

Abstract
Cell morphology has received considerable attention in recent years owing to its possible relationship with cell functions, including proliferation, differentiation, and migration. Recent evidence suggests that extracellular environments can also mediate cell functions, particularly cell adhesion. The aims of this study were to investigate the correlation between osteogenic differentiation activity and the morphology of rat mesenchymal stromal cells (MSCs), and to develop a method of estimating osteogenic differentiation capability of MSCs on biomaterials. We measured the attachment areas of MSCs on substrates with various types of surface after 2 h of seeding, and quantified the amount of osteocalcin secreted from MSCs after 3 weeks of culture under osteogenic differentiation conditions. MSCs with small attachment areas showed a high osteogenic differentiation activity. These findings indicate that cell attachment areas correlate well with the osteogenic differentiation activity of MSCs. They also suggest that the measurement of cell attachment areas is useful for estimating the osteogenic differentiation activity of MSCs and is a practical tool for applications of MSCs in regenerative medicine.
Keywords: mesenchymal stromal cells, proliferative activity, osteogenic differentiation activity, cell morphology, titanium, apatite, regenerative medicine
1. Introduction
Mesenchymal stromal cells (MSCs) are being used in regenerative medicine because of their capability to differentiate into a variety of mesenchymal lineages including osteoblasts, chondrocytes, adipocytes, neurons, and hepatocytes, depending on culture conditions [1–3]. Because MSCs are an effective tool in regenerative medicine [4], MSCs taken from a patient's bone marrow have been utilized for treating various diseases such as bone diseases [5–7]. For the osteogenic differentiation of patient's MSCs, about three weeks of cell culture is required before surgical implantation of the differentiated cells with the scaffold. The suitability of the cells as an implant from the viewpoint of osteogenic differentiation is determined only after the culture period. During the culture period, preparations and arrangements for the surgical operation are also carried out. The patient's valuable MSCs as well as the preparation and arrangement efforts would be wasted if the osteogenic differentiation turns out to be unsuccessful after the culture period. Therefore, early estimation of the osteogenic differentiation activity of patient's MSCs on a biomaterial is required to improve the clinical efficiency.
Through experience, we found that the osteogenic differentiation activity of MSCs and cell morphology vary among donors. Recent evidence suggests that extracellular environments can mediate cell functions, particularly the initial cell adhesion behavior that leads to a specific cell morphology, and cell proliferation and differentiation. Human MSCs with a small thickness show a high proliferation activity on tissue culture polystyrene (TCPS) [8]. Round rat MSCs show a higher osteogenic differentiation activity on ceramic sheets than those that are widely spread on polymer sheets after 2 weeks of culture with osteogenic media [9]. These findings suggest that the morphology and functions of MSCs are closely related to each other. However, most of the previous studies are comparisons of cell behaviors among scaffolds that are considerably different in chemical composition, which cannot exclude the direct effects of chemical factors on cell behavior. Therefore, the correlation between the morphology of MSCs on biomaterials and the osteogenic differentiation activity of MSCs is yet to be fully clarified.
The purposes of this study were to investigate the correlation between the morphology of MSCs on biomaterials and the osteogenic differentiation activity of MSCs cultured using scaffolds with slight differences in chemical composition, and to develop an early estimation method for predicting later osteogenic differentiation on biomaterials. Titanium (Ti) is a bioinert and biocompatible material that is widely used in substitutes for dental implants and fixation of fractures. The surface structure of Ti can be modified for cell culture by hydrolysis. Recently, we have reported that titanium screws can be coated with apatite layers using a supersaturated calcium phosphate solution, which improves the bone–screw interface strength and infection resistance in external skeletal fixation [10–12].
In this study, we prepared cell culture substrates of Ti with different surface properties by coating apatite layers in a calcium phosphate solution that is supersaturated with respect to apatite [10]. Then we measured the attachment areas of MSCs as an index of cell morphology 2 h after seeding and quantified the amount of osteocalcin in MSCs after 3 weeks of culture under osteogenic differentiation conditions.
2. Materials and methods
2.1. Reagents and cells
We used the following reagents: cell culture medium, minimum essential medium (MEM), and dexamethasone from Sigma-Aldrich Co.; fetal bovine serum (FBS) and a LIVE/DEAD viability/cytotoxicity assay kit from Invitrogen Co.; antibiotics (penicillin–streptomycin–amphotericin B suspension (×100)), β-glycerophosphate, phosphate-buffered saline (PBS), l-ascorbic acid 2-phosphate magnesium salt n-hydrate (ascorbic acid-2-phosphate), and trypsin-EDTA solution (0.25 w/v% trypsin 1 mmol l−1 EDTA·4Na solution with phenol red from Becton Dickinson and Company; and rat osteocalcin ELISA kit DS from DS Pharma Biomedical Co. Male rats, Fisher 344, were provided by Japan SLC Co. MSCs were extracted from the bone marrow of the rats.
2.2. Preparation of cell culture substrates
Four types of substrate were used for rat MSC culture in this study. Titanium plates (Nilaco Corporation; TI453512 99.5%, 11 × 11 × 1 mm3) were washed with acetone, ethanol, and ultrapure water for 15 min each using an ultrasonic cleaner; the plates were then dried at 40 °C for 24 h. Before the following experiments, they were sterilized using ethylene dioxide gas. Then, the plates were either immersed in 3 w/v% hydrogen peroxide solution (H2O2–Ti) and autoclaved, or they were heated at 300 °C for 2 h (300 °C-Ti) to form two types of titanium oxide layer.
2.3. Preparation of supersaturated calcium phosphate solutions for apatite coating
Supersaturated calcium phosphate solutions were prepared using infusion fluids clinically available in Japan under sterile conditions. Supersaturated calcium phosphate solutions were composed of Ringer's solution (Otsuka Pharmaceutical Co., Ltd, Japan), calcium chloride corrective injection (1 mEq ml−1, Otsuka Pharmaceutical) as Ca sources, Klinisalz® (I'rom Pharmaceutical Co., Ltd, Japan), dipotassium phosphate corrective injection (1 mEq ml−1; Otsuka Pharmaceutical) as P sources, and Bifil® (Ajinomoto Pharmaceutical Co., Ltd, Japan) as an alkalinizer (NaHCO3 solution). Chemical compositions of the solutions are shown in table 1. The Ca/P molar ratio of the supersaturated calcium phosphate solutions was set at 2.0.
Table 1.
Chemical compositions of supersaturated calcium phosphate solutions.
| Calcium-containing solution (mM) | Phosphorus-containing solution (mM) | NaHCO3 solution (mM) | Calcium phosphate solution (mM) | |||
|---|---|---|---|---|---|---|
| Ringer's solution | Calcium chloride corrective injection 1 (mEq ml−1) | Klinisalz | Dipotassium phosphate corrective injection 1 (mEq ml−1) | Bifil | ||
| Volume (ml) | 8.14 | 0.037 | 0.898 | 0.019 | 0.909 | 10.003 |
| Na+ | 147.00 | – | 45.00 | – | 166.00 | 138.79 |
| K+ | 4.00 | – | 25.00 | 1000.00 | – | 7.40 |
| Mg2+ | – | – | 2.5 | – | – | 0.22 |
| Ca2+ | 2.5 | 500.00 | – | – | – | 3.89 |
| Cl− | 156.00 | 1000.00 | 45.00 | – | – | 134.73 |
| HP2O4− | – | – | 10.00 | – | – | 0.90 |
| HPO42− | – | – | – | 500.00 | – | 0.95 |
| HCO3− | – | – | – | – | 166.00 | 15.09 |
| CH3COO− | – | – | 20.00 | – | – | 1.80 |
| Xylitol | – | – | 333.00 | – | – | 29.90 |
2.4. Apatite coating
Untreated Ti, H2O2–Ti, and 300 °C-Ti plates were immersed in the supersaturated calcium phosphate solution at 37 °C for 48 h to form an apatite (Ap) layer on them; hereafter, these samples are referred to as Ti-Ap, H2O2–Ti-Ap, and 300 °C-Ti-Ap, respectively. Untreated and uncoated Ti plates were also used as controls for the effect of Ap layer.
2.5. Surface analysis of cell culture substrates
Untreated Ti plate and apatite-coated plates were air-dried for 15 min at 25 °C. The cell culture substrate surfaces were characterized by thin-film x-ray diffraction (XRD, RINT-2500, Rigaku Co., Japan) analysis using CuKα x-rays. The air-dried samples were sputter-coated with gold and observed by scanning electron microscopy (SEM; Model XL30, FEI Japan Ltd., Japan).
2.6. Preparation of suspension of rat MSCs
Rat MSCs were isolated and their primary cultures were obtained as described previously [6]. Briefly, the rat bone marrow cells were obtained from the bone shafts of the femora of 7-week-old Fisher 344 male rats. Both ends of the femur epiphysis were cut, and bone marrow was flushed out using 10 ml of a standard medium through a 21-gauge needle. The cells were seeded onto a T-75 flask per femur. We used MEM containing 10% FBS and 1% antibiotics (100 units ml−1 penicillin, 0.1 mg ml−1 streptomycin, and 0.25 μg ml−1 amphotericin B). The bone marrow cells were cultured for 1 day in a humidified atmosphere of 95% air with 5% CO2 at 37 °C. The medium was replaced once to wash out all the floating cells. Then, adherent cells on the T-75 flask were initially cultured up to 80% confluence in T-75 flasks. The adherent cells were harvested using trypsin-EDTA solution (P-1; passage-1) to prepare a cell suspension.
2.7. Cell attachment and osteogenic differentiation studies
Six culture substrates of Ti-Ap, H2O2–Ti-Ap, 300 °C-Ti-Ap, or Ti were placed on each well of a 24-well TCPS plate. Among these substrates, three were used for measuring cell attachment areas while the remaining three substrates were used for the evaluation of osteogenic differentiation. Rat MSCs were seeded on each cell culture substrate at a density of 4 × 104 cells ml−1 (1 ml well−1). For measuring the attachment areas of live cells, cells were prestained using a LIVE/DEAD viability/cytotoxicity assay kit. The cells attached to the surface of each culture substrate were observed 2 h after seeding under an objective fluorescence microscope (Olympus, Model BX51). Live cells stained with calcein-AM appeared green owing to intracellular esterase activity, while dead cells stained with EthD-1 appeared red owing to plasma membrane disintegrity. The attachment area of live cells on each cell culture substrate was measured using Image-Pro® PLUS software (Media Cybernetics, Inc., Version 7.0). Cell attachment areas between 350 and 3500 μm2 were used for analysis. Features smaller than 350 μm2 were regarded as dust particles and discarded. Areas larger than 3500 μm2 were regarded as multiply connected cells and discarded too.
For the osteogenic differentiation study, the medium was supplemented with 10 nM dexamethasone, 10 mM β-glycerophosphate, and 0.28 mM ascorbic acid-2-phosphate. After confirmation of cell adhesion to the culture substrate, the cell differentiation experiment was started (day 0). The cells that adhered to the culture substrate were cultured for 21 days. The medium was renewed three times a week. The amount of osteocalcin secreted by the MSCs cultured on each culture substrate was measured after 21 days of culture. The 15 μl supernatant of the medium after the culture was collected from each well and diluted 100-fold with a dilution buffer in an appropriate vessel prior to the assay of intact osteocalcin using a rat osteocalcin ELISA kit DS. To determine the number of cells, the amount of total protein was determined after homogenizing the MSCs on each culture substrate after 21 days of culture. Total protein was quantified in accordance with the protocol of the manufacturer. Briefly, the medium of the MSCs on each culture substrate was replaced by 1 ml of TritonX-100 diluted 1000-fold with sterilized water, and MSCs were frozen for 20 min at −80 °C. After homogenizing the cells by pipetting, 100 μl of supernatant was taken from each well. The absorbance at 595 nm was measured using a Bio-Rad Protein Assay kit and a spectrophotometer. The amount of osteocalcin was normalized to that of total protein.
2.8. Statistical analysis
Cell attachment area and the amounts of osteocalcin and total protein are presented as average values with standard deviations. Two-sided Student's t-test was used to evaluate statistical significances among each group. P values less than 0.05 were considered statistically significant.
3. Results
3.1. Surface characterization of titanium plates
After immersion in the supersaturated calcium phosphate solution, apatite layers with different morphological, and chemical, and/or structural characteristics covered the surfaces of Ti, H2O2–Ti, and 300 °C-Ti plates. All the apatite layers mainly consisted of low-crystalline apatite in which octacalcium phosphate (OCP), amorphous calcium phosphate (ACP), and calcium carbonates might exist as indicated by the XRD patterns (figure 1) [11]. Within the range of ‘low-crystalline apatite’, the layers exhibited marked differences in chemical and structural characteristics as indicated by the differences in intensity and broadening of apatite peaks. The presence of apatite was determined using MDI JADE 5.0 software. It was confirmed by the close agreement between the JCPDS card No. 9–0432 and the intensities of the peaks for the apatite at 2θ = 26.0° and between 2θ = 30.0° and 35.0°.
Figure 1.
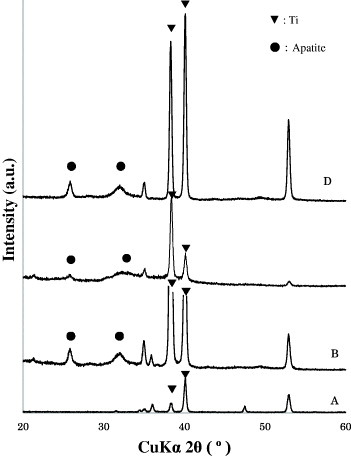
X-ray diffraction patterns of the top surfaces of (A) Ti, (B) Ti-Ap, (C) H2O2–Ti-Ap, and (D) 300 °C-Ti-Ap substrates.
The layers were also morphologically different depending on the pretreatment of the Ti plates. The apatite layer on Ti plates consisted of many minute plate-like crystals. The apatite layer on the Ti plates treated with H2O2 consisted of needle-like crystals. The apatite layer on the Ti plates treated at 300 °C consisted of somewhat densely aggregated tiny plate-like crystals (figure 2).
Figure 2.

Scanning electron micrographs of the top surfaces of (A) Ti-Ap, (B) H2O2–Ti-Ap, (C) 300 °C-Ti-Ap, and (D) Ti substrates. Bar: 2 μm.
3.2. Cell attachment areas and osteogenic differentiation of MSCs
We observed the attachment areas of MSCs by fluorescence microscopy 2 h after seeding and then measured them by image analysis. The image analysis showed that cell attachment areas were the largest on Ti and the smallest on 300 °C-Ti-Ap among the substrates tested (figure 3). Fluorescence microscopy images of MSCs on the culture substrates shown in figure 4 reveal widely spread cells with many cytoplasmic processes on Ti and Ti-Ap. On the other hand, the cells on 300 °C-Ti-Ap were round with few cytoplasmic processes.
Figure 3.

Cell attachment areas of live MSCs in early stage on (A) Ti, (B) Ti-Ap, (C) H2O2–Ti-Ap, and (D) 300 °C-Ti-Ap. Asterisks indicate significant differences (t-test: P < 0.05).
Figure 4.

Fluorescence microscopy images of MSCs on (A) Ti, (B) Ti-Ap, (C) H2O2–Ti-Ap, and (D) 300 °C-Ti-Ap substrates.
Figure 5 shows the amounts of osteocalcin secreted from the MSCs to culture media normalized by the total protein amount. The data in figure 5 show that the osteogenic differentiation activity of MSCs was the lowest on Ti and the highest on 300 °C-Ti-Ap.
Figure 5.

Amounts of osteocalcin/protein in MSCs after 3 weeks of culture on (A) Ti, (B) Ti-Ap, (C) H2O2–Ti-Ap, and (D) 300 °C-Ti-Ap substrates.
3.3. Correlation between osteogenic differentiation activity and attachment areas of rat MSCs
Cell attachment areas measured after 2 h culture were as follows: 696 ± 162 μm2 (300 °C-Ti-Ap), 770 ± 211 μm2 (H2O2–Ti-Ap), 1226 ± 335 μm2 (Ti-Ap), and 1231 ± 433 μm2 (Ti). The amounts of osteocalcin/total protein measured after 3-week cultures were as follows: 2987 ± 1237 ng mg−1 (300 °C-Ti-Ap), 2430 ± 222 ng mg−1 (H2O2–Ti-Ap), 1841 ± 245 ng mg−1 (Ti-Ap), and 1604 ± 519 ng mg−1(Ti). The square of the correlation coefficient between the cell attachment areas and osteocalcin/total protein was 0.9059. These results suggest that there is a correlation between the attachment area of rat MSCs and osteogenic differentiation activity (figure 6).
Figure 6.

Correlation between the cell attachment area in early stage and the osteogenic differentiation activity after 3 weeks of culture on (A) Ti, (B) Ti-Ap, (C) H2O2–Ti-Ap, and (D) 300 °C-Ti-Ap substrates.
4. Discussion
We have observed that the osteogenic differentiation activity of MSCs correlated well with the morphology of the cells in our previous work [8] and in this work. Recently, it has been reported that MSCs can maintain their expansion and differentiation capabilities by the forced expression of Sox2 or Nanog transcription factors [13]. The Sox2-expressing cells in the presence of basic fibroblast growth factor show higher proliferation and differentiation potentials and the cells are smaller than control cells. Chen et al [14] reported that the cell adhesion area on patterned surfaces affects cell growth and viability. Elastomeric substrates geometrically modulate cell morphology, resulting in the expression of specific functions of various types of cell [15, 16]. These results suggest that there is a general correlation between the MSC morphology and the potential for osteogenic differentiation activity. MSCs are promising candidates for applications in regenerative medicine because of their several differentiation potentials. Estimation of the osteogenic differentiation activity of MSCs is strongly needed for MSCs to be utilized in practical applications. The present findings indicate that the cell attachment area at a very early stage is an important property in estimating the osteogenic differentiation activity of MSCs.
A quick method and protocol for measuring cell attachment area was developed. In this study, different cell attachment areas were observed on various types of cell culture substrate. The advantages of the method are as follows: (i) it is easy to perform, (ii) an accurate prediction can be made within 2 h, and (iii) as many as 100 cells can be observed simultaneously. This method enables us to observe the projection area of a cell from top to bottom under a stereomicroscope. The accuracy of measurements increases with cell thickness, i.e., the thicker the cells, the more accurate the measurements. In this study, different cell attachment areas were measured on titanium coated by various types of apatite layers that had a slight difference in chemical composition within the classification of low-crystalline apatite.
Differences in osteogenic differentiation activity were observed for MSCs cultured on titanium coated by various types of apatite layers. Katsube et al [8] reported that the proliferative activity of human MSCs from different donors differs depending on the cell morphology on the same substrate (TCPS); however they did not evaluate osteogenic differentiation activity. In contrast, Tanaka et al [9] reported that the osteogenic differentiation activity of rat MSCs differs depending on the cell morphology on different substrates of ceramic sheets and polymer sheets. No previous study showed different osteogenic differentiation activities of MSCs depending on cell morphology on the same or similar substrates. In this study, different osteogenic differentiation activities of MSCs depending on cell morphology on substrates with similar chemical compositions were demonstrated.
In addition, the correlation between cell morphology and osteogenic differentiation activity was observed even among different substrates, i.e., between metallic and ceramic substrates. The correlation between cell attachment areas and osteogenic differentiation activity on ceramic layers roughly coincided with that on titanium. Titanium substrates had very different chemical compositions and surface roughness from those of the apatite layers. Therefore, it could be a general phenomenon that a small attachment area of MSCs is associated with a high osteogenic differentiation activity regardless of the substrate type.
This study indicates the benefits of evaluating cell morphology as an early prediction technique. When considering the clinical applications of MSCs, procedures for culture expansion and differentiation of MSCs are needed since the number of MSCs in bone marrow is extremely low. Such procedures implicate risks of bacterial/fungal contamination. To avoid these risks, biologically safe areas are required, such as a cell processing center (CPC) equipped with clean rooms where sufficiently careful and safe handling is possible. However, the establishment and maintenance of these facilities are costly. Currently, only autologous and noncryopreserved MSCs are used in clinical applications of MSCs in bone regeneration. Patients have to wait for about 4 weeks between the bone marrow aspiration and the surgical operation because about 4 weeks of culture at CPC is required for preparing the regenerative bone tissues. Unfortunately, we have experienced that some cultures of regenerative bone tissues could not be implanted to patients even after long culture because the osteogenic differentiation level was too low. To fully utilize the valuable MSCs from patients, the prediction of osteogenic differentiation level at a very early stage of culture is strongly required. In this context, the method developed in this study appears to be effective as an early prediction technique for osteogenic differentiation activity. However, one limitation of this work is worth noting: the samples were not reassessed once the evaluation was completed. Further studies are required.
References
- Pittenger M F, Mackey A M, Beck S C, Jaiswal R K, Douglas R, Mosca J D, Moorman M A, Simonetti D W, Craig S. and Marshak D R. Science. 1999;284:143. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Furuzono T, Kishida A. and Tanaka J. J. Mater. Sci. 2004;15:19. doi: 10.1023/B:JMSM.0000010093.39298.5a. [DOI] [PubMed] [Google Scholar]
- Bruder S P, Fink D J. and Caplan A I. J. Cell. Biochem. 1994;56:283. doi: 10.1002/jcb.240560303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan A I. and Bruder S P. Trends Mol. Med. 2001;7:259. doi: 10.1016/S1471-4914(01)02016-0. [DOI] [PubMed] [Google Scholar]
- Kotobuki N, Hirose M, Takakura Y. and Ohgushi H. Artif. Organs. 2004;28:33. doi: 10.1111/j.1525-1594.2004.07320.x. [DOI] [PubMed] [Google Scholar]
- Ohgushi H, Kotobuki N, Funaoka H, Machida H, Hirose M, Tanaka Y. and Takakura Y. Biomaterials. 2005;26:4654. doi: 10.1016/j.biomaterials.2004.11.055. [DOI] [PubMed] [Google Scholar]
- Morishita T, Honoki K, Ohgushi H, Kotobuki N, Matsushima A. and Takakura Y. Artif. Organs. 2006;30:115. doi: 10.1111/j.1525-1594.2006.00190.x. [DOI] [PubMed] [Google Scholar]
- Katsube Y, Hirose M, Nakamura C. and Ohgushi H. Biochem. Biophys. Res. Commun. 2008;368:256. doi: 10.1016/j.bbrc.2008.01.051. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Hirose M, Kotobuki N, Ohgushi H, Furuzono T. and Sato J. Mater. Sci. Eng. C. 2007;27:817. doi: 10.1016/j.msec.2006.09.019. [DOI] [Google Scholar]
- Mutsuzaki H, Ito A, Sakane M, Sogo Y, Oyane A. and Ochiai N. J. Biomed. Mater. Res. B. 2008;86:365. doi: 10.1002/jbm.b.31029. [DOI] [PubMed] [Google Scholar]
- Mutsuzaki H, Ito A, Sakane M, Sogo Y, Oyane A, Ebihara Y, Ichinose N. and Ochiai N. J. Mater. Sci. Mater. Med. 2007;18:1799. doi: 10.1007/s10856-007-3051-4. [DOI] [PubMed] [Google Scholar]
- Wang X, Ito A, Sogo Y, Li X. and Oyane A. J. Biomed. Mater. Res. A. 2010;92:1181. doi: 10.1002/jbm.a.32436. [DOI] [PubMed] [Google Scholar]
- Go M J, Takenaka C. and Ohgushi H. Exp. Cell Res. 2008;314:1147. doi: 10.1016/j.yexcr.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Chen C S, Mrksich M, Huang S, Whitesides G M. and Ingber D E. Science. 1997;276:1425. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Fu J, Wang Y K, Yang M T, Desai R A, Yu X, Liu Z. and Chen C S. Nature Methods. 2010;7:733. doi: 10.1038/nmeth.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber D E. Circ. Res. 2002;91:877. doi: 10.1161/01.RES.0000039537.73816.E5. [DOI] [PubMed] [Google Scholar]