peptide in DLPC with the peptide in cholesterol-free 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC; di14:0) bilayers. As compared to cholesterol-free DLPC, the dimer propensity increases in the longer-chain DMPC bilayer, again demonstrating the correlation between thickness and dimerization. Interestingly, the effect in DMPC (20% increase in dimers as compared to DLPC) is double that of DLPC +40 mol% cholesterol (10% increase). Experiments have shown that DLPC bilayers with 40 mol% cholesterol and cholesterol-free DMPC have approximately the same hydrophobic thicknesses [59,60]. Therefore, we expected that if hydrophobic thickness is the only driving force for increased dimerization, we would see a roughly equal effect in the two bilayers. Thus, the unexpectedly large increase in dimers in DMPC must reflect an additional driving force that supplements the hydrophobic thickness effect, as will be discussed below.
Molecular simulations suggest that membrane thickness controls DR5-L TM domain helical stability and Cys209 partition depth
We have shown that membrane cholesterol promotes ligand-induced DR5-L covalent dimerization; however, the biochemical and structural features of cholesterol-rich membrane domains that promote disulfide bond formation are unknown. You et al. investigated the disulfide bonding of the TM domain of FGFR3 in detergent and bilayers and suggested that dimerization tendency depends on the membrane environment [61]. Cholesterol causes bilayer thickening; therefore, to further explain cholesterol-dependent disulfide bonding, we ran replica exchange molecular dynamics (REMD) simulations to explore the differences in the preferred low-energy conformations of the DR5-L TM dimer in a thick (40 Å, corresponding to cholesterol-rich) versus a thin (32 Å, corresponding to cholesterol-depleted) bilayer. We previously simulated the disulfide-linked TM dimer to predict the structure and dynamicsofthe covalent dimer [21]. Here, instead, we simulated non-disulfide bonded monomers. We did this in order to explore the effect of membrane thickness on monomer structure and monomer– monomer interactions that may influence dimerization. We hypothesized that disparate bilayer thicknesses would give rise to differences in monomer–monomer interactions, tilt, and helicity and would suggest the basis by which the thicker membrane promotes disulfide bond formation. REMD is a powerful computational tool that allows a system to rapidly overcome free energy barriers and access low-energy conformational states while preserving ergodicity; however, our results should be interpreted with the caveat that the accessible conformational space of the isolated TM domains, which lack explicit membrane molecules, differs from that of the intact protein in situ.
We previously observed the formation and stability of the tightly packed GxxxG dimerization motif [33,34] in the disulfide-linked DR5 TM domain dimer, which is one helicalturn downstreamofCys209 [21]. Thismotif is likewise stable in the non-bonded dimer in both the 32-Å and 40-Å bilayers (Fig. 4). We expect that the presence of this motif stabilizes the dimer and thus promotes disulfide bond formation. Interestingly, the occurrence of the motif is more frequent in the thicker bilayer (Fig. 4a). It should be noted that while this bilayer thickness dependence was clearly observed in two of the four starting configurations, the difference was less convincing in the other two (Supplementary Fig. 3).
Fig. 4.

Membrane thickness affects DR5-L TM domain helical stability and Cys209 partition depth in molecular dynamics simulations. The DR5-L TM dimer was simulated using REMD. (a) The average GxxxG distance was plotted to identify the likely dimer conformation (<6 Å; dashed box). This dimer conformation was sampled in both the 40-Å (black) and 32-Å (blue) implicit bilayers representing cholesterol-rich and -poor domains, respectively. Subsequent analysis is conducted on these frames only. (b) The fraction of frames with helical secondary structure shows increased helix stability in the 40-Å bilayer. (c) A snapshot of the simulated peptide in its GxxxG dimer configuration is shown with C209 in red, the G(213)xxxG(217) motif as blue spheres, and W237 as a red sphere. (d) Trp237 (Cα atom) partitions directly outside the switching region in both the 40-Å (black) and 32-Å (blue) bilayers. Cys209 (sulfur atom) partitions at the switching region in the 32 Å-bilayer (blue) but deep within the low-dielectric region in the 40-Å bilayer (black). The positions of the switching regions of each bilayer are shown as dashed horizontal lines.
We then investigated three other possible thickness-dependent differences: helix tilt, helicity, and partition depth. Because our simulation of the isolated TM domain is able to access conformations that may not be sampled by the full-length protein or are otherwise not pertinent to the TM domain dimer (e.g., those in which the two helices are non-interacting), for the remainder of the analysis, we compared only those structures in which the GxxxG interface was present (Fig. 4a, boxed). The overall trends were the same if we considered all structures generated by the simulations. Bilayer thickness-dependent helix tilt and helical content due to hydrophobic mismatch (mismatch between the bilayer thickness and hydrophobic length of the TM domain) are often cited as determinants of TM domain behavior. We observed low tilt angles in both the 32-Å and 40-Å bilayers with little evidence of dependence on thickness (average tiltof 19° and 23°, respectively; Supplementary Fig. 3). We then used the DSSP (Define Secondary Structure of Proteins) algorithm [62,63] to calculate peptide secondary structure to determine whether a bilayer’s hydrophobic thickness influences helicity. Helicity for each residue is reported as the percentage of time in which the residue adopts a helical configuration. We found that the same residues were helical in both bilayers; however, helicity was increased by ~40% throughout the hydrophobic length of the peptide in the thicker bilayer (Fig. 4b). The most obvious explanation for an increase in helicity is that the interfacial (N-terminal), partially solvent-exposed residues in the thinner bilayer become more stably folded in the thicker bilayer in order to minimize their free energy. This offers an explanation for how the thicker bilayer favorably positions the two Cys209 residues for disulfide bond formation (by stabilizing them in the helices on the same helical face as the GxxxG motif).
The DR5 TM domain notably contains a Trp at residue 237 that partitions to the switching region of the bilayer (corresponding to the headgroup/acyl chain region of an explicit bilayer). Trp residues are well known to anchor TM helices in this region [64–69]. As shown in Fig. 4c and d, Trp237 partitions to the switching region in both systems. This causes Cys209 to partition to the switching region of the 32-Å bilayer but draws Cys209 deeper into the low-dielectric region (corresponding to the bilayer interior) of the 40-Å bilayer. Immersion of these interfacial residues in the hydrophobic region of the bilayer likely promotes their folding into a stable helix. A shift in TM domain partition depth has been previously suggested to influence dimerization tendency by altering the configuration or local environment of side chains and dimerization motifs [70]. The membrane interior has been shown experimentally to be unfavorable for thiol modification, and cysteine residues are hypothesized to make excursions to the extracellular solvent where they can undergo more rapid disulfide bonding [71]. This suggests that disulfide bonding would be favored in a narrow membrane, opposite of what we and others [58] have seen. Thus, we expect that the structural effect of a thicker bilayer on DR5-L conformation (increased helicity) is responsible for the increase in disulfide bonding. It should also be noted that a hydrophobic environment has been theorized to dramatically reduce the energy barrier of disulfide exchange [72] and that the burial of Cys209 in the bilayer may enhance the rate of disulfide bond formation in cholesterol-rich domains; however, in the absence of oxygen or catalyst in the bilayer interior, we expect the reaction rate to be substantially lower than in solvent.
Discussion
Our experimental results on the dimerization of the synthetic TM domain of DR5-L in DLPC versus DMPC lipid vesicles raise an intriguing question. DLPC and DMPC form bilayers with expected headgroup–head-group distances of 30.8 and 35.3 Å, respectively [59]. Neutron scattering experiments showed that the addition of 40 mol% cholesterol causes the bilayer to thicken by ~5.7 Å [60]. Thus, the DLPC bilayers with 40% cholesterol have roughly the same (or slightly greater) thickness as DMPC bilayers; however, we observed double the effect on dimer formation in the latter. Hydrophobic thickness, which cannot be the source of the observed difference in dimerization, is not the only factor that can alter the local hydrophobicity near the peptide. The helical secondary structure that favors disulfide bonding is promoted by the immersion of the TM domain in a hydrophobic environment. Our result for DMPC prompted the reevaluation of our previously published atomistic simulations comparing pure and cholesterol-containing bilayers [73]. Our simulations showed that cholesterol molecules increase the density of water in the headgroup region, presumably an inevitable and trivial consequence of the smaller hydroxyl (headgroups) of cholesterol. Thus, in cholesterol-containing bilayers, waters replace the polar lipid headgroups. As a consequence, the local thermodynamic environment, including the effective hydrophobicity, of the headgroup region of pure and cholesterol-containing bilayers is different, and this likely alters the driving force for helix formation, even if only mildly so. If one considers the difference in mobility and packing of waters in the cholesterol bilayer versus the large, bulky lipid headgroups in the cholesterol-free bilayer, then it is not unreasonable to speculate that the high entropy of the unfolded state should be more easily accommodated in the highly dynamic and more hydrated cholesterol-containing bilayer.
From a more biological perspective, cholesterol-dependent activation has been previously observed in a number of TNFRs, including DR5, when induced by TRAIL [47]. We have shown that cholesterol dependence is similarly a feature when DR5 is activated by agonistic antibody. Previously, Song et al. showed that DR5 in cholesterol-depleted membranes (in TRAIL-insensitive cells) triggered the NF-κB pathway rather than the apoptotic pathway [47]. We did not investigate whether mAb631 induces NF-κB activation in cholesterol-depleted cells; however, ongoing work will investigate this possibility. If mAb631 does activate the inflammatory pathway in cholesterol-depleted cells, it would suggest that DR5-L clusters that are not stabilized by disulfide bonding (Figs. 2b and 3) recruit different cytosolic machinery that activates the NF-κB pathway, either by favoring a different dimer structure or by altering the dimerization kinetics of the TM domain.
An additional possibility that we have not directly addressed is whether the depletion of cholesterol prevents the co-localization of the receptor with the intracellular signaling machinery [the death-inducing signaling complex (DISC)]. In the case of Fas-induced apoptosis, the components of the DR5 DISC are not constitutively present in the cholesterol-rich membrane fractions [45], and they have been shown to co-immunoprecipitate with DR5 in both cholesterol-rich and -poor fractions [47]. Therefore, the available evidence suggests that the co-localization of the DISC and DR5 is not purely cholesterol driven. Nonetheless, our data cannot fully rule out this possibility.
In conclusion, our findings support an evolving model of the interdependence of DR5 oligomerization and signaling. DR5 clusters are not random, unstructured aggregates whose high local concentration pushes signaling over a threshold. Instead, the constituent receptors of DR5 networks have a specific intermonomeric structure that supports the protein’s function. Specifically, while the formation of DR5-L networks does not require cholesterol, those networks only activate the caspase cleavage pathway if the DR5 TM domains undergo cholesterol-dependent dimerization. Given our previous results, we are currently investigating whether a similar requirement exists in the short isoform of DR5, which does not contain a disulfide linkage but does contain the GxxxG motif. To explain the effect of cholesterol, our computational modeling suggests that in thin bilayers, instability in the TM helix inhibits the covalent dimerization of DR5-L. When the receptor translocates into the cholesterol-rich and thicker bilayer, the TM domain is drawn deeper into the bilayer interior. While the burial of the reactive thiols in the hydrophobic acyl region of the membrane is expected to disfavor disulfide exchange, stabilization of the TM helices in a configuration that favors disulfide bond formation (Fig. 5) yields a net increase in the dimer fraction. The disulfide-linked DR5 dimer, subsumed within the ligand–receptor network that is immersed in the cholesterol-rich membrane fraction, can then undergo a transition to the active state as our previous results suggest [26,32].
Fig. 5.

Updated activation model for DR5 incorporating the bilayer structure. (a) Instability in the TM helix inhibits covalent dimerization of DR5 via Cys209 in thin bilayer fractions. (b) Translocation to thicker, cholesterol-rich membrane domains draws the cysteines into the bilayer interior, stabilizing the TM helices and driving disulfide bond formation.
Materials and Methods
Cell culture and reagents
Jurkat cells were cultured in RPMI-1640 media (HyClone) supplemented with 10% fetal bovine serum (FBS), 4 mM L-glutamine, penicillin, and streptomycin and were maintained between 105 and 106 per mL at 37 °C and 5% CO2 in a water-jacketed incubator. Jurkat cells are a leukemia T-lymphocyte that are confirmed to endogenously express DR5, are sensitive to agonist antibody, and are commonly used to investigate death receptor behavior, cholesterol-rich membrane domains, and apoptosis in cancer cells [21,48,52,57,74]. DR5 antibody agonist (mAb631) and fluorescent secondary antibody (NL557) were purchased from R&D Systems. DR5 antibody for Western blots was purchased from Cell Signaling Technologies.
DRM extraction
DRM and detergent-soluble membrane fractions were isolated as previously described [47,75,76]. Briefly, Jurkat cells (108 cells) were treated with DR5 agonist or control, resuspended in 550-μl TNE buffer [150 mM NaCl and 2 mM EDTA in 50 mM Tris–HCl (pH 7.4)], and homogenized 20 times using a 25 gauge needle. Homogenate (500 μl) was mixed with an equal volume of 2% Triton X-100 in TNE buffer supplemented with protease inhibitors (yielding a 1% Triton X-100 final concentration) and lysis proceeded for 30 min on ice. Samples were centrifuged at 10,000g for 5 min at 4 °C, and post-nuclear supernatants (1 ml) were mixed with 2 ml of 56% sucrose, transferred to a SW41 ultracentrifuge tube, and overlaid with 5 ml of 35% sucrose and 5 ml of 5% sucrose. All sucrose solutions were prepared in TNE buffer [76]. Step gradients were centrifuged at 39,800 rpm (271,000g) at 4 °C for 18 h using a SW41Ti rotor centrifuge. Gradients were fractionated into nine fractions of equal volume using a Gradient Station (BioComp Instruments). Protein content in each fraction was measured by bicinchroninic acid (BCA) assay (Pierce) and compared to a bovine serum albumin (BSA) standard curve. Cholesterol content in each fraction was measured using Amplex Red cholesterol assay (Invitrogen Life Technologies). DR5 migration into DRM fractions was analyzed by trichloroacetic acid (TCA) precipitation and Western blot analysis, using equal volumes of each fraction. Results are from three independent experiments.
Caspase-8 activity
Caspase-8 activity was measured as previously described [21]. Jurkat cells were treated with a DR5-specific antibody agonist at 1 μg/ml for 4 h at 37 °C. Caspase activity was measured using Casp-GLOW Red Active Caspase 8 staining kit (BioVision) according to manufacturer’s instructions. Fluorescent-labeled active caspase-8 was detected by flow cytometry (FACSCalibur) over 50,000 cells in a single experiment. Data was analyzed using FlowJo software (Tree Star, Inc.), where identical gating schemes were used to determine the percentage of caspase-8 active cells.
Cholesterol extraction
To extract the membrane cholesterol, we washed the Jurkat cells in phosphate buffered saline (PBS) and resuspended them in serum-free media (RPMI-1640). Cells were treated with 5 mM MβCD in serum-free RPMI for 30 min at 37 °C and then gently washed with PBS before stimulation with agonist.
Western blotting
Jurkat cells were washed with PBS and treated with DR5-specific antibody agonist for 1 h in PBS. In crosslinking experiments, cells were treated with 1 mM of the amine-reactive and membrane-impermeant crosslinker BS3 (Pierce) for 30 min at room temperature and then quenched with 20 mM Tris–HCl (pH 7.5) for 15 min at room temperature. Cells were pelleted by centrifugation and lysed in RIPA buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1% NP-40] supplemented with protease inhibitors and 10 mM iodoacetamide for 1–2 h at 4 °C. Total protein concentration of lysates was determined by BCA assay (Pierce), and equal amounts of total protein (~80 μg) were mixed with 4x NuPAGE sample buffer (in the absence of reducing agents), boiled for 10 min, and loaded on 4–12% Bis-Tris SDS-PAGE gel (Invitrogen). Proteins were transferred to nitrocellulose membrane and probed using an antibody against DR5 (D4E9; Cell Signaling) and anti-rabbit HRP-conjugated secondary antibody (GE Amersham) and were detected using ECL Plus (GE Amersham).
Determination of dimer fraction in vesicles
The 40-residue DR5-L sequence SSPGTPASPCS LSGIIIGVTVAAVVLIVAVFVSKSLLW-KK was synthesized by solid phase protein synthesis using a Symphony automated peptide synthesizer (Protein Technologies, Inc.) at a 50-μmol scale, using a sixfold excess of Fmoc amino acids (200 mM) relative to the Fmoc TentaGel Rink Amide Resin (0.19 meq/g; Protein Technologies, Inc.). The sequence was designed to match the one used in our computational modeling below. The cysteine at position 33 was mutated to serine to prevent the formation of antiparallel disulfide-linked dimers. Coupling was performed using 1:1:2 amino acid/HCTU/NMM in dimethyl formamide. The residues were double coupled. The final cleavage from the resin and side-chain protecting groups was performed using 82.5% trifluoroacetic acid, 5% phenol, 5% H2O, 5% thioanisole, and 2.5% EDT (Reagent K) at room temperature under nitrogen for 2 h. After cleavage, the peptide was precipitated with cold ether and collected by centrifugation. The crude peptide was purified by HPLC on a semi-preparative C8 column (Vydac 208TP, 300 Å, 250 × 10 mm, 10 μm particles) using a gradient from solvent A (0.1% TFA in H2O) to solvent B (0.1% TFA in 2-propanol) at 3 mL/min flow rate. Fractions containing peptides were lyophilized and stored at −20 °C. The purity of the peptide was established by analytical HPLC and SDS gel electrophoresis.
Vesicles were made following Ref. [58] using DLPC or DMPC. Crude DR5-L peptide dissolved in trifluoroethanol was combined with the indicated lipid and cholesterol ratios in chloroform in glass vials. The solvent was removed under nitrogen gas and the dried films were lyophilized overnight to complete drying. Films were rehydrated in 100 mM Tris, 0.2 M KCl, and 1 mM EDTA (pH 8.6) buffer to produce final concentrations of 20 μM peptide and 12.5 mM lipid/cholesterol. Mole fraction of cholesterol was varied between 0 and 0.4. Hydrated mixtures were then sonicated, and glutathione (GSSG/GSH ratio 0.25) was added to a final concentration of 1.5 mM. Samples were sonicated, incubated for 1 h at room temperature, frozenat −80°C, and thawed. This cycle was repeated hourly over 5 h. Disulfide exchangewas quenched by the addition of HCl to 0.12 M and the samples were then prepared for analysis by SDS-PAGE. 5 μL of sample was added to 5 μL of 5% SDS. We added 20 μL of Tricine sample buffer (Biorad 1610739), and 28 μL of each sample was run on 10–20% Tris–Tricine gels (Biorad 345-0068). Gels were stained using Coomassie blue, scanned, and analyzed densitometrically using the gel analysis plugin in ImageJ.
Confocal imaging and analysis
Jurkat cells were untreated or treated with 5 mM MβCD to deplete cholesterol as described above. Cell washes in subsequent steps were performed by centrifuging at 10g and gently resuspending in ice-cold PBS with 1% FBS. Cells were washed once and treated with mAb631 agonist at 5 μg/mL in PBS with 1% FBS for 30 min on ice. DR5 was removed by washing twice, and cells were resuspended with 5 μg/mL NorthernLights557 in PBS with 1% FBS and were then tumbled for 30 min at 4 °C. Cells were then washed twice, resuspended, and plated on poly-lysine-coated 35-mm glass-bottom culture dishes (MatTek Corporation) for imaging. Cells were allowed to settle before taking images.
Cells were imaged using an Olympus IX82 inverted microscope equipped with a FluoView FV1000 laser scanning confocal head with 543-nm laser excitation and 555- to 655-nm emission using a 100× (1.3NA) oil immersion objective lens. All images were acquired at the same laser intensity and resolution (1600 × 1600 pixel, 0.079 um/pixel edge).
Images were analyzed using MATLAB. Each image, containing 10–30 cells, was first smoothed using a low-pass 2D finite impulse response filter with a Gaussian window. Background intensity was subtracted for each image by selecting a region of the field containing no cells. Each cell border was selected manually from the light field image, and these points fit to an ellipse. A 30-pixel shell along this ellipse was isolated, avoiding the inclusion of any potential fluorescence in the cytoplasm (some cells showed limited internalized label). Angular intensity was then determined along the shell in 1° bins. The peak positions were determined using the MATLAB “findpeaks” command on an ideal low-pass filtered angular integration trace. The peak locations were used as parameters to fit the raw angular integration trace to a 1D mixed Gaussian distribution, which solved for the maximum intensity and standard deviation of each peak.
REMD of the long isoform DR5 TM domain
All-atom REMD was run using the MMTSB toolset [77]. DR5 is expressed as both long and short isoforms (DR5-S and DR5-L), where the long isoform is able to form disulfide-linked dimers and the short isoform is not. We used DR5-L here to expand on our finding that disulfide bonding in DR5 depends on membrane cholesterol. The starting structure of the DR5-L TM domain was an ideal helix using residues 200–239 (S S P G T P A S P C S L S G I I I G V T V A A V V L I-VAVFVCKSLLWKK). The single TM helix was duplicated to generate a dimer, and the two segments were started ~8 Å from each other. Simulations used the CHARMM 22 parameters [78,79] with CMAP correction [80] and the SHAKE algorithm [81]. The protein was centered in an implicit bilayer and modeled using the Generalized Born with simple switching function [82,83]. The hydrophobic thickness was set to 32 and 40 Å to model the cholesterol-poor and -rich domains, respectively, and the switching length was set to 0.6 Å. 16 replicates were run from each of 4 independent starting configurations at 300–600 K using the Nosé– Hoover method to control the temperature [84]. Each replicate was run for 10 ns (total of 640 ns for the 4 sets of 16 replicates) using a 2-fs time step. Replica exchange was attempted every 1000 steps with a success rate of ~20%. Structures from the 300-K window were used for analysis.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 GM107175. We would like to thank the University of Minnesota– University Imaging Center for confocal imaging resources. This work was carried out in part using the computing resources at the University of Minnesota Supercomputing Institute.
Abbreviations
- PBS
phosphate buffered saline
- FBS
fetal bovine serum
- DR5
death receptor 5
- α-DR5
anti-DR5 agonist antibody
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- TRAIL
TNF-related apoptosis-inducing ligand
- TM
transmembrane
- MβCD
methyl-β-cyclodextrin
- REMD
replica exchange molecular dynamics
- DRM
detergent-resistant membrane
- DISC
death-inducing signaling complex
- DLPC
1,2-dilauroyl-sn-glycero-3-phosphocholine
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
Appendix A. Supplementary Data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2016.10.001.
Footnotes
Edited by James Bowie
References
- 1.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 2.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–818. doi: 10.1126/science.277.5327.815. http://dx.doi.org/10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 3.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Schneider P, Tschopp J. Apoptosis induced by death receptors. Pharm Acta Helv. 2000;74:281–286. doi: 10.1016/s0031-6865(99)00038-2. [DOI] [PubMed] [Google Scholar]
- 5.Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci. 2002;27:19–26. doi: 10.1016/s0968-0004(01)01995-8. [DOI] [PubMed] [Google Scholar]
- 6.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. http://dx.doi.org/10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 7.Rowinsky EK. Targeted induction of apoptosis in cancer management: the emerging role of tumor necrosis factor-related apoptosis-inducing ligand receptor activating agents. J Clin Oncol. 2005;23:9394–9407. doi: 10.1200/JCO.2005.02.2889. http://dx.doi.org/10.1200/JCO.2005.02.2889. [DOI] [PubMed] [Google Scholar]
- 8.Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL) J Clin Oncol. 2008;26:3621–3630. doi: 10.1200/JCO.2007.15.7198. http://dx.doi.org/10.1200/JCO.2007.15.7198. [DOI] [PubMed] [Google Scholar]
- 9.Gerspach J, Pfizenmaier K, Wajant H. Therapeutic targeting of CD95 and the TRAIL death receptors. Recent Pat Anticancer Drug Discov. 2011;6:294–310. doi: 10.2174/157489211796957739. [DOI] [PubMed] [Google Scholar]
- 10.Yang A, Wilson NS, Ashkenazi A. Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Curr Opin Cell Biol. 2010;22:837–844. doi: 10.1016/j.ceb.2010.08.001. http://dx.doi.org/10.1016/j.ceb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Yada A, Yazawa M, Ishida S, Yoshida H, Ichikawa K, Kurakata S, et al. A novel humanized anti-human death receptor 5 antibody CS-1008 induces apoptosis in tumor cells without toxicity in hepatocytes. Ann Oncol. 2008;19:1060–1067. doi: 10.1093/annonc/mdn015. http://dx.doi.org/10.1093/annonc/mdn015. [DOI] [PubMed] [Google Scholar]
- 12.Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954–960. doi: 10.1038/91000. http://dx.doi.org/10.1038/91000. [DOI] [PubMed] [Google Scholar]
- 13.Rosevear HM, Lightfoot AJ, Griffith TS. Conatumumab, a fully human mAb against death receptor 5 for the treatment of cancer. Curr Opin Investig Drugs. 2010;11:688–698. [PubMed] [Google Scholar]
- 14.Georgakis GV, Li Y, Humphreys R, Andreeff M, O’Brien S, Younes M, et al. Activity of selective fully human agonistic antibodies to the TRAIL death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured lymphoma cells: induction of apoptosis and enhancement of doxorubicin- and bortezomib-induced cell death. Br J Haematol. 2005;130:501–510. doi: 10.1111/j.1365-2141.2005.05656.x. http://dx.doi.org/10.1111/j.1365–2141.2005.05656.x. [DOI] [PubMed] [Google Scholar]
- 15.Motoki K, Mori E, Matsumoto A, Thomas M, Tomura T, Humphreys R, et al. Enhanced apoptosis and tumor regression induced by a direct agonist antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 2. Clin Cancer Res. 2005;11:3126–3135. doi: 10.1158/1078-0432.CCR-04-1867. http://dx.doi.org/10.1158/1078-0432.CCR-04-1867. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Zhang X, Barrisford GW, Olumi AF. Lexatumumab (TRAIL-receptor 2 mAb) induces expression of DR5 and promotes apoptosis in primary and metastatic renal cell carcinoma in a mouse orthotopic model. Cancer Lett. 2007;251:146–157. doi: 10.1016/j.canlet.2006.11.013. http://dx.doi.org/10.1016/j.canlet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene. 2013;32:1341–1350. doi: 10.1038/onc.2012.164. http://dx.doi.org/10.1038/onc.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.81. http://dx.doi.org/10.1038/cdd.2014.81. [DOI] [PMC free article] [PubMed]
- 19.Adams C, Totpal K, Lawrence D, Marsters S, Pitti R, Yee S, et al. Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ. 2008;15:751–761. doi: 10.1038/sj.cdd.4402306. http://dx.doi.org/10.1038/sj.cdd.4402306 (doi:4402306 [pii]) [DOI] [PubMed] [Google Scholar]
- 20.Siegel RM, Muppidi JR, Sarker M, Lobito A, Jen M, Martin D, et al. SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol. 2004;167:735–744. doi: 10.1083/jcb.200406101. http://dx.doi.org/10.1083/jcb.200406101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valley CC, Lewis AK, Mudaliar DJ, Perlmutter JD, Braun AR, Karim CB, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces death receptor 5 networks that are highly organized. J Biol Chem. 2012;287:21,265–21,278. doi: 10.1074/jbc.M111.306480. http://dx.doi.org/10.1074/jbc.M111.306480 (doi:M111.306480 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akazawa Y, Mott JL, Bronk SF, Werneburg NW, Kahraman A, Guicciardi ME, et al. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology. 2009;136 doi: 10.1053/j.gastro.2009.02.071. http://dx.doi.org/10.1053/j.gastro.2009.02.071 (2365–2376.e1–7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan FK. Three is better than one: pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine. 2007;37:101–107. doi: 10.1016/j.cyto.2007.03.005. http://dx.doi.org/10.1016/j.cyto.2007.03.005 (doi:S1043-4666(07)00041-5 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozsoy HZ, Sivasubramanian N, Wieder ED, Pedersen S, Mann DL. Oxidative stress promotes ligand-independent and enhanced ligand-dependent tumor necrosis factor receptor signaling. J Biol Chem. 2008;283:23,419–23,428. doi: 10.1074/jbc.M802967200. http://dx.doi.org/10.1074/jbc.M802967200 (doi:M802967200 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009;457:1019–1022. doi: 10.1038/nature07606. http://dx.doi.org/10.1038/nature07606 (doi: nature07606 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis AK, Valley CC, Sachs JN. TNFR1 signaling is associated with backbone conformational changes of receptor dimers consistent with overactivation in the R92Q TRAPS mutant. Biochemistry. 2012;51:6545–6555. doi: 10.1021/bi3006626. http://dx.doi.org/10.1021/bi3006626. [DOI] [PubMed] [Google Scholar]
- 27.Donepudi M, Mac Sweeney A, Briand C, Grütter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell. 2003;11:543–549. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 28.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 29.Blanchard H, Kodandapani L, Mittl PR, Marco SD, Krebs JF, Wu JC, et al. The three-dimensional structure of caspase-8: an initiator enzyme in apoptosis. Structure. 1999;7:1125–1133. doi: 10.1016/s0969-2126(99)80179-8. [DOI] [PubMed] [Google Scholar]
- 30.Watt W, Koeplinger Ka, Mildner AM, Heinrikson RL, Tomasselli AG, Watenpaugh KD. The atomic-resolution structure of human caspase-8, a key activator of apoptosis. Structure. 1999;7:1135–1143. doi: 10.1016/s0969-2126(99)80180-4. [DOI] [PubMed] [Google Scholar]
- 31.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 32.Lewis AK, James ZM, McCaffrey JE, Braun AR, Karim CB, Thomas DD, et al. Open and closed conformations of the isolated transmembrane domain of death receptor 5 support a new model of activation. Biophys J. 2014;106:L21–L24. doi: 10.1016/j.bpj.2014.01.044. http://dx.doi.org/10.1016/j.bpj.2014.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix–helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. http://dx.doi.org/10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 34.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. http://dx.doi.org/10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 35.Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O’Connell M, Kelley RF, et al. Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell. 1999;4:563–571. doi: 10.1016/s1097-2765(00)80207-5. http://dx.doi.org/10.1016/S1097-2765(00)80207-5. [DOI] [PubMed] [Google Scholar]
- 36.Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. (doi:0092-8674(93)90132-A [pii]) [DOI] [PubMed] [Google Scholar]
- 37.Richter F, Liebig T, Guenzi E, Herrmann A, Scheurich P, Pfizenmaier K, et al. Antagonistic TNF receptor one-specific antibody (ATROSAB): receptor binding and in vitro bioactivity. PLoS One. 2013;8:e72156. doi: 10.1371/journal.pone.0072156. http://dx.doi.org/10.1371/journal.pone.0072156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Branschädel M, Aird A, Zappe A, Tietz C, Krippner-Heidenreich A, Scheurich P. Dual function of cysteine rich domain (CRD) 1 of TNF receptor type 1: conformational stabilization of CRD2 and control of receptor responsiveness. Cell Signal. 2010;22:404–414. doi: 10.1016/j.cellsig.2009.10.011. http://dx.doi.org/10.1016/j.cellsig.2009.10.011 (doi:S0898-6568(09)00328-3 [pii]) [DOI] [PubMed] [Google Scholar]
- 39.Fricke F, Malkusch S, Wangorsch G. Quantitative single-molecule localization microscopy combined with rule-based modeling reveals ligand-induced TNF-R1 reorganization toward higher-order. Histochem Cell Biol. 2014 doi: 10.1007/s00418-014-1195-0. http://link.springer.com/article/10.1007/s00418-014-1195-0 (accessed July 3, 2014) [DOI] [PubMed]
- 40.Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. http://dx.doi.org/10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 41.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. http://dx.doi.org/10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 42.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. http://dx.doi.org/10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 43.Cottin V, Doan JES, Riches DWH. Restricted localization of the TNF receptor CD120a to lipid rafts: a novel role for the death domain. J Immunol. 2002;168:4095–4102. doi: 10.4049/jimmunol.168.8.4095. [DOI] [PubMed] [Google Scholar]
- 44.Legler DF, Micheau O, Doucey MAA, Tschopp J, Bron C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity. 2003;18:655–664. doi: 10.1016/s1074-7613(03)00092-x. http://www.ncbi.nlm.nih.gov/pubmed/12753742. [DOI] [PubMed] [Google Scholar]
- 45.Scheel-Toellner D, Wang K, Singh R, Majeed S, Raza K, Curnow SJ, et al. The death-inducing signalling complex is recruited to lipid rafts in Fas-induced apoptosis. Biochem Biophys Res Commun. 2002;297:876–879. doi: 10.1016/s0006-291x(02)02311-2. http://www.ncbi.nlm.nih.gov/pubmed/12359234. [DOI] [PubMed] [Google Scholar]
- 46.Muppidi JR, Siegel RM. Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat Immunol. 2004;5:182–189. doi: 10.1038/ni1024. http://dx.doi.org/10.1038/ni1024. [DOI] [PubMed] [Google Scholar]
- 47.Song JH, Tse MC, Bellail A, Phuphanich S, Khuri F, Kneteman NM, et al. Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer Res. 2007;67:6946–6955. doi: 10.1158/0008-5472.CAN-06-3896. http://dx.doi.org/10.1158/0008-5472.CAN-06-3896. [DOI] [PubMed] [Google Scholar]
- 48.Min Y, Shi J, Zhang Y, Liu S, Liu Y, Zheng D. Death receptor 5-recruited raft components contributes to the sensitivity of Jurkat leukemia cell lines to TRAIL-induced cell death. IUBMB Life. 2009;61:261–267. doi: 10.1002/iub.166. http://dx.doi.org/10.1002/iub.166. [DOI] [PubMed] [Google Scholar]
- 49.Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood. 2007;109:711–719. doi: 10.1182/blood-2006-04-016824. http://dx.doi.org/10.1182/blood-2006-04-016824. [DOI] [PubMed] [Google Scholar]
- 50.Delmas D, Rébé C, Micheau O, Athias A, Gambert P, Grazide S, et al. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene. 2004;23:8979–8986. doi: 10.1038/sj.onc.1208086. http://dx.doi.org/10.1038/sj.onc.1208086. [DOI] [PubMed] [Google Scholar]
- 51.Mérino D, Lalaoui N, Morizot A, Schneider P, Solary E, Micheau O. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol. 2006;26:7046–7055. doi: 10.1128/MCB.00520-06. http://dx.doi.org/10.1128/MCB.00520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahammad S, Parmryd I. Cholesterol homeostasis in T cells. Methyl-beta-cyclodextrin treatment results in equal loss of cholesterol from Triton X-100 soluble and insoluble fractions. Biochim Biophys Acta. 2008;1778:1251–1258. doi: 10.1016/j.bbamem.2008.02.010. http://dx.doi.org/10.1016/j.bbamem.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Wang G, Wang X, Yu H, Wei S, Williams N, Holmes DL, et al. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat Chem Biol. 2013;9:84–89. doi: 10.1038/nchembio.1153. http://dx.doi.org/10.1038/nchembio.1153. [DOI] [PubMed] [Google Scholar]
- 54.Mongiat M, Ligresti G, Marastoni S, Lorenzon E, Doliana R, Colombatti A. Regulation of the extrinsic apoptotic pathway by the extracellular matrix glycoprotein EMILIN2. Mol Cell Biol. 2007;27:7176–7187. doi: 10.1128/MCB.00696-07. http://dx.doi.org/10.1128/MCB.00696-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richter C, Messerschmidt S, Holeiter G, Tepperink J, Osswald S, Zappe A, et al. The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol Cell Biol. 2012;32:2515–2529. doi: 10.1128/MCB.06458-11. http://dx.doi.org/10.1128/MCB.06458-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henkler F, Behrle E, Dennehy KM, Wicovsky A, Peters N, Warnke C, et al. The extracellular domains of FasL and Fas are sufficient for the formation of supramolecular FasL–Fas clusters of high stability. J Cell Biol. 2005;168:1087–1098. doi: 10.1083/jcb.200501048. http://dx.doi.org/10.1083/jcb.200501048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang L, Kaizuka Y, Hanagata N. Imaging of Fas–FasL membrane microdomains during apoptosis in a reconstituted cell–cell junction. Biochem Biophys Res Commun. 2012;422:298–304. doi: 10.1016/j.bbrc.2012.04.152. http://dx.doi.org/10.1016/j.bbrc.2012.04.152. [DOI] [PubMed] [Google Scholar]
- 58.Cristian L, Lear JD, DeGrado WF. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc Natl Acad Sci U S A. 2003;100:14,772–14,777. doi: 10.1073/pnas.2536751100. http://dx.doi.org/10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kucerka N, Liu Y, Chu N, Petrache HI, Tristram-Nagle S, Nagle JF. Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using X-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys J. 2005;88:2626–2637. doi: 10.1529/biophysj.104.056606. http://dx.doi.org/10.1529/biophysj.104.056606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gallová J, Uhríková D, Islamov A, Kuklin A, Balgavý P. Erratum: effect of cholesterol on the bilayer thickness in unilamellar extruded DLPC and DOPC liposomes: SANS contrast variation study (general physiology and biophysics (2004) vol. 23 (113–128)) Gen Physiol Biophys. 2004;23:263. [PubMed] [Google Scholar]
- 61.You M, Spangler J, Li E, Han X, Ghosh P, Hristova K. Effect of pathogenic cysteine mutations on FGFR3 transmembrane domain dimerization in detergents and lipid bilayers. Biochemistry. 2007;46:11,039–11,046. doi: 10.1021/bi700986n. http://dx.doi.org/10.1021/bi700986n. [DOI] [PubMed] [Google Scholar]
- 62.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. http://dx.doi.org/10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 63.Joosten RP, Te Beek TAH, Krieger E, Hekkelman ML, Hooft RWW, Schneider R, et al. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkq1105. http://dx.doi.org/10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grossfield A, Woolf TB. Interaction of tryptophan analogs with POPC lipid bilayers investigated by molecular dynamics calculations. Langmuir. 2002;18:198–210. http://dx.doi.org/10.1021/la0106485. [Google Scholar]
- 65.Landolt-Marticorena C, Williams KA, Deber CM, Reithmeier RA. Non-random distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J Mol Biol. 1993;229:602–608. doi: 10.1006/jmbi.1993.1066. http://dx.doi.org/10.1006/jmbi.1993.1066. [DOI] [PubMed] [Google Scholar]
- 66.Jacobs RE, White SH. The nature of the hydrophobic binding of small peptides at the bilayer interface: implications for the insertion of transbilayer helices. Biochemistry. 1989;28:3421–3437. doi: 10.1021/bi00434a042. (doi:2742845) [DOI] [PubMed] [Google Scholar]
- 67.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. http://dx.doi.org/10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 68.Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14,713–14,718. doi: 10.1021/bi980809c. http://dx.doi.org/10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 69.White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta. 1998;1376:339–352. doi: 10.1016/s0304-4157(98)00021-5. http://dx.doi.org/10.1016/S0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 70.Shahidullah K, Krishnakumar SS, London E. The effect of hydrophilic substitutions and anionic lipids upon the transverse positioning of the transmembrane helix of the ErbB2 (neu) protein incorporated into model membrane vesicles. J Mol Biol. 2010;396:209–220. doi: 10.1016/j.jmb.2009.11.037. http://dx.doi.org/10.1016/j.jmb.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Czerski L, Sanders CR. Thiol modification of diacylglycerol kinase: dependence upon site membrane disposition and reagent hydrophobicity. FEBS Lett. 2000;472:225–229. doi: 10.1016/s0014-5793(00)01457-5. http://dx.doi.org/10.1016/S0014-5793(00)01457-5. [DOI] [PubMed] [Google Scholar]
- 72.Fernandes PA, Ramos MJ. Theoretical insights into the mechanism for thiol/disulfide exchange. Chem A Eur J. 2004;10:257–266. doi: 10.1002/chem.200305343. http://dx.doi.org/10.1002/chem.200305343. [DOI] [PubMed] [Google Scholar]
- 73.Kucerka N, Perlmutter JD, Pan J, Tristram-Nagle S, Katsaras J, Sachs JN. The effect of cholesterol on short- and long-chain monounsaturated lipid bilayers as determined by molecular dynamics simulations and X-ray scattering. Biophys J. 2008;95:2792–2805. doi: 10.1529/biophysj.107.122465. http://dx.doi.org/10.1529/biophysj.107.122465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ko YG, Lee JS, Kang YS, Ahn JH, Seo JS. TNF-alpha-mediated apoptosis is initiated in caveolae-like domains. J Immunol. 1999;162:7217–7223. http://www.ncbi.nlm.nih.gov/pubmed/10358168. [PubMed] [Google Scholar]
- 75.Schuck S, Honsho M, Ekroos K, Shevchenko A, Simons K. Resistance of cell membranes to different detergents. Proc Natl Acad Sci U S A. 2003;100:5795–5800. doi: 10.1073/pnas.0631579100. http://dx.doi.org/10.1073/pnas.0631579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lingwood D, Simons K. Detergent resistance as a tool in membrane research. Nat Protoc. 2007;2:2159–2165. doi: 10.1038/nprot.2007.294. http://dx.doi.org/10.1038/nprot.2007.294. [DOI] [PubMed] [Google Scholar]
- 77.Feig M, Karanicolas J, Brooks CL., III . MMTSB Tool Set, MMTSB NIH Research Resource. The Scripps Research Institute; 2001. [Google Scholar]
- 78.Brooks B, Bruccoleri R. CHARMM: a program for macro-molecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. http://dx.doi.org/10.1002/jcc.540040211. [Google Scholar]
- 79.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulation. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. http://dx.doi.org/10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 80.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain X(1) and X(2) dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. http://dx.doi.org/10.1021/Ct3004000x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. http://dx.doi.org/10.1016/0021-9991(77)90098-5. [Google Scholar]
- 82.Im W, Feig M, Brooks CL. An implicit membrane generalized born theory forthe study of structure, stability, and interactions of membrane proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. http://dx.doi.org/10.1016/S0006-3495(03)74712–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Im W, Lee MS, Brooks CL. Generalized born model with a simple smoothing function. J Comput Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. http://dx.doi.org/10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 84.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J Chem Phys. 1994;101:4177–4189. http://dx.doi.org/10.1063/1.467468. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.