

Abstract
In response to myocardial infarction (MI), the wound healing response of the left ventricle (LV) consists of overlapping inflammatory, proliferative, and maturation phases; and the cardiac fibroblast is a key cell type involved in each phase. It has recently been appreciated that early post-MI, fibroblasts transform to a pro-inflammatory phenotype and secrete cytokines and chemokines as well as matrix metalloproteinases. Later post-MI, fibroblasts are activated to anti-inflammatory and pro-reparative phenotypes and generate anti-inflammatory and pro-angiogenic factors and extracellular matrix components that form the infarct scar. Additional studies are needed to systematically examine how fibroblast activation shifts over the time frame of MI response and how modulation at different activation stages could alter wound healing and LV remodeling in distinct ways. This review will summarize current fibroblast knowledge as a foundation to discuss existing knowledge gaps.
Keywords: computational models, extracellular matrix, fibroblast, inflammation, myocardial infarction, omics, big data
The cardiac fibroblast is a major player in cardiac remodeling
Almost 6 million Americans currently suffer from heart failure, and the high 5-year mortality rate of 50% is accompanied by an annual health care cost of >$34 billion [1]. In 70% of heart failure cases, myocardial infarction (MI, see Glossary) is the underlying etiology. Elucidating the molecular mechanisms whereby the post-MI left ventricle (LV) transitions to heart failure will help identify novel biomarkers and targets to prevent adverse remodeling and progression to heart failure.
Post-MI, an appropriate degree of fibrosis in the infarct zone is necessary to replace necrotic cardiomyocytes. Fibrosis occurs when there is a net increase in the rate of synthesis of the extracellular matrix (ECM). Insufficient fibrosis can lead to overabundant LV wall thinning; excessive fibrosis can lead to increased LV wall stiffness and decreased mechano-electric coupling to adversely impair cardiac physiology. The presence of abundant ECM may also decrease oxygen and nutrient availability to the myocardium, amplifying the pathological remodeling response [2].
The cardiac fibroblast is a key cellular component of post-MI LV remodeling, due to its location, numbers, and ECM scar generating capabilities. Cardiac fibroblasts are generally considered to have two distinctive phenotypic states: inactivated and activated (transdifferentiated to myofibroblasts). However, it is also possible that transient intermediary states also exist. In this review, we summarize the current literature to provide evidence that there likely exists a continuum of post-MI fibroblast activation profiles that range from pro-inflammatory and ECM-degrading to anti-inflammatory and ECM-building phenotypes. Exploring the relative contributions of each fibroblast subpopulation will provide insight into both mechanisms of LV remodeling as well as novel targets to inhibit or promote certain aspects of fibroblast physiology. Understanding how signals modulate fibroblast activation from the initial ischemic challenge through the scar formation period and beyond scar maturation will provide a foundation for future research. The post-MI remodeling literature is vast, yet few teams have attempted to consolidate results to focus comprehensively on the fibroblast at a systems biology level. Here, we summarize the cardiac fibroblast literature and highlight three knowledge gaps that remain in our understanding.
Knowledge gap 1. Cardiac fibroblast post-MI phenotypes have not been mapped
Fibroblast Origins
The first step to understand fibroblast roles is to understand their source and activation status. Cardiac fibroblasts have different sources in basal and stressed conditions. During development, the majority of cardiac fibroblasts originate from epicardial cells that undergo an epithelial-to-mesenchymal transition (EMT) [3, 4]. A minority of fibroblasts derive from endothelial cells or paired box 3-expressing cells [5]. In an unstressed adult heart, replacement of senescent and apoptotic fibroblasts is believed to occur through proliferation of existing resident fibroblasts. Following MI injury, cardiac myofibroblasts derive from multiple origins, including proliferation of resident cardiac fibroblasts, bone marrow-derived progenitors, epicardial cells, and endothelial cells [6, 7]. Although proliferation of the resident cardiac fibroblast is considered as the main source of myofibroblasts in the infarct, other sources contribute [8]. Bone marrow-derived myofibroblasts have been reported to contribute up to 24% of all myofibroblasts in the infarct area [9]. These myofibroblasts derived from bone marrow produce collagen and are involved in scar formation post-MI [9].
While epicardial cells are quiescent in the normal adult heart, MI can induce epicardial cell proliferation and stimulate their activation to form epicardium-derived cells (EPDCs) [10, 11]. EPDCs then differentiate into mesenchymal cells expressing fibroblast cell markers, including procollagen I, collagen III, fibronectin, and α-smooth muscle actin (SMA), suggesting that EPDCs differentiate to fibroblasts. Importantly, these EPDCs have been shown to reduce infarct size, facilitate angiogenesis, and preserve cardiac physiology by secreting paracrine factors [10].
Endothelial to mesenchymal transition (EndMT) is another potential source of myofibroblasts following MI. EndMT-derived fibroblasts have been reported to account for <1% of the total number of cells in the unstressed heart, but this number expands to about 25% in the ischemic heart [12]. Due to the limitations of current markers of fibroblasts and myofibroblasts, quantifications of source contributions can vary depending on the markers used. Fibrocytes, pericytes and monocyte precursors are reported to be potential sources of fibroblasts [7, 13, 14]. Whether these cells contribute to post-MI fibroblast population needs to be studied further.
Ali and colleagues have recently demonstrated that following pressure overload, there is a heterogeneous population of mouse cardiac fibroblasts that are derived from epicardium, endothelial cells, and paired box 3-expressing cells [5]. Of interest, regardless of cell source, the fibroblasts exhibit comparable proliferation and activation patterns, indicating the present environment dictates fibroblast phenotype. Bone marrow-derived or circulating cells do not appear to provide a source for fibroblasts in the pressure overload setting [5]. In line with the above findings, Moore-Morris and colleagues showed that fibroblasts do not derive from epicardial EMT, EndoMT, or hematopoietic cells in mouse pressure overload models [15]. Instead, pressure overload induces proliferation and activation of two resident fibroblast lineages, namely an epicardial-derived population and an endothelial-derived population. Similar studies are needed in the MI model, which is a much different pathophysiological model than pressure overload.
Cardiac fibrosis in mice with genetic hypertrophic cardiomyopathy (α-MHC719/+) is mediated by non-myocyte proliferation, which requires transforming growth factor (TGF)-β signaling [16]. Although the authors did not define exactly what the non-myocytes were, they are likely myofibroblasts. Hypertension or ischemia/reperfusion insults lead to differences in the expression of embryonic epicardial progenitor markers (e.g. Tcf21, Wt1, and Tb×18) and the localization of cardiac fibrosis [17]. Following ischemia/reperfusion, epicardial fibrosis is obvious in the thickened layer of subepicardial cells that express Tcf21, Wt1, and Tb×18. In contrast, perivascular fibrosis is dominant in hypertensive models with upregulated Tcf21 expression. The region of interstitial fibrosis in ischemia/reperfused and hypertensive hearts is similar, expressing Tcf21, Wt1, and Tb×18.
Fibroblast Markers
In spite of recent progress in the identification of multiple cardiac fibroblast markers, specificity still remains a key challenge [18, 19]. Table 1 provides a comprehensive list of cardiac fibroblast markers identified to date [18–22]. Unfortunately, all of these markers are also expressed by at least one other cell type. There remains a need to identify more selective fibroblast markers or identify the combination of markers to specifically define fibroblasts. It is also unclear whether cardiac fibroblasts express markers that can distinguish them from fibroblasts in other tissues.
Table 1.
| Markers | Location | Expressed by other cell type | Overexpressed in activated fibroblasts |
|---|---|---|---|
| α-smooth muscle actin | Intracellular | VSMCs, pericytes | Yes |
| Cadherin-9 | Cell surface | Neurons, tumor vasculature | Yes |
| CD248 (endosialin) | Cell surface | Endothelial cells, pericytes | Unknown |
| Collagen I (α1 and α2) | Secreted | VSMCs, epicardial cells | Yes |
| Connective tissue growth factor (CCN2) | Secreted | Hepatic stellate cells, epithelial cells | Yes |
| Discoidin domain receptor 2 | Cell surface | VSMCs, endothelial cells | Unknown |
| Fibronectin, ED-A splice variant | Secreted | Macrophages, endothelial cells | Yes |
| Embryonic smooth muscle myosin | Intracellular | Tumor cells, interstitial cells of Cajal | Yes |
| Fibroblast-activation protein-1 | Cell surface | Activated melanocytes | Yes |
| Fibroblast surface antigen | Cell surface | Monocytes/macrophages | Yes |
| Fibroblast specific protein-1 (S100A4) | Intracellular | VSMCs, leukocytes, cancer cells | Yes |
| Periostin | Secreted | VSMCs | Yes |
| Prolyl-4-hydroxylase | Intracellular | Endothelial cells, epithelial cells | Unknown |
| Platelet derived growth factor receptor α | Cell surface | Platelets | Yes |
| Scleraxis | Intracellular | A wide range of cell types | Unknown |
| Tcf21 (epicardin, Pod1, capsulin) | Intracellular | A wide range of cell types | Unknown |
| Thymus cell antigen-1 (CD90) | Cell surface | Leukocytes, endothelial cells, various progenitor cells | Unknown |
| Vimentin | Intracellular | Endothelial cells, VSMCs | Unknown |
VSMCs- vascular smooth muscle cells
The most commonly used marker to identify activated myofibroblasts is α-SMA, which is also highly abundant in vascular smooth muscle cells (VSMCs) and pericytes [23]. Vimentin is an intermediate filament protein and is another widely used fibroblast marker. Vimentin also labels endothelial cells and VSMCs [24]. Discoidin domain receptor (DDR) 2 represents a novel family of collagen-specific receptor tyrosine kinases. In addition to fibroblasts, DDR2 is also expressed in VSMCs and activated endothelial cells [25]. Fibroblast specific protein (FSP)-1 is a filament-associated calcium-binding protein involved in cell mobilization. Despite its name, FSP-1 is widely expressed in fibroblasts, leukocytes, and a number of cancer cell types [26]. Fibroblast activation protein (FAP)-1 is a homodimeric integral membrane gelatinase that belongs to the serine protease family. While FAP-1 has been used to distinguish fibroblasts from other cell types, it is present in activated melanocytes; thus FAP-1 can be considered fibroblast specific in the myocardium but not across the whole body [27]. Other proteins that have been used to identify cardiac fibroblasts or myofibroblasts include cadherin-9, CD248 (endosialin), collagen1α1, collagen1α2, connective tissue growth factor (CTGF, CCN2), ED-A splice variant of fibronectin, embryonic smooth muscle myosin, fibroblast surface antigen (FSA), prolyl-4-hydroxylase, scleraxis, and thymus cell antigen-1 (Thy-1, CD90) [18, 28–30]. While markers have been evaluated at the binary level (present or absent), whether marker expression changes in fibroblasts isolated from different in vivo stimulation conditions or times has not been evaluated.
Genetic lineage-tracing techniques have recently been used to identify and characterize fibroblast origins and fates. Commonly used markers include platelet derived growth factor receptor (PDGFR) α, collagen1α1, Tcf21, and periostin. PDGFRs are cell surface tyrosine kinase receptors and consist of PDGF -α and -β. PDGFR α signaling is required for epicardial EMT and the development of EPDCs and resident fibroblasts [20, 31]. A transgenic collagen1α1-green fluorescent protein reporter line has been developed to track collagen 1-positive cells [15, 20]. Tcf21, also known as epicardin/Pod1/capsulin, is a basic helix-loop-helix transcription factor. Acharya and colleagues have shown that Tcf21−/− hearts lack cardiac fibroblasts, indicating that Tcf21 is required for cardiac fibroblast cell fate determination [21]. Periostin is a secreted matricellular protein implicated in cellular adhesion and collagen organization. The periostin promoter is increasingly being used to target cardiac myofibroblasts in mice. Using lineage tracing analysis, Kanisicak and colleagues recently reported that the periostin genetic locus (PostnMCM) can be used to identify almost all subtypes of cardiac myofibroblasts [22]. Further, they demonstrated that periostin-expressing myofibroblasts in the heart were derived from resident Tcf21+ fibroblasts [22]. The role of this fibroblast cell subtype remains to be evaluated in the post-MI LV.
Knowledge gap 2. Post-MI cardiac fibroblast phenotypes have not been linked to cell physiology
Fibroblast Phenotypes
While the cardiac fibroblast is generically defined as a resident cell and the myofibroblast as a post-injury cell type, these definitions are loosely based on α-SMA expression. For the purpose of this review, we use the term fibroblast to encapsulate all fibroblast types that secrete ECM, including resident cardiac fibroblasts, proto-myofibroblasts, and myofibroblasts from any source, as well as fibroblast progenitor cells and fibrocytes. Figure 1 illustrates the proposed temporal evolution of post-MI fibroblast phenotypes. Post-MI, an overarching role of the myofibroblast is to synthesize ECM and actively participate in scar formation and deposition. In the mouse permanent occlusion MI model, myofibroblasts are activated within three days post-MI [32–36]. In terms of effects on fibroblast cell physiology, it is likely that post-MI there is an initial increase in migration and proliferation rates, followed by later cell cycle withdrawal, cessation of movement, and an increase in ECM production.
Figure 1. Temporal fibroblast phenotypes post-myocardial infarction (MI).
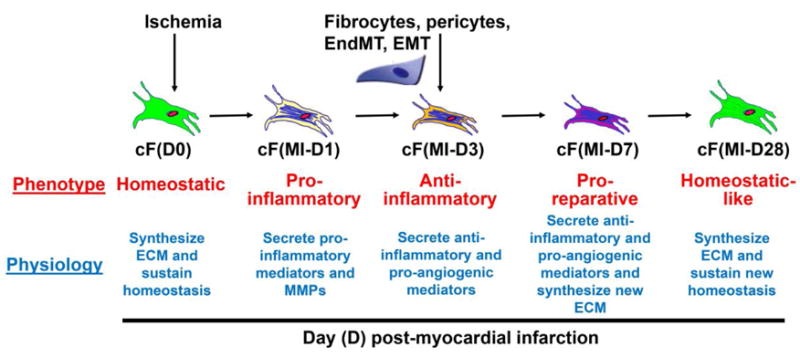
We propose that cardiac fibroblasts (cFs) exhibit distinct phenotypes at different time points post-MI. At day (D) 0 (basal condition), resident fibroblasts synthesize extracellular matrix (ECM) to maintain homeostasis. At day 1 post-MI, fibroblasts exhibit a pro-inflammatory subtype, secreting pro-inflammatory mediators and matrix metalloproteinases. At day 3, fibroblasts produce anti-inflammatory and pro-angiogenic mediators to facilitate the formation of granulation tissue. At day 7, fibroblasts are pro-reparative, generating anti-inflammatory and pro-angiogenic mediators and synthesizing new ECM. At day 28, fibroblasts return to a homeostatic-like phenotype to maintain post-reparative homeostasis. The different colors of the cells depict differential fibroblast phenotypes at each time point post-MI.
Inflammatory Phase
Emerging evidence indicate that fibroblasts exhibit different phenotypes and cell physiology at varying times post-MI [37–39]. Following MI, ischemia itself, as well as the increased inflammatory molecules released by dead cardiomyocytes both can activate fibroblasts to generate and release pro-inflammatory mediators and matrix metalloproteinases (MMPs) [37]. Interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and oncostatin-M have been shown to induce fibroblasts to an inflammatory phenotype, evidenced by secretion of cytokines and chemokines [40]. Reactive oxygen species stimulate an inflammatory response in fibroblasts as well [41–43]. IL-1β levels in the infarct area increase by 16-fold at day 1 post-MI, which is associated with enhanced migration and reduced proliferation of fibroblasts [44–46]. TNF-α is elevated in rat hearts after acute MI and is associated with increased cardiac fibroblast proliferation [47, 48]. In vitro, TNF-α induces fibroblast production of IL-1α, IL-1β, and IL-6 [49]. TNF-α also increases fibroblast migration but to a lesser extent compared to IL-1β [45]. Turner and colleagues have demonstrated that IL-1α can stimulate human fibroblasts to produce a wide range of pro-inflammatory cytokines (e.g. IL-1β, IL-6, and TNF-α) and chemokines (e.g. CXCL-1, 2, 5, 8) [40]. While the in vitro evidence is strong, in vivo studies remain to be performed to demonstrate the relative contribution of fibroblasts to the inflammatory response in the infarcted LV.
Proliferative Phase
The post-MI proliferative phase is characterized by the formation of granulation tissue composed of macrophages, myofibroblasts, new blood vessels, and newly synthesized ECM [50]. During this phase, fibroblasts are activated to myofibroblasts and express high concentrations of α-SMA, making them highly contractile. TGF-β1 is a major cytokine that induces transdifferentiation of fibroblasts into myofibroblasts and generation of ECM proteins. Smad2/3 forms part of downstream signaling pathway of TGF-β1. Smad3 deletion reduces collagen deposition in the infarcted heart, indicating an impact of TGF-β1/Smad signaling on myofibroblasts [51]. In fibroblasts isolated from lung, monocyte chemotactic protein (MCP)-1 was found to be involved in collagen secretion [52]. MCP-1 deficiency results in attenuated myofibroblast infiltration in the infarct [53, 54]. However the direct impact of MCP-1 on fibroblast activation remains unknown, as in vitro stimulation experiments have not been performed. While cytokines (e.g. IL-1 and cardiotrophin-1) and growth factors (e.g. TGF-β1 and fibroblast growth factor) can stimulate fibroblast migration into the infarct [45, 51, 55], the direct effects of these pro-fibrotic factors on scar formation after MI remain poorly defined.
In addition, MI induces changes in LV mechanical tension, which can activate fibroblasts to produce abundant ECM components [56–58]. Of note, the transcription factor scleraxis induces a myofibroblast phenotype that mimics the effects of stretch [59]. Moreover, the effect of stretch is attenuated in scleraxis null fibroblasts, indicating that scleraxis mediates stretch-induced fibroblast phenotypic conversion to myofibroblasts. Transient receptor potential canonical (TRPC) channels control Ca2+ influx in response to various stimuli, including mechanical signals and oxidative stress. TRPC3 triggers myofibroblast differentiation in atrial and renal fibroblasts through a nuclear factor of activated T cells- and extracellular signal-regulated kinases 1/2-dependent manner [60, 61]. Myocardin-related transcription factors (MRTFs), including MRTF-A (also known as MAL/MKL1/BSAC) and MRTF-B (MKL2), are ubiquitously expressed transcription factors. Following polymerization of filamentous-actin, MRTFs bind to transcription factor serum response factor (SRF) and induce α-SMA expression in vascular smooth muscle cells [62]. MRTFs have also been shown to modulate fibroblast phenotype. MRTF-A overexpression in fibroblasts or epithelial cells is sufficient to elicit a phenotypic switch to migratory and contractile myofibroblasts, an effect that can be blocked by pharmacological inhibition of MRTF-A signaling [63, 64]. However, whether these mechanical tension and signals are involved in post-MI fibroblast activation remains to be investigated.
Maturation phase
The post-MI maturation phase is characterized by scar formation and is accompanied by the increased production of anti-inflammatory cytokines (e.g. IL-10) and profibrotic factors (e.g. TGF-β1) [65]. In this phase, inflammation has receded, and myofibroblasts are the main players [66, 67]. Angiotensin II can increase fibroblast proliferation and upregulate the expression of collagen, fibronectin, and integrins [68]. Our group has recently shown that ECM fragments called matrikines (matricryptins) can regulate fibroblast physiology, thus affecting cardiac repair post-MI [69]. It has been shown that myofibroblasts can remain active in the infarct region, even up to 18 years post-MI in humans [70, 71]. Therefore, how long the maturation phase lasts is highly variable at the individual level, and the continual persistence of fibroblasts post-MI is likely required to maintain the new homeostasis.
Several potential key drivers of fibroblast activation post-MI include IL-1α/β, TGF-β1, collagen, fibronectin, osteopontin, thrombospondin-1, and secreted protein acidic and rich in cysteine (SPARC) as well as mechanical signals (e.g. scleraxis, TRPC, and MRTF/SRF, Table 2). However, the complete list of factors that involve fibroblast activation post-MI still need to be identified and the importance of individual factors ranked. The need to identify makers to distinguish fibroblast physiological phenotypes during early inflammatory and scar formation responses represents an important existing knowledge gap. The phenotypes of cardiac fibroblasts across the spectrum, from time 0 before MI (day 0) through day 28 post-MI (and longer) have not been mapped in detail. Figure 2 illustrates the construction of a fibroblast signaling module, with signaling pathways beginning at the extracellular-cell membrane interface and ending with effects on fibroblast cell and LV tissue physiology. A work flow that seamlessly and synergistically integrates knowledge on the varying phenotypes will enable knowledge gaps 1 and 2 to be filled. Using big data approaches will help to harness this attempt by exploring which of the many variables that can be measured need to be included to define the phenotype [72].
Table 2.
Potential key drivers of fibroblast activation post-MI [83].
| Cytokines and growth factors | Interleukin-1α/β, Transforming growth factor-ß1, connective tissue growth factor, angiotensin II |
| ECM | Collagen, fibronectin |
| Matricellular proteins | Osteopontin, periostin, tenascin-C, thrombospondin 1, secreted protein acidic and rich in cysteine |
| Mechanical signals | Scleraxis, transient receptor potential canonical channels, myocardin-related transcription factors, serum response factor |
Figure 2. Fibroblast signaling module.
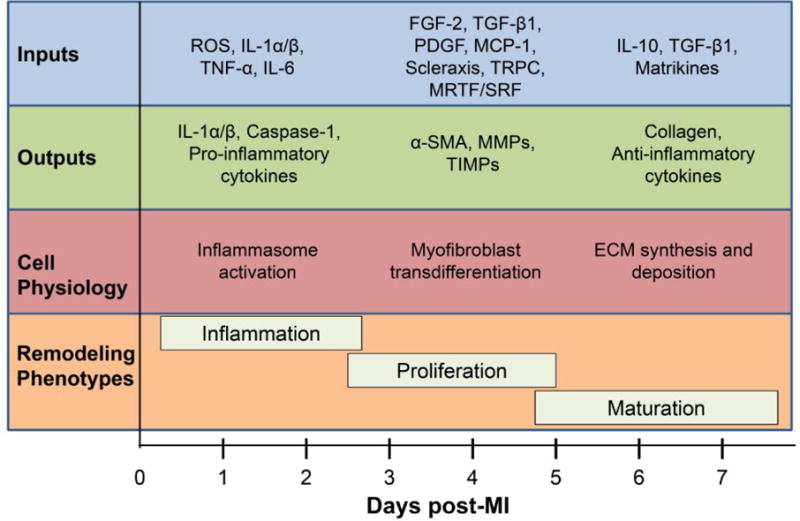
The signaling pathways begin at the extracellular-cell membrane interface, where cytokines, chemokines, growth factors, extracellular matrix (ECM) proteins, and mechanical signals act as inputs and transmit signals through cell surface receptors. While the exact signals operational post-MI have not been fully mapped, in vitro evidence indicates that these signals culminate in the release and activation of a wide variety of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs), cytokines, and growth factors. This cascade leads to multiple effects on a variety of cell physiology variables to modulate all three phases of cardiac remodeling.
Knowledge gap 3. Computational models simulating post-MI LV fibroblast activation are not available
In silico fibroblast relevant models have been developed, including a model of fibroblast-myocyte coupling, fibroblast changes that increase arrhythmia potential, and models of specific signaling pathways in fibroblasts [73–76]. Nim and colleagues have recently built a very extensive curated pathway map of cardiac fibroblasts (CARFMAP; http://visionet.erc.monash.edu.au/CARFMAP), an interactive map stemming from the current biomedical literature and utilizing a software-assisted manual data collection approach [77]. The CARFMAP allows users to explore the large body of fibroblast literature in various interactive ways. From expression profiles of mouse cardiac and tail fibroblasts, the authors have applied CARFMAP to characterize cardiac fibroblast pathways [77]. Building on this infrastructure to include details of post-MI fibroblast activation will provide a highly useful resource.
The Saucerman lab has developed cellular level models of fibroblast activation and kinetics that have application to post-MI cardiac remodeling [78, 79]. However, a more complete computational map that includes additional activation parameters, signaling network details, and phenotypic information is needed. Building these models requires building an initial framework to ensure the establishment of optimum models. Cardiac fibroblast activation has been defined mostly in terms of in vitro single stimulus responses; there is a need to understand how fibroblasts respond to mixed stimuli [7, 23, 32]. While most models end at gene expression as the outputs, models incorporating gene and protein expression, cell physiology, cell-cell interactions, and cell-matrix interactions are lacking. Logic-based diagrams of signaling pathways can be used to frame cell responses at different activation stages with flexibility to allow orchestration across inputs [80].
Initial models can be developed based on already known spatial and temporal stages of fibroblast phenotype shifts. Initial models could focus on short term in vitro treatment of fibroblasts with different factors known to affect fibroblast activation and function (e.g. IL-1α/β, TGF-β1, and fibronectin) [44]. The Turner lab has demonstrated that stimulating human cardiac fibroblasts to IL-1α upregulates the expression of IL-6, IL-8, MMP-3, and collagen III and downregulates α-SMA and CTGF [81]. In comparison, TGF-β1 significantly elevates α-SMA, CTGF, collagen1α1 and collagen3α1 expression, with minor effects on IL-6, IL-8, and MMP-3. Co-stimulation with both IL-1α and TGF-β1 synergistically increases MMP-3 content. While TGF-β1 has no significant effect on IL-1α-induced IL-6 or IL-8 levels, IL-1α inhibits the TGF-β1 stimulated increase in α-SMA and blocks gel contraction [81]. These data suggest that IL-1α can inhibit myofibroblast formation induced by TGF-β1.
The benefit of using already known inputs is that the previously defined activation patterns of these inputs can guide the model to specific downstream gene, secreted protein, and cell physiology outputs. Conversion of these results into logic-based differential equations will provide a window for simulations with other novel players that may be identified to build on the existing framework. The in silico integration of complex datasets can help define key trigger point responses; coupled to bioinformatics analysis, this approach provides a more comprehensive evaluation. Fibroblast computational models generated for this purpose can be used in future to understand both short-term and long-term cardiac remodeling patterns post-MI, which would allow predictions of new therapeutic interventions to be tested, validated, and refined.
Concluding Remarks
Future studies addressing the knowledge gaps discussed in this review will expand our understanding of fibroblast activation and the roles these cells play post-MI and provide new knowledge necessary to improve our current pharmaceutical arsenal (see Outstanding Questions). One point that needs to be considered while answering the knowledge gaps is if the assumption of cell homogeneity at each day post-MI holds true. This assumption is based on results in the pressure overload model, where current environment defines activation status. Because pressure overload pathology progresses on a much slower time frame than MI, the possibility exists that carryover of subtypes is likely during the inflammatory and reparative phases of MI. For example, day 1 post-MI environment activated fibroblasts that have a pro-inflammatory phenotype and day 3 post-MI environment activated fibroblasts that have a proliferative phenotype are likely to both be present in the day 3 post-MI LV. Pulse chase experiments are needed to determine the percentage contributions of cells from current and past times.
Fibroblast sub-types may oscillate between homogenous and heterogeneous pools of phenotypes, depending on what stage post-MI is being evaluated. We would expect day 1 and day 14 post-MI fibroblasts to be more homogeneous than cells from times in between these two very early and late times. In addition, we assume homogeneity in cell response across individuals, when in actuality individuals likely arrive at specific response stages at varying times. It is possible that an individual at day 3 post-MI could display a day 1 average phenotype, as an illustration of this concept. Evaluation of the full time course and exploring responses across individuals will allow assessment of the effects of cell heterogeneity and individual variability on the system.
In addition, future studies are needed to determine to what degree in vitro stimulated resident fibroblasts mimic the post-MI in vivo scenario. Other cell types such as macrophages, neutrophils, cardiomyocytes, or endothelial cells may play pertinent roles in affecting post-MI fibroblast phenotype and the resultant fibrosis and remodeling. Considerations for cell-cell communication are also needed when interventions are examined. For example, stimulation with one input may affect a fibroblast only culture differently than it affects a conglomerate of cell types in a co-culture experiment. Since many permutations of stimulation experiments are possible, computational models that allow effects of targeted interventions on a high-throughput scale will help explore cell cross-talk in a broad scale, allowing wet lab validation experiments to be performed in a more focused and efficient manner. Experiments where control cardiac fibroblasts are isolated and stimulated in vitro and experiments where cardiac fibroblasts are isolated from post-MI LV and examined ex vivo are warranted. While culture conditions need to be taken into consideration, cardiac fibroblasts are easily amendable to these evaluations.
Excessive cardiac fibrosis can impair LV wall compliance and exacerbate LV dysfunction post-MI. Though not primarily targeting fibrosis, current therapeutic agents for patients with MI, including angiotensin converting enzyme inhibitors, angiotensin AT1 receptor blockers, aldosterone antagonists, β blockers, and statins have secondary anti-fibrotic effects that are beneficial to outcomes [82]. Cardiac fibroblasts, being the major ECM source, represent a novel intervention target. With the emerging knowledge on fibroblast activation, phenotype, and physiology, it is highly expected that pharmacological agents specifically targeting fibroblast phenotypes could provide favorable effects.
In summary, establishing the post-MI fibroblast phenotype, signaling patterns, and response network will provide mechanistic insight into how fibroblasts regulate cardiac remodeling post-MI and help us identify novel intervention targets to improve on current pharmacological treatment strategies. Developing computational models that incorporate the fibroblast phenotype continuum will aid in the progress of this goal.
Trends.
The cardiac fibroblast is a major regulator of cardiac wound healing and remodeling following myocardial infarction (MI).
Published fibroblast markers, including vimentin, discoidin domain receptor 2, and fibroblast specific protein-1, lack selectivity and are expressed by other cell types.
Different fibroblast phenotypes, namely pro-inflammatory and pro-reparative subtypes, have recently been described following MI.
Key drivers of fibroblast activation have not been totally mapped post-MI and likely include a variety of cytokines, growth factors, extracellular matrix components, and mechanical signals.
Conversion of experimental results into logic-based differential equations will provide a window for simulations to explore permutations of interactions on a large scale and identify novel pharmacological targets.
Outstanding Questions.
What are the phenotypes and heterogeneity of cardiac fibroblasts over the myocardial infarction time course?
What are the best activation markers that can distinguish sub-populations of fibroblasts and myofibroblasts based on gene expression and phenotypic properties?
What is the effect of cellular cross-talk on fibroblast activation?
What are the mechanisms whereby cytokine and growth factor signaling pathways, ECM components, and mechanical signals regulate fibroblast activation?
Can we develop in silico and in vitro models that sufficiently simulate post-MI in vivo fibroblast activation patterns to more efficiently identify novel pharmacological targets?
Acknowledgments
We acknowledge support from the American Heart Association under Award Number 15SDG22930009, by the Canadian Institutes of Health Research Operating Grant under Award Number MOP-136862, by the National Heart, Lung, and Blood Institute and the National Institute of General Medical Sciences of the National Institutes of Health under Award Numbers HL075360, HL129823, HL051971, GM104357, GM114833, and GM115428, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under Award Number 5I01BX000505. The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Heart Association, the Canadian Institutes of Health Research, the National Institutes of Health, or the Veterans Administration.
Glossary
- Alpha-Smooth Muscle Actin (α-SMA)
A highly conserved protein involved in cell motility, structure, and integrity. Highly expressed in vascular smooth muscle cells and myofibroblasts
- Angiotensin II
A peptide hormone that causes vasoconstriction and increases blood pressure
- Cardiac remodeling
Changes in size, shape, structure, and function of the heart following injury
- Connective tissue growth factor (CTGF)
CTGF, also known as CCN2, is a matricellular protein that belongs to the CCN family of extracellular matrix-associated heparin-binding proteins
- Discoidin domain receptor (DDR)
A tyrosine kinase receptor that plays a role in cell to cell communication, and that is activated by collagen
- Epicardium-derived cells (EPDCs)
Following injury, epicardial cells undergo epithelial-mesenchymal transition to form EPDCs, which can become fibroblasts and smooth muscle cells
- Epithelial/endothelial-mesenchymal transition (EMT/EndMT)
A biologic process that allows an epithelial or endothelial cell to undergo multiple biochemical changes that enable it to assume a mesenchymal cell phenotype, which includes elevated migratory capacity, invasiveness, enhanced resistance to apoptosis, and increased generation of ECM components
- Extracellular matrix (ECM)
Molecules secreted by cells that assemble to provide structural and biochemical support to the surrounding cells
- Fibrosis
The scarring and hardening of tissues characterized by the accumulation of excessive extracellular matrix
- Fibroblast activation protein (FAP)-1
FAP1 is a member of the serine protease family. It is involved in the regulation of fibroblast growth or epithelial-mesenchymal transition
- Fibroblast specific protein (FSP)-1
FSP-1, also known as S100A4, is a cytoplasmic protein expressed by a number of cell types, including fibroblasts, smooth muscles cells, and leukocytes
- Interleukin (IL)-1α or β
A classic pro-inflammatory cytokine that is secreted by immune cells
- Left ventricle (LV)
LV is a large chamber in the heart that collects and expels blood received from the left atrium towards the peripheral beds within the body
- Matrix metalloproteinase (MMP)
A calcium-dependent zinc-containing endopeptidase that degrades extracellular matrix proteins
- Monocyte chemoattractant protein (MCP)-1
MCP-1, also known as CCL2, is a chemokine that recruits monocytes, memory T cells, and dendritic cells to sites of inflammation
- Myocardial infarction (MI)
The irreversible death of heart muscle secondary to prolonged lack of oxygen supply
- Myocardin-related transcription factors (MRTFs)
MRTFs, including MRTF-A and MRTF-B, are transcriptional coactivators that physically associate with the MADS box transcription factor, serum response factor, and synergistically activate transcription
- Myofibroblast
An activated fibroblast with smooth muscle cell contractile properties
- Reactive oxygen species (ROS)
Radical and non-radical oxygen species formed by the partial reduction of oxygen
- Secreted protein acidic and rich in cysteine (SPARC)
SPARC, also named osteonectin or BM-40, is a highly conserved, multifunctional glycoprotein
- Serum response factor (SRF)
SRF is a highly conserved and widely expressed transcription factor. SRF binds and activates promoters harboring the DNA element CC(A/T)6GG (CArG box)
- Transforming growth factor (TGF)-β1
A secreted protein that promotes the conversion of fibroblasts into activated myofibroblasts
- Transient receptor potential canonical (TRPC) proteins
TRPC proteins are a family of plasma membrane calcium-permeable channels. TRPC proteins can be activated by various stimuli and act as cellular sensors in mammals
- Tumor necrosis factor (TNF)-α
A classic pro-inflammatory cytokine that is secreted by immune cells
- Vascular smooth muscle cells (VSMCs)
VSMCs provide structural integrity to blood vessels and regulate the lumen size by contracting and relaxing dynamically in response to vasoactive stimuli
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
None.
References
- 1.Writing Group, M. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Travers JG, et al. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol. 1996;174:221–232. doi: 10.1006/dbio.1996.0068. [DOI] [PubMed] [Google Scholar]
- 4.Cai CL, et al. A myocardial lineage derives from Tb ×18 epicardial cells. Nature. 2008;454:104–108. doi: 10.1038/nature06969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali SR, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115:625–635. doi: 10.1161/CIRCRESAHA.115.303794. [DOI] [PubMed] [Google Scholar]
- 6.Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac fibroblasts. J Mol Cell Cardiol. 2014;70:2–8. doi: 10.1016/j.yjmcc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, et al. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Archiv: European journal of physiology. 2014;466:1113–1127. doi: 10.1007/s00424-014-1463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yano T, et al. Intracardiac fibroblasts, but not bone marrow derived cells, are the origin of myofibroblasts in myocardial infarct repair. Cardiovasc Pathol. 2005;14:241–246. doi: 10.1016/j.carpath.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 9.van Amerongen MJ, et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol. 2008;214:377–386. doi: 10.1002/path.2281. [DOI] [PubMed] [Google Scholar]
- 10.Zhou B, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121:1894–1904. doi: 10.1172/JCI45529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz-Villalba A, et al. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol. 2015;65:2057–2066. doi: 10.1016/j.jacc.2015.03.520. [DOI] [PubMed] [Google Scholar]
- 12.Aisagbonhi O, et al. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech. 2011;4:469–483. doi: 10.1242/dmm.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fligny C, Duffield JS. Activation of pericytes: recent insights into kidney fibrosis and microvascular rarefaction. Curr Opin Rheumatol. 2013;25:78–86. doi: 10.1097/BOR.0b013e32835b656b. [DOI] [PubMed] [Google Scholar]
- 14.Trial J, et al. Th1/M1 conversion to th2/m2 responses in models of inflammation lacking cell death stimulates maturation of monocyte precursors to fibroblasts. Front Immunol. 2013;4:287. doi: 10.3389/fimmu.2013.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore-Morris T, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teekakirikul P, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braitsch CM, et al. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J Mol Cell Cardiol. 2013;65:108–119. doi: 10.1016/j.yjmcc.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurose H, Mangmool S. Myofibroblasts and inflammatory cells as players of cardiac fibrosis. Arch Pharm Res. 2016;39:1100–1113. doi: 10.1007/s12272-016-0809-6. [DOI] [PubMed] [Google Scholar]
- 19.Krenning G, et al. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinto AR, et al. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acharya A, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanisicak O, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, et al. Extracellular matrix and fibroblast communication following myocardial infarction. J Cardiovasc Transl Res. 2012;5:848–857. doi: 10.1007/s12265-012-9398-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mork C, et al. Regulation of vimentin expression in cultured human mammary epithelial cells. Differentiation. 1990;43:146–156. doi: 10.1111/j.1432-0436.1990.tb00441.x. [DOI] [PubMed] [Google Scholar]
- 25.Vogel WF, et al. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal. 2006;18:1108–1116. doi: 10.1016/j.cellsig.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Sugimoto H, et al. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 27.Rettig WJ, et al. Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer Res. 1993;53:3327–3335. [PubMed] [Google Scholar]
- 28.Orlandini M, Oliviero S. In fibroblasts Vegf-D expression is induced by cell-cell contact mediated by cadherin-11. J Biol Chem. 2001;276:6576–6581. doi: 10.1074/jbc.M009573200. [DOI] [PubMed] [Google Scholar]
- 29.Serini G, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frangogiannis NG, et al. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 31.Smith CL, et al. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res. 2011;108:e15–26. doi: 10.1161/CIRCRESAHA.110.235531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Squires CE, et al. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39:699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 33.Turner NA, Porter KE. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis & tissue repair. 2013;6:5. doi: 10.1186/1755-1536-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daskalopoulos EJB, Blankesteijn WM. Myofibroblasts in the Infarct Area: Concepts and Challenges. Microsc Microanal. 2012;18:35–49. doi: 10.1017/S143192761101227X. [DOI] [PubMed] [Google Scholar]
- 35.van den Borne SW, et al. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 36.van den Borne SWM, et al. Mouse strain determines the outcome of wound healing after myocardial infarction. Cardiovasc Res. 2009;84:273–282. doi: 10.1093/cvr/cvp207. [DOI] [PubMed] [Google Scholar]
- 37.van Nieuwenhoven FA, Turner NA. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascul Pharmacol. 2013;58:182–188. doi: 10.1016/j.vph.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner NA. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs) J Mol Cell Cardiol. 2016;94:189–200. doi: 10.1016/j.yjmcc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Turner NA, et al. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol. 2009;297:H1117–1127. doi: 10.1152/ajpheart.00372.2009. [DOI] [PubMed] [Google Scholar]
- 41.Park SK, et al. Hydrogen peroxide is a novel inducer of connective tissue growth factor. Biochem Biophys Res Commun. 2001;284:966–971. doi: 10.1006/bbrc.2001.5058. [DOI] [PubMed] [Google Scholar]
- 42.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 43.Lijnen P, et al. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens. 2006;24:757–766. doi: 10.1097/01.hjh.0000217860.04994.54. [DOI] [PubMed] [Google Scholar]
- 44.Hwang MW, et al. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol. 2001;38:1546–1553. doi: 10.1016/s0735-1097(01)01591-1. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell MD, et al. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol Heart Circ Physiol. 2007;292:H1139–1147. doi: 10.1152/ajpheart.00881.2005. [DOI] [PubMed] [Google Scholar]
- 46.Palmer JN, et al. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest. 1995;95:2555–2564. doi: 10.1172/JCI117956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobs M, et al. Tumor necrosis factor-alpha at acute myocardial infarction in rats and effects on cardiac fibroblasts. J Mol Cell Cardiol. 1999;31:1949–1959. doi: 10.1006/jmcc.1999.1007. [DOI] [PubMed] [Google Scholar]
- 48.Peng J, et al. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–1126. doi: 10.1161/01.res.0000047090.08299.d5. [DOI] [PubMed] [Google Scholar]
- 49.Turner NA, et al. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res. 2007;76:81–90. doi: 10.1016/j.cardiores.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Jourdan-Lesaux C, et al. Extracellular matrix roles during cardiac repair. Life Sci. 2010;87:391–400. doi: 10.1016/j.lfs.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobaczewski M, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gharaee-Kermani M, et al. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 53.Frangogiannis NG, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 54.Dewald O, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 55.Freed DH, et al. Role of myosin light chain kinase in cardiotrophin-1-induced cardiac myofibroblast cell migration. Am J Physiol Heart Circ Physiol. 2011;301:H514–522. doi: 10.1152/ajpheart.01041.2010. [DOI] [PubMed] [Google Scholar]
- 56.Dobaczewski M, et al. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eckes B, et al. Mechanical tension and integrin alpha 2 beta 1 regulate fibroblast functions. J Investig Dermatol Symp Proc. 2006;11:66–72. doi: 10.1038/sj.jidsymp.5650003. [DOI] [PubMed] [Google Scholar]
- 58.van Putten S, et al. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. 2016;93:133–142. doi: 10.1016/j.yjmcc.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 59.Roche PL, et al. Role of scleraxis in mechanical stretch-mediated regulation of cardiac myofibroblast phenotype. Am J Physiol Cell Physiol. 2016;311:C297–307. doi: 10.1152/ajpcell.00333.2015. [DOI] [PubMed] [Google Scholar]
- 60.Harada M, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saliba Y, et al. Evidence of a Role for Fibroblast Transient Receptor Potential Canonical 3 Ca2+ Channel in Renal Fibrosis. J Am Soc Nephrol. 2015;26:1855–1876. doi: 10.1681/ASN.2014010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lighthouse JK, Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol. 2016;91:52–60. doi: 10.1016/j.yjmcc.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elberg G, et al. MKL1 mediates TGF-beta1-induced alpha-smooth muscle actin expression in human renal epithelial cells. Am J Physiol Renal Physiol. 2008;294:F1116–1128. doi: 10.1152/ajprenal.00142.2007. [DOI] [PubMed] [Google Scholar]
- 64.O’Connor JW, Gomez EW. Cell adhesion and shape regulate TGF-beta1-induced epithelial-myofibroblast transition via MRTF-A signaling. PloS one. 2013;8:e83188. doi: 10.1371/journal.pone.0083188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833:945–953. doi: 10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cleutjens JP, et al. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 67.Dewald O, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Booz GW, Baker KM. Molecular signalling mechanisms controlling growth and function of cardiac fibroblasts. Cardiovasc Res. 1995;30:537–543. [PubMed] [Google Scholar]
- 69.Lindsey ML, et al. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-myocardial Infarction by Promoting Scar Formation and Angiogenesis. J Am Coll Cardiol. 2015;66:1364–1374. doi: 10.1016/j.jacc.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Willems IE, et al. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 71.Hermans KC, et al. The Janus face of myofibroblasts in the remodeling heart. J Mol Cell Cardiol. 2016;91:35–41. doi: 10.1016/j.yjmcc.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 72.Scruggs SB, et al. Harnessing the heart of big data. Circ Res. 2015;116:1115–1119. doi: 10.1161/CIRCRESAHA.115.306013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie Y, et al. Cardiac alternans induced by fibroblast-myocyte coupling: mechanistic insights from computational models. Am J Physiol Heart Circ Physiol. 2009;297:H775–784. doi: 10.1152/ajpheart.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McDowell KS, et al. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys J. 2011;101:1307–1315. doi: 10.1016/j.bpj.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhan HQ, et al. Fibroblast proliferation alters cardiac excitation conduction and contraction: a computational study. J Zhejiang Univ Sci B. 2014;15:225–242. doi: 10.1631/jzus.B1300156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sachse FB, et al. A model of electrical conduction in cardiac tissue including fibroblasts. Ann Biomed Eng. 2009;37:874–889. doi: 10.1007/s10439-009-9667-4. [DOI] [PubMed] [Google Scholar]
- 77.Nim HT, et al. CARFMAP: A Curated Pathway Map of Cardiac Fibroblasts. PloS one. 2015;10:e0143274. doi: 10.1371/journal.pone.0143274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zeigler AC, et al. Computational modeling of cardiac fibroblasts and fibrosis. J Mol Cell Cardiol. 2016;93:73–83. doi: 10.1016/j.yjmcc.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zeigler AC, et al. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol. 2016;94:72–81. doi: 10.1016/j.yjmcc.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raza S, et al. Construction of a large scale integrated map of macrophage pathogen recognition and effector systems. BMC Syst Biol. 2010;4:63. doi: 10.1186/1752-0509-4-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Nieuwenhoven FA, et al. Combined effects of interleukin-1alpha and transforming growth factor-beta1 on modulation of human cardiac fibroblast function. Matrix Biol. 2013;32:399–406. doi: 10.1016/j.matbio.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 82.Minicucci MF, et al. Heart failure after myocardial infarction: clinical implications and treatment. Clin Cardiol. 2011;34:410–414. doi: 10.1002/clc.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dobaczewski M, et al. The Extracellular Matrix Modulates Fibroblast Phenotype and Function in the Infarcted Myocardium. J Cardiovasc Transl Res. 2012;5:837–847. doi: 10.1007/s12265-012-9406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]