


Abstract
Autoinhibition enables spatial and temporal regulation of cellular processes by coupling protein activity to surrounding conditions, often via protein partnerships or signaling pathways. We report the molecular basis of DNA-binding autoinhibition of ETS transcription factors ETV1, ETV4 and ETV5, which are often overexpressed in prostate cancer. Inhibitory elements that cooperate to repress DNA binding were identified in regions N- and C-terminal of the ETS domain. Crystal structures of these three factors revealed an α-helix in the C-terminal inhibitory domain that packs against the ETS domain and perturbs the conformation of its DNA-recognition helix. Nuclear magnetic resonance spectroscopy demonstrated that the N-terminal inhibitory domain (NID) is intrinsically disordered, yet utilizes transient intramolecular interactions with the DNA-recognition helix of the ETS domain to mediate autoinhibition. Acetylation of selected lysines within the NID activates DNA binding. This investigation revealed a distinctive mechanism for DNA-binding autoinhibition in the ETV1/4/5 subfamily involving a network of intramolecular interactions not present in other ETS factors. These distinguishing inhibitory elements provide a platform through which cellular triggers, such as protein–protein interactions or post-translational modifications, may specifically regulate the function of these oncogenic proteins.
INTRODUCTION
Autoinhibition occurs in diverse proteins and allows for spatiotemporal modulation of biological processes in response to various inputs such as signaling pathways and macromolecular interactions (1). This self-dampening behavior can influence the equilibria between the active and inactive states of proteins by serving as the integration point for post-translational modifications and protein partnerships. Partaking in alternative intramolecular and intermolecular interactions is often the key attribute for an autoinhibitory element (2,3). Both structured regions with dynamic character and intrinsically disordered regions (IDRs) can be effective inhibitory elements (4). Conformational flexibility, and even disorder, allows for distinct and adaptable recognition of intramolecular interfaces, as well as surfaces on diverse interacting proteins (5).
The ETS gene family, which encodes 28 human transcription factors, has provided a model system to develop an understanding of autoinhibition of sequence-specific DNA binding (6). The conserved ETS domain is autoinhibited in several family members, yet by different mechanisms. For example, a serine-rich IDR allosterically inhibits DNA binding of ETS1 through transient phosphorylation-enhanced interactions with the structured ETS domain and flanking N- and C-terminal inhibitory α-helices (7,8). In contrast, a single flanking C-terminal α-helix sterically inhibits DNA binding of ETV6 (9–11). In the biological context, autoinhibition of a particular ETS factor provides distinct routes to specific regulation, such as via post-translational modifications (8,12) and protein-protein interactions (13–16). Collectively, this has led to the hypothesis that divergent modes of autoinhibition involving regions flanking the ETS domain help enable specific gene regulation by individual ETS factors (6).
The involvement of the ETS genes of the ERG and ETV1/4/5 (also known as PEA3) subfamilies in prostate cancer motivated our goal to expand the mechanistic understanding of autoinhibition to these ETS factors. Chromosomal rearrangements involving the ERG and ETV1/4/5 subfamilies are observed in the majority of prostate cancer tumors (17–19). There is aberrant expression of these full-length, or nearly full-length, ETS proteins upon rearrangement with a prostate-specific or a constitutively expressed promoter (20). In addition, ETV1 and ETV4 mediate PI3-kinase and Ras signaling pathways, resulting in aggressive and metastatic disease phenotypes (21,22). Previous studies suggested that the ETV1/4/5 subfamily also displays autoinhibition of DNA binding (13,23,24); however, detailed characterization, including structural mapping of inhibitory elements and mechanistic insights are lacking. We propose that a full understanding of the autoinhibition of ERG and ETV1/4/5 and its regulation by cellular processes will enable insights into the roles of these factors in prostate cancer progression and provide windows of opportunity for targeted therapeutic interventions.
In this study we describe the molecular basis of DNA-binding autoinhibition in the ETV1/4/5 subfamily of ETS factors. Using ETV4 as a model for this subfamily, we found that inhibitory domains reside both N- and C-terminal of the ETS domain and cooperate to inhibit DNA binding. Crystal structures identified the C-terminal inhibitory domain (CID) as an α-helix that packs against the ETS domain and perturbs the relative positioning of its DNA-recognition helix. Nuclear magnetic resonance (NMR) spectroscopy demonstrated that the N-terminal inhibitory domain (NID) is an IDR that transiently interacts with the ETS domain and the CID. Lysine acetylation of the NID relieves autoinhibition, likely through disruption of these intramolecular interactions. Mutational analyses revealed specific intramolecular linkages among the regulatory elements. From these findings, we propose a model for autoinhibition in the ETV1/4/5 subfamily in which structured and disordered regions regulate the DNA-recognition helix.
MATERIALS AND METHODS
Expression plasmids
Human ETV1, ETV4, ETV5, ERG and FLI1 cDNAs corresponding to full-length or truncated proteins were cloned into the bacterial expression vector pET28 (Novagen) using standard sequence and ligation independent cloning strategies (25). Point mutations were introduced into the ETV4 plasmid using the QuikChange site-directed mutagenesis protocol (Stratagene). For acetylation studies, codons encoding Lys226 or Lys260 in the full-length ETV4 gene were mutated to an amber codon (UAG), and the natural amber stop codon was mutated to an opal codon (UGA). Mutated ETV4 cDNA was then cloned from the pET28 plasmid into a pCDF plasmid (kind gift from Dr. Jason Chin) for expression (26).
Expression and purification of proteins
All proteins were produced in Escherichia coli (λDE3) cells. Uninhibited ETS factor DNA-binding domains (DBDs) and the ETV1/4/5 fragments not containing the NID were efficiently expressed into the soluble fraction. Cultures of 1 L Luria broth (LB) were grown at 37°C to OD600 ∼ 0.7–0.9, induced with 1 mM isopropyl-β -D-thiogalactopyranoside (IPTG) and grown at 30°C for ∼ 3 h. To produce isotopically enriched proteins, expression was carried out using M9 minimal media supplemented with 3 g/l (13C6, 99%)-D-glucose and/or 1 g/l (15N, 99%)-NH4Cl.
Harvested cells were resuspended in 25 mM Tris pH 7.9, 1 M NaCl, 5 mM imidazole, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 2 mM 2-mercaptoethanol (BME) and 1 mM phenylmethanesulfonylfluoride (PMSF). Cells were lysed by sonication and centrifuged at 125,000 × g for at least 30 min at 4°C. After centrifugation, the soluble supernatants were loaded onto a Ni2+ affinity column (GE Biosciences) and eluted over a 5–500 mM imidazole gradient. Fractions containing purified protein were pooled, combined with ∼ 1 U thrombin/mg of purified protein and dialyzed overnight at 4°C into 25 mM Tris pH 7.9, 10% glycerol (v:v), 1 mM EDTA, 50 mM KCl and 1 mM dithiothreitol (DTT). After centrifugation at 125,000 × g and 4°C, the soluble fraction was loaded onto a SP-sepharose cation exchange column (GE Biosciences) and eluted over a 50–1000 mM KCl gradient. Fractions containing the ETS proteins were loaded onto a Superdex 75 gel filtration column (GE Biosciences) in 25 mM Tris pH 7.9, 10% glycerol (v:v), 1 mM EDTA, 300 mM KCl and 1 mM DTT. Eluted fractions were analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). The final, purified proteins were then concentrated on a 10-kDa molecular weight cut-off (MWCO) Centricon device, snap-frozen with liquid nitrogen, and stored at -80°C in single-use aliquots for subsequent EMSA studies.
Full-length ETS factors and ETV4 truncations containing the NID generally expressed more efficiently in the insoluble fraction using an autoinduction protocol (27). Briefly, bacteria in 250 mL of autoinduction media were grown in 4 L flasks at 37°C to an OD600 ∼ 0.6 – 1. The temperature was then reduced to 30°C and cultures were grown for another ∼ 12 – 24 hr. Final OD600 values were typically ∼ 6 – 12, indicating robust autoinduction. Harvested cells were resuspended as described above, sonicated and centrifuged at 31,000 x g for 15 min at 4°C. The soluble fraction was discarded and this procedure was repeated with the pellet / insoluble fraction twice more to rinse the inclusion bodies. The final insoluble fraction was resuspended with 25 mM Tris pH 7.9, 1 M NaCl, 0.1 mM EDTA, 5 mM imidazole, 2 mM BME, 1 mM PMSF, and 6 M urea. After sonication and incubation for ∼ 1 hr at 4°C, the sample was centrifuged at 125,000 x g for at least 30 min at 4°C. The soluble fraction was loaded onto a Ni2+ affinity column (GE Biosciences) and refolded by immediately switching to a buffer with the same components as above, except lacking urea. After elution with 5 – 500 mM imidazole, the remaining purification steps using ion-exchange and size-exclusion chromatography were performed as described above. However, a Q-sepharose anion-exchange column was used instead of a SP-sepharose cation-exchange column due to differing isoelectric points of the desired proteins.
Acetyllysine was incorporated into defined locations in the amino acid sequence and acetylated full-length ETV4 proteins were expressed according to a published protocol (26). Briefly, expression was induced with IPTG, as described above, but in the presence of 10 mM acetyllysine, 20 mM nicotinamide, and a plasmid expressing an amber tRNA that has been mutated to recognize acetyllysine. Acetylated proteins were purified as outlined above for unacetylated full-length ETV4.
ETV4 proteins prepared for NMR spectroscopy were purified using protocols slightly different from above. Harvested cells were resuspended in 50 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, 6 M guanidinium HCl, pH 7.4 and lysed by at least one round of freeze/thaw, followed by passage 5 times through an EmulsiFlex-C5 homogenizer at 10 kPa, and finally, 15 min of sonication. The cell lysate was spun down by centrifuging at 25,000 x g for 1 hr at 4°C. The supernatant containing ETV4 was then loaded onto Ni2+ affinity column (GE Biosciences), washed with 30 mM imidazole and eluted with 1000 mM imidazole and 6 M guanidinium HCl. Eluted fractions containing the desired protein were dialyzed against 3 L of refolding buffer (50 mM sodium phosphate, 1 M NaCl, 2 mM DTT and 1 mM EDTA, pH 7.5) at 4°C overnight. The His6-tag of the refolded proteins was cleaved by adding 1 U of thrombin/mg or tobacco etch virus protease (TEV) at a TEV/protein ratio of 1/200 (w/w). The mixture was loaded onto another Ni2+ affinity column, and the flow-through containing the tag-free ETV4 fragment was concentrated using a 3 kDa MWCO Centricon device to 2 mL. Size exclusion chromatography with Superdex 75 was used for a last purification step. Eluted fractions were assessed using SDS-PAGE and those containing the purified protein were pooled and dialyzed against NMR sample buffer (20 mM sodium phosphate, 200–1000 mM NaCl, 2 mM DTT, 0.1 mM EDTA, pH 6.5).
Protein concentrations were determined by measuring the absorbance at 280 nm using predicted ε280 values, or at 595 nm after mixing 20 μl of protein with 1 ml of Bio-Rad Protein Assay Dye Reagent (diluted 1:5 in deionized water) and comparing to a bovine serum albumin standard curve. Molecular weights for each ETS protein were predicted using the Peptide Property Calculator (Northwestern University) or using the ExPASY web server (28).
Expressed protein ligation and purification
The DNA encoding ETV4 ETS domain and CID (residues 337–436) was sub-cloned into bacterial expression vector pEM5B (kind gift from Dr. Pierre Barraud, Université Paris Descartes) between XhoI and BamHI restriction sites. This enabled the addition of the required cysteine and TEV cleavage site (ENLYFQC) preceding the ETS domain, as described for the segmental labeling and expressed protein ligation protocol (29). The protein construct was expressed in LB media (unlabeled) or M9 media (15N-labeled), purified under denaturing conditions, and refolded as described above. The protein was concentrated to 0.3 mM and stored in the inactive reaction buffer (50 mM HEPES, 200 mM NaCl, 0.1 mM TCEP, pH 7).
The DNA encoding ETV4 NID (165–336) was sub-cloned into pEM9B (kind gift from Dr. Pierre Barraud) between NdeI and SapI restriction sites. The pEM9B expression vector also encodes a C-terminal Mxe GyrA intein. Nine additional amino acids (GGGHM preceding and GSSC following the NID) were introduced as a result of cloning and to enable protein ligation. The protein construct was expressed in LB media (unlabeled) or M9 media (15N-labeled), cells were harvested and resuspended in native buffer (50 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, pH 7.4) and lysed by cell homogenization and sonication, as described above. The supernatant containing the desired protein was purified first by loading onto the Ni2+ affinity column, washed by 30 mM imidazole and eluted with 1000 mM imidazole. The protein was concentrated to 0.5 mM and stored in the inactive reaction buffer (50 mM HEPES, 200 mM NaCl, 0.1 mM TCEP, pH 7).
Purified protein samples containing the 15N-labeled ETV4 ETS domain and CID and unlabeled ETV4 NID were mixed in a 1:2 molar ratio. The reaction was activated by adding 100 mM 2-mercaptoethanesulfonate (MESNA) and TEV protease at a TEV/protein ratio of 1/200 (w/w). The reaction mixture was incubated at 16°C for 5 days. Time points were collected and analyzed on SDS-PAGE to monitor the ligation efficiency. TEV protease-cleaved products and intein self-cleaved products were purified on a chitin column equilibrated with 50 mM HEPES, 200 mM NaCl, pH 7. The flow-through of the chitin column containing the ligated product was purified on either ion-exchange chromatography (Mono Q) equilibrated with 50 mM HEPES pH 7 and eluted with 0–1000 mM NaCl gradient, and/or size exclusion chromatography (Superdex 75) equilibrated with NMR sample buffer (20 mM sodium phosphate, 200 mM NaCl, 2 mM DTT, 0.1 mM EDTA, pH 6.5). Fractions containing the final product were verified by SDS-PAGE and MALDI-ToF mass spectrometry on a Voyager-DE STR (Applied Biosystems) with a sinapinic acid matrix. The final product was dialyzed against NMR buffer. For the ligation reaction using 15N-labeled ETV4 NID and unlabeled ETV4 ETS domain and CID, equal molar ratio were mixed (100 μM) to minimize aggregation due to highly concentrated ETV4 (337–436). The reaction was initiated and the final product was purified and confirmed, as described above.
Segmental isotope labeling using sortase A
The DNA encoding the Sortase A peptidase was a kind gift from Dr. Michael Sattler (Institute of Structural Biology, Helmholtz Zentrum München). A mutation of Gly to Ala immediately after the TEV cleavage site was made to optimize ligation efficiency. The plasmids encoding Sortase A and ETV4 (337-436) were transformed into E. coli (λDE3) cells for protein expression at 37°C with 1 mM IPTG induction. Sortase A was purified by Ni+2 affinity and size exclusion chromatography. 15N-labeled ETV4 (337–436) with an N-terminal glycine was prepared as described above, dialyzed into 50 mM Tris, 150 mM NaCl, pH 8.0, concentrated to 0.1 mM and stored at −80°C. ETV4 (165–336) was sub-cloned into pET28a between NdeI and Xhol restriction sites, with modification in the C-terminal to include the Sortase recognition sequence LPQTG plus a C-terminal His6-tag. ETV4 (165–336) was expressed and purified using the same protocol as for ETV4 (337–436), except without thrombin digestion. Both Sortase A and ETV4 (165–336) were dialyzed into 50 mM Tris, 150 mM NaCl, pH 8.0 and concentrated to 0.5 mM for storage at −80°C. The ligation reaction was carried out as previously described (30). Briefly, the 15N-labeled ETV4 fragment, the unlabeled ETV4 fragment and Sortase A were combined in a 2:6:1 molar ratio. The mixture was centifuged at 2000 x g in a 3 kDa MWCO Centricon device with reaction buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10 mM CaCl2) at 20°C for 4 h. The ligated product was purified by passing twice through a Ni+2 affinity column with thrombin cleavage after the first purification step, as described above. The final Sortase ligated sample of unlabeled ETV4 (165–336) linked to 15N-labeled ETV4 (337–436) was verified by MALDI-ToF mass spectrometry and SDS-PAGE.
Electrophoretic mobility shift assays (EMSA)
DNA-binding assays of ETS factors utilized a duplexed 27-bp oligonucleotide with a consensus ETS binding site: 5΄-TCGACGGCCAAGCCGGAAGTGAGTGCC-3΄ (arbitrarily assigned as ‘top’ strand) and 5΄-TCGAGGCACTCACTTCCGGCTTGGCCG-3΄ (‘bottom’ strand). Boldface GGAA indicates the core ETS binding site motif. Each of these oligonucleotides, at 2 μM as measured by absorbance at 260 nM on a NanoDrop 1000 (Thermo Scientific), were labeled with [γ-32P] ATP using T4 polynucleotide kinase at 37°C for ∼30–60 min. After purification over a Bio-Spin 6 chromatography column (Bio-Rad), the oligonucleotides were incubated at 100°C for ∼5 min, and then cooled to room temperature over 1–2 h. The DNA for EMSAs was diluted to 1 × 10−12 M and held constant, whereas protein concentrations ranged approximately six orders of magnitude. For full binding isotherms, the exact concentration range was chosen according to the KD of particular protein fragments. Protein concentrations were determined after thawing each aliquot of protein, using the Protein Assay Dye Reagent. Equivalent starting amounts (0.2 μg) of each protein utilized on a given day were run on an SDS-PAGE gel to confirm their relative concentrations. The binding reactions were incubated for 45 min at room temperature in a buffer containing 25 mM Tris pH 7.9, 0.1 mM EDTA, 60 mM KCl, 6 mM MgCl2, 200 μg/ml BSA, 10 mM DTT, 2.5 ng/μl poly(dIdC) and 10% (v:v) glycerol, and then resolved on an 8% (w:v) native polyacrylamide gel at room temperature. The 32P-labeled DNA was quantified on dried gels by phosphorimaging on a Typhoon Trio Variable Mode Imager (Amersham Biosciences). Equilibrium dissociation constants (KD) were determined by nonlinear least squares fitting of the total protein concentration [P]t at each titration point versus the fraction of DNA bound ([PD]/[D]t) to the equation [PD]/[D]t = 1/[1 + KD/[P]t)] using Kaleidagraph (v. 3.51; Synergy Software). The DNA, running at the position of free DNA, is unbound and by comparing the DNA signal in this position, [D], to the DNA signal in lanes with no binding at the lowest protein concentrations, [D]t, we calculated the fraction of bound DNA, [PD]/[D]t = 1 - [D]/[D]t. Due to the low concentration of total DNA, [D]t, in all reactions relative to the KD, the total protein concentration is a valid approximation of the free, unbound protein concentration [P] = [P]t. Reported KD values represent the mean of at least three independent experiments and the standard error of the mean. We used Microsoft Excel (v.14.6.5 for Mac 2011) to perform two-tailed, heteroscedastic t-tests in order to compare the KD values of different proteins. This method provided the same P-values as using a Welch's t-test.
Although there were deviations between the EMSA titration data and the non-linear regression at higher protein concentrations, this had essentially no influence on calculated KD values. For comparison we fit all data using a one-site specific binding model with a Hill coefficient (Prism, version 6), while treating the maximum fraction of DNA bound as a variable. Use of this alternative approach, which eliminated the deviation between the data and the curve fit at the higher protein concentrations, led to KD values that were uniformly stronger by about 1.3–1.7-fold, depending on the protein tested, with Hill coefficients ranging from 0.9 to 1.1. Importantly, all fold-inhibition values changed <1.5-fold, and all conclusions made from the binding experiments were supported by either curve fitting approach.
To test protein activity, binding reactions with known concentrations of radiolabeled duplex DNA (as described above) were titrated against a fixed concentration of protein corresponding to ∼ 50-fold greater than the KD value of each individual protein, as previously described (31). All proteins analyzed demonstrated high activity levels (>95 %), indicating that the autoinhibition of DNA binding observed for the larger protein species is not due to a large fraction of the protein being totally inactive in DNA binding.
Partial proteolysis
For tryptic digestion studies, 20 μl of full length (FL) ETV4 at 20 μM was incubated with 1.5–450 ng of trypsin (Sigma) in a buffer containing 25 mM Tris pH 7.9, 10 mM CaCl2, and 1 mM DTT. After 2 min of incubation, the reaction was quenched with 1 % (v:v) acetic acid (final volume). The resulting samples were analyzed by SDS-PAGE and ESI-MS (total mixture analyzed), and used for EMSA studies.
Crystallization and structure determination
Purified proteins were dialyzed overnight in 10 mM Tris pH 7.9 and 50 mM NaCl, and then concentrated to 5 mg/ml. Crystals were grown by vapor diffusion in sitting drops of 2:1 protein:reservoir (v:v). CID-inhibited ETV1 (332–435) was crystallized against a reservoir of 30% (w:v) PEG 5000 monomethyl ether, 0.1 M MES sodium salt and 0.2 M ammonium sulfate at pH 6.5 and 20°C. CID-inhibited ETV4 (337–441) was crystallized against a reservoir of 1 M diammonium phosphate and 0.1 M sodium acetate at pH 4.5 and 20°C. Uninhibited ETV5 (364–457) was crystallized against a reservoir of 0.2 M diammonium phosphate and 20% PEG 3350 at pH 5.0 and 4°C.
Crystals were immersed briefly in mother liquor containing 20% glycerol, and then cryocooled by plunging into liquid nitrogen. Diffraction data were collected on a Q315 CCD using Stanford Synchrotron Radiation Lightsource (SSRL) beamline 7-1 with X-rays at 1.0000 Å (ETV1 and ETV4) or 1.1271 Å (ETV5). The resulting data were integrated and scaled using HKL2000 (32). Phases were determined by molecular replacement with Phaser-MR (33) using the ETS domain of ETS1 (1MD0.pdb) as a search model. Models were built with COOT (34) and refined with PHENIX (35). PyMOL (Schrödinger, LLC) was used to render molecular structure figures.
Model geometries were analyzed by MolProbity (36) within PHENIX. For ETV1 (1.4 Å resolution data), 87.5% of residues have favorable backbone dihedrals and 12.5% fall into allowed regions. Residues 332–333 and 435 were not visible in the electron density. For ETV4 (1.1 Å), 91.8% of residues have favorable backbone dihedrals and 8.2% of residues fall into allowed regions. Residues 337–339 and 337–441 were not visible in the electron density. For ETV5 (1.8 Å), 87.7% of residues have favorable backbone dihedrals and 12.3% of residues fall into allowed regions. Residues 364–365 were not visible in the electron density. X-ray crystallography data collection and refinement statistics are provided in Supplementary Table S1. Structural coordinates have been deposited in the RCSB Protein Data Bank under ID codes 5ILS (ETV1), 5ILU (ETV4) and 5ILV (ETV5).
Circular dichroism spectroscopy
Aliquots of frozen ETV4 NID (165–336), expressed and purified as described above, were thawed, dialyzed overnight into 20 mM sodium phosphate, 50 mM NaCl, pH 7.9, and diluted to 25 μM concentration. CD spectra were recorded at 4°C through the wavelength range of 190–260 nm with a 1 nm wavelength step. A baseline reference, consisting of buffer only, was subtracted from the CD spectra. Three scans were collected in series and averaged after visually verifying their consistency. Data were converted to molar ellipticity as described (37).
NMR spectroscopy
NMR data were recorded at 25°C on cryoprobe-equipped 500, 600 and 850 MHz Bruker Avance III spectrometers. Proteins were in NMR sample buffer (plus 10% lock D2O) with 1 M NaCl for spectral assignments and with 200 mM NaCl for all other experiments. The elevated ionic strength reduced slow aggregation over long-term measurements. Data were processed and analyzed using NMRpipe (38) and Sparky (39). Signals from mainchain and sidechain 1H, 13C and 15N nuclei were assigned by standard multi-dimensional heteronuclear correlation experiments, including 15N-HSQC, HNCO, HN(CA)CO, CBCA(CO)NH and HNCACB (40). Amide 1H/2H hydrogen exchange (HX), after transfer into ∼99% D2O NMR sample buffer via a Sephadex G25 spin column and CLEANEX-PM 1H/1H HX measurements were recorded using 850 MHz NMR spectrometer and analyzed as described previously (10,41). The two approaches detect slow (minutes-days) and fast (seconds) timescale exchange, respectively. Initially, HX measurements were carried out at pH 6.5 and 25°C with uninhibited ETV4 (328–430). However, a more complete set of data were obtained for CID-inhibited ETV4 (337–436) via 1H/2H exchange measurements at pH 5.7 and 20°C and CLEANEX-PM 1H/1H exchange at pH 7.5 and 25°C. Protection factors were calculated as ratio of the predicted HX rate constant for each amide in an unstructured polypeptide with the sequence of ETV4 versus the corresponding measured HX rate constant. The predicted values, corrected for pH, temperature and isotope effects, were obtained with the program Sphere (http://landing.foxchase.org/research/labs/roder/sphere/) (42). The protection factors for the two proteins, studied under several conditions, were merged and reported using the combined name ETV4 (328–436) (Figure 5C). For amides with HX quantitated under more than one condition, the highest protection factor is shown.
Figure 5.

The CID perturbs the dynamic DNA-recognition helix H3. (A) Overlaid 15N-HSQC spectra of uninhibited ETV4 (328–430; red), and CID-inhibited ETV4 (313–446; purple). Selected assignments are indicated. Despite minor differences in the boundaries of these constructs, they bind to DNA with similar affinities as the previously described uninhibited and CID-inhibited fragments (Supplementary Figure S5A). (B) Amide chemical shift perturbations (Δδ = [(ΔδH)2 + (0.2ΔδN)2 ]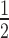 ) for corresponding residues in the spectra of (A) are plotted as a histogram and mapped onto the crystal structure of ETV4. Perturbed residues with Δδ > 0.025 ppm (dashed line), are highlighted in red on the structure. (C) Amide HX protection factors of ETV4 (328–436), are plotted as a histogram and mapped onto the crystal structure of ETV4 using spheres with the indicated size/color scale. Although ETV4 (328–436) is a combined name to denote data merged from HX studies of uninhibited ETV4 (328–430) and CID-inhibited ETV4 (337–436), the additional six C-terminal residues, which form the full CID, did not measurably change the protection factors of amides within the ETS domain. Missing values correspond to prolines, residues with unassigned or overlapped NMR signals and residues exchanging too slowly to be measured with CLEANEX-PM (sec timescale), yet too fast to measured via 1H/2H exchange (greater than hours).
) for corresponding residues in the spectra of (A) are plotted as a histogram and mapped onto the crystal structure of ETV4. Perturbed residues with Δδ > 0.025 ppm (dashed line), are highlighted in red on the structure. (C) Amide HX protection factors of ETV4 (328–436), are plotted as a histogram and mapped onto the crystal structure of ETV4 using spheres with the indicated size/color scale. Although ETV4 (328–436) is a combined name to denote data merged from HX studies of uninhibited ETV4 (328–430) and CID-inhibited ETV4 (337–436), the additional six C-terminal residues, which form the full CID, did not measurably change the protection factors of amides within the ETS domain. Missing values correspond to prolines, residues with unassigned or overlapped NMR signals and residues exchanging too slowly to be measured with CLEANEX-PM (sec timescale), yet too fast to measured via 1H/2H exchange (greater than hours).
RESULTS
DNA binding by ETV1, ETV4 and ETV5 is autoinhibited
We initially sought to determine the magnitude of autoinhibition in the ERG and ETV1/4/5 subfamilies of ETS factors. Toward this aim, we measured the DNA binding affinities (equilibrium dissociation constant, KD) of the full-length proteins and nearly-minimal DBDs for ERG, FLI1, ETV1, ETV4 and ETV5 (Figure 1A). Robust autoinhibition was observed in ETV1, ETV4, and ETV5, with the full-length proteins displaying ∼10 to 30-fold weaker binding than their minimal DBDs (Figure 1B–D and Supplementary Table S2). These levels of autoinhibition are comparable to those previously reported for ETS1 (8) and ETV6 (9). In contrast, ERG and its subfamily member FLI1 displayed modest 2- to 3-fold autoinhibition, as also previously reported (43). Interestingly, the KD values cluster in a pattern that reflects their subfamily phylogenetic classifications (Figure 1E) (44). The ∼100-fold range of KD values for ETS DBDs suggests that there are key differences that influence DNA binding despite the high overall sequence conservation in the ETS domain of these factors. Additionally, the known inhibitory elements from ETS1 and ETV6 are not conserved in the ETV1/4/5 or ERG subfamilies; the poor sequence conservation outside of the ETS domain among these factors indicates that the mechanism of autoinhibition is likely different for the ETV1/4/5 and ERG subfamilies (Supplementary Figure S1). Based on the larger magnitude of autoinhibition observed with ETV1, ETV4, and ETV5, as compared to ERG and FLI1, we focused on the ETV1/4/5 subfamily for mechanistic studies.
Figure 1.

Autoinhibition in the ERG and ETV1/4/5 subfamilies. (A) Schematic of full-length protein, FL, and nearly minimal DNA-binding domain, DBD, for ETV4. Based on the sequences of all ETS factors, the conserved ETS domain, ED, is noted in red. (B) Representative examples of EMSA gels for ETV4 FL or DBD with a double-stranded DNA duplex containing a core ETS binding site. (C) Binding isotherms for ETV4 FL and DBD. Data points and error bars refer to the mean and the standard error of the mean with four replicates for each protein. See ‘Materials and Methods’ section for details. (D) Fold inhibition of ERG, FLI1, ETV1, ETV4 and ETV5, calculated as KD (FL or DBD) / KD (DBD). ETS1 (8)# and ETV6 (9)$ data are included for comparison. Mean and standard error of the mean from at least three replicates are plotted; ‘**’ and ‘***’ indicate P < 0.01 and P < 0.001, respectively. See Supplementary Table S2 for KD values and numbers of replicates. (E) KD values of FL versus DBD for each of the ETS factors tested. The dashed diagonal line represents no autoinhibition [i.e. KD (FL) = KD (DBD)].
We chose ETV4 as a model factor to further investigate autoinhibition in the ETV1/4/5 subfamily. Partial proteolysis aided the design of truncation boundaries for mapping inhibitory elements (Supplementary Figure S2A–C). We found that the predominant trypsin-resistant fragment, spanning amino acids 165–484, retained levels of autoinhibition comparable to full-length ETV4 (Figure 2A and Supplementary Table S3). Subsequent deletion studies revealed that amino acid residues both N- and C-terminal of the ETS domain inhibit DNA binding independently, but also act cooperatively to yield higher than additive levels of inhibition (Figure 2B). Hereafter, these regions will be denoted as the NID and the CID, whereas the nearly-minimal DBD will be denoted as an uninhibited species. We hypothesized that the ETV1/4/5 NID and CID function through direct interactions with the ETS domain and/or with each other to cooperatively inhibit DNA binding (Figure 2C).
Figure 2.

NID and CID cooperate to inhibit ETV4 DNA binding. (A) Fold inhibition of the ETV4 fragments with mean and standard error of the mean displayed. Fold inhibition calculated as KD (fragment) / KD (337–430). ‘*’, ‘**’, And ‘***’ indicate P < 0.05, P < 0.01 and P < 0.001, respectively. See Supplementary Table S3 for KD values and numbers of replicates. (B) ΔΔG = RT ln [KD ETV4 inhibited fragment / KD ETV4 (337–430)] measured for fragments containing the NID (165–430), the CID (337–436) or both (165–436). The dotted line indicates the sum of the ΔΔG values for 165–430 and 337–436. (C) Schematic of ETV4 autoinhibition depicting cooperative inhibitory contributions from both the NID and CID, cyan. The ETS domain (ED) is noted in red.
CID interactions perturb the DNA-recognition helix H3 to mediate autoinhibition
To elucidate the mechanism of autoinhibition by the CID, we used X-ray crystallography to determine the structures of the partially inhibited fragments of ETV1 (332–435) and ETV4 (337–441). These proteins contain both the ETS domain and the CID, as mapped in ETV4. Their structures were very similar with a root mean square deviation (RMSD) of 0.16 Å for backbone alignment of their ETS domains (Figure 3A and B; Supplementary Figure S3A). The CID includes an α-helix, termed H4, which packs on one face of the ETS domain. In ETV4, Ala426 and Leu430 in H4 lie in a hydrophobic groove along the ETS domain in proximity to the conserved residues Trp344 from H1, Ile407 from the loop between H3 and β-strand S3, and Phe420 from S4 (Figure 3C). Homologous residues had similar interactions in ETV1. Replacing Leu430 with an alanine resulted in a reduction in autoinhibition (activation in DNA binding), whereas mutation to methionine, the homologous amino acid in ETV1 and ETV5, did not affect DNA binding (Figure 3D). These structural and functional data demonstrated that the CID inhibits DNA binding through intramolecular contacts between H4 and the ETS domain, mediated in part by a leucine or methionine in this helix.
Figure 3.

The CID inhibits DNA binding through hydrophobic contacts between α-helix H4 and the ETS domain. (A) Schematic of ETS domain, H1-H3 and S1-S4, and α-helix H4 of ETV1, ETV4 and ETV5. ETS domain, red; inhibitory elements, cyan; α-helices, cylinders; β-strands, arrows. (B) Cartoon representations of the closely aligned structures for the ETS domain and CID of ETV1 (332–435) and ETV4 (337–441). Displayed in stick format are Ala426 and Leu430 from α-helix H4 in ETV4, and the analogous Ala420 and Met424 from ETV1, as well as the conserved amino acids in the ETS domain that form a hydrophobic cluster. Numbering for homologous amino acids and endpoints denoted as ETV1/ETV4. (C) Portions of the ETV1, left, and ETV4, right, structures in van der Waals sphere format to show hydrophobic interactions between the ETS domain and H4. There is clear evidence for two conformations of the δ1-methyl of Ile407 in the electron density map of ETV4. (D) Fold inhibition of ETV4 FL in its wild-type form, WT (n = 35) or with point mutations L430A (n = 11) or L430M (n = 3). ‘*’ Indicates P < 0.05.
Based on the crystal structures of CID-inhibited ETV1 and ETV4, we noted that the uninhibited, minimal DBD fragments used for demonstrating autoinhibition in ETV1, ETV4 and ETV5 were predicted to have a shorter or possibly unfolded helix H4 (Supplementary Figure S3B). As with ETV4, loss of these homologous residues in ETV1 and ETV5 also activated DNA binding (Figure 4A). Therefore, an intact and full-length H4 is a necessary and conserved feature of the CID.
Figure 4.

Interactions between the CID and the ETS domain affect DNA-recognition helix H3 positioning. (A) Equilibrium dissociation constant, KD, values for uninhibited ETV1 (residues 332–425; n = 6), ETV4 (337–430; n = 25) and ETV5 (364–457; n = 4) versus CID-inhibited ETV1 (332–430; n = 7), ETV4 (337–436; n = 23) and ETV5 (364–463; n = 7). ‘*’, ‘**’ and ‘***’ indicate P < 0.05, P < 0.01 and P < 0.001, respectively. (B) Crystal structure of uninhibited ETV5 (364–457), showing the truncated H4 and the same selected sidechains as in Figure 3. (C) H3 positioning from CID-inhibited ETV4 (gray), uninhibited ETV5 (red) and ETV4 bound to DNA (pink, 4UUV.pdb) (45). Structures were aligned to DNA-bound ETV4 across the entire protein sequence (See Supplementary Figure S3C). Met457 of ETV5, the homologous residue to Leu430 in ETV4, is not in frame due to the repositioning of H4 in the uninhibited ETV5 crystal structure. (D) Comparison of KD values for ETV4 FL in its WT form (n = 35), or with point mutations L430A (n = 11), I407A (n = 4) or both I407A and L430A (n = 4). ‘*’ Indicates P < 0.05 and ‘n.s.’ indicates P > 0.05. Fold difference for KD values are relative to WT ETV4 FL.
To understand the structural nature of the residues mapped to H4 within the context of uninhibited ETV1, ETV4 and ETV5, we attempted to crystalize these fragments with success for ETV5 (364–457) (Figure 4B). Despite the deletion of amino acids mapped to the intact H4, the α-helix is retained, albeit truncated. However, the shorter H4 is rotated ∼60° away from the ETS domain relative to the position of the full-length H4 in ETV1 and ETV4. This alternate position is accommodated in the crystal by intermolecular contacts between the truncated H4 and the ETS domain of a neighboring molecule (Supplementary Figure S4). With H4 in this alternate position, Met457 is unable to form the intramolecular inhibitory contacts with the ETS domain observed for the homologous Met424 and Leu430 in the CID-inhibited structures of ETV1 and ETV4, respectively, potentially explaining the loss of autoinhibition of this fragment (compare Figures 3B and 4B). In conclusion, the relief of autoinhibition by the partial truncation of H4 and by disruption of an intramolecular contact between H4 and the ETS domain demonstrated the role of H4 in autoinhibition. In addition, while the folding of truncated helix H4 and its alternate position is potentially a consequence of crystallization, we propose that this repositioning reflects an intrinsic mobility of the CID. This idea is supported by NMR spectroscopy studies, presented below.
To further our structural studies of ETV1/4/5, we compared our crystal structures of the uninhibited ETV5 with a truncated H4 to that of the CID-inhibited ETV1 and ETV4 with a full-length H4. In comparison to the highly similar CID-inhibited ETV1 and ETV4 structures (backbone RMSD of 0.16 Å), the ETS domain from uninhibited ETV5 was distinct with RMSD values of 0.79 Å and 0.72 Å when aligned to ETV1 and ETV4, respectively (Supplementary Figure S3A). Closer examination of subsections of the ETS domain revealed that the differences between uninhibited and CID-inhibited structures were most pronounced around the DNA-recognition helix H3, as well as β-strands S3 and S4. Visually, the backbone of the C-terminal half of the DNA-recognition helix H3 is shifted about 2 Å between the inhibited and uninhibited structures, relative to the rest of the ETS domain (Figure 4C). Further comparison with the previously reported structure of ETV4 in complex with DNA (45) demonstrated that in the DNA-bound form, H3 of ETV4 is also shifted to a similar position as observed for uninhibited ETV5 (Figure 4C and Supplementary Figure S3C). We speculate that in the ETV1/4/5 subfamily, the active state of a DNA-bound ETS domain requires this shift of H3 and, thus, matches the conformation of uninhibited ETV5 determined by X-ray crystallography.
Having observed the activation of the ETV4 mutant L430A (Figure 3D) and the variable positioning of the DNA-recognition helix H3 in our crystal structures (Figure 4C), we hypothesized that helix H4 inhibits DNA binding by modulating H3 through an interaction between Leu430 in H4 and Ile407 in the H3-S3 loop. We tested this postulate by mutating Ile407 and Leu430 to alanine, separately and in combination. The ETV4 mutant I407A had a reduction in DNA-binding affinity compared to the wild-type protein indicating that I407A contributes to DNA binding. Importantly, the I407A mutation also abrogated the activating nature of L430A in the double mutant I407A/L430A (Figure 4D). We conclude that H3 and the CID are coupled through the Ile407-Leu430 interaction and propose that CID-mediated autoinhibition functions by shifting a conformational equilibrium of H3 toward a state that is less competent for DNA binding.
Dynamic features of CID autoinhibition mechanism
To further investigate the CID mechanism of autoinhibition, we utilized NMR spectroscopy to compare uninhibited and CID-inhibited species (Figure 5A). Although differing slightly at their N- and C-terminal boundaries, the two ETV4 fragments displayed the same affinities for DNA as the corresponding species discussed above (Supplementary Figure S5A). Based on mainchain chemical shifts, residues from the truncated H4 in the uninhibited ETV4 (328–430) and the full-length H4 in the CID-inhibited ETV4 (313–46) both adopted folded α-helical conformations under solution conditions. However, relative to ETV4 (313–446), the C-terminal residues in the shorter H4 of ETV4 (328–430) exhibited reduced chemical shift-derived helical propensities and increased mobility as detected by amide 15N relaxation measurements (Supplementary Figure S5B and data not shown). Nevertheless, these NMR spectroscopic studies indicate that the truncated α-helix H4 observed by X-ray crystallography is not an artefact of the crystallization process (Figure 4B).
A comparison of the 1NH and 15N chemical shifts of the uninhibited and CID-inhibited species demonstrated that amides near the C-terminal end of H1 and H3, and throughout H4 were most affected by the presence of the full-length versus truncated H4 (Figure 5B). The amino acids in H3 that showed chemical shift perturbations match closely to those undergoing the backbone realignment observed in the comparison of the crystal structures of CID-inhibited ETV1 and ETV4 versus uninhibited ETV5. Thus, the interaction between H4 and the ETS domain, as well as the H4-dependent perturbations of H3, observed in the crystal structures are also retained in solution.
Additional NMR-monitored amide HX experiments were used to probe the dynamics of CID-inhibited ETV4 (Figure 5C). Residues within H1, H2 and the β-sheet displayed relatively large protection factors (>104), indicating that they form the stable core of the ETS domain. In contrast, residues preceding the ETS domain and in loop regions had lower protection factors, as expected based on their solvent exposure and lack of any persistent hydrogen-bonded secondary structure. Most interestingly, many residues in the DNA-recognition α-helix H3 and in the inhibitory CID α-helix H4, displayed intermediate protection factors (100–10,000) indicative of conformational dynamics to sample partially unfolded states detectable by HX. Similar behavior is observed with the DNA-recognition and inhibitory helices of ETS1 (8,12) and ETV6 (10,11). These NMR experiments extend our hypothesis that the CID autoinhibitory mechanism involves a conformational equilibrium involving interactions between helices H3 and H4, by demonstrating the dynamic nature of the helices.
Inhibitory properties of the NID map to intrinsically disordered sequences
As the next step toward a mechanistic understanding of ETV4 autoinhibition, we investigated the NID using biophysical approaches. Crystallization of fragments of ETV4 encompassing the NID was unsuccessful, potentially reflecting the predicted disordered nature of the NID (Supplementary Figure S2D). Consistent with this prediction, the 15N-HSQC spectrum of the isolated ETV4 NID (165–336) displayed limited 1HN chemical shift dispersion (Figure 6A). An analysis of its assigned mainchain 1H, 13C and 15N chemical shifts confirmed that the NID predominantly samples random coil conformations and thus is an IDR (Supplementary Figure S6) (46,47). Circular dichroism spectroscopy added additional evidence for the overall disordered character of the NID (Supplementary Figure S7).
Figure 6.

The NID is intrinsically disordered. (A) Both panels show the 15N-HSQC spectrum of the isolated ETV4 NID (165–336) in red. The right panel also shows the overlapped spectrum of the 15N-labled NID intein-ligated to the unlabeled ETS domain and CID (337-436) in blue. Consistent with its limited 1HN dispersion, analysis of the assigned mainchain chemical shifts (1HN, 15N, 13Cα, 13Cβ, 13CO) of the isolated NID with the algorithms CSI 2.0 (46) and δ2D (47) confirms that it is an IDR (Supplementary Figure S6). Comparisons of 15N-HSQC amide chemical shift perturbations (Δδ = [(ΔδH)2 + (0.2ΔδN)2 ] ), (B), and relative peak intensities, (C), for the NID alone versus attached to the ETS domain and CID. The simple peak intensity ratios were not normalized for differences in sample concentration and spectral acquisition times. Amides broadly localized near the N-terminus of the NID showed small chemical shift and relative intensity perturbations due to the ETS domain and CID ‘in cis’. The 15N-HSQC spectrum of the intein-ligated species was assigned by comparison with that of the isolated NID, and red bars indicate amide signals that could not be confidently identified in both proteins. Missing histogram bars correspond to prolines and unassigned amides.
), (B), and relative peak intensities, (C), for the NID alone versus attached to the ETS domain and CID. The simple peak intensity ratios were not normalized for differences in sample concentration and spectral acquisition times. Amides broadly localized near the N-terminus of the NID showed small chemical shift and relative intensity perturbations due to the ETS domain and CID ‘in cis’. The 15N-HSQC spectrum of the intein-ligated species was assigned by comparison with that of the isolated NID, and red bars indicate amide signals that could not be confidently identified in both proteins. Missing histogram bars correspond to prolines and unassigned amides.
Many IDRs, while disordered in isolation, take on a more structured character in the presence of a binding partner through a coupled ‘folding and binding’ mechanism (48). Therefore, we asked whether the NID is still disordered in the presence of the ETS domain and CID. Using intein and sortase ligation technologies, we covalently linked the 15N-labeled NID (165–336) to unlabeled ETS domain and CID of ETV4 (337–436). We confirmed that this ligated fragment displayed comparable autoinhibition to the native protein fragment (Supplementary Figure S8). The NID spectrum retained limited 1HN chemical shift dispersion, indicating the lack of any detectable structure induced upon covalent-linkage to the ETS domain and CID (Figure 6A). Although small changes in the chemical shifts or relative intensities of the 1HN-15N signals from amides localized near the N-terminus of the NID were observed (Figure 6B and C), no segment of residues interacted with the ETS domain and/or CID of ETV4 with sufficient affinity to adopt a persistent conformation detectable by NMR spectroscopy. Thus, whether isolated or ‘attached in cis’, the NID was observed to be predominantly disordered.
We next interrogated which regions of the NID are important for inhibition. A truncation series indicated that the progressive inclusion of residues N-terminal to the ETS domain provided progressively greater autoinhibition of DNA binding (Supplementary Figure S9). The region spanning residues 203–287 contributed the largest effect of autoinhibition, but other regions of the NID also contributed toward the overall inhibitory effect. The lack of a clear boundary for the inhibitory residues is consistent with the intrinsic disorder of the NID and the absence of any identifiable cluster of residues that interact strongly with the ETS domain and CID.
Intramolecular interactions of NID with the ETS domain and CID
To better define the possible interactions of the NID with the ETS domain and CID, we also compared the 15N-HSQC spectra of an ETV4 fragment containing the ETS domain and the CID only (337–436), with one also containing the NID (165–436) (Figure 7A). As described above, for spectral simplification, an unlabeled NID was joined by intein technology to a 15N-labeled fragment containing the ETS domain and CID. In addition to the expected changes at the N-terminus of H1, the ligated NID weakly perturbed the 1HN-15N signals of amides in H2, the C-terminal region of H3 and the surface-exposed face of the CID (Figure 7B and C). These data suggest that the NID may inhibit DNA binding by transiently interacting with the DNA-recognition helix H3 and/or by interacting with, and reinforcing the inhibitory position of, the CID.
Figure 7.

The NID interacts with the CID and the DNA-recognition helix H3. (A) Overlaid 15N-HSQC spectra of 15N-labeled ETV4 ETS domain and CID alone (337–436, red), and with the unlabeled NID (165–336, blue) joined via intein ligation. Selected peaks are labeled. (B and C) The amide chemical shift perturbations, (Δδ = [(ΔδH)2 + (0.2 ΔδN)2 ] ), resulting from the ligated NID are displayed in histogram format and mapped onto the structure of ETV4 ETS domain and CID (blue, Δδ > 0.025 ppm; gray, Δδ < 0.025 ppm, prolines, or residues with unassigned NMR signals). (D) Fold difference of KD values between full length (FL: residues 1–484) and CID-inhibited (ED + CID: 337–436) ETV4 for wild-type proteins (WT) or proteins with both Y401N and Y403G mutations. Bars and error bars represent the mean and the standard error of the mean for the following number of replicates: FL Y401N/Y403G, 6; ED + CID Y401N/Y403G, 5. ‘*’ indicates P < 0.05. See Supplementary Figure S10 for binding isotherms.
), resulting from the ligated NID are displayed in histogram format and mapped onto the structure of ETV4 ETS domain and CID (blue, Δδ > 0.025 ppm; gray, Δδ < 0.025 ppm, prolines, or residues with unassigned NMR signals). (D) Fold difference of KD values between full length (FL: residues 1–484) and CID-inhibited (ED + CID: 337–436) ETV4 for wild-type proteins (WT) or proteins with both Y401N and Y403G mutations. Bars and error bars represent the mean and the standard error of the mean for the following number of replicates: FL Y401N/Y403G, 6; ED + CID Y401N/Y403G, 5. ‘*’ indicates P < 0.05. See Supplementary Figure S10 for binding isotherms.
The largest chemical shift perturbations, besides those near the N-terminus ligation site, were observed for Tyr401 and Tyr403 on the DNA-recognition helix H3. Therefore, we tested whether these tyrosine residues are functionally important for autoinhibition by serving as an interaction site within the ETS domain for the NID. Tyr401 and Tyr403 were mutated to asparagine and glycine, respectively, in the CID-inhibited fragment of ETV4 (337–436). These alternative residues were chosen due to their presence in homologous positions in other ETS factors, suggesting that there would be less chance of structural perturbations. However, due to their position at the DNA interface, mutation of both residues substantially impaired DNA binding (Supplementary Figure S10A). Importantly, full-length ETV4 and the CID-inhibited fragment of ETV4 with Y401N and Y403G substitutions had identical affinity for DNA, indicating a loss of NID-mediated inhibition (Figure 7D and Supplementary Figure S10A). These data indicate that Tyr401 and Tyr403 are required for NID-mediated inhibition and may serve as part of a transiently occupied interface. Due to the role of these two tyrosines in direct contact to DNA, this NID intramolecular interaction may be acting sterically by masking the DNA interface.
Another potential NID-interaction site mapped to the surface of the CID. However, mutation of several glutamate or phenylalanine residues along the surface-exposed face of the CID (E423K, E425K or F428A/F432A) did not affect NID-mediated inhibition (Supplementary Figure S10B). Therefore, two possibilities exist to explain the NMR spectroscopically detected perturbation of the CID by the NID. The potential NID–CID interface may be sufficiently broad such that it is resilient to individual mutations, or the CID and the NID indirectly interact via the direct interactions between the NID and H3, and between H3 and the CID.
We took another approach to detect intramolecular interaction by following a biological regulatory activity. Widespread acetylation of lysine residues activates the DNA binding of ETV4, and two known sites of acetylation, Lys226 and Lys260, reside within the NID (49–51). Therefore, we tested whether acetylation of these residues is sufficient for activating ETV4 DNA binding. Acetylation of either Lys226 or Lys260, independently, resulted in a decrease of DNA binding autoinhibition by 2.8- or 1.6-fold, respectively (Figure 8A and B). First, we hypothesized that the positive charge of these lysine residues may be important for inhibition. However, mutation of Lys226 and Lys260 to glutamate or glutamine failed to recapitulate the activating nature by acetylation of these residues (Supplementary Figure S11). Next we tested whether hydrophobic forces provided intramolecular interactions between the NID and the ETS domain and CID, such that the added bulk of acetyllysine might disrupt such interactions formed by nearby aromatic residues in the NID. Conserved aromatic residues that are located proximally to Lys226 in the NID were substituted with alanines, singly and in combination. However, this mutagenesis did not activate DNA binding (Supplementary Figure S11). Therefore, while the exact nature of the inhibiting residues within the NID remains unclear, several lines of evidence support the occurrence of intramolecular interactions that might include a steric mechanism of DNA-binding autoinhibition.
Figure 8.

Acetylation of Lys226 or Lys260 relieves NID-dependent autoinhibition. (A) Binding isotherms for FL ETV4 in its unacetylated form (No Ac; black), and acetylated at Lys226 (top, red) or Lys260 (bottom, red). Data points and error bars correspond to the mean and standard error of the mean from four replicates. (B) Quantification of fold inhibition relative to uninhibited ETV4 (337–430) as in Figures 1D and 2A. The DNA binding of ETV4 Lys226Ac (KD, 30 ± 6 × 10−11 M) and ETV4 Lys260Ac (KD, 51 ± 3 × 10−11 M) was inhibited 5 ± 1-fold and 8 ± 1-fold, respectively, whereas, unmodified ETV4 was inhibited 14 ± 2-fold. ‘***’ Indicates P < 0.001.
DISCUSSION
Mechanistic model of autoinhibition
Here, we demonstrated that members of the ETV1/4/5 subfamily of ETS factors have regions N- and C-terminal of their ETS domains that act together to impinge upon helix H3 and inhibit DNA binding (Figure 9). We propose that the CID allosterically shifts the conformational equilibrium of the DNA-recognition helix H3 toward a state less competent for binding. In contrast, the NID works, at least in part, in a steric manner to occlude the DNA binding interface, thus also requiring a conformational change for DNA binding.
Figure 9.

ETV1/4/5 subfamily factors are in equilibrium between forms that are more or less competent for binding to DNA. The N-terminal inhibitory domain (NID) and C-terminal inhibitory domain (H4) inhibit the ETS domain (red oval) of ETV1/4/5 subfamily factors. H4 ‘pushes’ the DNA-recognition helix (H3) toward a position that is less competent for DNA binding. The NID makes direct contact with the DNA-recognition helix to sterically inhibit DNA binding, and may also reinforce the inhibitory position of H4. Acetylation of lysine residues in the NID partially relieves autoinhibition, likely by disrupting NID interactions with the ETS domain. USF1 relieves ETV4 autoinhibition (13), and we speculate that interactions with other factors, such as the DNA binding factors AR or AP1, may also regulate ETV4 DNA-binding activity.
The CID functions by influencing the position of helix H3, as supported by structural and mutational analysis. Amide HX experiments revealed that the CID helix H4 and the DNA-recognition helix H3 are both dynamic. These helices also exist in distinct conformations in crystallographic structures. Importantly, the uninhibited and the DNA-bound conformations of H3 match one another, but are distinct from the CID-inhibited conformation (45). We propose that the dynamic nature of helices H3 and H4, detected by HX measurements, reflect the sampling of these multiple conformations. Direct interactions between Ile407-Leu430 couple these two helices allowing the CID to ‘push’ H3 toward a state with lower affinity for DNA binding (Figure 10A).
Figure 10.

Autoinhibition in ETS family of transcription factors (ETS domain, red; inhibitory elements, cyan). (A) Structural and molecular elements of ETV1/4/5 subfamily autoinhibition. The CID is an α-helix, H4, that interacts with the ETS domain primarily through Leu430 (ETV4 numbering). In particular, Leu430 interacts with Ile407 to influence the positioning of H3 (right inset). The NID, cyan dotted line, is intrinsically disordered and interacts via multiple regions with H3 of the ETS domain, as well as the CID. Tyr401 and Tyr403 of H3 are required for NID-mediated autoinhibition (left inset). (B) Previous examples of structurally characterized autoinhibited ETS factors: ETS1 (7,8,12,14), ERG (43) and ETV6 (9–11). Dashed cyan lines refer to the disordered NID of ETV1/4/5 or the SRR of ETS1.
The NID is predominantly intrinsically disordered and inhibits the ETS domain through interactions with helix H3, and possibly with the CID. These interactions are weak and transient as evidenced by the small NMR spectral perturbations accompanying ligation of the NID, the lack of detectable structural changes in the NID due to the presence of the ETS domain or CID, and the contribution of multiple regions of the NID to autoinhibition. However, multiple weak (or ‘fuzzy’) interactions (52) may lead to the overall inhibitory effect of the NID on the ETS domain and/or CID of ETV4. Tyr401 and Tyr403 in H3 are critical for NID-mediated autoinhibition and these residues directly contact DNA base pairs, suggesting that the NID sterically occludes part of the DNA binding interface. The NID also influences the CID, either through direct interaction that utilizes a broad interface or indirectly through the composite of the NID-H3 and H3-CID interactions. The tyrosine residues from H3 are not conserved in all ETS domains, and the sequence and positioning of CID helix H4 is unique to the ETV1/4/5 subfamily; therefore, the putative NID-interaction interface is specific to the DBD of this subfamily (Figure 10A). Based on these collective findings, we propose that ETV1, ETV4 and ETV5 are in a dynamic equilibrium between conformations with different competencies for binding to DNA and that the NID and CID shift the equilibrium toward the less competent state (Figure 9).
The binding affinities of ETV1/4/5 fragments with either amino or carboxyl truncations suggests that the NID and CID work cooperatively, rather than additively, to inhibit DNA binding. Broadly speaking, this cooperation is supported by our structural and mutational data as the NID and the CID both impact the same location of the ETS domain, including the C-terminal portion of the DNA-recognition helix H3. In contrast to CID inhibition, there is insufficient understanding of the NID to ascertain the full basis of its inhibitory effects. Nevertheless, we speculate that in addition to a simple steric mechanism, as evidenced by direct perturbations of the DNA binding interface, the NID also reinforces the CID-driven conformation of helix H3. This is consistent with a cooperative mechanism of autoinhibition and the NID-induced perturbations of residues in both the ETS domain and the CID, detected by NMR spectroscopy.
Autoinhibition in ETS family of transcription factors
The characterization of autoinhibition in the ETV1/4/5 subfamily adds to the diversity of molecular mechanisms utilized in inhibiting DNA binding by ETS factors (Figure 10B). The DNA binding of ETS1 is allosterically inhibited by an α-helical module that flanks the ETS domain and by an IDR termed the serine-rich region (SRR). (7,8). Partial unfolding of the inhibitory module is linked to DNA binding (53,54). In contrast, a single C-terminal α-helix sterically inhibits the DNA-binding interface of ETV6 and unfolds to allow DNA binding (9–11). An α-helix and an IDR are reported to allosterically inhibit the DNA binding of ERG (43). Although these inhibitory elements show no sequence similarity (Supplementary Figure S1) and are structurally distinct between ETS factors (Figure 10), in all of these cases helix H4 interacts with a conserved hydrophobic surface on the ETS domain (Supplementary Figure S12). As this interaction is important for coupling the CID to the DNA-recognition helix H3 in ETV4, this conserved hydrophobic contact may reflect a common mechanism of inhibition among ETS factors. Beyond this conserved contact, the diversity of appended helices likely facilitates distinct intramolecular interactions with inhibitory IDRs and intermolecular interactions with unique protein partners.
Although the inhibitory domains of ETV1/4/5 are distinct from the previously characterized examples of ETS1, ETV6 and ERG, the cooperation of inhibitory elements is most reminiscent of ETS1 autoinhibition. Four α-helices flanking the ETS domain of ETS1 provide a small 2-fold level of inhibition (31), and this autoinhibition is reinforced to ∼20-fold by the SRR (8,12). As is the case for the proposed interaction between the NID and the ETS domain/CID of ETV4, the dynamic SRR also interacts transiently with both the flanking inhibitory α-helices of ETS1 and its ETS domain. Furthermore, tyrosine and phenylalanine residues, amino acids that are usually depleted within IDRs (55), are present in the SRR of ETS1 (56) and in the NID of ETV1, ETV4 and ETV5. However, in ETS1, these aromatic residues reside in a repeating Ser-(Tyr/Phe)-Asp pattern, and these repeats are key to the transient interactions that respond to signaling-induced phosphorylation and mediate inhibition. Such a repeat unit is not observed in the NID of ETV1/4/5, and mutation of aromatic residues in the NID of ETV4 did not influence autoinhibition. Therefore, although inhibitory IDRs are present in ETS1 and ETV1/4/5, these IDRs appear to modulate DNA binding via different intramolecular interactions with their corresponding ETS domains.
Divergent biological pathways and protein partnerships regulate the inhibitory elements of the ETS factors. Serine phosphorylation of ETS1, which targets the Ser-(Tyr/Phe)-Asp repeat in the SRR, enhances the DNA-binding autoinhibition (8,56). In contrast, serine phosphorylation of ETV1 does not impact autoinhibition (57). Conversely, the relief of ETV4 autoinhibition by lysine acetylation, reported here, has not been observed for ETS1, ETV6 or ERG. Similarly, disparate protein partnerships regulate the DNA-binding autoinhibition of ETS1 and ETV1/4/5. For example, RUNX1 (15,58) and PAX5 (14) counter ETS1 autoinhibition, and USF1 relieves ETV4 autoinhibition (13). The RUNX1-ETS1 partnership results in ETS1-specific regulation at ETS-RUNX composite sites in T-cells (59), suggesting that the specific regulation of DNA-binding autoinhibition for an individual ETS factor can form the basis for that factor's unique biological function.
With a mechanistic foundation now in place for ETV1/4/5 autoinhibition, potential regulatory routes may be discovered, thus, providing insight into how these factors function in prostate cancer. We identified one possible route of ETV4 DNA binding activation through acetylation of lysines in the NID. Interestingly, the expression of p300, one of the acetyltransferases that modifies ETV1/4/5 factors (49,50), correlates with prostate cancer progression and high levels of p300 are prognostic of biochemical recurrence in prostate cancer patients (60,61). Additionally, protein-protein partnerships may regulate the DNA binding of ETV1/4/5 factors. Investigating the effect on ETV1/4/5 DNA-binding autoinhibition by other transcription factors that bind to proximal genomic sites and act in the prostate will be of particular interest. Besides USF1 (13), candidates include the AP1 factors (44) and the androgen receptor (22,62–64). Finally, inhibitory IDRs, and the signaling pathways that regulate them, are a potential therapeutic target as they differ between ETS factors and could provide factor-specific interventions. Despite the difficulty in rationally inhibiting IDRs (65), recent successes suggest that IDRs are a tractable small-molecule target (66–69).
Autoinhibition as a route to transcription factor specificity
Many transcription factors are encoded by gene families and share a conserved DBD (70). Disparate roles in development and disease indicate that individual transcription factors carry out specific functions and are not completely redundant with all other factors from the same family (6,71). ETS factors, as a prototype for investigating this conundrum, have provided insight into how such specificity could have evolved. Previous work had established that ETS factors have distinct inhibitory domains that are specific to an individual factor, or subfamily of factors (7–10,43). Cellular triggers can specifically regulate an individual ETS factor by integrating different signaling pathways or protein partnerships into these distinctive inhibitory features (14–16,58,59). We have extended this knowledge by describing an additional inhibitory module in the ETV1/4/5 subfamily with a distinct mechanism of inhibition and mode of cellular regulation. The divergent inhibitory domains of the four subclasses of ETS factors studied to date contact a conserved region on the ETS DBD. Thus, subfamily-specific α-helices that flank the ETS domain serve as ‘adapters’ that generate unique intra- and intermolecular interaction surfaces. Subfamily-specific IDRs interact with these surfaces to inhibit DNA binding via steric and/or allosteric mechanisms. These unstructured regions also provide diverse sites of post-translational modification that can inhibit or activate a factor in response to cellular regulation. Thus, the modest sequence variability among related DBDs could have been leveraged during evolution to enhance biological specificity by the coordinated divergence of structured inhibitory regions that flank the DBD and more distal inhibitory IDRs.
ACCESSION NUMBERS
Structural coordinates have been deposited in the RCSB Protein Data Bank under ID codes 5ILS (ETV1), 5ILU (ETV4) and 5ILV (ETV5).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Chris Hill and Dr Michael Kay for access to, and helpful discussions on, X-ray crystallography and circular dichroism, respectively. We acknowledge Dr Jason Chin, Dr Pierre Barraud and Dr Michael Sattler for providing plasmids for incorporating acetyllysine residues, intein ligation and sortase ligation, respectively. We thank Niraja Bhachech for assistance with protein purification and members of the Graves and McIntosh labs for helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM38663 to B.J.G.]; Canadian Cancer Society Research Institute (CCSRI) [2011–700772 to L.P.M.]; Canadian Institutes of Health Research (CIHR) [MOP-136834 to L.P.M.]; CIHR; Canada Foundation for Innovation; British Columbia Knowledge Development Fund; UBC Blusson Fund; Michael Smith Foundation for Health Research; Huntsman Cancer Institute/Huntsman Cancer Foundation and the Howard Hughes Medical Institute (to B.J.G.); National Cancer Institute, National Institutes of Health [P30CA042014 to Huntsman Cancer Institute]; National Institute of General Medical Sciences, National Institutes of Health [P50 GM082545 to Chris Hill and Wes Sundquist]; U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences [DE-AC02-76SF00515]; DOE Office of Biological and Environmental Research; National Institutes of Health, National Institute of General Medical Sciences [P41GM103393]. Funding for open access charge: National Institutes of Health [R01GM38663].
Conflict of interest statement. None declared.
REFERENCES
- 1.Pufall M.A., Graves B.J.. Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 2002; 18:421–462. [DOI] [PubMed] [Google Scholar]
- 2.Kim A.S., Kakalis L.T., Abdul-Manan N., Liu G.A., Rosen M.K.. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. 2000; 404:151–158. [DOI] [PubMed] [Google Scholar]
- 3.Morreale A., Venkatesan M., Mott H.R., Owen D., Nietlispach D., Lowe P.N., Laue E.D.. Structure of Cdc42 bound to the GTPase binding domain of PAK. Nat. Struct. Biol. 2000; 7:384–388. [DOI] [PubMed] [Google Scholar]
- 4.Trudeau T., Nassar R., Cumberworth A., Wong E.T., Woollard G., Gsponer J.. Structure and intrinsic disorder in protein autoinhibition. Structure. 2013; 21:332–341. [DOI] [PubMed] [Google Scholar]
- 5.Wright P.E., Dyson H.J.. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015; 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollenhorst P.C., McIntosh L.P., Graves B.J.. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011; 80:437–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee G.M., Donaldson L.W., Pufall M.A., Kang H.S., Pot I., Graves B.J., McIntosh L.P.. The structural and dynamic basis of Ets-1 DNA binding autoinhibition. J. Biol. Chem. 2005; 280:7088–7099. [DOI] [PubMed] [Google Scholar]
- 8.Pufall M.A., Lee G.M., Nelson M.L., Kang H.S., Velyvis A., Kay L.E., McIntosh L.P., Graves B.J.. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005; 309:142–145. [DOI] [PubMed] [Google Scholar]
- 9.Green S.M., Coyne H.J. 3rd, McIntosh L.P., Graves B.J.. DNA binding by the ETS protein TEL (ETV6) is regulated by autoinhibition and self-association. J. Biol. Chem. 2010; 285:18496–18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coyne H.J. 3rd, De S., Okon M., Green S.M., Bhachech N., Graves B.J., McIntosh L.P.. Autoinhibition of ETV6 (TEL) DNA binding: appended helices sterically block the ETS domain. J. Mol. Biol. 2012; 421:67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De S., Chan A.C., Coyne H.J. 3rd, Bhachech N., Hermsdorf U., Okon M., Murphy M.E., Graves B.J., McIntosh L.P.. Steric mechanism of auto-inhibitory regulation of specific and non-specific DNA binding by the ETS transcriptional repressor ETV6. J. Mol. Biol. 2014; 426:1390–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee G.M., Pufall M.A., Meeker C.A., Kang H.S., Graves B.J., McIntosh L.P.. The affinity of Ets-1 for DNA is modulated by phosphorylation through transient interactions of an unstructured region. J. Mol. Biol. 2008; 382:1014–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenall A., Willingham N., Cheung E., Boam D.S., Sharrocks A.D.. DNA binding by the ETS-domain transcription factor PEA3 is regulated by intramolecular and intermolecular protein.protein interactions. J. Biol. Chem. 2001; 276:16207–16215. [DOI] [PubMed] [Google Scholar]
- 14.Garvie C.W., Pufall M.A., Graves B.J., Wolberger C.. Structural analysis of the autoinhibition of Ets-1 and its role in protein partnerships. J. Biol. Chem. 2002; 277:45529–45536. [DOI] [PubMed] [Google Scholar]
- 15.Shrivastava T., Mino K., Babayeva N.D., Baranovskaya O.I., Rizzino A., Tahirov T.H.. Structural basis of Ets1 activation by Runx1. Leukemia. 2014; 28:2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiina M., Hamada K., Inoue-Bungo T., Shimamura M., Uchiyama A., Baba S., Sato K., Yamamoto M., Ogata K.. A novel allosteric mechanism on protein-DNA interactions underlying the phosphorylation-dependent regulation of Ets1 target gene expressions. J. Mol. Biol. 2015; 427:1655–1669. [DOI] [PubMed] [Google Scholar]
- 17.Tomlins S.A., Rhodes D.R., Perner S., Dhanasekaran S.M., Mehra R., Sun X.W., Varambally S., Cao X., Tchinda J., Kuefer R. et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005; 310:644–648. [DOI] [PubMed] [Google Scholar]
- 18.Tomlins S.A., Mehra R., Rhodes D.R., Smith L.R., Roulston D., Helgeson B.E., Cao X., Wei J.T., Rubin M.A., Shah R.B. et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006; 66:3396–3400. [DOI] [PubMed] [Google Scholar]
- 19.Helgeson B.E., Tomlins S.A., Shah N., Laxman B., Cao Q., Prensner J.R., Cao X., Singla N., Montie J.E., Varambally S. et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008; 68:73–80. [DOI] [PubMed] [Google Scholar]
- 20.Tomlins S.A., Laxman B., Dhanasekaran S.M., Helgeson B.E., Cao X., Morris D.S., Menon A., Jing X., Cao Q., Han B. et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007; 448:595–599. [DOI] [PubMed] [Google Scholar]
- 21.Aytes A., Mitrofanova A., Kinkade C.W., Lefebvre C., Lei M., Phelan V., LeKaye H.C., Koutcher J.A., Cardiff R.D., Califano A. et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E3506–E3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baena E., Shao Z., Linn D.E., Glass K., Hamblen M.J., Fujiwara Y., Kim J., Nguyen M., Zhang X., Godinho F.J. et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013; 27:683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laget M.P., Defossez P.A., Albagli O., Baert J.L., Dewitte F., Stehelin D., de Launoit Y.. Two functionally distinct domains responsible for transactivation by the Ets family member ERM. Oncogene. 1996; 12:1325–1336. [PubMed] [Google Scholar]
- 24.Bojovic B.B., Hassell J.A.. The PEA3 Ets transcription factor comprises multiple domains that regulate transactivation and DNA binding. J. Biol. Chem. 2001; 276:4509–4521. [DOI] [PubMed] [Google Scholar]
- 25.Li M.Z., Elledge S.J.. SLIC: a method for sequence- and ligation-independent cloning. Methods Mol. Biol. 2012; 852:51–59. [DOI] [PubMed] [Google Scholar]
- 26.Neumann H., Peak-Chew S.Y., Chin J.W.. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008; 4:232–234. [DOI] [PubMed] [Google Scholar]
- 27.Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005; 41:207–234. [DOI] [PubMed] [Google Scholar]
- 28.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A.. Walker JM. Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook, Humana Press. 2005; Totowa: Humana Press; 571–607. [Google Scholar]
- 29.Barraud P., Allain F.H.. Solution structure of the two RNA recognition motifs of hnRNP A1 using segmental isotope labeling: how the relative orientation between RRMs influences the nucleic acid binding topology. J. Biomol. NMR. 2013; 55:119–138. [DOI] [PubMed] [Google Scholar]
- 30.Freiburger L., Sonntag M., Hennig J., Li J., Zou P., Sattler M.. Efficient segmental isotope labeling of multi-domain proteins using Sortase A. J. Biomol. NMR. 2015; 63:1–8. [DOI] [PubMed] [Google Scholar]
- 31.Jonsen M.D., Petersen J.M., Xu Q.P., Graves B.J.. Characterization of the cooperative function of inhibitory sequences in Ets-1. Mol. Cell. Biol. 1996; 16:2065–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z., Minor W.. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 33.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenfield N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006; 1:2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 39.Goddard T.D., Kneller D.G.. Sparky. 1999; 3rd edn, University of California: San Francisco. [Google Scholar]
- 40.Sattler M., Schleucher J., Griesinger C.. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Mag. Reson. Spect. 1999; 34:93–158. [Google Scholar]
- 41.Hwang T.L., van Zijl P.C., Mori S.. Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR. 1998; 11:221–226. [DOI] [PubMed] [Google Scholar]
- 42.Bai Y., Milne J.S., Mayne L., Englander S.W.. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993; 17:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Regan M.C., Horanyi P.S., Pryor E.E. Jr, Sarver J.L., Cafiso D.S., Bushweller J.H.. Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13374–13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hollenhorst P.C., Ferris M.W., Hull M.A., Chae H., Kim S., Graves B.J.. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011; 25:2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooper C.D., Newman J.A., Aitkenhead H., Allerston C.K., Gileadi O.. Structures of the Ets protein DNA-binding domains of transcription factors Etv1, Etv4, Etv5, and Fev: determinants of DNA binding and redox regulation by disulfide bond formation. J. Biol. Chem. 2015; 290:13692–13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hafsa N.E., Wishart D.S.. CSI 2.0: a significantly improved version of the Chemical Shift Index. J. Biomol. NMR. 2014; 60:131–146. [DOI] [PubMed] [Google Scholar]
- 47.Camilloni C., De Simone A., Vranken W.F., Vendruscolo M.. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry. 2012; 51:2224–2231. [DOI] [PubMed] [Google Scholar]
- 48.Dyson H.J., Wright P.E.. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002; 12:54–60. [DOI] [PubMed] [Google Scholar]
- 49.Goel A., Janknecht R.. Acetylation-mediated transcriptional activation of the ETS protein ER81 by p300, P/CAF, and HER2/Neu. Mol. Cell. Biol. 2003; 23:6243–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo B., Panagiotaki N., Warwood S., Sharrocks A.D.. Dynamic modification of the ETS transcription factor PEA3 by sumoylation and p300-mediated acetylation. Nucleic Acids Res. 2011; 39:6403–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim H.J., Kim S.H., Yu E.J., Seo W.Y., Kim J.H.. A positive role of DBC1 in PEA3-mediated progression of estrogen receptor-negative breast cancer. Oncogene. 2015; 34:4500–4508. [DOI] [PubMed] [Google Scholar]
- 52.Fuxreiter M. Fuzziness: linking regulation to protein dynamics. Mol. Biosyst. 2012; 8:168–177. [DOI] [PubMed] [Google Scholar]
- 53.Petersen J.M., Skalicky J.J., Donaldson L.W., McIntosh L.P., Alber T., Graves B.J.. Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an alpha helix. Science. 1995; 269:1866–1869. [DOI] [PubMed] [Google Scholar]
- 54.Desjardins G., Okon M., Graves B.J., McIntosh L.P.. Conformational dynamics and the binding of specific and nonspecific DNA by the autoinhibited transcription factor Ets-1. Biochemistry. 2016; 55:4105–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams R.M., Obradovi Z., Mathura V., Braun W., Garner E.C., Young J., Takayama S., Brown C.J., Dunker A.K.. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001; 6:89–100. [DOI] [PubMed] [Google Scholar]
- 56.Desjardins G., Meeker C.A., Bhachech N., Currie S.L., Okon M., Graves B.J., McIntosh L.P.. Synergy of aromatic residues and phosphoserines within the intrinsically disordered DNA-binding inhibitory elements of the Ets-1 transcription factor. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:11019–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu J., Janknecht R.. Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J. Biol. Chem. 2002; 277:42669–42679. [DOI] [PubMed] [Google Scholar]
- 58.Goetz T.L., Gu T.L., Speck N.A., Graves B.J.. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor alpha2. Mol. Cell. Biol. 2000; 20:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hollenhorst P.C., Shah A.A., Hopkins C., Graves B.J.. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 2007; 21:1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Debes J.D., Sebo T.J., Lohse C.M., Murphy L.M., Haugen D.A., Tindall D.J.. p300 in prostate cancer proliferation and progression. Cancer Res. 2003; 63:7638–7640. [PubMed] [Google Scholar]
- 61.Isharwal S., Miller M.C., Marlow C., Makarov D.V., Partin A.W., Veltri R.W.. p300 (histone acetyltransferase) biomarker predicts prostate cancer biochemical recurrence and correlates with changes in epithelia nuclear size and shape. Prostate. 2008; 68:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Massie C.E., Adryan B., Barbosa-Morais N.L., Lynch A.G., Tran M.G., Neal D.E., Mills I.G.. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007; 8:871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Y., Chi P., Rockowitz S., Iaquinta P.J., Shamu T., Shukla S., Gao D., Sirota I., Carver B.S., Wongvipat J. et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med. 2013; 19:1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shin S., Kim T.D., Jin F., van Deursen J.M., Dehm S.M., Tindall D.J., Grande J.P., Munz J.M., Vasmatzis G., Janknecht R.. Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81. Cancer Res. 2009; 69:8102–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y., Cao H., Liu Z.. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. 2015; 24:688–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hammoudeh D.I., Follis A.V., Prochownik E.V., Metallo S.J.. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009; 131:7390–7401. [DOI] [PubMed] [Google Scholar]
- 67.Krishnan N., Koveal D., Miller D.H., Xue B., Akshinthala S.D., Kragelj J., Jensen M.R., Gauss C.M., Page R., Blackledge M. et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014; 10:558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pop M.S., Stransky N., Garvie C.W., Theurillat J.P., Hartman E.C., Lewis T.A., Zhong C., Culyba E.K., Lin F., Daniels D.S. et al. A small molecule that binds and inhibits the ETV1 transcription factor oncoprotein. Mol. Cancer Ther. 2014; 13:1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z., Boskovic Z., Hussain M.M., Hu W., Inouye C., Kim H.J., Abole A.K., Doud M.K., Lewis T.A., Koehler A.N. et al. Chemical perturbation of an intrinsically disordered region of TFIID distinguishes two modes of transcription initiation. Elife. 2015; 4:e07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vaquerizas J.M., Kummerfeld S.K., Teichmann S.A., Luscombe N.M.. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 2009; 10:252–263. [DOI] [PubMed] [Google Scholar]
- 71.Rezsohazy R., Saurin A.J., Maurel-Zaffran C., Graba Y.. Cellular and molecular insights into Hox protein action. Development. 2015; 142:1212–1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.