

Abstract
Cardiac microvascular endothelial cells (CMECs) extensively secrete cytokines during myocardial ischemia/reperfusion injury (MIRI). Tongxinluo (TXL) has been demonstrated to preserve the function of the endothelium and myocardium against MIRI. This study was designed to identify alterations in the paracrine function of CMECs under hypoxia/reoxygenation (H/R) conditions and assess its modulation by TXL. CMECs were exposed to different concentrations of TXL for 30 min and then subjected to hypoxia and reoxygenation for 12 and 2 h, respectively. Apoptosis was measured to determine the optimal TXL concentration. Protein antibody arrays were used to assess changes in cytokines secreted into conditioned medium by CMECs. A Gene Ontology (GO) analysis was applied to interpret the functional implications of changes in cytokines. TXL inhibited CMEC apoptosis in a concentration-dependent manner after H/R, reaching peak efficacy at a concentration of 800 μg/ml. H/R significantly altered 33 cytokines, and TXL (800 μg/ml) changed the levels of 121 different cytokines compared with the H/R group. Among these cytokines, 10 that were increased by H/R were decreased by TXL, five that were decreased by H/R were increased by TXL, and eight that were attenuated by H/R were further decreased by TXL. Insulin-like growth factor binding protein-1 was up-regulated by H/R and was further increased by TXL. Significantly altered factors were found to be involved in cell proliferation, growth and differentiation, as well as chemotaxis and transport. TXL inhibited the apoptosis of CMECs and modulated their paracrine function in MIRI.
Keywords: Cytokine, paracrine, cardiac microvascular endothelial cells, myocardial ischemia/reperfusion injury, Tongxinluo
Introduction
Myocardial ischemia/reperfusion injury (MIRI) remains an unresolved problem in the era of coronary revascularization therapy for acute myocardial infarction. MIRI accounts for almost 50% of the final necrosis size and leads to many acute complications, such as reperfusion arrhythmias, myocardial stunning, and the no-reflow phenomenon [1]. A notable feature of myocardial reperfusion injury is microcirculation dysfunction [2,3]. Cardiac microvascular endothelial cells (CMECs), the fundamental building blocks of the myocardial microcirculation, are the primary cells in the myocardium, and their function is jeopardized in MIRI [4]. The initiation of MIRI upon restoration of blood flow contact with the myocardium sets in motion a pathophysiological process involving complex interactions among hematocytes, endothelial cells, and cardiomyocytes. CMECs, which compose the microcirculatory tract, serve as a functional interface between flowing blood and cardiomyocytes. In this capacity, they communicate with both cardiomyocytes and blood cells. However, how this interaction is changed in MIRI is poorly understood.
Cytokines, acting as mediators or messengers, play important roles in the interactions among different types of cells, in cellular responses to the environment, and in the maintenance of cellular and tissue homeostasis. Cytokine synthesis and secretion are modulated in most pathophysiological processes, such as cancer and cardiovascular diseases, including MIRI. CMECs secrete a considerable range of cytokines that participate in regulating not only the metabolism and activity of cardiomyocytes, but also the communication with leukocytes [5,6], which may be modulated during the process of ischemia/reperfusion injury. Cytokine modulation reflects interactions among cells. It has been shown that some individual factors secreted by CMECs, such as neuregulin-1β (NRG-1β), are modulated in MIRI [7,8]. To date, however, how the cytokine profile or the paracrine function of CMECs is regulated in MIRI has remained unclear.
Tongxinluo (TXL), a traditional Chinese medicine made from 12 different herbals and available in capsule form, was approved for treating cardiocerebral vascular diseases by the State Food and Drug Administration (SFDA) of China in 1996. Results of high-performance liquid chromatography and gas chromatography analyses indicate that the major ingredients of TXL include peoniflorin, ginsenoside Rg1, ginsenoside Rb1, jujuboside A, jujuboside B, isoborneol and borneol [9]. In our previous studies, we found that TXL reduced myocardial no-reflow and reperfusion injury by inhibiting the apoptosis of CMECs and preserving endothelial function [10-12]. Diverse ingredients in TXL were speculated to have multiple synergetic or antagonistic effects on the cytokine profile of CMECs. However, it is still unclear whether TXL regulates the paracrine function of CMECs and, if so, how the mixture ultimately changes the cytokine profile. To address these unanswered questions, we employed an in vitro CMEC model of ischemia/reperfusion in conjunction with human cytokine antibody arrays to assess cytokine modulation and its regulation by TXL.
Materials and methods
Cell culture and the hypoxia/reoxygenation model
CMECs from healthy humans were purchased from ScienCell Research Laboratories (San Diego, CA) and cultured as instructed. The cells were grown in complete Endothelial Cell Medium (ECM) containing 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement (ECGS), and 1% penicillin/streptomycin at 37°C with 5% CO2. When cells reached ~90% confluence, they were detached using 0.25% trypsin-EDTA (Invitrogen) and subcultured at a ratio of 1:3. Cells in passage 3 were washed with phosphate-buffered saline (PBS) and then exposed to different treatments in serum-free DMEM for 30 min. Thereafter, cells were exposed to hypoxia by incubating in a sealed, hypoxic GENbox jar fitted with a catalyst (Bio-Me’rieux) to scavenge free oxygen for 12 h, and then transferred to normoxic conditions for a 2-h reoxygenation period, as previously described [13]. Oxygen tension in the medium was measured using an anaer indicator (BioMe’rieux).
Preparation of the TXL solution
A solution of TXL ultrafine powder (Lot Number: 071201; Shijiazhuang Yiling Pharmaceutical Co., Shijiazhuang, China) was prepared as described in our previous study [12]. Briefly, after dissolving TXL powder in serum-free Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Grand Island, USA), the mixture was sonicated for 30 min and then centrifuged at 2000×g for 15 min. The supernatant was filtered, and the precipitate was dried at 60°C to allow calculation of an accurate weight of the dissolved ingredients. The solution was adjusted to a final concentration of 2000 μg/ml by adding DMEM and then stored at -20°C. The TXL solution was adjusted to different concentrations (100, 200, 400, 800, 1200 μg/ml) by gradually adding DMEM before addition to cultures for 30 min and exposure to hypoxia/reoxygenation (H/R).
Assessment of apoptosis
Cell apoptosis was quantitatively determined using an Annexin V-FITC/PI Kit (Biosea Biotechnology, Beijing), according to the manufacturer’s instructions. Briefly, cells from different experimental groups were collected and resuspended in 500 μl of buffer. Five microliters of Annexin V solution was added to the cell suspension, and the mixture was incubated in the dark for 15 min at room temperature. Thereafter, 5 μl of propidium iodide (PI) was added, and at least 15,000 cells were acquired by flow cytometry using a FACSCalibur System (Becton-Dickinson) and analyzed with Flowjo software (version 7.6.1). The cells were segregated into four quadrants: viable cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic cells (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+). Apoptotic rates were calculated by summing the early and late apoptotic quadrants.
Human cytokine antibody array
Medium from the control group, the H/R group, and the TXL group at the experimentally determined best working concentration was collected after H/R and centrifuged. The concentrations of cytokines in the supernatant were then quantified using a Quantibody Human Cytokine Antibody Array 7000 kit (Catalog No. QAH-CAA-7000; RayBiotech, Norcross, GA, USA). This array consists of 320 different antibodies spotted in quadruplicate onto eight slide chips. The cytokine profile of each group was determined using three independent biological samples. Samples in each array were assayed simultaneously using a sandwich enzyme-linked immunosorbent assay (ELISA) procedure. The signal was acquired by fluorescence detection and quantified, and the relative expression levels of cytokines were determined by comparison between groups. Experiments were performed by CapitalBio Technology (Beijing, China) following the manufacturer’s instructions.
Gene ontology analysis
The Gene Ontology (GO) project provides a controlled vocabulary for describing gene and gene product attributes in any organism (http://www.geneontology.org). GO assignments are categorized into three domains: biological process, cellular component, and molecular function. Fisher’s exact test was used to determine whether the overlap between the differentially expressed factors list and the GO annotation list was greater than would be expected by chance alone. The p-value denotes the significance of GO term enrichment in the set of differentially expressed factors. The lower the p-value, the more significant the GO Term (a p-value ≤ 0.05 is recommended).
Statistical analysis
The experimental data are presented as the means of each condition ± SEM. A one-way analysis of variance (ANOVA) was used to compare more than two groups, and a paired Student’s t-test was performed for comparisons of two conditions (SPSS for Windows version 16.0). A P-value < 0.05 (two sided) was considered statistically significant. The original data were standardized using IQR methods, and pooled analysis of the data was implemented using R method.
Results
TXL protects CMECs from H/R-induced apoptosis
We adopted an in vitro model of endothelial reperfusion injury (2 h of hypoxia followed by 12 h of reoxygenation) as described previously [13,14] to induce the apoptosis of CMECs. To investigate whether TXL inhibited apoptosis, we exposed CMECs to different concentrations of TXL (100, 200, 400, 800, 1200 μg/ml) for 30 min before H/R. Flow cytometric assays (Figure 1) demonstrated that H/R exposure significantly increased the apoptotic rate of CMECs compared with controls (35.15% ± 0.78% vs. 5.90% ± 0.96%, P < 0.0001). TXL attenuated H/R-induced apoptosis in a concentration-dependent manner, beginning at a concentration of 200 μg/ml. TXL achieved its maximum anti-apoptotic efficacy at 800 μg/ml, a concentration that reduced the apoptosis rate after H/R exposure to 14.64% ± 2.23% (P = 0.001; Figure 1).
Figure 1.
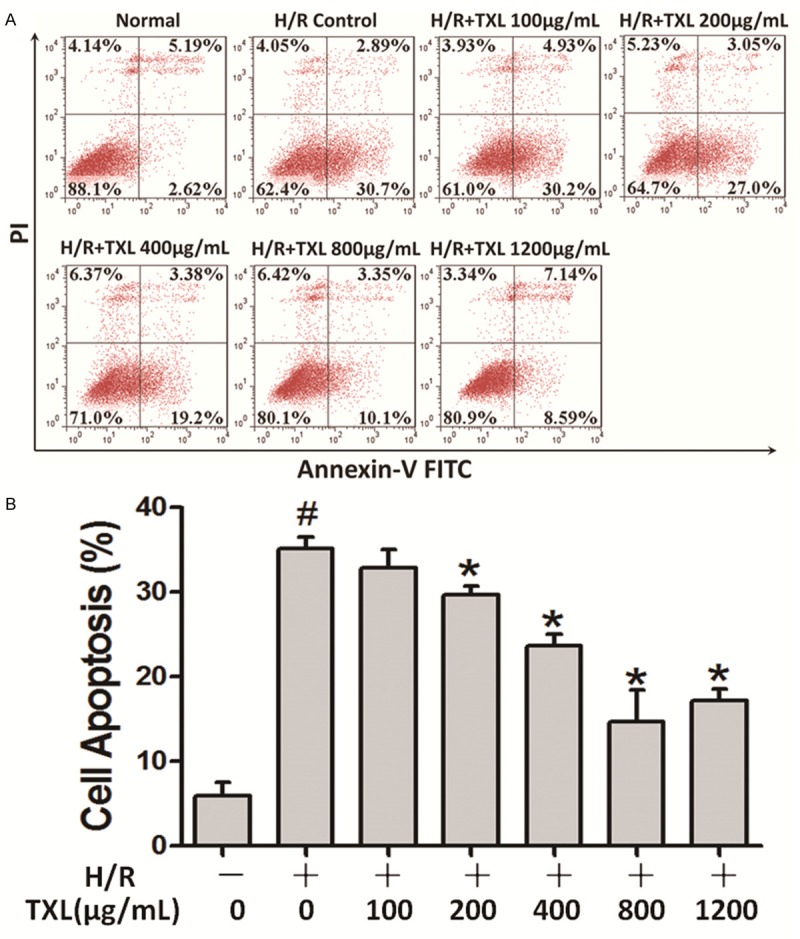
TXL inhibits H/R-induced apoptosis of CMECs in a concentration-dependent manner. A. Representative scatterplots of flow cytometry results showing apoptotic quadrants. B. Histogram of CMEC apoptosis rates at different TXL concentrations. The values represent means ± SEM. Each group was biologically repeated three times (#P < 0.05 versus the normal group; *P < 0.05 versus the H/R group).
H/R exposure and TXL treatment alters the cytokine profile of CMECs
To investigate the paracrine function of CMECs after H/R and determine how it is modulated by TXL, we used a human cytokine antibody array to assess changes in secreted cytokines in culture supernatants. TXL was used at its optimal anti-apoptotic concentration of 800 μg/ml. The results of a cluster analysis are shown in Figure 2. To identify meaningful changes in the cytokine profile, we selected factors that were significantly altered (fold-change > 1.5; P-value < 0.05) as previously described [15], in comparisons between the H/R group and the control group and/or the H/R + TXL group and the H/R group. Compared with the control group, 33 different cytokines were significantly changed by H/R, including 19 that were down-regulated and 14 that were up-regulated. Among these factors, angiopoietin-like 4 (AGNPTL-4) was the most up-regulated (3.99-fold, P = 9.04×10-5; Figure 3A), and urokinase-type plasminogen activator (uPA) was the most down-regulated (0.23-fold, P = 5.31×10-5; Figure 3A). Treatment with TXL significantly changed 121 factors compared with the H/R group, including 28 that were up-regulated and 93 that were down-regulated. Among these factors, granulocyte colony-stimulating factor (G-CSF) was the most up-regulated (38.01-fold, P = 4.07×10-4; Figure 3B) and bone morphogenetic protein-2 (BMP-2) was the most down-regulated (0.02-fold, P = 1.12×10-4; Figure 3B).
Figure 2.
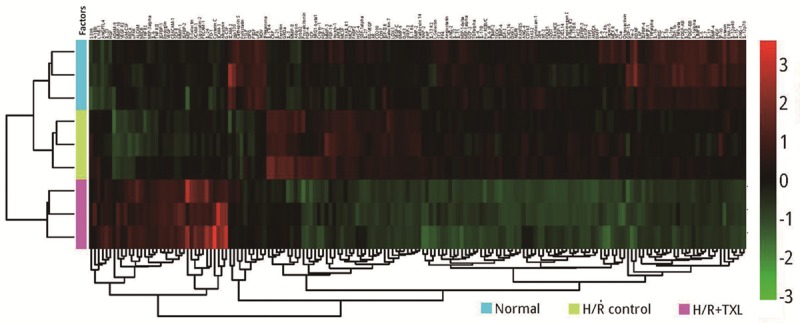
Results of a cluster analysis based on samples and all significantly altered factors (fold-change > 1.5; P < 0.05). The color gradient represents the range of cytokine levels, as indicated on the right. Each group was biologically repeated three times.
Figure 3.
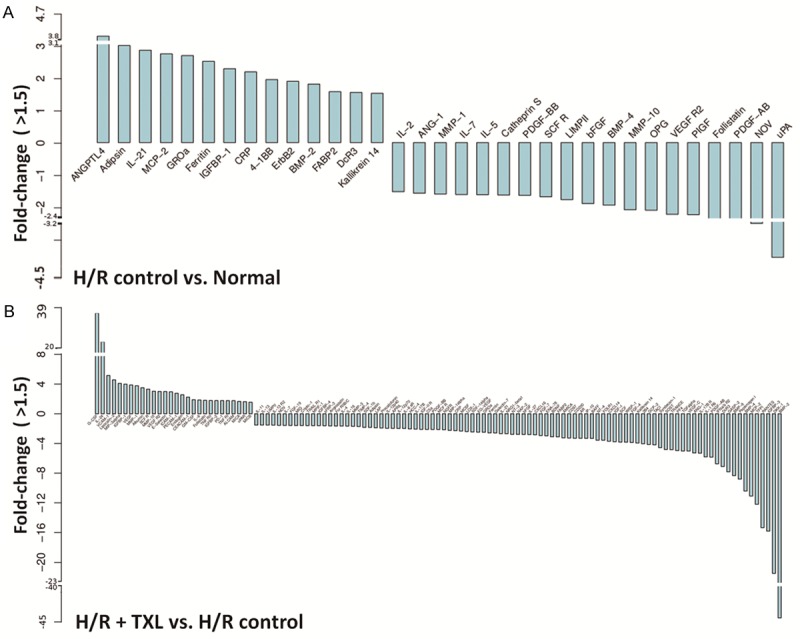
Cytokines that were significantly differentially expressed in pairwise comparisons. (fold-change > 1.5; P < 0.05). A. Histogram of fold-changes of factors in the H/R group compared with the normal group. B. Histogram of fold-changes of factors in the TXL group compared with the H/R group.
Twenty-four factors were significantly changed in comparisons between the H/R group and the control group as well as those between the TXL group and the H/R group, as shown in Figure 4A. Among these factors, 10 that were increased in the H/R group were decreased by TXL, five that were decreased in the H/R group were increased by TXL, and eight that were down-regulated by H/R were further reduced by TXL. Notably, insulin-like growth factor binding protein-1 (IGFBP-1) was among the factors that were up-regulated by H/R and further increased by TXL.
Figure 4.
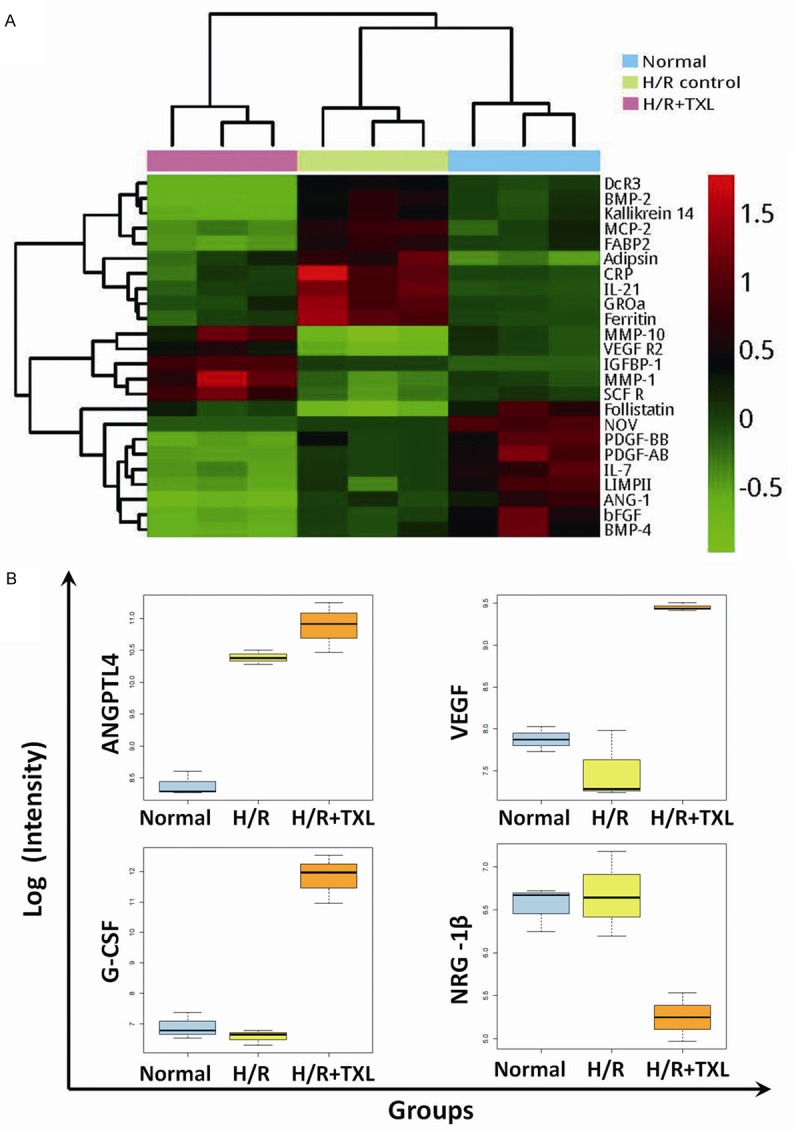
Notable differentially expressed factors revealed by pairwise comparisons. A. Color gradient representation of 24 factors that were significantly changed in comparisons of the H/R group versus the control group and of the TXL group versus the H/R group. B. Boxplots of ANGPTL-4, G-CSF, VEGF, and NRG-1β expression in different groups.
Some factors that were previously shown to participate in the myocardial reperfusion injury process were also identified by the protein arrays; however, the observed changes were shown in comparisons either between the H/R group and the control group, or between the TXL group and the H/R group. For example, the level of ANGPTL-4 was significantly changed only in comparisons of the H/R group with the normal group. Also notable, vascular endothelial growth factor (VEGF) and G-CSF were increased by TXL, and NRG-1β was decreased by TXL. (Figure 4B).
GO analysis of significantly altered cytokines
As previously reported, some growth factors might participate in the protection against myocardial ischemia/reperfusion injury [16]. Thus, we used a GO analysis to identify the functions of significantly altered factors identified in cytokine antibody arrays. The GO project provides a controlled vocabulary for describing gene and gene product attributes in any organism (http://www.geneontology.org). Using this controlled vocabulary, we analyzed ‘Biological processes’ and ‘molecular functions’ associated with these factors and identified a number of relevant terms, including positive and negative regulation of cell proliferation or differentiation, cell growth, chemotaxis, transport, vasculogenesis, and positive and negative regulation of angiogenesis. The classification of these factors by different functions is shown in Table 1.
Table 1.
GO analysis of differentially expressed cytokines in conditioned media
| GO TERM/GO ID | H/R vs. Normal | H/R vs. Normal | TXL vs. H/R | TXL vs. H/R |
|---|---|---|---|---|
| Up-regulated | Down-regulated | Up-regulated | Down-regulated | |
| Positive regulation of cell proliferation GO:0008284 | IL-21 | VEGF R2; IL-7; PDGF-AB; IL-2; bFGF; SCF R; PDGF-BB | IL-24; G-CSF; VEGF R2; VEGF; SCF R; GM-CSF; IL-6 | VEGF-C; PDGF-AB; IGF-II R; bFGF; APRIL; IL-21; AR; IL-6 sR; NT-3; PDGF-BB; IL-7; EG-VEGF; ENA-78; FGF-19; MCSF; FGF-4; IGF-I; IL-11; BAFF; FGF-7; cripto-1; IP-10 |
| Negative regulation of cell proliferation GO:0008285 | GROα; BMP-2; 4-1BB | bFGF; BMP-4 | IL-24; IL-6; TNF RII; TNF RI | IGFBP-3; BMP-2; IL-1 R6; BMP-4; bFGF; IL-6 sR; MPIF-1; GROα; sFRP-3; angiostatin; IGF-I |
| Regulation of cell growth GO:0001558 | ErbB2; IGFBP-1 | bFGF; IL-2; NOV | IGFBP-1; IGFBP-2; VEGF | bFGF; sFRP-3; BMP-9; NRG-1β; IGFBP-3; IGFBP-4; NOV; IL-17B R; CXCL16; WISP-1 |
| Positive regulation of cell differentiation GO:0045597 | IL-21; BMP-2 | bFGF; IL-2; IL-5; IL-7; VEGF R2; BMP-4 | GM-CSF; IL-6; VEGF R2; TNF RI; TNF RII; VEGF | MCSF; bFGF; sFRP-3; BMP-9; NRG-1β; IGF-I; IGFBP-3; IL-4; IL-6 sR; IL-7; IL-12p40; IL-12p70; IL-13 R2; IL-13; SCF; NGFR; NT-3; PF4; angiostatin; IL-21; BDNF; BMP-2; BMP-4; VEGF-C; procalcitonin; TRANCE |
| Negative regulation of cell differentiation GO:0045596 | CRP; BMP-2 | SCF R; BMP-4; follistatin | IL-6; SCF R; TNF RI; TNF RII; follistatin | CRP; FGF-4; DKK-1; sFRP-3; IFNα/β R2; IGF-I; IL-4; IL-6 sR; IL-13 R2; IL-13; NGFR; PF4; BDNF; MIP-1α; BMP-2; BMP-4; procalcitonin |
| Chemotaxis GO:0006935 | GROα; MCP-2 | uPA; bFGF | uPAR; MIP-3α | CXCL16; RANTES; bFGF; MIP-1α; IL-4; MCP-2; MPIF-1; GROα; ENA-78; MIG; C5a; GCP-2; CXCL14; IP-10; HCC-4 |
| Transport GO:0006810 | CRP; adipsin; ferritin; 4-1BB; MCP-2 | bFGF; follistatin; ANG-1; IL-2; IL-5; SCF R; PDGF-AB; PDGF-BB; NOV; BMP-4; LIMPII | GM-CSF; G-CSF; follistatin; albumin; ICAM-1; IL-6; SCF R; lipocalin-2; PECAM-1; PD-1; E-selectin; TNF RI; TNF RII; VEGF | BAFF; AMICA; CRP; adipsin; bFGF; FGF-7; DKK-1; FOLR1; ferritin; ANG-1; NRG-1β; IFNα/β R2; IGF-I; IGF-II R; IL-1 R6; IL-4; IL-6 sR; IL-11; IL-12p40; IL-12p70; IL-13 R2; IL-13; IP-10; AR; NGFR; NT-3; PDGF-AB; PDGF-BB; PF4; NAP-2; angiostatin; CXCL16; NOV; BDNF; MIP-1α; RANTES; MCP-2; BMP-4; C5a; VEGF-C; procalcitonin; TRANCE; LIMPII; FGF-19 |
| Vasculogenesis GO:0001570 | VEGF R2 | VEGF R2; VEGF; endoglin | BMP-9; cripto-1 | |
| Positive regulation of angiogenesis GO:0045766 | ANGPTL4 | VEGF R2; bFGF | VEGF R2; VEGF | BMP-9; IL-1 R6; bFGF; C5a |
| Negative regulation of angiogenesis GO:0016525 | BMP-9; PF4; IP-10 | |||
| Angiogenesis GO:0001525 | ANGPTL4 | VEGF R2 | VEGF R2; VEGF; CEACAM-1 | VEGF-C; EG-VEGF; TGFα; angiostatin |
Discussion
In this study, we used a previous model of CMEC ischemia/reperfusion injury to illuminate changes in the cytokine profile after H/R and its modulation by TXL. Our results indicated that 1) H/R significantly altered cytokine secreted by CMECs, and TXL further changed the cytokine profile sharply; and 2) the functions of differentially secreted CMEC-derived factors mainly centered on cell proliferation, cell growth, cell differentiation, chemotaxis, and transport. To our knowledge, this is the first study to investigate alterations in the CMEC cytokine profile under ischemia/reperfusion conditions.
CMECs are the principal cells in the myocardium, and outnumber cardiomyocytes by approximately 2-fold [4]. They not only constitute the pathway through which the microcirculation supplies nutrients and oxygen, they also play a unique role as a barrier against inflammatory cells and toxins. Notably, CMECs act through their paracrine functions to differentially affect cardiomyocytes in the MIRI process. For example, factors secreted by these cells, such as interleukins (ILs), tumor necrosis factor-α (TNF-α), monocyte chemotactic protein (MCP), endothelin (ET) and angiotensin-II (Ang-II), aggravate myocardial inflammatory responses [1,4,6]. But CMECs also protect cardiomyocytes by secreting protective factors, such as nitric oxide (NO) and NRG-1β [17,18].
In pairwise comparisons, we identified 24 secreted factors that were significantly changed by ischemia/reperfusion injury and TXL. A few studies have previously reported some of these myocardial reperfusion injury-related factors, notably including GROα (growth-related oncogene α) also called chemokine (C-X-C motif) ligand 1 (CXCL1). CXCL1 is a chemoattractant secreted by ischemic tissue that induces early inflammatory responses, including neutrophil infiltration [19-21]. Previous studies have demonstrated that some therapeutics used for myocardial preservation share the mechanism of down-regulating CXCL1 and subsequently limiting leukocyte infiltration [22,23]. Some of the factors we found to be changed have not previously been reported in the context of MIRI, although the effects of these factors on inflammatory responses or cell apoptosis have been described in other studies. For example, MCP-2, a chemoattractant protein that recruits monocytes, lymphocytes, basophils and eosinophils, is secreted by microvascular endothelial cells under conditions of infection or inflammation [24]. Decoy receptor 3 (DCR3), which is barely detectable in normal tissue but is elevated under conditions of infection or stress, is known to modulate immune functions of monocytes and macrophages [25-27]. DCR3 has also been shown to induce apoptosis in rodent RAW 264.7 cells [28]. Inflammation and apoptosis are recognized as key mechanisms in myocardial reperfusion injury [29]. Consistent with their known roles, these three inflammation or apoptosis-related factors-CXCL1, MCP-2 and DCR3-were up-regulated by H/R and down-regulated by TXL in our study. A GO term analysis indicated chemotaxis as a common function of many altered factors, implying that the dysfunctional endothelium in MIRI attracts white blood cells. Notably, TXL inhibited the secretion of many chemotactic factors. This suggests that TXL enhances microvascular endothelial cell barrier function, possibly accounting for the alleviation of inflammatory cell leakage outside the microvasculature produced by TXL observed in our previous animal study [11].
ANGPTL-4, which we found to be up-regulated after H/R, has been shown to inhibit the no-reflow phenomenon and maintain vascular integrity [30]. Thus, the elevation of ANGPTL-4 in response to MIRI might be part of a protective mechanism initiated by CMECs. ANGPTL-4 levels also showed a trend toward further increases with TXL treatment. Although this latter effect did not reach statistical significance, the possible modulation of ANGPTL-4 by TXL warrants further study. Furthermore, TXL sharply increased the levels of secreted VEGF [31,32] and G-CSF [33], which are known to ameliorate MIRI, further helping to explain the protective role of TXL.
Some findings in our study were unanticipated. NRG-1β has been previously identified as a protective factor secreted by CMECs experiencing reperfusion injury [7]. In accord with this previous study, we found that H/R significantly increased NRG-1β; unexpectedly however, TXL decreased it. Two additional factors-kallikerein-14 (KLK14) and BMP-2-followed a similar trend. Delivery of the KLK14 gene has been shown to attenuate myocardial reperfusion injury by inhibiting apoptosis of cardiomyocytes and by activating Akt-glycogen synthase kinase-3 (GSK3) and Akt-Bad signaling pathways [34,35]; hence, KLK14 seems to be protective. BMP-2 has shown therapeutic potential in individuals with myocardial ischemia by improving contractility of cardiomyocytes in a phosphatidylinositol 3-kinase (PI3K)-dependent manner [36] and by inhibiting the death of cardiomyocytes through phosphorylation of Smad1/5/8 proteins [37]. Nevertheless, our study indicated that TXL decreased KLK14 secretion, and we found the H/R increased the level of BMP-2, whereas TXL decreased it.
We also found unexpectedly that a few factors that protect against MIRI, such as platelet-derived growth factor (PDGF) [38], basic fibroblast growth factor (bFGF) [39-41] and angiopoietin-1 (ANG-1) [42-44], were down-regulated by H/R and further reduced by TXL. Hausenloy and Yellon have proposed the concept of a reperfusion injury salvage kinase (RISK) pathway, which posits that growth factors protect against ischemia/reperfusion injury by activating pro-survival kinase signaling cascades, including PI3K/Akt and p42/p44 extracellular signal-regulated kinases (ERK1/2) [16]. PDGF, bFGF and ANG-1 exert their protective effect, at least in part, by promoting the RISK pathway. Moreover, KLK14 and BMP2, as well as NRG-1β [45], activate PI3K/Akt during MIRI. A GO term analysis showed that most factors that affect cell proliferation, differentiation, and growth were down-regulated by TXL. However, we previously demonstrated that TXL inhibited MIRI by activating both PI3K/Akt and MEK/ERK pathways [12,46]. These observations suggest that TXL activates RISK pathways directly rather than indirectly by promoting growth factors that, in turn, activate RISK cascades.
We further found that IL-21, fatty acid-binding protein (FABP) and C-reactive protein (CRP) were elevated after H/R and reduced by TXL. In accord with these findings, previous studies showed that IL-21 and FABP were up-regulated in some non-cardiac organs after reperfusion injury. Specifically, IL-21 is elevated in hepatic or nephritic tissues [47,48], and FAPB is elevated during intestinal reperfusion injury [49-51]. We also found that TXL attenuated the secretion of BMP-4. BMP-4 induces apoptosis by activating the JNK/MAP kinase pathway, and inhibiting the BMP-4 signaling pathway has been shown to alleviate MIRI [52]. From this, we infer that the reduction in BMP-4 levels by TXL is one of the protective mechanisms against MIRI. Our study was limited in its ability to demonstrate changes in well-known injurious factors, such as TNF-α, ET, and Ang-II.
In this study, we used protein arrays to investigate alterations in cytokines secreted by CMECs. A protein array is a mid-throughput method for simultaneously studying a series of protein, but it is only able to identify changes in preestablished factors. Thus, whether additional unknown factors participate in the MIRI process currently remains unclear. In addition, we chose only a single time point for illuminating alterations in the cytokine profile; thus, time-dependent trends of altered factors remain to be revealed by further research.
Conclusions
We found that TXL inhibited H/R-induced apoptosis of CMECs in a concentration-dependent manner, reaching its peak efficacy at 800 μg/ml. H/R significantly affected CMEC paracrine functions, which were further modulated by TXL. ANGPTL-4 was up-regulated by H/R, and its possible regulation by TXL requires further investigation. TXL increased VEGF and C-GSF and decreased NRG-1β. The levels of most factors that affect chemotaxis or cell growth were attenuated by TXL.
Acknowledgements
This study was supported by grants from the National Basic Research Program (973 Program) of China (No. 2012CB518602) and National Natural Science Foundation of China (No. 81603425, No. 81500270, No. 81370223, No. 81573957 and No. 81300157) and Beijing Talents Fund (No. 2016000021469G221).
Disclosure of conflict of interest
None.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial Reperfusion Injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the “dark side” of reperfusion. Circulation. 2009;120:2105–2112. doi: 10.1161/CIRCULATIONAHA.108.814640. [DOI] [PubMed] [Google Scholar]
- 3.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 4.Singhal AK, Symons JD, Boudina S, Jaishy B, Shiu YT. Role of endothelial cells in myocardial ischemia-reperfusion injury. Vasc Dis Prev. 2010;7:1–14. doi: 10.2174/1874120701007010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brutsaert DL. Cardiac Endothelial-Myocardial Signaling: Its Role in Cardiac Growth, Contractile Performance, and Rhythmicity. Physiol Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 6.Rodrigues SF, Granger DN. Role of blood cells in ischaemia-reperfusion induced endothelial barrier failure. Cardiovasc Res. 2010;87:291–299. doi: 10.1093/cvr/cvq090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hedhli N, Huang Q, Kalinowski A, Palmeri M, Hu X, Russell RR, Russell KS. Endothelium-Derived Neuregulin Protects the Heart Against Ischemic Injury/Clinical Perspective. Circulation. 2011;123:2254–2262. doi: 10.1161/CIRCULATIONAHA.110.991125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuramochi Y, Cote GM, Guo X, Lebrasseur NK, Cui L, Liao R, Sawyer DB. Cardiac Endothelial Cells Regulate Reactive Oxygen Species-induced Cardiomyocyte Apoptosis through Neuregulin-1β/erbB4 Signaling. J Biol Chem. 2004;279:51141–51147. doi: 10.1074/jbc.M408662200. [DOI] [PubMed] [Google Scholar]
- 9.Chen WQ, Zhong L, Zhang L, Ji XP, Zhao YX, Zhang C, Jiang H, Wu YL, Zhang Y. Chinese medicine tongxinluo significantly lowers serum lipid levels and stabilizes vulnerable plaques in a rabbit model. J Ethnopharmacol. 2009;124:103–110. doi: 10.1016/j.jep.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Li XD, Yang YJ, Geng YJ, Jin C, Hu FH, Zhao JL, Zhang HT, Cheng YT, Qian HY, Wang LL, Zhang BJ, Wu YL. Tongxinluo reduces myocardial no-reflow and ischemia-reperfusion injury by stimulating the phosphorylation of eNOS via the PKA pathway. Am J Physiol Heart Circ Physiol. 2010;299:H1255–H1261. doi: 10.1152/ajpheart.00459.2010. [DOI] [PubMed] [Google Scholar]
- 11.Cheng YT, Yang YJ, Zhang HT, Qian HY, Zhao JL, Meng XM, Luo FL, Wu YL. Pretreatment with Tongxinluo protects porcine myocardium from ischaemia/reperfusion injury through a nitric oxide related mechanism. Chin Med J. 2009;122:1529–1538. [PubMed] [Google Scholar]
- 12.Cui H, Li X, Li N, Qi K, Li Q, Jin C, Zhang Q, Jiang L, Yang Y. Induction of autophagy by Tongxinluo through the MEK/ERK pathway protects human cardiac microvascular endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc Pharmacol. 2014;64:180–190. doi: 10.1097/FJC.0000000000000104. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z, Hu Z, Lu Z, Cai S, Gu X, Zhuang H, Ruan Z, Xia Z, Irwin MG, Feng D, Zhang L. Differential microRNA profiling in a cellular hypoxia reoxygenation model upon posthypoxic propofol treatment reveals alterations in autophagy signaling network. Oxid Med Cell Longev. 2013;2013:378484. doi: 10.1155/2013/378484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu HY, Gao YH, Wang ZY, Xu B, Wu AM, Xing YW, Liu B, Lou LX, Chen LX. Astragalus Polysaccharide Suppresses the Expression of Adhesion Molecules through the Regulation of the p38 MAPK Signaling Pathway in Human Cardiac Microvascular Endothelial Cells after Ischemia-Reperfusion Injury. Evid Based Complement Alternat Med. 2013;2013:280493. doi: 10.1155/2013/280493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S, Peng L, Tang Y, Zhang L, Guo W, Zou X, Peng X. Hypoxia of PC-3 prostate cancer cells enhances migration and vasculogenesis in vitro of bone marrow-derived endothelial progenitor cells by secretion of cytokines. Oncol Rep. 2013;29:2369–2377. doi: 10.3892/or.2013.2363. [DOI] [PubMed] [Google Scholar]
- 16.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 17.Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK, Warltier DC, Kersten JR, Pratt PF Jr. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. 2011;51:803–811. doi: 10.1016/j.yjmcc.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, Mummidi S. Activation of Intrinsic and Extrinsic Proapoptotic Signaling Pathways in Interleukin-18-mediated Human Cardiac Endothelial Cell Death. J Biol Chem. 2004;279:20221–20233. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 19.Sager R, Haskill S, Anisowicz A, Trask D, Pike MC. GRO: a novel chemotactic cytokine. Adv Exp Med Biol. 1991;305:73–77. doi: 10.1007/978-1-4684-6009-4_9. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–2407. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 21.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–R28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 22.Mersmann J, Berkels R, Zacharowski P, Tran N, Koch A, Iekushi K, Dimmeler S, Granja TF, Boehm O, Claycomb WC, Zacharowski K. Preconditioning by toll-like receptor 2 agonist Pam3CSK4 reduces CXCL1-dependent leukocyte recruitment in murine myocardial ischemia/reperfusion injury. Crit Care Med. 2010;38:903–909. doi: 10.1097/CCM.0b013e3181ce50e6. [DOI] [PubMed] [Google Scholar]
- 23.Al-Amran F, Shahkolahi M. Oxytocin ameliorates the immediate myocardial injury in rat heart transplant through downregulation of neutrophil-dependent myocardial apoptosis. Transplant Proc. 2013;45:2506–2512. doi: 10.1016/j.transproceed.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 24.Struyf S, Proost P, Vandercappellen J, Dempe S, Noyens B, Nelissen S, Gouwy M, Locati M, Opdenakker G, Dinsart C, Van Damme J. Synergistic up-regulation of MCP-2/CCL8 activity is counteracted by chemokine cleavage, limiting its inflammatory and anti-tumoral effects. Eur J Immunol. 2009;39:843–857. doi: 10.1002/eji.200838660. [DOI] [PubMed] [Google Scholar]
- 25.Chang YC, Hsu TL, Lin HH, Chio CC, Chiu AW, Chen NJ, Lin CH, Hsieh SL. Modulation of macrophage differentiation and activation by decoy receptor 3. J Leukoc Biol. 2004;75:486–494. doi: 10.1189/jlb.0903448. [DOI] [PubMed] [Google Scholar]
- 26.Tai SK, Chang HC, Lan KL, Lee CT, Yang CY, Chen NJ, Chou TY, Tarng DC, Hsieh SL. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J Immunol. 2012;188:2464–2471. doi: 10.4049/jimmunol.1101101. [DOI] [PubMed] [Google Scholar]
- 27.Lin WW, Hsieh SL. Decoy receptor 3: a pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochem Pharmacol. 2011;81:838–847. doi: 10.1016/j.bcp.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Cheng CP, Sheu MJ, Sytwu HK, Chang DM. Decoy receptor 3 suppresses RANKL-induced osteoclastogenesis via down-regulating NFATc1 and enhancing cell apoptosis. Rheumatology (Oxford) 2013;52:609–622. doi: 10.1093/rheumatology/kes343. [DOI] [PubMed] [Google Scholar]
- 29.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galaup A, Gomez E, Souktani R, Durand M, Cazes A, Monnot C, Teillon J, Le Jan S, Bouleti C, Briois G, Philippe J, Pons S, Martin V, Assaly R, Bonnin P, Ratajczak P, Janin A, Thurston G, Valenzuela DM, Murphy AJ, Yancopoulos GD, Tissier R, Berdeaux A, Ghaleh B, Germain S. Protection Against Myocardial Infarction and No-Reflow Through Preservation of Vascular Integrity by Angiopoietin-Like 4. Circulation. 2012;125:140–149. doi: 10.1161/CIRCULATIONAHA.111.049072. [DOI] [PubMed] [Google Scholar]
- 31.Guzman MJ, Crisostomo PR, Wang M, Markel TA, Wang Y, Meldrum DR. Vascular endothelial growth factor improves myocardial functional recovery following ischemia/reperfusion injury. J Surg Res. 2008;150:286–292. doi: 10.1016/j.jss.2007.12.772. [DOI] [PubMed] [Google Scholar]
- 32.Poynter JA, Manukyan MC, Wang Y, Brewster BD, Herrmann JL, Weil BR, Abarbanell AM, Meldrum DR. Systemic pretreatment with dimethyloxalylglycine increases myocardial HIF-1alpha and VEGF production and improves functional recovery after acute ischemia/reperfusion. Surgery. 2011;150:278–283. doi: 10.1016/j.surg.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Kang S, Yang Y, Li CJ, Gao R. Effectiveness and tolerability of administration of granulocyte colony-stimulating factor on left ventricular function in patients with myocardial infarction: A meta-analysis of randomized controlled trials. Clin Ther. 2007;29:2406–2418. doi: 10.1016/j.clinthera.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad. 14-3-3 signaling pathways. J Biol Chem. 2005;280:8022–8030. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida H, Zhang JJ, Chao L, Chao J. Kallikrein gene delivery attenuates myocardial infarction and apoptosis after myocardial ischemia and reperfusion. Hypertension. 2000;35:25–31. doi: 10.1161/01.hyp.35.1.25. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh-Choudhury N, Abboud SL, Chandrasekar B, Ghosh Choudhury G. BMP-2 regulates cardiomyocyte contractility in a phosphatidylinositol 3 kinase-dependent manner. FEBS Lett. 2003;544:181–184. doi: 10.1016/s0014-5793(03)00507-6. [DOI] [PubMed] [Google Scholar]
- 37.Ebelt H, Hillebrand I, Arlt S, Zhang Y, Kostin S, Neuhaus H, Muller-Werdan U, Schwarz E, Werdan K, Braun T. Treatment with bone morphogenetic protein 2 limits infarct size after myocardial infarction in mice. Shock. 2013;39:353–360. doi: 10.1097/SHK.0b013e318289728a. [DOI] [PubMed] [Google Scholar]
- 38.Edelberg JM, Lee SH, Kaur M, Tang L, Feirt NM, McCabe S, Bramwell O, Wong SC, Hong MK. Platelet-derived growth factor-AB limits the extent of myocardial infarction in a rat model: feasibility of restoring impaired angiogenic capacity in the aging heart. Circulation. 2002;105:608–613. doi: 10.1161/hc0502.103672. [DOI] [PubMed] [Google Scholar]
- 39.Cuevas P, Carceller F, Lozano RM, Crespo A, Zazo M, Gimenez-Gallego G. Protection of rat myocardium by mitogenic and non-mitogenic fibroblast growth factor during post-ischemic reperfusion. Growth Factors. 1997;15:29–40. doi: 10.3109/08977199709002110. [DOI] [PubMed] [Google Scholar]
- 40.Cuevas P, Reimers D, Carceller F, Martinez-Coso V, Redondo-Horcajo M, Saenz de Tejada I, Gimenez-Gallego G. Fibroblast growth factor-1 prevents myocardial apoptosis triggered by ischemia reperfusion injury. Eur J Med Res. 1997;2:465–468. [PubMed] [Google Scholar]
- 41.Xiao J, Lv Y, Lin S, Jin L, Zhang Y, Wang X, Ma J, Hu K, Feng W, Cai L, Li X, Tan Y. Cardiac protection by basic fibroblast growth factor from ischemia/reperfusion-induced injury in diabetic rats. Biol Pharm Bull. 2010;33:444–449. doi: 10.1248/bpb.33.444. [DOI] [PubMed] [Google Scholar]
- 42.Lee SW, Won JY, Lee HY, Lee HJ, Youn SW, Lee JY, Cho CH, Cho HJ, Oh S, Chae IH, Kim HS. Angiopoietin-1 protects heart against ischemia/reperfusion injury through VE-cadherin dephosphorylation and myocardiac integrin-beta1/ERK/caspase-9 phosphorylation cascade. Mol Med. 2011;17:1095–1106. doi: 10.2119/molmed.2011.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Luo D, Liao X, He J, Liu C, Yang C, Ma H. Ang-(1-7) offers cytoprotection against ischemia-reperfusion injury by restoring intracellular calcium homeostasis. J Cardiovasc Pharmacol. 2014;63:259–264. doi: 10.1097/FJC.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 44.Yu H, Wang P, An P, Xue Y. Recombinant human angiopoietin-1 ameliorates the expressions of ZO-1, occludin, VE-cadherin, and PKCalpha signaling after focal cerebral ischemia/reperfusion in rats. J Mol Neurosci. 2012;46:236–247. doi: 10.1007/s12031-011-9584-5. [DOI] [PubMed] [Google Scholar]
- 45.Odiete O, Hill MF, Sawyer DB. Neuregulin in Cardiovascular Development and Disease. Circ Res. 2012;111:1376–1385. doi: 10.1161/CIRCRESAHA.112.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang JQ, Wu K, Jia ZH, Liu C, Ding J, Huang SN, Yin PP, Wu XC, Wei C, Wu YL, Wang HY. Chinese medicine Tongxinluo modulates vascular endothelial function by inducing eNOS expression via the PI-3K/Akt/HIF-dependent signaling pathway. J Ethnopharmacol. 2011;133:517–523. doi: 10.1016/j.jep.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 47.Feng M, Li G, Qian X, Fan Y, Huang X, Zhang F, Lu L. IL-17A-producing NK cells were implicated in liver injury induced by ischemia and reperfusion. Int Immunopharmacol. 2012;13:135–140. doi: 10.1016/j.intimp.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Cicora F, Roberti J, Lausada N, Gonzalez P, Guerrieri D, Stringa P, Cicora P, Vasquez D, Gonzalez I, Palti G, Intile D, Raimondi C. Donor preconditioning with rabbit anti-rat thymocyte immunoglobulin ameliorates ischemia reperfusion injury in rat kidney transplantation. Transpl Immunol. 2012;27:1–7. doi: 10.1016/j.trim.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Schellekens DH, Grootjans J, Dello SA, van Bijnen AA, van Dam RM, Dejong CH, Derikx JP, Buurman WA. Plasma Intestinal Fatty Acid-Binding Protein Levels Correlate With Morphologic Epithelial Intestinal Damage in a Human Translational Ischemia-reperfusion Model. J Clin Gastroenterol. 2014;48:253–260. doi: 10.1097/MCG.0b013e3182a87e3e. [DOI] [PubMed] [Google Scholar]
- 50.Uitterdijk A, Sneep S, van Duin RW, Krabbendam-Peters I, Gorsse-Bakker C, Duncker DJ, van der Giessen WJ, van Beusekom HM. Serial measurement of hFABP and high sensitivity Troponin I post-PCI in STEMI: how fast and accurate can Myocardial Infarct Size and No-Reflow be predicted? Am J Physiol Heart Circ Physiol. 2013;305:H1104–H1110. doi: 10.1152/ajpheart.00447.2013. [DOI] [PubMed] [Google Scholar]
- 51.Cordwell SJ, Edwards AV, Liddy KA, Moshkanbaryans L, Solis N, Parker BL, Yong AS, Wong C, Kritharides L, Hambly BD, White MY. Release of Tissue-specific Proteins into Coronary Perfusate as a Model for Biomarker Discovery in Myocardial Ischemia/Reperfusion Injury. J Proteome Res. 2012;11:2114–2126. doi: 10.1021/pr2006928. [DOI] [PubMed] [Google Scholar]
- 52.Pachori AS, Custer L, Hansen D, Clapp S, Kemppa E, Klingensmith J. Bone morphogenetic protein 4 mediates myocardial ischemic injury through JNK-dependent signaling pathway. J Mol Cell Cardiol. 2010;48:1255–1265. doi: 10.1016/j.yjmcc.2010.01.010. [DOI] [PubMed] [Google Scholar]