

Abstract
Sulfur‐containing gaseous signal molecules including hydrogen sulphide and sulfur dioxide were previously recognized as toxic gases. However, extensive studies have revealed that they can be generated in the cardiovascular system via a sulfur‐containing amino acid metabolic pathway, and have an important role in cardiovascular physiology and pathophysiology. Ion channels are pore‐forming membrane proteins present in the membrane of all biological cells; their functions include the establishment of a resting membrane potential and the control of action potentials and other electrical signals by conducting ions across the cell membrane. Evidence has now accumulated suggesting that the sulfur‐containing gaseous signal molecules are important regulators of ion channels and transporters. The aims of this review are (1) to discuss the recent experimental evidences in the cardiovascular system regarding the regulatory effects of sulfur‐containing gaseous signal molecules on a variety of ion channels, including ATP‐sensitive potassium, calcium‐activated potassium, voltage‐gated potassium, L‐ and T‐type calcium, transient receptor potential and chloride and sodium channels, and (2) to understand how the gaseous signal molecules affect ion channels and cardiovascular diseases.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- 3MPST
3‐mercaptopyruvate sulfurtransferase
- AAT
aspartate aminotransferase
- BKCa
big‐conductance calcium‐activated potassium channel
- CBS
cystathionine‐β‐synthase
- CFTR
cystic fibrosis transmembrane conductance regulator
- CSE
cystathionine‐γ‐lyase
- ICa,L
L‐type calcium channel currents
- IKCa
intermediate‐conductance calcium‐activated potassium channel
- KATP
ATP‐sensitive potassium channel
- KCa
calcium‐activated potassium channel
- Kir
inwardly rectifying potassium channel
- Kv
voltage‐gated potassium channel
- NCX
sodium calcium exchanger
- SHR
spontaneously hypertensive rat
- SKCa
small‐conductance calcium‐activated potassium channel
- SUR
sulfonylurea receptor
- TRP
transient receptor potential
- TRPA1
transient receptor potential ankyrin 1
- WKY
Wistar–Kyoto
Introduction
NO was the first gaseous signal molecule to be identified in the cardiovascular system and its pivotal roles in many physiological and pathological processes have been elucidated (Ignarro et al., 1987; Palmer et al., 1987). Later on, sulfur‐containing gaseous signal molecules, hydrogen sulphide (H2S) and sulfur dioxide (SO2), were identified (Hosoki et al., 1997; Wang, 2002; Du et al., 2008; Jin et al., 2010; Liu et al., 2010). Subsequent studies revealed that H2S and SO2 had a wide range of biological functions as well as broader effects on the cardiovascular system under both physiological and pathophysiological conditions (Wang, 2003; Du et al., 2008; Liu et al., 2012; Polhemus and Lefer, 2014; Meng et al., 2015). The sulfur‐containing gaseous signal molecules collectively have the following basic characteristics: (1) endogenous generation via a controllable enzymatic reaction by metabolism of sulfur‐containing amino acids in the body; (2) small molecular weight, so can freely pass through the cell membrane and rapidly diffuse; (3) clear cellular and molecular targets; and (4) widely involved in the homeostatic control and regulation of the cardiovascular system as well as the pathogenesis of cardiovascular diseases (Barañano et al., 2001; Li and Moore, 2007; Wang et al., 2010). Recent studies demonstrated that sulfur‐containing gaseous signal molecules regulate a variety of ion channels, which has a great impact on the physiology and pathology of the cardiovascular system.
Ion channels represent a family of important membrane proteins that conduct ions across the membrane, thereby controlling the intracellular ion content, membrane potential and action potentials. The abnormal function and structure of ion channels can lead to a variety of cardiovascular diseases (Ackerman, 1998; Navedo et al., 2010; Hsiao et al., 2013; George, 2014; Wang et al., 2016). More importantly, endogenous sulfur‐containing gaseous signal molecules play an important role in the pathogenesis of cardiovascular diseases by targeting ion channels (Wang, 2003; Brampton and Aaronson, 2016).
This review discusses the recent experimental evidences for the regulatory effects of H2S and SO2 on ion channels and their significance for cardiovascular diseases.
Hydrogen sulphide
Generation and metabolism of hydrogen sulphide in cardiovascular tissue
H2S is a colourless gas smelling of rotten eggs. At the end of the 1990s, H2S was found to be endogenously produced in the sulfur‐containing amino acid metabolic pathway in mammals. Subsequently, H2S was found to participate in regulating the physiological functions of the circulatory system (Zhao and Wang, 2002; Ji et al., 2008; Wei et al., 2010; Li et al., 2013; Quan et al., 2015; Wei et al., 2015). Exploring the biological effects of endogenous H2S has become a hot issue in life science and medicine (Wang, 2003; Tang et al., 2006; Skovgaard and Olson, 2012; Vandiver and Snyder, 2012).
The key enzymes for producing endogenous H2S include cystathionine‐γ‐lyase (CSE), cystathionine‐β‐synthase (CBS) and 3‐mercaptopyruvate sulfurtransferase (3MPST), with CBS and CSE dependent on L‐cystine as a substrate and pyridoxal phosphate as a coenzyme to catalyse the production of H2S. 3MPST catalyses the generation of H2S by using β‐mercapto‐pyruvic acid as a substrate. CBS primarily catalyses the homocysteine and cysteine condensation reaction to form cystathionine while releasing H2S. CSE catalyses the cleavage reaction of cystine to thiocysteine, which spontaneously degrades to cysteine and H2S (Shibuya et al., 2009; Gong et al., 2011).
H2S has good lipophilicity and hydrophilicity and freely passes through cell membranes. When H2S is dissolved in water, it can be partially hydrated into H2S and sulfur ions. H2S is in two forms in mammals, namely, physical dissolution of H2S gas and chemical forms of HS−; the latter combined with hydrogen ions in vivo can generate H2S, which forms a dynamic balance in the body. This condition is conducive to maintaining the H2S content in vivo and the stability of pH in the internal environment (Liu et al., 2012). Most of the endogenous H2S is excreted in the form of thiosulfate and sulfate produced by oxidative metabolism in mitochondria, and a small portion is converted to methyl mercaptan and methyl sulfide by methylation in cytoplasmic solution. H2S in the plasma is cleared by methaemoglobin. Its metabolites can be excreted in the kidneys, intestines and lungs within 24 h.
Hydrogen sulphide regulates the cardiovascular system by targeting the ion channels
ATP‐sensitive potassium channel (KATP)
KATP is a receptor‐dependent potassium channel, and its activity can be significantly inhibited with an increase in intracellular ATP concentration. The KATP channel consists of the inwardly rectifying potassium channel (Kir) and sulfonylurea receptors (SURs). The two Kir subtypes, Kir6.1 and Kir6.2, form the ion pore of the KATP channel. SUR also has two subtypes, SUR1 and SUR2 [SUR2A (ABCC8) and SUR2B (ABCC9)]. The combination of different Kir subunits and SUR subunits forms the diversity of KATP molecular structure, which determines the complex function of the KATP channel in different tissues. In cardiomyocytes, the KATP channel consists of Kir6.2 and SUR2A, whereas in vascular smooth muscle cells (VSMCs), it is mainly composed of Kir6.1 and SUR2B.
The KATP channel mediates the vasodilatation induced by H2S in cardiovascular tissues, such as the thoracic aorta (Zhao et al., 2001), mesenteric artery (Cheng et al., 2004) and coronary artery (Casalini et al., 2014). Zhao et al. (2001) found that H2S was a KATP channel opener and that H2S (2.8 and 14 μmol·kg−1) induced a transient decrease in mean arterial blood pressure (12.5 ± 2.1 and 29.8 ± 7.6 mmHg) in anaesthetized rats. Pinacidil (2.8 μmol·kg−1) mimics but the KATP channel blocker glibenclamide inhibits the hypotensive effect. The currents of the KATP channel in VSMCs were recorded and found to be amplified by H2S (300 μM) from 85.8 ± 12.4 to 149.0 ± 21.4 pA. H2S hyperpolarizes the cellular membrane from −35.7 to −53.3 mV, and this effect is reversed by glibenclamide. Furthermore, after inhibition of H2S, whole‐cell KATP currents are reduced (Tang et al., 2005). Sun et al. (2015) found the expression of Kir6.1 and SUR2B was significantly down‐regulated in the aortas of spontaneously hypertensive rats (SHRs), and this effect was reversed by treatment with NaHS, a H2S donor. As a KATP channel opener, pinacidil dose‐dependently relaxes rat thoracic artery rings, but in aortic rings of SHRs, the vasorelaxant effects are attenuated as compared with those of Wistar–Kyoto (WKY) rats. NaHS at 100 μM improved the vasorelaxant response in aortic rings of SHR. These studies suggest that H2S, by targeting the KATP channel, is involved in the pathogenesis of hypertension.
The protective role of H2S was shown in ischaemia–reperfusion injury (Gross and Fryer, 1999). As a protective gaseous signal molecule, H2S has a negative inotropic action and glibenclamide can, in part, block this effect of H2S (Geng et al., 2004). In rat models of myocardial ischaemia–reperfusion injury, Johansen et al. (2006) found that NaHS (1 μM) limited the infarct size from 41.0 ± 2.6 to 20.2 ± 2.1% of the risk zone; pretreatment with glibenclamide abolished this effect. More electrophysiological findings revealed that in cardiomyocytes, an H2S donor at 40 μM increased the opening probability of the KATP channel from 0.07 ± 0.03 to 0.15 ± 0.08 and a NaHS donor at 100 μM increased the opening probability from 0.07 ± 0.03 to 0.36 ± 0.15. This effect was abolished by the KATP blocker glibenclamide (Zhang et al., 2007). In addition, the mitochondrial KATP channel (mitoKATP) was shown to mediate this cardioprotective effect of H2S, as a specific blocker of the mitoKATP channel, 5‐hydroxydecanoate, inhibited this effect of H2S (Sivarajah et al., 2009). These studies suggest that H2S, by targeting the KATP channel, is involved in protecting the myocardium against injury.
The potential mechanisms by which H2S acts on the KATP channel have been clarified. Jiang et al. (2010) demonstrated that H2S activates the rvKir6.1⁄rvSUR1 channel by sulfhydryl modification of Cys6 and Cys26 of the rvSUR1 subunit; the major sulfhydration site was Cys43. Mustafa et al. (2011) showed that H2S promotes cell hyperpolarization and vasorelaxation and this is associated with an enhancement of Kir6.1 activity induced by modifying sulfhydration, which subsequently reduces Kir 6.1‐ATP binding and increases Kir6.1 and PIP2 binding. Other studies showed that in cultured VSMCs, H2S stimulates FOXO1 and FOXO3a nuclear translocation and promotes FOXO1 or FOXO3a binding to the Kir6.1 and SUR2B gene promoters, thereby increasing the protein expression of the KATP channel and relaxing artery rings (Sun et al., 2015).
Calcium‐activated potassium channel (KCa)
The KCa channel is a kind of voltage and calcium‐sensitive channel and widely exists in excitable cell membranes of mammals. It is divided into three types according to electrophysiological properties and conductance levels: big‐conductance KCa (BKCa), intermediate‐conductance KCa (IKCa) and small‐conductance KCa (SKCa).
Sheng et al. (2013) showed that under identical voltage‐clamp conditions, NaHS at 100 μM reduced the peak current density of BKCa, I to and I Kir channels in human atrial fibroblasts to attenuate atrial fibroblast proliferation and moderate myofibroblast differentiation. Na2S (10 μM) hyperpolarized pressurized arterioles from −30.3 to −47.9 mV, which returned to −39.0 mV after the application of iberiotoxin, a selective KCa channel blocker. This result suggests that the KCa channel mediates the vasodilatation induced by H2S in cerebral arteries (Liang et al., 2012).
Cheng et al. (2004) observed that the vasorelaxant effects of H2S in rat mesenteric artery were blocked by charybdotoxin/apamin, with the EC50 changing from 22.5 ± 1.1 to 141.9 ± 21.5 μM. Furthermore, H2S can hyperpolarize endothelial cells, and neither glibenclamide nor iberiotoxin can block this effect. However, the combination of blockers of the IKCa/SKCa channel, charybdotoxin/apamin, abolished this effect. Moreover, in human artery endothelial cells, the hyperpolarization induced by H2S was diminished by TRAM‐34, an IKCa channel blocker, but was unchanged by glibenclamide or iberiotoxin treatment (Mustafa et al., 2011). These studies suggest that the IKCa/SKCa channel is the possible target of H2S in the cardiovascular system.
L‐type calcium channel
The L‐type calcium channel, one subtype of voltage‐dependent calcium channel, is widely distributed in VSMCs as well as cardiomyocytes, and has a characteristically slow activation time, high activation voltage and long opening time (Catterall, 2000). The L‐type calcium channel has four subtypes – Cav1.1, Cav1.2, Cav1.3 and Cav1.4 – and regulates rapid depolarization and plateau formation and maintenance.
Zhao and Wang (2002) revealed that the vasorelaxation of rat aortic rings induced by H2S (600 μM) was decreased by 59.3 ± 8.3% by treatment with nifedipine, an antagonist of the L‐type calcium channel, which suggests that the L‐type calcium channel mediates the relaxant effects of H2S.
Sun et al. (2008) investigated the possible mechanism for the negative inotropic effects of H2S in cardiomyocytes. Electrophysiological measurements were used to record isolated cardiomyocyte L‐type calcium channel currents (I Ca,L) in WKY rats and SHRs. NaHS at 100 μM decreased peak I Ca,L by 15.00 ± 2.08% in WKY cardiomyocytes and by 19.61 ± 2.18% in SHR cardiomyocytes, which suggests the inhibitory role of H2S in calcium release from cardiomyocytes. Pre‐incubation with NaHS (100 μM) enhanced cell survival to 78 ± 3% in H9c2 cells treated with H2O2 and decreased resting intracellular calcium [Ca2+]i content, which was mimicked by nifedipine treatment, which suggests that H2S inhibits the L‐type calcium channel to enhance cell survival (Avanzato et al., 2014).
With regard to the potential mechanisms by which H2S targets the L‐type calcium channel, Mustafa et al. (2009) and Li et al. (2011b) indicated that H2S regulates the activity of proteins by modifying cysteine residues via S‐sulfhydration. A whole‐cell voltage‐clamp technique was used to obtain more electrophysiological evidence. Negative inotropic effects by H2S on cardiac function were attenuated by diamide, an oxidant sulfhydryl modifier, and the inhibitory effect on the peak amplitude of I Ca,L was reversed by dithiothreitol, a reducing modifier of the sulfhydryl group. These observations suggest that H2S inhibits the cardiomyocyte L‐type calcium channel to produce a negative inotropic effect, and the main action site might be the sulfhydryl group in the L‐type calcium channel (Zhang et al., 2012).
T‐type calcium channel
The T‐type calcium channel has the following electrophysiological characteristics: fast activation, lower activation voltage, fast inactivation and short opening time. It includes three subtypes – Cav3.1, Cav3.2 and Cav3.3 – and participates in maintaining the ventricular action potential plateau and regulates the initiation and contraction of myocardium (Triggle, 1998). Unlike the L‐type calcium channel, in the T‐type calcium channel, the organic calcium antagonist and Bay k8644 are ineffective, but it can be blocked by the inorganic calcium blocker Ni2+.
Avanzato et al. (2014) found that the inhibitory effect of H2S on resting [Ca2+]i content in H9c2 cells was abolished by pretreatment with nifedipine and Ni2+ combined as compared with nifedipine alone. This result suggests that both the T‐type and L‐type calcium channels are inhibited by H2S.
Sekiguchi et al. (2014) demonstrated that H2S enhanced Cav3.2 T‐type calcium channel function in HEK293 cells. The Cav3.2 T‐type calcium channel is also involved in the carotid body response to hypoxia mediated by H2S, as NaHS has been shown to increase [Ca2+]i content in glomus cells and enhance the activity of carotid body, and this effect was attenuated in glomus cells and carotid bodies of Cav3.2‐knockout mice (Makarenko et al., 2015).
Sodium calcium exchanger (NCX)
NCX is a bidirectional transporter and consists of three subtypes: NCX1, NCX2 and NCX3. NCX1 is the main subtype present in cardiomyocytes. The ratio of Na+/Ca2+ exchangers was three Na+ exchangers to one Ca2+ exchanger in cardiomyocytes. The currents of Na+/Ca2+ exchangers are divided into inward Na+/Ca2+, outward Na+/Ca2+ and dual‐ward Na+/Ca2+ exchange currents.
Moccia et al. (2011) used NaHS (250 μM) to stimulate Ca2+ inflow in 80% of rat aortic endothelial cells, which mimics the effects in adult human dermal microvascular endothelial cells. To investigate the possible role of NCX in the Ca2+ current induced by NaHS, the authors found that KB‐R7943 (20 μM), a selective inhibitor of the NCX, reduced the effect by 25.4%. The amplitude of the Ca2+ response induced by NaHS was small after the application of KB‐R7943. Collectively, these results suggest a novel target of H2S in rat aortic endothelial cells, and it plays a significant role in Ca2+ inflow and is involved in NaHS‐induced angiogenesis.
To explore the effects of H2S on NCX in cardiomyocytes, Pan et al. (2008) used caffeine to keep the ryanodine receptor open and prevent the sarcoplasmic reticulum from sequestering Ca2+. The cells were pretreated with NaHS to elicit the Ca2+ inflow current and the half‐decay time and 90% decay time (t50 and t90) of [Ca2+]I content were measured. The t50 and t90 of the decayed [Ca2+]i transients were shortened by NaHS, and the effects were reversed by chelerythrine, a selective PKC inhibitor. The observations confirmed that H2S promotes Ca2+ inflow via NCX in a PKC‐dependent manner.
Markova et al. (2014) found increased mRNA and protein levels and the activity of NCX1 as well as concentration of cAMP were enhanced in GYY4137 (a H2S donor)‐treated HeLa cells. The levels of β1‐ and β3‐adrenoceptors were also up‐regulated after GYY4137 treatment. This GYY4137‐induced increase was eliminated when NCX1 was silenced. To test the relationship between β‐adrenoceptors and NCX1 in the GYY4137‐induced apoptosis, they confirmed that a mutual interaction between β1‐ and β3‐adrenoceptors and NCX1 mediates the stimulant effects of H2S on apoptosis.
Transient receptor potential (TRP) channel
The TRP channel widely exists in the mammalian brain, spinal cord, heart, kidney and other tissues. The channel can form functional homo‐tetramers or hetero‐tetramers, thus playing a role in signal transduction. According to the homology, the TRP ion channel superfamily has seven subgroups, including TRPA, TRPC, TRPML, TRPM, TRPP, TRPN and TRPV. Most of the TRP channels are nonselective cationic channels; TRPM4 and TRPM5 are monovalent cationic channels; and TRPV5 and TRPV6 have very high selectivity for calcium.
Intensive research showed that a TRP channel mediates the regulatory effects of H2S on neuronal and synaptic activity (Abe and Kimura, 1996; Nagai et al., 2004; Kimura, 2013). White et al. (2013) observed that the NaHS (300 μM) induced vasodilatation of small mesenteric arteries could not be blocked by a non‐specific potassium‐channel inhibitor, tetraethylammonium (TEA), glibenclamide or XE991, a voltage‐gated potassium channel (Kv) channel blocker. However, the chloride channel blocker 4,4′‐diisothiocyano‐2,2′‐stilbenedisulfonic acid (DIDS) abolished this effect, but 5‐nitro‐2‐(3‐phenylpropylamino)‐benzoate (NPPB) and anthracere‐9‐carboxylic acid had no effect. To explore the potential mechanisms of this NaHS‐induced vasodilatation, capsaicin was found to abolish this NaHS‐induced vasodilatation, but it was not affected by capsazepine or a TRPV1 blocker, but was blocked by HC‐030031, a TRPA1 channel blocker. These findings indicate that H2S activates the TRPA1 channel, which stimulates the release of sensory neurotransmitters and then mediates the vasodilatation in rat mesenteric arteries.
Chloride channel
Chloride ion is the most abundant anion inside or outside cells, and its transmembrane transport is very important. The chloride channel is widely distributed in various tissues, and its functions include inhibiting cell excitability, promoting depolarization after repolarization and maintaining the resting membrane potential of cells. The chloride channel is divided into three subtypes according to its protein structure: voltage‐dependent chloride ion channel, cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel and ligand‐gated chloride channel.
Lee et al. (2007) examined whether H2S affects the intracellular pH (pHi) of VSMCs and found that the H2S donor NaHS dose‐dependently decreased pHi, which was attenuated by DIDS, a blocker of the Cl−/HCO3 − exchanger, but was not affected by inhibition of the Na+/H+ exchanger or Ca2+‐ATPase. Then, the activity of Cl−/HCO3 − exchanger was detected by NH4Cl perfusion and was found to be enhanced by NaHS by approximately 60%. Similar results were found in rat aortic rings: DIDS blocked the diastolic effects of NaHS on aortic rings. These data suggest that H2S decreases pHi via activation of VSMC Cl−/HCO3 − exchangers and relaxed rat aortic rings.
A chemical hypoxia‐induced cell injury model was used to investigate the possible role of CFTR in the cardioprotective effects of H2S on cardiomyocytes. NaHS (400 μM) protected H9c2 cells against the toxicity of CoCl2 and increased the cell survival rate from 48.3 ± 2.1 to 65.4 ± 0.6%. NPPB, an inhibitor of the CFTR chloride channel, which had no effect on myocardial viability, attenuated the protective effects of NaHS, with a decrease in survival rate of cardiomyocytes from 65.4 ± 0.6 to 56.3 ± 1.8%. These results suggest that the CFTR chloride channel might be involved in the protective effects of H2S on cardiomyocytes against hypoxia‐mediated injury (Li et al., 2009b).
Electrophysiological evidence was provided by Malekova et al. (2009); they observed current changes of the single chloride channel in cardiomyocyte lysosomal vesicles incorporated into a bilayer lipid membrane in rats. NaHS decreased the open probability of the chloride channel by increasing the channel closing time but had no effect on the amplitude, conductance or mean opening time (Malekova et al., 2009). These results reveal that H2S has the potential to inhibit the chloride channel and this could be ones of its mechanisms of action.
Sulfur dioxide
Generation and metabolism of sulfur dioxide in cardiovascular tissues
Similar to NO, CO and H2S, SO2 is known to be one of the air pollutants and is present in industrial waste gases. However, SO2 is also generated endogenously. Biochemical studies have shown that the enzymatic reaction of sulfur‐containing amino acid metabolic pathways with methionine can produce SO2 (Singer and Kearney, 1956). In recent years, a complete endogenous SO2 pathway has been detected in the cardiovascular system and found to play an important role in cardiovascular physiology and pathophysiology (Zhao et al., 2008; Sun et al., 2010; Wang et al., 2011; Li et al., 2011a). Therefore, endogenous SO2 is expected to be a new cardiovascular signal molecule after NO, CO and H2S (Li et al., 2009a; Liu et al., 2010).
In the body, cysteine is oxidized by cysteine dioxygenase to form cysteinesulfinate, which generates β‐sulfonyl pyruvate by transamination under the action of aspartate aminotransferase (AAT), and β‐sulfonyl pyruvate further spontaneously decomposes into SO2 and pyruvate. In addition, the oxidation of H2S is one way to generate endogenous SO2. Endogenous SO2 is metabolized to produce sulfites, which are further oxidized by sulfite oxidase to sulfate and excreted in the urine. Endogenous SO2 can be detected in rat plasma, myocardium and vascular tissues (Du et al., 2008). As the key enzyme for endogenous SO2 production, AAT is a pyridoxal phosphate‐dependent transaminase that catalyses the transamination of aspartate to α‐ketoglutarate to form oxaloacetate and glutamate and participate in its reverse reaction. Cysteinesulfinate, similar to the structure of aspartic acid, can be considered an analogue of aspartic acid and is catalysed by AAT via a transamination reaction to produce β‐sulfonated pyruvic acid, by which SO2 is produced. AAT is divided into two subtypes: AAT1 exists in the cytoplasm, and AAT2 exists in mitochondria. The activity of AAT and the mRNA expression of AAT1 and AAT2 is detected in the myocardium and vascular tissues of rats.
Regulatory effects of sulfur dioxide on ion channels in cardiovascular system
ATP‐sensitive potassium channel (KATP)
Isolated aortic rings of rats were used to examine the vascular electrophysiological changes in the presence of SO2. The relaxant reactivity of aortic rings induced by SO2 was observed after precontraction with noradrenaline (NA), and the SO2 derivatives Na2SO3/NaHSO3, with a mol ratio of 3:1, concentration‐dependently relaxed aortic rings. Inhibiting the production of endogenous SO2 by use of hydroxamate enhanced the contractile response of the aortic rings to NA. The relaxant effects of low‐doses of SO2 derivatives (≤4 mM) were inhibited by glibenclamide, a blocker of the KATP channel, so, in part, the relaxant effects of SO2 on rat aortic rings are mediated by the KATP channel (Du et al., 2006). Wang et al. (2009) found that SO2 endothelium‐dependently and ‐independently relaxed rat aortic rings. The diastolic effects induced by SO2 derivatives at a low dose (0.5 and 1 mM) were attenuated by the removal of endothelium, and a similar phenomenon was observed in endothelium‐intact rings after the application of L‐NAME, a non‐selective NOS inhibitor. Hence, the NOS pathway is likely to be involved in the endothelium‐dependent diastolic effects of SO2. However, a higher concentration of SO2 derivatives relaxed aortic rings significantly, and this effect was not inhibited by removal of the endothelium. High K+ (60 mM) inhibited the relaxation of endothelium‐denuded aortic rings to SO2 derivatives (2 mM), and this effect was blocked by glibenclamide (3 μM). Incubation with SO2 or SO2 derivative solution or exposure to SO2 at 14 mg·m−3 increased the expression of the KATP channel subunits Kir6.1, Kir6.2 and SUR2B in rat aortas in vitro (Zhang et al., 2014; 2016). These findings confirm that SO2 relaxes vessels by activating the KATP channel.
The negative inotropic action of SO2 was confirmed by the decreased left‐ventricular pressure and ±dp/dtmax, heart rate and coronary flow in isolated perfused rat hearts (Zhang and Meng, 2012; Zhang et al., 2015). The effects of SO2 were blocked in part by the administration of glibenclamide (Zhang and Meng, 2012). Zhang et al. (2015) examined the effect of SO2 on KATP channel expression and found that Kir6.2 and SUR2A mRNA and protein levels were increased on exposure to SO2 (14 mg·m−3). These findings indicate that the KATP channel participates in the SO2‐induced negative inotropic effects.
Calcium‐activated potassium channel (KCa)
While investigating the endothelium‐independent mechanisms involved in the relaxing effects of SO2 on rat aortic rings, Wang et al. (2009) found that the relaxation of aortic rings, after removal of the endothelium, induced by SO2 derivatives (2 mM) was inhibited by K+ (60 mM). Application of TEA, a blocker of KCa channel, decreased the SO2‐induced relaxation of rings, suggesting that the KCa channel in smooth muscle cells mediates the relaxing effects of SO2 on rat aortic rings (Wang et al., 2009). TEA attenuated the vasorelaxant effects of SO2 on endothelium‐intact and ‐denuded rings. The endothelium‐dependent vasorelaxation to low concentrations of SO2 was blocked in part by iberiotoxin (Zhang and Meng, 2009; Meng et al., 2012). Also the protein expression of the channel subunits α and β1 in the BKCa channel of the rat aorta was up‐regulated by SO2 application (Zhang et al., 2014; Zhang et al., 2016). These data suggest that the BKCa channel mediates the SO2‐induced relaxation of vessels.
Voltage‐gated potassium channel (Kv)
The Kv channel, the most complex subtype of the potassium channel superfamily, is essential in maintaining the physiological function of excitable cells. Nie and Meng (2005a) used whole‐cell patch‐clamp assay to explore the impact of SO2 on the voltage‐gated potassium current in isolated cardiomyocytes in adult rats. SO2 (10 μM) elicited both I to and I K1, with increased peak current amplitude of I to by approximately 37% and I K1 by approximately 26%, which suggests that I to and I K1 are probably involved in the biological effects of SO2 on cardiomyocytes.
L‐type calcium channel
Du et al. (2006) observed that SO2 has relaxant properties in rat aortic rings. The vasoconstrictor responses induced by NA were enhanced by inhibition of SO2. Nicardipine, a blocker of the L‐type calcium channel, attenuated the contractile effects of NA and significantly blocked the vasorelaxant effects of SO2, which confirmed that the L‐type calcium channel is involved in the vasorelaxant effects of SO2. SO2 at a physiological concentration was used to relax rat aortic rings slightly. Pretreatment with nicardipine prevented the vasoconstriction induced by Bay K8644 and blocked the SO2‐induced vasorelaxation of NA‐precontracted aortic rings (Du et al., 2008). CaCl2 (0.01–10 mM) induced a vasoconstriction of endothelium‐denuded aortic rings perfused with 60 mM K+ and Ca2+‐free solution, and this effect was dose‐dependently reduced by SO2. However, the vasoconstriction of endothelium‐denuded aortic rings to CaCl2 was abolished by nifedipine, a blocker of the L‐type calcium channel (Wang et al., 2009). Similar results were observed in rat isolated aortic rings pretreated with nifedipine, and the relaxant effects of SO2 in NA‐precontracted rings were inhibited (Zhang and Meng, 2009; Meng et al., 2012). Further investigations showed that the protein expression of subunits Cav1.2 and Cav1.3 was increased on exposure to SO2 at 14 mg·m−3 or after incubation with SO2 in solution (1500 μM) (Zhang et al., 2014; 2016). These observations indicate that the L‐type calcium channel mediates the vasorelaxant effects of SO2 in rat aorta.
The I Ca,L channel in rat cardiomyocytes was measured by a whole‐cell patch‐clamp assay. SO2 derivatives activated the L‐type calcium channel by reducing the fast and slow time constants of inactivation, promoting the recovery of I Ca,L from inactivation and shifting the channel to more positive potentials (Nie and Meng, 2006). In rat isolated, perfused hearts, SO2 inhibits the cardiac function. These negative inotropic effects of SO2 on the myocardium were blocked by pretreatment with nicardipine (Zhang et al., 2008). To investigate whether SO2 acts on ventricular cardiomyocytes via the L‐type calcium channel, Zhang et al. (2011) used the whole‐cell patch‐clamp method to detect the I Ca,L channel in rat cardiomyocytes. SO2 derivatives depressed the peak amplitudes of I Ca,L, and I Ca,L was decreased in response to SO2, which suggests that SO2 protects cardiomyocytes during cardiovascular diseases by inhibiting the L‐type calcium channel. Zhang and Meng (2012) also confirmed the role of the L‐type calcium channel in the negative inotropic action of gaseous SO2 and its derivatives. The expression of Cav1.2 and Cav1.3 channels in rat hearts was detected after exposure to SO2 at 14 mg·m−3 for 30 days. SO2 inhibited the mRNA and protein expression of Cav1.2 and Cav1.3 in rat hearts (Zhang et al., 2015). Collectively, SO2, by up‐regulating the expression and activity of the L‐type calcium channel, has a negative inotropic effect on cardiomyocytes.
Sodium channel
Sodium currents (I Na) in isolated cardiomyocytes were measured by a whole‐cell patch‐clamp assay. SO2 derivatives activated I Na dose‐dependently, shortened their activation and inactivation time and accelerated their recovery (Nie and Meng, 2005b). The activating effects of SO2 on I Na were significantly enhanced by diethyldithiocarbamate (Wei and Meng, 2008). These findings suggest that SO2 activates the sodium channel, which plays an important role in the cardiovascular system.
Conclusions and perspectives
Extensive studies have confirmed the important roles of the sulfur‐containing gaseous signal molecules H2S and SO2, as a result of their regulatory effects on ion channels, in cardiovascular physiology and the development of cardiovascular diseases. Tables 1 and 2 and Figures 1 and 2 illustrate the important roles of H2S and SO2 in regulating the ion channels and their associated pathways as well as cardiovascular function. However, the molecular mechanisms by which H2S and SO2 target the ion channels have not been fully elucidated. Also, the interaction between H2S and SO2 and their possible integrated action on ion channels are unclear. Further studies are needed to reveal the mechanisms responsible for the effect of H2S and SO2 on ion channels. The potential integrated action of H2S and SO2 and even among the gaseous signal molecules on the structure and function of ion channels are also worthy of investigation. Furthermore, the effect of a single gaseous signal molecule, H2S or SO2, on all the ion channels merits further studies for better understanding the mechanisms of cardiovascular physiology and pathologies, which is of great importance for providing new targets and strategies for treating cardiovascular diseases.
Table 1.
Biological effects of H2S on ion channels in the cardiovascular system
| Molecule | Concentration | Ion channel | Effect | Disease | Reference |
|---|---|---|---|---|---|
| H2S gas | 0.1 μM‐10 mM | KATP ↑ | Vasorelaxant effects | – | Cheng et al. (2004) |
| 10 μM‐10 mM | KCa ↑ | – | Cheng et al. (2004) | ||
| 600 μM | KATP ↑ | – | Zhao et al. (2001) | ||
| 600 μM | L‐type calcium channel ↑ | – | Zhao and Wang (2002) | ||
| 50 μM | KATP ↑ | Electrophysiological evidence | – | Cheng et al. (2004) | |
| 100 μM | KATP ↑ | – | Jiang et al. (2010) | ||
| 300 μM | KATP ↑ | – | Zhao et al. (2001) | ||
| 300 μM | KATP ↑ | – | Tang et al. (2005) | ||
| H2S donor (NaHS) | 90 μmol·kg−1 | KATP ↑ | Vasorelaxant effects | Hypertension | Sun et al. (2015) |
| 100 μM | KATP ↑, IKCa ↑, SKCa ↑ | – | Mustafa et al. (2011) | ||
| 300 μM | TRPA1 ↑ | – | White et al. (2013) | ||
| > 300 μM | KATP ↑ | – | Casalini et al. (2014) | ||
| 1 mM | Cl−/HCO3 − exchangers ↑ | – | Lee et al. (2007) | ||
| 2.8 μmol·kg−1, 40 μM | KATP ↑ | Negative inotropic effects | – | Geng et al. (2004) | |
| 1 μM | KATP ↑ | Cardioprotective effects | Myocardial ischaemia–reperfusion injury | Johansen et al. (2006) | |
| 40 μM | KATP ↑ | Myocardial ischaemia–reperfusion injury | Zhang et al. (2007) | ||
| 100 μM | NCX ↑ | – | Pan et al. (2008) | ||
| 100 μM | L‐type calcium channel ↓T‐type calcium channel ↓ | – | Avanzato et al. (2014) | ||
| 400 μM | CFTR chloride channel ↑ | – | Li et al. (2009b) | ||
| 3 mg·kg−1 | mitoKATP ↑ | Myocardial ischaemia–reperfusion injury | Sivarajah et al. (2009) | ||
| 50 μM | T‐type calcium channel ↑ | Enhance the activity of carotid body | – | Makarenko et al. (2015) | |
| 100 μM | BKCa ↓, I to ↓, I Kir ↓ | Inhibit proliferation and differentiation of atrial fibroblasts | – | Sheng et al. (2013) | |
| 250 μM | NCX ↑, KATP ↑ | Promote angiogenesis | – | Moccia et al. (2011) | |
| 40, 100 μM | KATP ↑ | Electrophysiological evidences | – | Zhang et al. (2007) | |
| 100 μM | L‐type calcium channel ↓ | – | Sun et al. (2008) | ||
| 100 μM | chloride channel ↓ | – | Malekova et al. (2009) | ||
| 100 μM | L‐type calcium channel ↓ | – | Zhang et al. (2012) | ||
| 1.5 mM | T‐type calcium channel ↑ | – | Sekiguchi et al. (2014) | ||
| H2S donor (GYY4137) | 10 μM | NCX1 ↑ | Stimulate apoptosis | – | Markova et al. (2014) |
| H2S donor (Na2S) | 10 μM | KCa ↑ | Vasorelaxant effects | – | Liang et al. (2012) |
| 0.1–0.3 mM | T‐type calcium channel↑ | Electrophysiological evidences | – | Sekiguchi et al. (2014) |
I to, transient outward potassium current; I Kir, inward rectifier potassium current. ↑ stands for activation; ↓ stands for inhibition.
Table 2.
Biological effects of SO2 on ion channels in the cardiovascular system
| Molecule | Concentration | Ion channel | Effect | Disease | Reference |
|---|---|---|---|---|---|
| SO2 gas | 14 mg·m−3 | KATP ↑, BKCa ↑, L‐type calcium channel ↓ | Vasorelaxant effects | – | Zhang et al. (2016) |
| 30, 300 μM | BKCa ↑ | – | Zhang and Meng (2009) | ||
| 1500 μM | KATP ↑ | – | Zhang et al. (2014) | ||
| 1500 μM | KATP ↑, L‐type calcium channel ↓ | – | Zhang and Meng (2009) | ||
| 14 mg·m−3 | KATP ↑, L‐type calcium channel ↓ | Negative inotropic effects | – | Zhang et al. (2015) | |
| 1000 μM | KATP ↑, L‐type calcium channel ↓ | – | Zhang and Meng (2012) | ||
| SO2 derivative (Na2SO3/NaHSO3) | 1500 μM | KATP ↑, BKCa ↑, L‐type calcium channel↓ | Vasorelaxant effects | – | Zhang et al. (2014) |
| 2 mM | KATP ↑, KCa ↑ | – | Wang et al. (2009) | ||
| ≤ 4 mM | KATP ↑ | – | Du et al. (2006) | ||
| 2–8 mM | L‐type calcium channel ↓ | – | Du et al. (2006) | ||
| 6 mM | L‐type calcium channel ↓ | – | Du et al. (2008) | ||
| 10 μM | L‐type calcium channel ↑ | Negative inotropic effects | – | Zhang et al. (2008) | |
| 300, 1000 μM | KATP ↑, L‐type calcium channel ↓ | – | Zhang and Meng (2012) | ||
| 10 μM | I to ↑, I K1 ↑ | Electrophysiological evidence | – | Nie and Meng (2005a) | |
| 10 μM | sodium channel ↑ | – | Wei and Meng (2008) | ||
| 1–200 μM | sodium channel ↑ | – | Nie and Meng (2005b) | ||
| 2–100 μM | L‐type calcium channel ↑ | – | Nie and Meng (2006) | ||
| 50, 100, 500, 1000 μM | L‐type calcium channel ↓ | – | Zhang et al. (2011) | ||
| SO2 derivative (NaHSO3) | 400 μM | BKCa ↑ | Vasorelaxant effects | – | Meng et al. (2012) |
| 2000, 4000 μM | KATP ↑, L‐type calcium channel ↓ | – |
I to, transient outward potassium current; I K1, inward rectifier potassium current; ↑ stands for activation, ↓ stands for inhibition.
Figure 1.
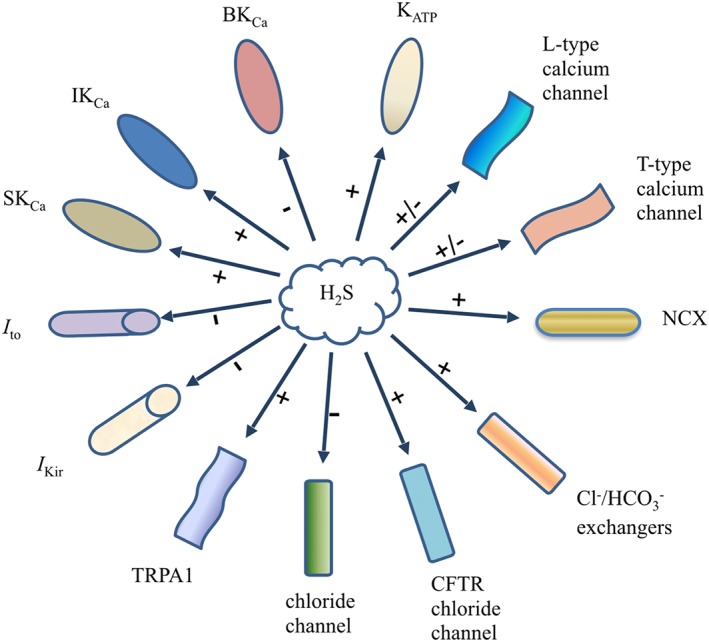
Effects of H2S on ion channels in the cardiovascular system. I to, transient outward potassium current; I Kir, inward rectifier potassium current. + stands for activation; − stands for inhibition.
Figure 2.
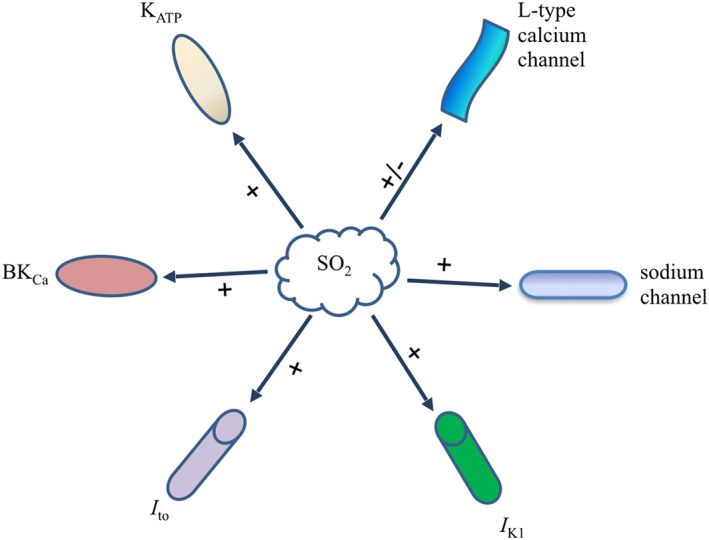
Effects of SO2 on ion channels in the cardiovascular system. I to, transient outward potassium current; I K1, inward rectifier potassium current; + stands for activation, − stands for inhibition.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c,d,e).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This paper was supported by the National Natural Science Foundation of China (nos 91439110 and 81400311), Bejing Natural Science Foundation (no. 7171010), the Major Basic Research Development Program of China (no. 2013CB933801) and National Youth Top‐notch Talent Support Program.
Yu, W. , Jin, H. , Tang, C. , Du, J. , and Zhang, Z. (2018) Sulfur‐containing gaseous signal molecules, ion channels and cardiovascular diseases. British Journal of Pharmacology, 175: 1114–1125. doi: 10.1111/bph.13829.
Contributor Information
Junbao Du, Email: junbaodu1@126.com.
Zhiren Zhang, Email: zhirenz@yahoo.com.
References
- Abe K, Kimura H (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman MJ (1998). The long QT syndrome: ion channel diseases of the heart. Mayo Clin Proc 73: 250–269. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The concise guide to PHARMACOLOGY 2015/16: Other ion channels. Br J Pharmacol 172: 5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015e). The concise guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzato D, Merlino A, Porrera S, Wang R, Munaron L, Mancardi D (2014). Role of calcium channels in the protective effect of hydrogen sulfide in rat cardiomyoblasts. Cell Physiol Biochem 33: 1205–1214. [DOI] [PubMed] [Google Scholar]
- Barañano DE, Ferris CD, Snyder SH (2001). Atypical neural messengers. Trends Neurosci 24: 99–106. [DOI] [PubMed] [Google Scholar]
- Brampton J, Aaronson PI (2016). Role of hydrogen sulfide in systemic and pulmonary hypertension: cellular mechanisms and therapeutic implications. Cardiovasc Hematol Agent Med Chem 14: 4–22. [DOI] [PubMed] [Google Scholar]
- Casalini ED, Goodwill AG, Owen MK, Moberly SP, Berwick ZC, Tune JD (2014). Contribution of hydrogen sulfide to the control of coronary blood flow. Microcirculation 21: 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2000). Structure and regulation of voltage‐gated Ca2+ channels. Ann Rev Cell Dev Biol 16: 521–555. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K, Wang R (2004). Hydrogen sulfide‐induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 287: H2316–H2323. [DOI] [PubMed] [Google Scholar]
- Du SX, Zhang CY, Jin HF, Du JB, Tang CS (2006). Vasorelaxant effect of sulfur dioxide derivatives on isolated aortic rings of rats and its mechanisms. Beijing Da Xue Xue Bao 38: 581–585. [PubMed] [Google Scholar]
- Du SX, Jin HF, Bu DF, Zhao X, Geng B, Tang CS et al. (2008). Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol Sin 29: 923–930. [DOI] [PubMed] [Google Scholar]
- Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J et al. (2004). H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun 313: 362–368. [DOI] [PubMed] [Google Scholar]
- George AL Jr (2014). Recent genetic discoveries implicating ion channels in human cardiovascular diseases. Curr Opin Pharmacol 15: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong QH, Shi XR, Hong ZY, Pan LL, Liu XH, Zhu YZ (2011). A new hope for neurodegeneration: possible role of hydrogen sulfide. J Alzheimers Dis 24: 173–182. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Fryer RM (1999). Sarcolemmal versus mitochondrial ATP‐sensitive K+ channels and myocardial preconditioning. Circ Res 84: 973–979. [DOI] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237: 527–531. [DOI] [PubMed] [Google Scholar]
- Hsiao PY, Tien HC, Lo CP, Juang JM, Wang YH, Sung RJ (2013). Gene mutations in cardiac arrhythmias: a review of recent evidence in ion channelopathies. Appl Clin Genet 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G (1987). Endothelium‐derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A 84: 9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Pang QF, Xu G, Wang L, Wang JK, Zeng YM (2008). Exogenous hydrogen sulfide postconditioning protects isolated rat hearts against ischemia‐reperfusion injury. Eur J Pharmacol 587: 1–7. [DOI] [PubMed] [Google Scholar]
- Jiang B, Tang G, Cao K, Wu L, Wang R (2010). Molecular mechanism for H(2)S‐induced activation of K(ATP) channels. Antioxid Redox Signal 12: 1167–1178. [DOI] [PubMed] [Google Scholar]
- Jin HF, Du JB, Tang CS (2010). Frontiers in research reviews: the pathophysiological significance of sulfur‐containing gases: introduction simple molecules play complicated roles. Clin Exp Pharmacol Physiol 37: 743–744. [DOI] [PubMed] [Google Scholar]
- Johansen D, Ytrehus K, Baxter GF (2006). Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia‐reperfusion injury – evidence for a role of KATP channels. Basic Res Cardiol 101: 53–60. [DOI] [PubMed] [Google Scholar]
- Kimura H (2013). Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem Int 63: 492–497. [DOI] [PubMed] [Google Scholar]
- Lee SW, Cheng Y, Moore PK, Bian JS (2007). Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochem Biophys Res Commun 358: 1142–1147. [DOI] [PubMed] [Google Scholar]
- Li JP, Yang CT, Yang ZL, Liao XX, Wang LC, Huang X et al. (2009b). Roles of CFTR Cl− channels in hydrogen sulfide‐induced cardioprotection and cell proliferation in H9c2 cells. Chin J Pathophysiol 25: 1070–1075. [Google Scholar]
- Li L, Liu D, Bu D, Chen S, Wu J, Tang C et al. (2013). Brg1‐dependent epigenetic control of vascular smooth muscle cell proliferation by hydrogen sulfide. Biochim Biophys Acta 1833: 1347–1355. [DOI] [PubMed] [Google Scholar]
- Li L, Moore PK (2007). An overview of the biological significance of endogenous gases: new roles for old molecules. Biochem Soc Trans 35: 1138–1141. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P, Moore PK (2011b). Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol 51: 169–187. [DOI] [PubMed] [Google Scholar]
- Li W, Tang C, Jin H, Du J (2011a). Regulatory effects of sulfur dioxide on the development of atherosclerotic lesions and vascular hydrogen sulfide in atherosclerotic rats. Atherosclerosis 215: 323–330. [DOI] [PubMed] [Google Scholar]
- Li X, Bazer FW, Gao H, Jobgen W, Johnson GA, Li P et al. (2009a). Amino acids and gaseous signaling. Amino Acids 37: 65–78. [DOI] [PubMed] [Google Scholar]
- Liang GH, Xi Q, Leffler CW, Jaggar JH (2012). Hydrogen sulfide activates Ca2+ sparks to induce cerebral arteriole dilatation. J Physiol 590: 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Jin H, Tang C, Du J (2010). Sulfur dioxide: a novel gaseous signal in the regulation of cardiovascular functions. Mini Rev Med Chem 10: 1039–1045. [DOI] [PubMed] [Google Scholar]
- Liu YH, Lu M, Hu LF, Wong PT, Webb GD, Bian JS (2012). Hydrogen sulfide in the mammalian cardiovascular system. Antioxid Redox Signal 17: 141–185. [DOI] [PubMed] [Google Scholar]
- Makarenko VV, Peng YJ, Yuan G, Fox AP, Kumar GK, Nanduri J et al. (2015). Cav3.2 T‐type Ca2+ channels in H₂S‐mediated hypoxic response of the carotid body. Am J Physiol Cell Physiol 308: C146–C154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malekova L, Krizanova O, Ondrias K (2009). H(2)S and HS(−) donor NaHS inhibits intracellular chloride channels. Gen Physiol Biophys 28: 190–194. [DOI] [PubMed] [Google Scholar]
- Markova J, Hudecova S, Soltysova A, Sirova M, Csaderova L, Lencesova L et al. (2014). Sodium/calcium exchanger is upregulated by sulfide signaling, forms complex with the β1 and β3 but not β2 adrenergic receptors, and induces apoptosis. Pflugers Arch 466: 1329–1342. [DOI] [PubMed] [Google Scholar]
- Meng G, Ma Y, Xie L, Ferro A, Ji Y (2015). Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br J Pharmacol 172: 5501–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Yang Z, Li J, Zhang Q (2012). The vasorelaxant effect and its mechanisms of sodium bisulfite as a sulfur dioxide donor. Chemosphere 89: 579–584. [DOI] [PubMed] [Google Scholar]
- Moccia F, Bertoni G, Pla AF, Dragoni S, Pupo E, Merlino A et al. (2011). Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr Pharm Biotechnol 12: 1416–1426. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK et al. (2009). H2S signals through protein S‐sulfhydration. Sci Signal 2: ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK et al. (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Tsugane M, Oka J, Kimura H (2004). Hydrogen sulfide induces calcium waves in astrocytes. FASEB J 18: 557–559. [DOI] [PubMed] [Google Scholar]
- Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD et al. (2010). Increased coupled gating of L‐type Ca2+ channels during hypertension and Timothy syndrome. Circ Res 106: 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie A, Meng Z (2005a). Sulfur dioxide derivative modulation of potassium channels in rat ventricular myocytes. Arch Biochem Biophys 442: 187–195. [DOI] [PubMed] [Google Scholar]
- Nie A, Meng Z (2005b). Study of the interaction of sulfur dioxide derivative with cardiac sodium channel. Biochim Biophys Acta 1718: 67–73. [DOI] [PubMed] [Google Scholar]
- Nie A, Meng Z (2006). Modulation of L‐type calcium current in rat cardiac myocytes by sulfur dioxide derivatives. Food Chem Toxicol 44: 355–363. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S (1987). Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature 327: 524–526. [DOI] [PubMed] [Google Scholar]
- Pan TT, Neo KL, Hu LF, Yong QC, Bian JS (2008). H2S preconditioning‐induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am J Physiol Cell Physiol 294: C169–C177. [DOI] [PubMed] [Google Scholar]
- Polhemus DJ, Lefer DJ (2014). Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res 114: 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan X, Luo H, Liu Y, Xia H, Chen W, Tang Q (2015). Hydrogen sulfide regulates the colonic motility by inhibiting both L‐type calcium channels and BKCa channels in smooth muscle cells of rat colon. PLoS One 10: e0121331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi F, Miyamoto Y, Kanaoka D, Ide H, Yoshida S, Ohkubo T et al. (2014). Endogenous and exogenous hydrogen sulfide facilitates T‐type calcium channel currents in Cav3.2‐expressing HEK293 cells. Biochem Biophys Res Commun 445: 225–229. [DOI] [PubMed] [Google Scholar]
- Sheng J, Shim W, Wei H, Lim SY, Liew R, Lim TS et al. (2013). Hydrogen sulphide suppresses human atrial fibroblast proliferation and transformation to myofibroblasts. J Cell Mol Med 17: 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K et al. (2009). 3‐Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal 11: 703–714. [DOI] [PubMed] [Google Scholar]
- Singer TP, Kearney EB (1956). Intermediary metabolism of L‐cysteinesulfinic acid in animal tissues. Arch Biochem Biophys 61: 397–409. [DOI] [PubMed] [Google Scholar]
- Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E et al. (2009). Anti‐apoptotic and anti‐inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I⁄ R. Shock 31: 267–274. [DOI] [PubMed] [Google Scholar]
- Skovgaard N, Olson KR (2012). Hydrogen sulfide mediates hypoxic vasoconstriction through a production of mitochondrial ROS in trout gills. Am J Physiol Regul Integr Comp Physiol 303: R487–R494. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Huang Y, Zhang R, Chen Q, Chen J, Zong Y et al. (2015). Hydrogen sulfide upregulates KATP channel expression in vascular smooth muscle cells of spontaneously hypertensive rats. J Mol Med (Berl) 93: 439–455. [DOI] [PubMed] [Google Scholar]
- Sun Y, Tian Y, Prabha M, Liu D, Chen S, Zhang R et al. (2010). Effects of sulfur dioxide on hypoxic pulmonary vascular structural remodeling. Lab Invest 90: 68–82. [DOI] [PubMed] [Google Scholar]
- Sun YG, Cao YX, Wang WW, Ma SF, Yao T, Zhu YC (2008). Hydrogen sulphide is an inhibitor of L‐type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res 79: 632–641. [DOI] [PubMed] [Google Scholar]
- Tang CS, Li XH, Du JB (2006). Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. Curr Vasc Pharmacol 4: 17–22. [DOI] [PubMed] [Google Scholar]
- Tang G, Wu L, Liang W, Wang R (2005). Direct stimulation of KATP channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol Pharmacol 68: 1757–1764. [DOI] [PubMed] [Google Scholar]
- Triggle DJ (1998). The physiological and pharmacological significance of cardiovascular T‐type, voltage‐gated calcium channels. Am J Hypertens 11: 80S–87S. [DOI] [PubMed] [Google Scholar]
- Vandiver M, Snyder SH (2012). Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med (Berl) 90: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, Zhu W, Kanter RJ, Silva JR, Honeywell C, Gow RM et al. (2016). A novel NaV1.5 voltage sensor mutation associated with severe atrial and ventricular arrhythmias. J Mol Cell Cardiol 92: 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MJ, Cai WJ, Zhu YC (2010). Mechanisms of angiogenesis: role of hydrogen sulphide. Clin Exp Pharmacol Physiol 37: 764–771. [DOI] [PubMed] [Google Scholar]
- Wang R (2002). Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 16: 1792–1798. [DOI] [PubMed] [Google Scholar]
- Wang R (2003). The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal 5: 493–501. [DOI] [PubMed] [Google Scholar]
- Wang XB, Huang XM, Ochs T, Li XY, Jin HF, Tang CS et al. (2011). Effect of sulfur dioxide preconditioning on rat myocardial ischemia/reperfusion injury by inducing endoplasmic reticulum stress. Basic Res Cardiol 106: 865–878. [DOI] [PubMed] [Google Scholar]
- Wang YK, Ren AJ, Yang XQ, Wang LG, Rong WF, Tang CS et al. (2009). Sulfur dioxide relaxes rat aorta by endothelium‐dependent and ‐independent mechanisms. Physiol Res 58: 521–527. [DOI] [PubMed] [Google Scholar]
- Wei H, Meng Z (2008). Enhancing effects of diethyldithiocarbamate on increase of sodium channel by sulfur dioxide derivatives in ventricular myocytes. Cell Biol Int 32: 1143–1149. [DOI] [PubMed] [Google Scholar]
- Wei H, Zhang R, Jin H, Liu D, Tang X, Tang C et al. (2010). Hydrogen sulfide attenuates hyperhomocysteinemia‐induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxid Redox Signal 12: 1079–1091. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhang B, Cheng L, Chi M, Deng L, Pan H et al. (2015). Hydrogen sulfide induces neuroprotection against experimental stroke in rats by down‐regulation of AQP4 via activating PKC. Brain Res 1622: 292–299. [DOI] [PubMed] [Google Scholar]
- White BJ, Smith PA, Dunn WR (2013). Hydrogen sulphide‐mediated vasodilatation involves the release of neurotransmitters from sensory nerves in pressurized mesenteric small arteries isolated from rats. Br J Pharmacol 168: 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Bai Y, Yang Z, Tian J, Meng Z (2015). Effect of sulfur dioxide inhalation on the expression of KATP and L‐Ca(2+) channels in rat hearts. Environ Toxicol Pharmacol 39: 1132–1138. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Bai Y, Yang Z, Tian J, Meng Z (2016). The molecular mechanism of the effect of sulfur dioxide inhalation on the potassium and calcium ion channels in rat aortas. Hum Exp Toxicol 35: 418–427. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Meng Z (2009). The vasodilator mechanism of sulfur dioxide on isolated aortic rings of rats: involvement of the K+ and Ca2+ channels. Eur J Pharmacol 602: 117–123. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Meng Z (2012). The negative inotropic effects of gaseous sulfur dioxide and its derivatives in the isolated perfused rat heart. Environ Toxicol 27: 175–184. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Tian J, Bai Y, Lei X, Li M, Yang Z et al. (2014). Effects of gaseous sulfur dioxide and its derivatives on the expression of KATP, BKCa and L‐Ca(2+) channels in rat aortas in vitro. Eur J Pharmacol 742: 31–41. [DOI] [PubMed] [Google Scholar]
- Zhang R, Sun Y, Tsai H, Tang C, Jin H, Du J (2012). Hydrogen sulfide inhibits L‐type calcium currents depending upon the protein sulfhydryl state in rat cardiomyocytes. PLoS One 7: e37073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RY, Du JB, Sun Y, Chen S, Tsai HJ, Yuan L et al. (2011). Sulfur dioxide derivatives depress L‐type calcium channel in rat cardiomyocytes. Clin Exp Pharmacol Physiol 38: 416–422. [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Du JB, Tian Y, Geng B, Tang CS, Tang XY (2008). Effects of sulfur dioxide on cardiac function of isolated perfusion heart of rat. Zhonghua Yi Xue Za Zhi 88: 830–834. [PubMed] [Google Scholar]
- Zhang Z, Huang H, Liu P, Tang C, Wang J (2007). Hydrogen sulfide contributes to cardioprotection during ischemia‐reperfusion injury by opening KATP channels. Can J Physiol Pharmacol 85: 1248–1253. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang R (2002). H2S‐induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 283: H474–H480. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R (2001). The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 20: 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jin HF, Tang CS, Du JB (2008). Effects of sulfur dioxide, on the proliferation and apoptosis of aorta smooth muscle cells in hypertension: experiments with rats. Zhonghua Yi Xue Za Zhi 88: 1279–1283. [PubMed] [Google Scholar]