

Introduction
Helen Ollendorff-Curth (Fig. 1) was one of the first female pioneers in academic dermatology and the study of genodermatoses, and her research contributions continue to resonate today. Born in 1899 into a successful Jewish family in Breslau, Germany (now Wroclaw, Poland), she grew up under the guidance of her mother, Paula, a women’s advocate in her own right. Paula was an elected city councilor and social advocate who worked to establish supportive housing for single women and victims of domestic abuse (Burgdorf and Hoenig, 2013).
Fig. 1.

Helen Ollendorff-Curth. Reprinted from Burgdorf and Scholz (2004), with permission from Elsevier.
Ollendorff-Curth’s early life was not without hardship. She lost her father at a young age and, tragically, only two of her three siblings lived to adulthood. Nevertheless, determined to pursue a career in medicine, Ollendorff-Curth flourished while attending several prestigious German universities, rotating in Breslau, Freiburg, and Munich. This was an extraordinary accomplishment given that she was a Jewish woman, a demographic that was part of an extreme minority in the profession at the time (Burgdorf and Hoenig, 2013, Burgdorf and Scholz, 2004).
When she graduated from medical school in 1923 (Bader and Shipman, 2015), the rise of the Third Reich and the dawn of World War II proved to be pivotal in Ollendorff’s career. Growing anti-Semitism in Germany catalyzed her immigration to the United States where she, along with her husband, Rudolf Wilhelm Paul Curth, would form a longstanding professional relationship with Columbia University (Burgdorf and Hoenig, 2013, Burgdorf and Scholz, 2004).
Of particular interest, Ollendorff-Curth lent both her maiden and married names to four distinct eponymous entities still in use in academic dermatology. First, the Ollendorff probe sign refers to the exquisite tenderness of papules found on the palms, plantar surfaces, face, flexural surfaces, and trunk of patients with secondary syphilis when touched gently during an examination probe (James et al., 2011). The probe sign, which helps distinguish syphilitic from non-syphilitic lesions, was originally published as a component of Ollendorff-Curth’s medical school thesis, which earned her top honors (Burgdorf and Scholz, 2004). Later in her career and before emigrating from Germany, Ollendorff-Curth—as she was known after her marriage—would further define the “Curth criteria” in her seminal work on acanthosis nigricans for the diagnosis of paraneoplastic dermatoses. The “Curth criteria” were defined as concurrent onset with primary neoplasm, parallel development/resolution with primary neoplasm, specificity between tumor type and cutaneous eruption type, and significant statistical and genetic association between dermatosis and malignancy (Bader and Shipman, 2015, Thiers et al., 2009). During this time, she also provided key insight into the classification of acanthosis nigricans, insisting upon differences in disease course and age of onset between the benign and malignant forms of dermatosis. Benign is often early-onset and genetically-determined, while the malignant form is associated with underlying neoplasm (Curth, 1952).
Ollendorff-Curth also earned the distinction of being eponymously commemorated in two separate genodermatoses: Buschke-Ollendorff Syndrome (BOS) under her maiden name, and Ichthyosis hystrix, Curth-Macklin (IHCM) type under her married name (Burgdorf and Hoenig, 2013). The current article provides an update on these two conditions and shows how modern genetic analysis of affected patients has both improved our understanding of the disease pathophysiology and honored the legacy of one of the first female leaders of academic dermatology.
Buschke-Ollendorff Syndrome
After studying in multiple universities in Germany and earning her medical degree, Ollendorff-Curth moved to Berlin in 1924 and further cultivated her academic career under the tutelage of Abraham Buschke, an internationally renowned professor of dermatology (Burgdorf and Scholz, 2004). Under his mentorship, Ollendorff-Curth first described and defined disseminated dermatofibrosis lenticularis, later known as BOS, in 1928 (Burgdorf and Hoenig, 2013, Burgdorf and Scholz, 2004, Woodrow et al., 2001).
BOS is a rare autosomal dominant connective tissue disease, characterized by the formation of elastic and collagenous nevi associated with a spectrum of osseous findings (Woodrow et al., 2001). Dermatologic findings include the development of painless, skin-colored, and often coalescent papules in either an asymmetric or symmetric distribution (Fig. 2). Histological analysis characteristically reveals an increased volume of large, interlacing elastin fibrils (Yadegari et al., 2010). Associated osseous findings vary and typically increase with age. A common associated manifestation is osteopoikilosis, a benign sclerosing bone dysplasia that appears as multiple, well-circumscribed, ovoid opacities seen on plain radiographs of the epiphyses and metaphyses of long bones and the pelvis, and trabeculated small bones of the distal extremities (Fig. 3) (Roberts et al., 1993). These bone islands are small foci of dense remodeled cortical bone with a lamellar structure (Lagier et al., 1984). Some patients may present with melorheostosis, a sclerosing bone dysplasia stemming from abnormal angiogenesis and bone proliferation that characteristically appears as an irregular exophytic cortical hyperostosis resembling dripping candle wax on plain radiography (Fig. 4) (Bansal, 2008). Unlike osteopoikilosis, which is typically an asymptomatic incidental finding, melorheostosis is often painful and may result in severe joint contractures (Gutierrez et al., 2015). Some authors have reported additional, although less frequent, skeletal manifestations of BOS, including otosclerosis (Pope et al., 2016, Schnur et al., 1994), congenital spinal stenosis (Pope et al., 2016, Schnur et al., 1994), scoliosis (Pope et al., 2016), and craniosynostosis (Reid et al., 2008).
Fig. 2.

Painless, skin-colored, coalescent papules characteristic of Buschke-Ollendorff syndrome.
Fig. 3.
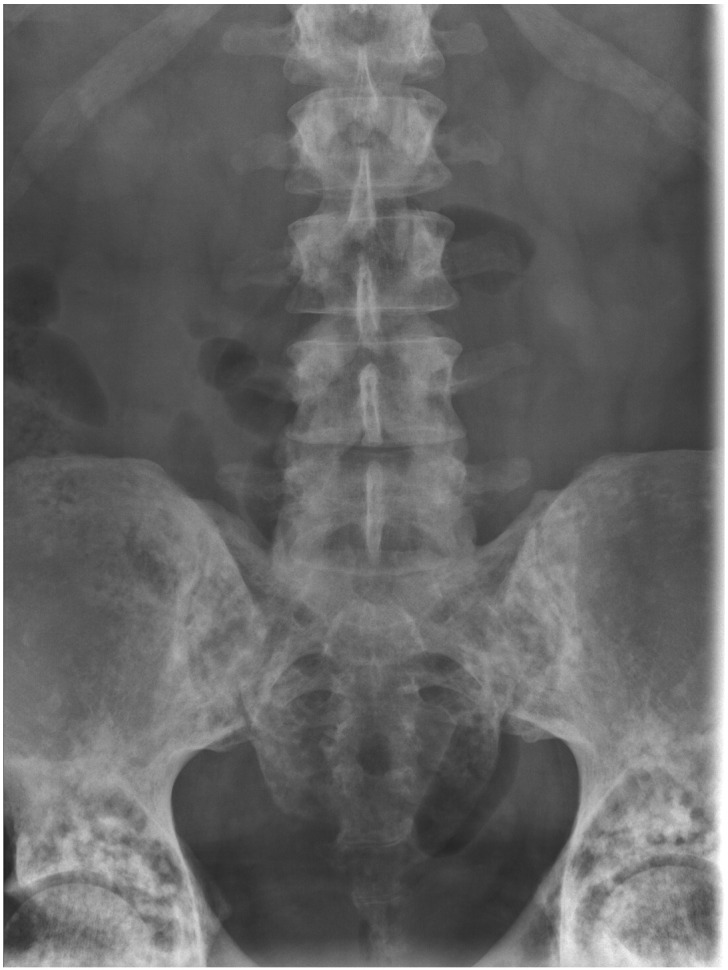
Osteopoikilosis, a benign sclerosing bone dysplasia, affecting the lumbar spine and pelvis.
Fig. 4.
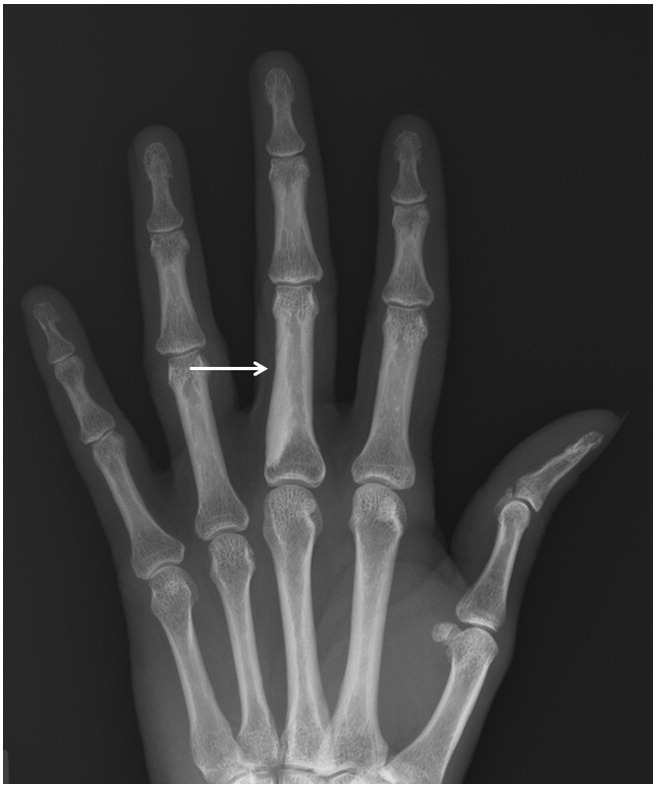
Melorheostosis involving the ulnar aspect of the third proximal phalanx. Note that it takes on the appearance of dripping candle wax.
Recent genome-wide linkage analysis studies showed the underlying pathogenesis of the dermatologic and skeletal findings originally described by Ollendorff. BOS is an autosomal dominant disease, described in several series in the literature as recurring in families (Hellemans et al., 2004, Kawamura et al., 2005, Yadegari et al., 2010, Zhang et al., 2009). The causative mutation is believed to be in the gene encoding the LEM domain-containing protein 3 (LEMD3), an inner nuclear membrane protein, which, via interactions through Smad proteins, plays a key role in the regulation of transforming growth factor beta (TGF-β) and bone morphogenic protein (Hellemans et al., 2004, Zhang et al., 2009). Abnormal function of mutant LEMD3 leads to unmitigated TGF-β signaling and consequent increased steady-state levels of elastin messenger RNA (mRNA) and elastin accumulation in the dermis (Woodrow et al., 2001). Of note, increased TGF-β signaling has also been associated with other fibrotic skin disorders (Hellemans et al., 2004, Saito et al., 2001, Mori et al., 2003) and hypertrophic scarring (Korekawa et al., 2012). The skeletal findings in BOS may be directly attributed to loss of LEMD3 function, as increased activity of both bone morphogenic protein and TGF-β have been implicated in increased bone formation in a variety of sclerosing bone disorders (de Vernejoul and Kornak, 2010, Hellemans et al., 2004). To date, 125 pathogenic mutations of the LEMD3 gene have been detected (Kratzsch et al., 2016), which cause the skeletal and dermatologic findings often encountered in BOS through a multitude of different mechanisms, including nonsense-mediated decay of LEMD3 mRNA (Burger et al., 2010) and nonsense-mediated obliteration of the functional domains of the protein (Yuste-Chaves et al., 2011).
In the majority of cases, no specific treatment is indicated because the skin papules are often small and painless (Pope et al., 2016). There is also no significant increase in mortality. However, early and accurate diagnosis of BOS in patients with concomitant elastic nevi and benign osteopoikilosis is paramount in avoiding unnecessary workup and patient anxiety for the incidental discovery of bone islands, which may appear as sclerotic neoplastic disease. Moreover, in patients diagnosed with melorheostosis, active pain management, bisphosphonate administration, and surgical correction may be indicated in the most severe cases (Moulder and Marsh, 2006, Slimani et al., 2013, Takashima et al., 2016).
Ichthyosis Hystrix, Curth-Macklin Type
While flourishing under Buschke’s mentorship, Ollendorff-Curth met fellow dermatologist Rudolf Wilhelm Paul Curth, who joined the department in 1925. Their relationship rapidly evolved from a professional collaboration to friendship and finally to love, and they were married on New Year’s Eve in 1927 (Fig. 5) (Burgdorf and Scholz, 2004). The couple immigrated to the United States in 1931 where they opened a medical practice in New York City and formed a long-standing association with Columbia University (Burgdorf and Hoenig, 2013, Burgdorf and Scholz, 2004). Ollendorff-Curth continued her landmark work in the study of genodermatoses and began collaborating in 1954 with fellow female academic medical geneticist, Madge Thurlow Macklin (Fig. 6). A luminary in her own right who originally coined the term medical genetics, Macklin conducted compelling research on the contribution of genetic factors to the development of many diseases, such as cancer and mental illness, and advocated strongly for the inclusion of genetics studies in medical school curricula (Comfort, 2006).
Fig. 5.

Helen Ollendorff-Curth and Rudolf Wilhelm Paul Curth. Reprinted from Burgdorf and Scholz (2004), with permission from Elsevier.
Fig. 6.

Madge Thurlow Macklin (center). Acc. 90-105 - Science Service, Records, 1920s-1970s, Smithsonian Institution Archives.
In 1954, Ollendorff-Curth and Macklin played a pivotal role in the first description of a form of ichthyosis hystrix (Bader and Shipman, 2015, Ollendorff Curth et al., 1972). Referred to as IHCM, this extremely rare genodermatosis has the unique distinction of being one of the first syndromes named after two women (Burgdorf and Hoenig, 2013, Burgdorf and Scholz, 2004). Different clinical variants of Ichthyosis hystrix, characterized by bilateral, widespread development of gray-dark brown, hyperkeratotic verrucae, scales, and plaques, have been recognized in the literature (Fig. 7) (James et al., 2011, Mehta et al., 2015). In IHCM, hyperkeratotic plaques may coalesce over flexor and extensor surfaces, including palmar and plantar surfaces, causing painful, bleeding fissures. Unlike epidermolytic hyperkeratosis, a similar condition, lesions in IHCM do not demonstrate erythroderma, skin fragility, or blistering (Mehta et al., 2015). Light microscopy of involved skin demonstrates keratin intermediate filaments aggregated into continuous peripheral shells, perinuclear vacuoles, and binuclear keratinocytes. Electron microscopy, however, shows no keratin clumping, suggesting that expression of abnormal keratin plays a central role in pathogenesis (Niemi et al., 1990, Sprecher et al., 2001).
Fig. 7.

Severe palmoplantar keratoderma often seen in Ichthyosis hystrix, Curth-Macklin type. Photo courtesy Leslie Castelo-Soccio, MD, PhD. Children’s Hospital of Philadelphia.
Although sporadic cases have been reported in the literature (Mehta et al., 2015, Yusuf et al., 2009, Brusasco et al., 1994), IHCM is typically described using Ollendorff-Curth’s original description (Ollendorff-Curth et al., 1972) as an autosomal dominant condition recurring in two families (Niemi et al., 1990, Sprecher et al., 2001). More recent genetic linkage studies have revealed that various mutations in the KRT1 gene, which encodes keratin 1, play a central role in disease pathogenesis (Fonseca et al., 2013, Ishida-Yamamoto et al., 2003, Kubo et al., 2011, Richardson et al., 2006, Sprecher et al., 2001). Wild-type keratin 1, a component of the intermediate filament cytoskeleton in the suprabasal epidermis, normally heterodimerizes via weak hydrophobic and hydrogen bonding interactions with keratin 10 (Richardson et al., 2006) and enables crosslinking between the cornified cell envelope and the intracellular matrix to contribute to the barrier integrity of the skin (Roth et al., 2012). The ability of keratin to dimerize and crosslink is believed to be dependent on the presence of glycine loops present in the variable tail domain of keratin 1. Many of the reported mutations implicated in clinically established IHCM diminish the number of glycine loops present in the variable tail domain of the translated protein (Fonseca et al., 2013, Ishida-Yamamoto et al., 2003, Kubo et al., 2011, Richardson et al., 2006, Sprecher et al., 2001), resulting in abnormal interactions between the cell envelope and intracellular matrix. Consequently, this leads to abnormal keratinization and the clinical phenotype characterized by hyperkeratotic plaques and skin fissures.
Treatment options for this rare condition are limited. Although no specific treatments for IHCM exist, systemic retinoids can be of limited benefit (Kanerva et al., 1984) but must be prescribed with caution in children due to the potential risk of skeletal toxicity (Sethuraman et al., 2016). Topical emollients, calcipotriol ointment, and liarozole cream appear to result in some therapeutic benefit, but evaluation of long-term efficacy and safety is needed (Hernandez-Martin et al., 2013).
Conclusion
Commemorated with four distinct eponyms, Helen Ollendorff-Curth contributed greatly to the field of dermatology and the study of genodermatoses. In her final years, she was diagnosed with Alzheimer’s dementia and after her death in 1982, her brain was donated to science to study the disease (Burgdorf and Scholz, 2004). Ollendorff-Curth was survived by her husband, who passed away from carcinoma of the breast only 2 years later. Their daughter, Elisabeth Susanne Marie Curth, would go on to graduate from Smith College and earn a master’s degree from Teachers College at Columbia University.
Ollendorff-Curth’s impact on academic dermatology remains evident today. Current genetic studies have shed a powerful light on the pathogenesis of these conditions and established compelling avenues for future research to further understand the intricate disease mechanisms and help affected patients. Moreover, her legacy continues after her death through the many distinguished trainees Ollendorff-Curth influenced throughout her life, including Drs. Robert W. Goltz, Robert K. Gorlin, and David R. Bickers. These and countless other trainees continue to conduct their own groundbreaking research and serve as academic leaders, committed educators, and master clinicians. The legacy of Helen Ollendorff-Curth as one of dermatology’s first and most influential female academicians will surely not be forgotten.
References
- Bader E, Shipman AR. The women behind the names: dermatology eponyms named after women. Int J Womens Dermatol. 2015;1:157–160. doi: 10.1016/j.ijwd.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A. The dripping candle wax sign. Radiology. 2008;246(2):638–640. doi: 10.1148/radiol.2462050537. [DOI] [PubMed] [Google Scholar]
- Brusasco A, Cavalli R, Cambiaghi S, Tadini G, Berti E, Caputo R. Ichthyosis Curth-Macklin: a new sporadic case with immunohistochemical study of keratin expression. Arch Dermatol. 1994;130(8):1077–1079. [PubMed] [Google Scholar]
- Burgdorf WH, Hoenig LJ. A female pioneer doubly recognized. JAMA Dermatol. 2013;149(5):527. doi: 10.1001/jamadermatol.2013.2777. [DOI] [PubMed] [Google Scholar]
- Burgdorf WH, Scholz A. Helen Ollendorff Curth and William Curth: from Breslau and Berlin to Bar Harbor. J Am Acad Dermatol. 2004;51(1):84–89. doi: 10.1016/j.jaad.2003.12.035. [DOI] [PubMed] [Google Scholar]
- Burger B, Hershkovitz D, Indelman M, Kovac M, Galambos J, Haeusermann P. Buschke-Ollendorff syndrome in a three-generation family: influence of a novel LEMD3 mutation to tropoelastin expression. Eur J Dermatol. 2010;20(6):693–697. doi: 10.1684/ejd.2010.1051. [DOI] [PubMed] [Google Scholar]
- Comfort N. "Polyhybrid heterogeneous bastards": promoting medical genetics in America in the 1930s and 1940s. J Hist Med Allied Sci. 2006;61(4):415–455. doi: 10.1093/jhmas/jrl001. [DOI] [PubMed] [Google Scholar]
- Curth HO. Significance of acanthosis nigricans. AMA Arch Derm Syphilol. 1952;66(1):80–100. doi: 10.1001/archderm.1952.01530260083009. [DOI] [PubMed] [Google Scholar]
- de Vernejoul MC, Kornak U. Heritable sclerosing bone disorders: presentation and new molecular mechanisms. Ann N Y Acad Sci. 2010;1192:269–277. doi: 10.1111/j.1749-6632.2009.05244.x. [DOI] [PubMed] [Google Scholar]
- Fonseca DJ, Rojas RF, Vergara JI, Rios X, Uribe C, Chavez L. A severe familial phenotype of Ichthyosis Curth-Macklin caused by a novel mutation in the KRT1 gene. Br J Dermatol. 2013;168(2):456–458. doi: 10.1111/j.1365-2133.2012.11181.x. [DOI] [PubMed] [Google Scholar]
- Gutierrez D, Cooper KD, Mitchell AL, Cohn HI. Novel somatic mutation in LEMD3 splice site results in Buschke-Ollendorff syndrome with polyostotic melorheostosis and osteopoikilosis. Pediatr Dermatol. 2015;32(5):e219–e220. doi: 10.1111/pde.12634. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004;36(11):1213–1218. doi: 10.1038/ng1453. [DOI] [PubMed] [Google Scholar]
- Hernandez-Martin A, Aranegui B, Martin-Santiago A, Garcia-Doval I. A systematic review of clinical trials of treatments for the congenital ichthyoses, excluding ichthyosis vulgaris. J Am Acad Dermatol. 2013;69(4):544–549.e8. doi: 10.1016/j.jaad.2013.05.017. [DOI] [PubMed] [Google Scholar]
- Ishida-Yamamoto A, Richard G, Takahashi H, Iizuka H. In vivo studies of mutant keratin 1 in ichthyosis hystrix Curth-Macklin. J Invest Dermatol. 2003;120(3):498–500. doi: 10.1046/j.1523-1747.2003.12064.x. [DOI] [PubMed] [Google Scholar]
- James WD, Elston DM, Berger TG, Andrews GC. 11th ed. Saunders Elsevier; London: 2011. Andrews' diseases of the skin: clinical dermatology. [Google Scholar]
- Kanerva L, Karvonen J, Oikarinen A, Lauharanta J, Ruokonen A, Niemi KM. Ichthyosis hystrix (Curth-Macklin). Light and electron microscopic studies performed before and after etretinate treatment. Arch Dermatol. 1984;120(9):1218–1223. doi: 10.1001/archderm.120.9.1218. [DOI] [PubMed] [Google Scholar]
- Kawamura A, Ochiai T, Tan-Kinoshita M, Suzuki H. Buschke-Ollendorff syndrome: three generations in a Japanese family. Pediatr Dermatol. 2005;22(2):133–137. doi: 10.1111/j.1525-1470.2005.22209.x. [DOI] [PubMed] [Google Scholar]
- Korekawa A, Nakano H, Toyomaki Y, Takiyoshi N, Rokunohe D, Akasaka E. Buschke-Ollendorff syndrome associated with hypertrophic scar formation: a possible role for LEMD3 mutation. Br J Dermatol. 2012;166(4):900–903. doi: 10.1111/j.1365-2133.2011.10691.x. [DOI] [PubMed] [Google Scholar]
- Kratzsch J, Mitter D, Ziemer M, Kohlhase J, Voth H. Identification of a novel point mutation in the LEMD3 gene in an infant with Buschke-Ollendorff syndrome. JAMA Dermatol. 2016 doi: 10.1001/jamadermatol.2016.0350. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Urano Y, Matsuda R, Ishigami T, Murao K, Arase S. Ichthyosis hystrix, Curth-Macklin type: a new sporadic case with a novel mutation of keratin 1. Arch Dermatol. 2011;147(8):999–1001. doi: 10.1001/archdermatol.2011.217. [DOI] [PubMed] [Google Scholar]
- Lagier R, Mbakop A, Bigler A. Osteopoikilosis: a radiological and pathological study. Skelet Radiol. 1984;11(3):161–168. doi: 10.1007/BF00349489. [DOI] [PubMed] [Google Scholar]
- Mehta S, Agarwal US, Agarwal N. A sporadic case of Ichthyosis Curth Macklin: rare presentation of a rare disease. Indian J Dermatol. 2015;60(5):522. doi: 10.4103/0019-5154.164439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Chen SJ, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003;48(7):1964–1978. doi: 10.1002/art.11157. [DOI] [PubMed] [Google Scholar]
- Moulder E, Marsh C. Soft tissue knee contracture of the knee due to melorheostosis, treated by total knee arthroplasty. Knee. 2006;13(5):395–396. doi: 10.1016/j.knee.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Niemi KM, Virtanen I, Kanerva L, Muttilainen M. Altered keratin expression in ichthyosis hystrix Curth-Macklin. A light and electron microscopic study. Arch Dermatol Res. 1990;282(4):227–233. doi: 10.1007/BF00371641. [DOI] [PubMed] [Google Scholar]
- Ollendorff-Curth H, Allen FH, Jr., Schnyder UW, Anton-Lamprecht I. Follow-up of a family group suffering from ichthyosis hystrix type Curth-Macklin. Humangenetik. 1972;17(1):37–48. doi: 10.1007/BF01789598. [DOI] [PubMed] [Google Scholar]
- Pope V, Dupuis L, Kannu P, Mendoza-Londono R, Sajic D, So J. Buschke-Ollendorff syndrome: a novel case series and systematic review. Br J Dermatol. 2016;174(4):723–729. doi: 10.1111/bjd.14366. [DOI] [PubMed] [Google Scholar]
- Reid EM, Baker BL, Stees MA, Stone SP. Buschke-Ollendorff syndrome: a 32-month-old boy with elastomas and craniosynostosis. Pediatr Dermatol. 2008;25(3):349–351. doi: 10.1111/j.1525-1470.2008.00680.x. [DOI] [PubMed] [Google Scholar]
- Richardson ES, Lee JB, Hyde PH, Richard G. A novel mutation and large size polymorphism affecting the V2 domain of keratin 1 in an African-American family with severe, diffuse palmoplantar keratoderma of the ichthyosis hystrix Curth-Macklin type. J Invest Dermatol. 2006;126(1):79–84. doi: 10.1038/sj.jid.5700025. [DOI] [PubMed] [Google Scholar]
- Roberts NM, Langtry JA, Branfoot AC, Gleeson J, Staughton RC. Case report: osteopoikilosis and the Buschke-Ollendorff syndrome. Br J Radiol. 1993;66(785):468–470. doi: 10.1259/0007-1285-66-785-468. [DOI] [PubMed] [Google Scholar]
- Roth W, Kumar V, Beer HD, Richter M, Wohlenberg C, Reuter U. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J Cell Sci. 2012;125(Pt 22):5269–5279. doi: 10.1242/jcs.116574. [DOI] [PubMed] [Google Scholar]
- Saito T, Kinoshita A, Yoshiura K, Makita Y, Wakui K, Honke K. Domain-specific mutations of a transforming growth factor (TGF)-beta 1 latency-associated peptide cause Camurati-Engelmann disease because of the formation of a constitutively active form of TGF-beta 1. J Biol Chem. 2001;276(15):11469–11472. doi: 10.1074/jbc.C000859200. [DOI] [PubMed] [Google Scholar]
- Schnur RE, Grace K, Herzberg A. Buschke-Ollendorff syndrome, otosclerosis, and congenital spinal stenosis. Pediatr Dermatol. 1994;11(1):31–34. doi: 10.1111/j.1525-1470.1994.tb00070.x. [DOI] [PubMed] [Google Scholar]
- Sethuraman G, Marwaha RK, Challa A, Yenamandra VK, Ramakrishnan L, Thulkar S. Vitamin D: a new promising therapy for congenital ichthyosis. Pediatrics. 2016;137(1) doi: 10.1542/peds.2015-1313. [DOI] [PubMed] [Google Scholar]
- Slimani S, Nezzar A, Makhloufi H. Successful treatment of pain in melorheostosis with zoledronate, with improvement on bone scintigraphy. BMJ Case Rep. 2013;2013 doi: 10.1136/bcr-2013-009820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher E, Ishida-Yamamoto A, Becker OM, Marekov L, Miller CJ, Steinert PM. Evidence for novel functions of the keratin tail emerging from a mutation causing ichthyosis hystrix. J Invest Dermatol. 2001;116(4):511–519. doi: 10.1046/j.1523-1747.2001.01292.x. [DOI] [PubMed] [Google Scholar]
- Takashima S, Fujita Y, Suzuki S, Saito N, Shinkuma S, Nomura T. RNA recognition motif of LEMD3 as a key player in the pathogenesis of Buschke-Ollendorff syndrome. J Dermatol Sci. 2016;81(3):205–208. doi: 10.1016/j.jdermsci.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59(2):73–98. doi: 10.3322/caac.20005. [DOI] [PubMed] [Google Scholar]
- Woodrow SL, Pope FM, Handfield-Jones SE. The Buschke-Ollendorff syndrome presenting as familial elastic tissue naevi. Br J Dermatol. 2001;144(4):890–893. doi: 10.1046/j.1365-2133.2001.04152.x. [DOI] [PubMed] [Google Scholar]
- Yadegari M, Whyte MP, Mumm S, Phelps RG, Shanske A, Totty WG. Buschke-Ollendorff syndrome: absence of LEMD3 mutation in an affected family. Arch Dermatol. 2010;146(1):63–68. doi: 10.1001/archdermatol.2009.320. [DOI] [PubMed] [Google Scholar]
- Yuste-Chaves M, Canueto J, Santos-Briz A, Ciria S, Gonzalez-Sarmiento R, Unamuno P. Buschke-Ollendorff syndrome with striking phenotypic variation resulting from a novel c.2203C > T nonsense mutation in LEMD3. Pediatr Dermatol. 2011;28(4):447–450. doi: 10.1111/j.1525-1470.2010.01206.x. [DOI] [PubMed] [Google Scholar]
- Yusuf SM, Mijinyawa MS, Maiyaki MB, Mohammed AZ. Ichthyosis hystrix Curth-Macklin type in an African girl. Int J Dermatol. 2009;48(12):1343–1345. doi: 10.1111/j.1365-4632.2007.03291.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Castori M, Ferranti G, Paradisi M, Wordsworth BP. Novel and recurrent germline LEMD3 mutations causing Buschke-Ollendorff syndrome and osteopoikilosis but not isolated melorheostosis. Clin Genet. 2009;75(6):556–561. doi: 10.1111/j.1399-0004.2009.01177.x. [DOI] [PubMed] [Google Scholar]