

SUMMARY
Inactivating mutations in the thyroid hormone (TH) transporter Monocarboxylate transporter 8 (MCT8) cause severe psychomotor retardation in children. Animal models do not reflect the biology of the human disease. Using patient-specific induced pluripotent stem cells (iPSCs), we generated MCT8- deficient neural cells that showed normal TH-dependent neuronal properties and maturation. However, the blood-brain barrier (BBB) controls TH entry into the brain, and reduced TH availability to neural cells could instead underlie the diseased phenotype. To test potential BBB involvement, we generated an iPSC-based BBB model of MCT8 deficiency, and we found that MCT8 was necessary for polarized influx of the active form of TH across the BBB. We also found that a candidate drug did not appreciably cross the mutant BBB. Our results therefore clarify the underlying physiological basis of this disorder, and they suggest that circumventing the diseased BBB to deliver active TH to the brain could be a viable therapeutic strategy.
In Brief
Vatine et al. show that human iPSC-based modeling can pinpoint the origin of a neuronal disorder in the brain as a defect in transport of thyroid hormone across the blood-brain barrier, rather than in the neurons themselves.
Graphical Abstract
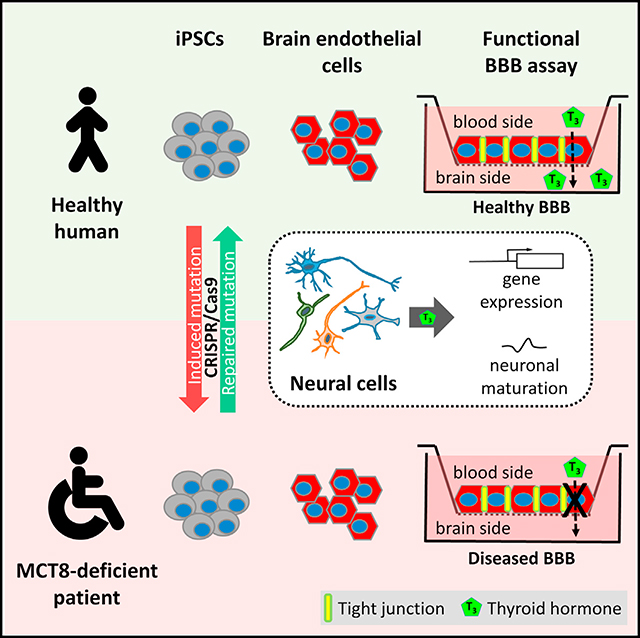
INTRODUCTION
The solute carrier family 16, member 2 (SLC16A2) gene, located on the X chromosome, encodes monocarboxylate transporter 8 (MCT8), a specific membrane transporter of thyroid hormone (TH) (Friesema et al., 2003) broadly expressed in the central nervous system (CNS). Inactivating mutations of MCT8 in males cause serum TH abnormalities and severe neuropsychomotor impairments (Dumitrescu et al., 2004; Friesema et al., 2004). The thyroid phenotype includes elevated serum levels of total and free L-3,3′,5-triiodothyronine (T3) and reduced serum levels of L-3,3′,5,5′-tetraiodothyronine (thyroxine or T4), as well as hypermetabolism. This condition, named the Allan-Herndon-Dudley syndrome (AHDS), was initially described in patients with sex-linked mental retardation (Allan et al., 1944). The one published post-mortem analysis of a MCT8-deficient fetus revealed a decrease in T3 and T4 contents in the cerebral cortex (López-Espíndola et al., 2014), indicating that impaired transport of TH into the CNS may underlie AHDS. However, the exact mechanisms remain poorly understood.
Various animal models of the disease have been generated (Dumitrescu et al., 2006; Trajkovic et al., 2007; Vatine et al., 2013; Zada et al., 2014). Intriguingly, two Mct8 knockout (KO) mouse strains closely recapitulate the thyroid phenotype, but they fail to display neurological or behavioral abnormalities (Dumitrescu et al., 2006; Trajkovic et al., 2007). When these mice were made hypothyroid at the neonatal stage, the exogenous supplementation of T4, but not T3, was able to rescue a mild phenotype in the cerebellum (Ceballos et al., 2009). This suggests that T4, but not T3, may cross the mouse MCT8-deficient blood-brain barrier (BBB) through the presence of a mouse-specific T4 transporter that is absent in human. Several transgenic models were subsequently generated to identify the mouse-specific TH transporter. These included KO models of Mct10 (Slc16a10) (Müller et al., 2014), L-type amino acid transporter (Lat2 and Slc7a8) (Núñez et al., 2014), and Organic anion-transporting polypeptide 1c1 (Oatp1c1 and SlcO1c1), either as independent KO or with MCT8 (Mayerl et al., 2012, 2014). While most of these models did not exhibit the desired neurological phenotype, the Mct8/Oatp1c1 double KO displayed a severe form of CNS-specific hypothyroidism, including decreased myelination and abnormal neuronal differentiation (Mayerl et al., 2014). Interestingly, Oatp1c1 is highly expressed in endothelial cells of the rodent BBB (Roberts et al., 2008), but not the human BBB (Uchida et al., 2011), suggesting a crucial differential regulation of TH transporters among species.
MCT8-deficient patient fibroblasts and human cell lines expressing wild-type or mutant MCT8 have been used to analyze TH transport (Visser et al., 2009) and gene expression (Visser et al., 2010). While these cell models showed some features of MCT8 deficiency, they do not harbor the characteristics of affected neural or endothelial cells that are required for a relevant human cell-based disease model of AHDS. Technologies to reprogram adult cells back to a pluripotent state, termed induced pluripotent stem cells (iPSCs) (Takahashi and Yamanaka, 2006; Yu et al., 2007), provide an unprecedented platform for studying human genetic disorders (Ebert et al., 2009; Mattis et al., 2015). Moreover, the development of protocols to differentiate human pluripotent cells into neural and brain endothelial cells permits the generation of disease-in-a-dish models for human MCT8 deficiency. These models using relevant brain cell types could potentially close the gap between inadequate animal models and the human AHDS condition.
Here we report the results from iPSCs generated from patients with MCT8 mutations, along with associated controls and isogenic cell lines engineered using the CRISPR/Cas9 system (Li et al., 2013). MCT8-deficient iPSC-derived neural cells showed diminished TH uptake, however, they still displayed normal T3-dependent neuronal maturation. This suggested that, rather than MCT8 transporter deficiency, reduced TH availability to neural cells may underlie the neuronal-related phenotypes of AHDS. As the BBB controls TH entry into the brain, iPSC-derived brain microvascular endothelial cells were also used to model the diseased MCT8-deficient human BBB. We demonstrate that T3 transport across the BBB is dependent on MCT8, likely driving the decreased concentrations of T3 observed in MCT8-deficient brains. In summary, this study provides the first iPSC-based disease model of the human BBB, and it shows that the diseased BBB may be the primary obstacle for T3 transport into the brain, providing both a critical mechanistic insight and a new tool for future drug development.
RESULTS
MCT8-Deficient Patient iPSCs Can Be Differentiated into Neural Cells
Inactivating mutations in the TH transporter have been studied in animal models, but these models do not precisely represent the human condition (Heuer and Visser, 2013). To study MCT8 deficiency in a human model, fibroblasts from two severely affected male patients with MCT8 deficiency (CS58iMCT8 and CS01iMCT8) were reprogrammed into iPSCs (Figure S1). Control lines used were CS03iCTR (parental line of CS01iMCT8), CS02iCTR, CS00iCTR, and CS14iCTR (see Table S1 for additional information). In addition, isogenic lines in which MCT8 mutations were induced (CS03iCTRmut) or corrected (CS01iMCT8cor) using the CRISPR/Cas9 system were generated (Figure S2; Table S2).
MCT8 deficiency leads to critical changes in neural function that manifest in psychomotor retardation (Dumitrescu et al., 2004; Friesema et al., 2004). To investigate impairments in human neural cells caused by MCT8 deficiency, patient and control iPSCs were differentiated into neural cells (Figure 1A). The iPSC colonies were maintained in suspension as free-floating spheres, previously termed EZ-spheres (Ebert et al., 2013; Shelley et al., 2014). EZ-spheres contained Nestin+ neural progenitor cells, which could be differentiated into neural cultures expressing βIII-Tubulin+ neurons and glial fibrillary acidic protein (GFAP)+ astrocytes that expressed MCT8 (Figure 1B). Western blot analysis confirmed MCT8 expression in the CS03iCTR control line as well as in the CS01iMCT8 line that expressed mutated, nonfunctional MCT8 protein and its corrected isogenic line (CS01iMCT8cor), but not in the CS03iCTRmut line that was engineered to express a truncated MCT8 protein (Figure 1C). RNA sequencing (RNA-seq) analysis on differentiated neural cells revealed that these cells not only expressed MCT8 but also other TH transporters, LAT1 and LAT2, and low levels of OATP1C1 (Figure 1D), with similar levels in control and MCT8-deficient cells. In addition, assessing iodothyronine deiodinase mRNA levels revealed that DIO3, but not DIO1 and DIO2, was expressed (Figure 1E), suggesting that these cells can inactivate T3 and T4.
Figure 1. Properties of iPSC-Derived MCT8-Deficient Neural Cells.

(A) Scheme for differentiating iPSCs to neural cultures composed of neural progenitors, astrocytes, and neurons.
(B) Immunocytochemistry on CS03iCTR control line demonstrates that βIII-Tubulin neurons (red) and GFAP glia (red) produce MCT8 protein (green), with a DAPI nuclei stain (20× magnification).
(C) Western blot confirms MCT8-deficient and functional MCT8 neural cultures produce MCT8, while the mutant isogenic line does not produce detectable MCT8. GAPDH housekeeping protein used as a loading control.
(D and E) RNA-seq analysis shows RNA expression of (D) TH transporters and (E) deiodinases (DIO) in MCT8-deficient and functional MCT8 neural cultures. Data are expressed as fragments per kilobase of transcript per million mapped reads (FPKM). There were no significant differences among lines.
(F-I) Accumulation or clearance rates of 1 nM radiolabeled T3 or T4 were quantified in MCT8-deficient and control neural cultures. Levels are expressed as percent of cellular T3 or T4 normalized to protein amount per minute. (F) T3 uptake (ANOVA, n = 5, p < 0.0077), (G) T3 efflux (ANOVA, n = 5, p < 0.0479), (H) T4 uptake (ANOVA, n = 4, p < 0.0099), and (I) T4 efflux (ANOVA, n = 3, p < 0.0309) were all significantly reduced in MCT8-deficient neural cells compared to control neural cells.
(J) qRT-PCR quantification of T3-induced KLF9 gene expression showed a significant linear relationship between mRNA expression and T3 concentrations (at 0.1 to 1 nM) for both the healthy control (CS03iCTR, r = 0.99122, p < 0.0001) and MCT8-deficient (CS58iMCT8, r = 0.98346, p < 0.0001) neural cells. No statistically significant differences were observed in linear regression across cell lines (ANOVA).
(K) qRT-PCR quantification of T3-induced HR mRNA showed a significant linear relationship between mRNA expression and T3 concentrations (at 0.1 to 1 nM) for both the healthy control (CS03iCTR, r = 0.93436, p < 0.0001) and MCT8-deficient (CS58iMCT8, r = 0.94338, p < 0.0001) neural cells. No statistically significant differences were observed in linear regression across cell lines (ANOVA).
See also Figures S1–S3 and Tables S1 and S2.
MCT8-Deficient Neural Cells Have Reduced TH Transport but Normal T3-Induced Transcription
MCT8 deficiency may affect neural function by reducing the cells’ ability to transport TH. As such, TH transport in and out of iPSC-derived neural cells was quantified using radiolabeled TH. Notably, compared to both control lines, the MCT8-deficient cell lines exhibited a significant 5-fold decrease in T3 uptake (p < 0.005; Figure 1F) and a 5-fold decrease in T3 efflux (p < 0.05; Figure 1G), suggesting that MCT8 plays an important role in the transport of T3 in and out of neural cells. A suggested major source of T3 in the brain is achieved through conversion of T4 to T3 in astrocytes by deiodination (Heuer and Visser, 2013). Therefore, the role of MCT8 in T4 transport in neural cells was also tested. Compared to both control lines, the MCT8-deficient lines exhibited a 3-fold decrease in uptake (p < 0.0001; Figure 1H) and a 4-fold decrease in efflux (p < 0.0002; Figure 1I). Together, these results show that functional MCT8 expression in neural cells is crucial for the bidirectional transport of THs.
The main mechanism of TH action is achieved through transcriptional regulation of T3-inducible genes, such as transcription factor Kruppel-like factor 9 (KLF9) and transcriptional co-repressor Hairless (HR) (Dugas et al., 2012). KLF9 and HR transcription in neural cells was T3 induced in a dose-dependent manner in concentrations equal to or higher than 0.1 nM in differentiated control neural cultures (Figures 1J and 1K). Unexpectedly, MCT8-deficient neural cells exhibited similar T3 dose-dependent gene expression, despite their significantly reduced T3 uptake. This suggests that sufficient T3 was able to cross the cell membrane over 48 hr, even without a functional MCT8, to drive normal T3-dependent transcription.
MCT8-Deficient Neural Cells Maintain Normal Growth Rates, Differentiation Profiles, and T3-Dependent Neuronal Maturation
THs affect cell development, through the regulation of proliferation and apoptosis rates, as well as neuronal and glial differentiation (Bernal, 2000; Williams, 2008). Assessment of neural cultures with the proliferation marker Ki67 and a Tunel assay showed that control and MCT8-deficient neural cells had similar proliferation and apoptosis rates (Figures S3A and S3B). We next investigated whether the reduced transport of TH in the MCT8-deficient cell lines would alter the differentiation potential of EZ-spheres into neurons and astrocytes. Surprisingly, the differentiated neural cell cultures of MCT8-deficient cells and control cells were similar, with both lines showing ~45% Nestin+ neural progenitors, ~10% neurons, and ~15% astrocytes (Figure 2A). Collectively, these results indicate that, at least at this early stage of neural development, the lack of a functional MCT8 does not significantly affect proliferation, apoptosis, or neural cell differentiation.
Figure 2. MCT8-Deficient Cells Have Normal Differentiation and T3-Dependent Neuronal Maturation.

(A) Quantifying immunocytochemistry-stained cells showed that MCT8-deficient and control iPSCs had similar neural differentiation potential into Nestin+ progenitor cells, GFAP+ astrocytes and βIII-Tubulin+ neurons. Error bars represent SD.
(B) Scheme for differentiating iPSCs into neural cultures, in the presence or absence of T3 (100 nM) throughout 30–40 days.
(C) Mean of total spikes/5 min in the MCT8 intact (control) CS02iCTR, CS03iCTR and CS01iMCT8cor lines differentiated in the presence of T3 (solid line) was significantly higher than in the absence of T3 (dashed line) (ANOVA, n = 3, p < 0.0384).
(D) Mean of total spikes/5 min in the CS01iMCT8, CS58iMCT8 and CS03iCTRmut MCT8-deficient lines differentiated in the presence of T3 (solid line) was significantly higher than in the absence of T3 (dashed line) (ANOVA, n = 3, p < 0.014).
(E) Mean of total bursts/5 min in the MCT8 intact (control) lines in the presence of T3 (solid line) was significantly higher than in the absence of T3 on the third week of differentiation (t test, n = 3, p < 0.01).
(F) Mean of total bursts/5 min in the MCT8-deficient lines in the presence of T3 (solid line) was not significantly higher than in the absence of T3 (dashed line).
(G) Volcano plot shows differences of expression patterns in healthy control cells in response to T3 treatment. The dotted lines represent 1.5-fold differentially expressed genes by paired Student’s t test followed by Benjamini Hochberg correction for multiple comparisons at a false discovery rate (FDR) of 0.1. See Tables S3 and S4 for complete list.
We hypothesized that, instead of neural differentiation, neuronal maturation may be altered in MCT8-deficient cells. To study T3 effects on neuronal maturation in an MCT8-dependent manner, control and MCT8-deficient neural cultures were further matured for 30–40 days, rather than only 14 days, in the presence or absence of T3 (Figure 2B). After 3–4 weeks in culture, both control and MCT8-deficient neurons that were differentiated in the presence of T3 displayed a significant increase in spontaneous activity, recorded as spikes per minute by a multi-electrode array (MEA) system, compared to cultures without T3 (p < 0.038 [Figure 2C] and p < 0.014 [Figure 2D]). Moreover, the recorded bursts per minute were also significantly increased by T3 in control cells (p < 0.01; Figure 2E), and they showed a trend in MCT8-deficient cells (Figure 2F). These results demonstrate, for the first time in human neurons, that T3 contributes to neuronal maturation. Importantly, although MCT8 deficiency leads to significantly reduced uptake of T3, chronic exposure to T3 in the media permits sufficient transport of T3 in MCT8-deficient neural cells and subsequent normal neuron maturation.
We next explored what gene clusters may drive the T3-dependent neuronal maturation observed at day 30, when cells displayed a peak in spontaneous neuronal activity. Global, unsupervised hierarchical gene clustering showed that genes did not segregate by T3 and/or MCT8 expression (Figure S4A). To determine whether the T3-based neuronal maturation was correlated with any changes in T3-induced genes, global gene expression was compared in control cells differentiated in the presence or absence of T3 (Figure 2G). This approach revealed a list of ~200 genes that were differentially expressed after T3 exposure, including 70 upregulated and 40 downregulated by 1.5-fold or more (Tables S3 and S4, respectively). Ingenuity pathway analysis of the differentially regulated genes revealed pathways that are regulated by T3 in human neural cells (Figure S4B), some of which may correlate with neuronal maturation.
Human Brain Endothelial Cells Can Be Differentiated from Control and MCT8-Deficient iPSCs
A primary role of the BBB is to regulate the passage of molecules from the blood into the brain. Therefore, we hypothesized that, in MCT8 deficiency, TH transport from the blood into the brain may be disrupted, leading to a lack of neuronal maturation. Murine Mct8 regulates the transport of T3 across the BBB (Ceballos et al., 2009; Mayerl et al., 2014), but the role of MCT8 at the human BBB has yet to be investigated. Analyzing RNA-seq data for MCT8 gene expression in the human brain (Zhang et al., 2016) demonstrated similar levels in purified human endothelial cells, astrocytes, and neurons, and to a lesser extent in mature oligodendrocytes, and it was not detected in microglia (Figure 3A). Immunohistochemistry confirmed MCT8 expression in blood vessel-like structures in the post-mortem adult human healthy cerebellum, a brain region that is tightly regulated by TH signaling during development (Heuer and Mason, 2003) (Figure 3B). Furthermore, immunofluorescence determined that MCT8 was co-localized with the BBB Glucose transporter 1 (GLUT1) and expressed in Purkinje cells (Figure 3C).
Figure 3. Human Brain Microvascular Endothelial Cells Express MCT8.

(A) MCT8 RNA expression in immunopanned purified human cells from the Barres lab database. Data are expressed as FPKM.
(B) Immunohistochemisty of post-mortem cerebellum from a healthy human shows that MCT8 (purple) is expressed in microvessels in the granular cell layer (GCL), purkinje cell layer (PCL) and in the molecular layer (ML). White arrows denote microvessel-like structures. No antibody was used as a negative control (20× magnification).
(C) Immunofluorescence shows MCT8 protein (green) within Glucose transporter 1 (GLUT1, red)-expressing BMECs (white arrows), and within purkinje cells (yellow arrows). Scale bar, 10 μm.
(D) Scheme to differentiate iPSCs into BMECs. Colonies are differentiated for 6 days in unconditioned media (UM) and then expanded for 2 days in endothelial cell media (ECM). Differentiated cultures contain mixed colonies of neural and endothelial cells. Colonies are replated onto collagenIV/fibronectin-coated tissue culture plates or Transwell inserts at day 8 of differentiation, yielding purified iBMEC monolayers.
(E) Immunocytochemistry shows that purified iBMEC monolayers produce MCT8 (green) and GLUT1 (red) (20× magnification).
(F) Western blot confirms purified iBMECs produce MCT8. GAPDH housekeeping protein was used as a loading control.
(G and H) RNA-seq analysis shows RNA expression of (G) TH transporters and (H) deiodinases in purified human BMECs (green bar) and purified iBMECs. Data are expressed as FPKM. No significant differences were found between MCT8-deficient and control iBMECs. Error bars represent SD.
Since the human BBB expressed MCT8, it was important to explore the potential role of MCT8 deficiency on human BBB function. For this, iPSCs were differentiated into brain microvascular endothelial cells (iBMECs) using previously described methods (Lippmann et al., 2012, 2014) (Figure 3D). Similar to the post-mortem human brain and purified brain endothelial cells, iBMECs expressed both MCT8 and GLUT1 (Figure 3E), demonstrating the relevance of iBMECs to the study of MCT8 function. Western blot confirmed that MCT8 was expressed in iBMECs that were differentiated from the CS03iCTR control line, as well as the CS01iMCT8 diseased line and its corrected isogenic line, but not in the CS03iCTRmut cells (Figure 3F). Global RNA-seq analysis of control and MCT8-deficient iBMECs generated from control and MCT8-deficient iPSC lines revealed that all lines were highly similar on a transcriptome-wide basis (Figure S5A), indicating the robustness of the iBMEC differentiation protocol. In addition, iBMECs expressed a similar set of key BBB genes compared with cultured primary human BMECs and a majority of those expressed by acutely purified human brain endothelial cells (88%; Figure S5B; Table S5), indicating the relevance of iPSC-derived BMECs for studying the human BBB. Further, principal component analysis revealed that MCT8 was not a segregating factor in iBMEC differentiation (Figure S5C). At the individual gene level, the expression profile of TH transporters showed that, in addition to MCT8, iBMECs expressed LAT2, MCT10, and high levels of LAT1, and that these transporters were expressed uniformly in iBMECs derived from the various iPSC lines (Figure 3G). Importantly, OATP1C1 was not expressed, as expected in a human-based model. In addition, iBMECs expressed DIO2 and higher levels of DIO3, but not DIO1 (Figure 3H). This overall expression pattern of TH-related genes mirrors the expression in purified human BMECs, indicating that iBMECs are well suited to studying MCT8 deficiency.
Although transcriptomic analyses suggested a high similarity in control and MCT8-deficient iBMECs, we wanted to further assess whether key iBMEC phenotypes were affected by MCT8 deficiency. The physical barrier presented by the BBB limits paracellular molecular diffusion, and it is provided by tight junctions (TJs) between adjacent BMECs (Lippmann et al., 2012, 2014; Pardridge, 2012). All iPSC lines could generate purified PECAM1+/GLUT1+ iBMEC monolayers with proper junctional localization of TJ proteins CLAUDIN5 and OCCLUDIN (Figure 4A). Moreover, RNA-seq showed that the TJ gene expression was similar across control and MCT8-deficient iBMECs (Figure 4B). Next, the ability of iBMEC monolayers plated on a Transwell system to form functionally tight barriers to passive diffusion was investigated by measuring transendothelial electrical resistance (TEER). Results showed tight monolayers, with average TEER values of ~1,500 Ω × cm2, with no statistically significant differences in TEER among the lines (Figure 4C). These findings were supported by measuring statistically indistinguishable paracellular permeability using a fluorescein tracer and monitoring apical to basolateral flux (Figures 4D and 4E). Given the robust physical barrier formed by iBMECs, we analyzed the transport barrier phenotype in the form of drug efflux transporter activity. The iBMECs derived from each iPSC line expressed BBB drug efflux transporters P-glycoprotein (P-gp/ABCB1), breast cancer-resistant protein (BCRP/ABCG2), and multidrug resistance-associated protein 1 (MRP1/ABCC1) (Figure S6A), with similar expression levels between lines (Figure S6B). Moreover, the efflux transporter function in iBMECs was tested by measuring active efflux through the pumps using the fluorescent substrates (1) rhodamine 123 for P-gp, (2) FL-prazosin for BCRP, and (3) carboxymethyl-2′,7’-dichlorofluorescein diacetate (DCFDA) for MRP, in the presence or absence of the respective specific inhibitors PSC833 (P-gp), Ko143 (BCRP), and MK571 (MRP), in an accumulation assay. Increased accumulation in the presence of inhibitors showed that functional efflux was similar across the control and MCT8-deficient iBMECs lines, correlating with indistinguishable transporter expression levels (Figure S6C). In addition, the proliferation marker Ki67 and a Tunel assay showed that control and MCT8-deficient iBMECs had similar proliferation and apoptosis rates (Figures S6D and S6E).
Figure 4. Characterization of Patient-Derived iBMECs.

(A) Immunocytochemistry shows the BBB markers PECAM1, GLUT1, CLAUDIN5, and OCCLUDIN in healthy control (CS03iCTR and CSi00CTR) and in MCT8-deficient (CS01iMCT8 and CS58iMCT8) iBMECs (20× magnification).
(B) RNA-seq analysis depicting RNA expression of BBB marker GLUT1 and TJ molecules in control and MCT8-deficient iBMECs. No statistically significant differences were observed across cell lines.
(C) Quantification of transendothelial electrical resistance (TEER) showed no statistically significant differences between control (CS03iCTR, CS00iCTR, and CS01iMCT8cor) and MCT8-deficient (CS01iMCT8, CS58iMCT8, and CS03iCTRmut) iBMECs.
(D) Schematic of the iBMEC monolayers in a Transwell system. Fluorescent or radiolabeled molecules are added to the apical (blood) side, and the diffusion is monitored on the basolateral (brain) side over time.
(E) Quantification of the paracellular permeability of sodium fluorescein showed no statistically significant differences between control (CS03iCTR, CS00iCTR, and CS01iMCT8cor) and MCT8-deficient (CS01iMCT8, CS58iMCT8, and CS03iCTRmut) iBMECs. Error bars represent SD.
See also Figures S5 and S6 and Table S5.
Collectively, these results demonstrate the ability to obtain robust, functional iBMECs derived from both healthy controls and MCT8-deficient subjects, and they suggest that MCT8 and/or TH are not necessary for the induction and maturation of BBB properties during differentiation.
MCT8-Deficient iBMECs Are Defective in T3 Transport
As iBMECs from control and MCT8-deficient patients exhibited similar BBB genotypes and phenotypes, we next investigated whether TH transport across the MCT8-deficient iBMECs was impacted. Measuring the accumulation of radiolabeled T3 showed that MCT8-deficient iBMECs had a significant reduction in T3 uptake rate compared to control iBMECs (Figure 5A; p < 0.0001), suggesting that MCT8 functions in transporting T3 into iBMECs. Similar to the neural cells, MCT8-deficient iBMECs exhibited control-like T3 dose-dependent gene expression, despite their significantly reduced T3 uptake (Figure S7). T4 uptake was also significantly impaired in MCT8-deficient iBMECs (Figure 5B; p < 0.0001). To test whether MCT8-driven effects on iBMEC uptake could influence the transport of THs across the BBB, the apical to basolateral transport of radiolabeled T3 or T4 across iBMEC monolayers was measured in a Transwell system. Notably, MCT8-deficient cells displayed a significant decrease in overall T3 permeability (Figure 5C; p < 0.0001), while the transport of T4 across MCT8-deficient iBMECs was not significantly affected (Figure 5D). The net diffusion rates of T3 and T4 (i.e., the trans-iBMEC component) were assessed by correcting for paracellular diffusion using a fluorescein tracer. Corrected permeability indicated that the net trans-iBMEC permeability of T3 was completely abolished across MCT8-deficient iBMECs (Figure 5E; p < 0.0001), while T4 permeability was not reduced compared to controls (Figure 5F). These results demonstrate that MCT8 is critical for the transport of T3, but not T4, across the BBB model.
Figure 5. MCT8 Role in the Transport of THs across the BBB.

(A) Quantifying uptake rate of radiolabeled T3 (1 nM) showed that MCT8-deficient iBMECs (CS01iMCT8 and CS58iMCT8) displayed significantly lower uptake rates compared to control iBMECs (CS00iCTR and CS03iCTR) (ANOVA, n = 4, p < 0.0138).
(B) Measuring T4 (1 nM) uptake rate into iBMECs showed that MCT8-deficient iBMECs displayed significantly lower uptake rates than control iBMECs (ANOVA, n = 4, p < 0.0001).
(C) Measuring T3 transport across iBMECs showed that the permeability of radiolabeled T3 (1 nM) across MCT8-deficient iBMECs was significantly lower than control cells (ANOVA, n = 4, p < 0.0208).
(D) Measuring T4 (1 nM) transport across iBMECs showed that the permeability of 1 nM radiolabeled T4 across iBMECs was not significantly different across cell lines.
(E) After correcting for paracellular diffusion using a fluorescein tracer, the net permeability or trans-iBMEC component of radiolabeled T3 was abolished with MCT8-deficient cells and significantly differed from the T3 transport across control cells (ANOVA, n = 4, p < 0.0358).
(F) The net permeability of radiolabeled T4 across MCT8-deficient iBMECs was not significantly different from controls. Error bars represent SD.
See also Figure S7.
Given the striking impact of MCT8 deficiency on T3 transport, a mass spectrometry (MS) approach was developed in order to simultaneously monitor T3 and T4 transport across the BBB. The iPSCs from healthy controls, MCT8-deficient patients, and the engineered isogenic lines were differentiated into iBMECs and plated as monolayers on Transwells. T3 and T4 were added together to the apical side, and their transport to the basolateral side was monitored by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Recapitulating the radiolabeled T3 assay findings, T3 transport in MCT8-deficient cell lines was significantly lower than in the cell lines expressing nonmutated MCT8 (Figure 6A). Notably, these findings extended to the engineered MCT8-corrected line (CS01iMCT8cor), which displayed restored T3 transport, and to the MCT8-mutated line (CS03CTRmut), which now displayed deficient T3 transport. This highlights that no other transporter present in human iBMECs can effectively compensate for the lack of MCT8. MCT8-deficient cells again did not show compromised T4 transport (Figure 6B).
Figure 6. TH Transport across Patient-Derived and Isogenic-Derived iBMECs by LC-MS/MS.

(A) The apical-to-basolateral transport of T3 at 1 nM across MCT8-deficient lines (CS58iMCT8, CS01iMCT8, and CS03CTRmut, denoted by “b”) was significantly lower than in cells containing a functional MCT8 (CS00iCTR, CS03iCTR and CS01iMCT8cor, denoted by “a”) (ANOVA, n = 4, p < 0.0001). Pairwise comparisons showed significant differences between the isogenic lines CS03iCTR and CS03iCTRmut (p < 0.0012) and CS01iMCT8 and CS01iMCT8cor (p < 0.0001).
(B) The apical-to-basolateral transport of T4 at 1 nM across MCT8-deficient lines was not significantly different than transport across cells containing a functional MCT8.
(C) Measuring T3 efflux from iBMECs showed that MCT8-deficient cells (b) displayed significantly lower efflux rates than cells with a functional MCT8 (a) (n = 4, ANOVA, p < 0.0001). Pairwise comparisons showed significant differences between the isogenic lines CS03iCTR and CS03iCTRmut (p < 0.0027), CS01iMCT8 and CS01iMCT8cor (p < 0.0053).
(D) Measuring T4 efflux from iBMECs showed that MCT8-deficient cells (b) displayed significantly lower efflux rates than cells with a functional MCT8 (a) (n = 4, ANOVA, p < 0.001). Pairwise comparisons showed significant differences between the CS01iMCT8 and the CS01iMCT8cor isogenic lines.
(E) Scheme of experimental design. Transport from the apical (blood) side to the basolateral (brain) side corresponds to the forward (Fwd) direction. Reverse (Rev) direction corresponds to the basolateral to apical transport.
(F) Testing the bidirectional transport of 1 nM T3 across iBMECs showed a statistically significant difference between Fwd and Rev directions in functional MCT8 lines (CS02iCTR, p < 0.03; CS03iCTR, p < 0.03; and CS01iMCT8cor, p < 0.0002) and in CS58iMCT8 (p < 0.01). No significant differences were observed across cell lines in the Rev direction.
(G) DITPA (100 nM) transport across iBMECs did not differ between MCT8-deficient and control lines and was ~100-fold lower compared to T3 transport in control lines. Error bars represent SD.
Combining this novel MS-based assay with the BBB disease model can be used to further examine MCT8 functions, for instance, the effect of MCT8 on T3 and T4 efflux from iBMECs. Saturating iBMECs with T3 and T4 and measuring the efflux rates by LC-MS/MS demonstrated that both T3 and T4 efflux was significantly decreased in MCT8-deficient cells (Figures 6C and 6D), suggesting that MCT8 can also function in the efflux of both THs from iBMECs. Next, bidirectional transport of T3 across iBMECs was tested by comparing the apical-to-basolateral transport (forward [Fwd] direction) with the basolateral-to-apical transport (reverse [Rev] direction) (Figure 6E). While T3 transport in the reverse direction was low and similar across all control and MCT8-deficient lines, the presence of MCT8 in controls or engineered controls clearly led to increased transport in the forward direction compared with the MCT8-deficient lines (Figure 6F; p < 0.0001). These results indicate that MCT8 mediates the polarized influx of T3 across iBMECs in the blood-to-brain direction.
MCT8-Deficient Patient iBMECs Provide a Novel Tool to Assess Transport of Novel Drugs across the BBB
The characterized MCT8-deficient iBMECs display well-developed TJs and high TEER that serve to mute nonspecific paracellular transport, as would be the case in vivo. In addition, the MCT8-deficient iBMECs display a specific disruption of trans-iBMEC T3 transport. Combined with the MS approach, these characteristics provide an ideal platform for screening drugs that could either compensate for T3 deficiency or, alternatively, influence T3 transport at the BBB. Diiodothyropropionic acid (DITPA), currently under clinical investigation in MCT8-deficient patients (Verge et al., 2012), provides rescue of some of the thyroid phenotypes. However, it remains unclear whether this TH analog can improve neurological deficits. For an effective neurological treatment, the drug must be able to cross the human BBB. As such, we used our LC-MS/MS platform to measure BBB permeability of DITPA in healthy control and MCT8-deficient iBMEC monolayers. DITPA was shown to cross control and MCT8-deficient iBMECs, albeit with very low permeability that was more than 100-fold lower than that of T3 (Figure 6G). This suggests that DITPA transport is MCT8 independent and at a level that can be ascribed to paracellular diffusion. Collectively, these data show that iBMEC monolayers provide a unique platform to test the ability of TH analogs and future therapeutics to cross the diseased, MCT8-deficient human BBB.
DISCUSSION
Psychomotor retardation, as seen in AHDS, is critically related to MCT8 deficiency. Due to the lack of appropriate human models and the limited availability of MCT8-deficient post-mortem samples, many studies have addressed this disease using transgenic animals, but their TH transporter phenotype does not represent the human phenotype (Dumitrescu et al., 2006; Trajkovic et al., 2007). As a result, no successful treatment exists for the devastating neurological components of AHDS. The advent of human iPSCs now provides a promising resource to study AHDS through the use of patient-derived cells (Mattis and Svendsen, 2011).
Since MCT8 deficiency is primarily a neurological disorder, this study initially focused on the role of MCT8 in iPSC-derived neural cultures containing neural progenitors, neurons, and astrocytes. The validity of this novel human cell model was supported by the expression of MCT8, along with additional TH transporters LAT1, LAT2, MCT10, and lower levels of OATP1C1, as well as the main human cerebral deidodinase DIO3 (Kaplan, 1984). Radiolabeled transport experiments showed that the uptake and efflux of both T3 and T4 were significantly impaired in MCT8-deficient neural cells, indicating that MCT8 is responsible for the bulk of the TH transport across neural cell membranes. However, a low level of T3 and T4 transport was still observed in MCT8-deficient cells, which may be attributed to the expression of the additional transporters LAT1, LAT2, and OATP1C1 (Heuer and Visser, 2013; Wittmann et al., 2015).
As TH’s main mechanism of action is achieved through transcriptional regulation of T3-induced genes (Wu and Koenig, 2000), HR and KLF9 expression was assessed. As predicted, control cells showed elevated mRNA levels of both genes in response to T3. Intriguingly, despite the reduced T3 transport, MCT8-deficient cells showed similar dose-dependent T3-induced gene expression. Furthermore, neural cultures of MCT8-deficient and control cells displayed similar levels of differentiation, apoptosis, and proliferation. These indistinguishable T3-linked phenotypes in MCT8-deficient cells were unexpected, and they suggest that chronic exposure to high levels of T3 in culture medium can overcome the lack of MCT8, possibly through the function of additional TH transporters. This highlights that in vitro cell models with exposure to relatively high T3 concentrations (0.1–100 nM) do not fully recapitulate the in vivo conditions with lower picomolar levels of free THs in the bloodstream.
Subsequent investigation of TH-dependent differences at later stages of neural development demonstrated that chronic T3 exposure had a prominent effect on neuronal maturation in the form of increased spontaneous neuronal activity. These results align with several findings in MCT8 deficiency, including reduced synaptophysin expression in the post-mortem human brain (López-Espíndola et al., 2014) and neuronal deficiencies observed in the Mct8/Oatp1c1 double KO (dKO) mouse (Mayerl et al., 2014) and zebrafish Mct8 mutant model (Zada et al., 2014). These findings also extend beyond the scope of MCT8 deficiency, as neuronal maturation is a limiting factor in iPSC models of neurodegenerative diseases (Ho et al., 2016). The effect of T3 on neural gene expression revealed a novel set of T3-regulated genes in disease-relevant human cells, and it suggests pathways that can be directly affected by T3 exposure.
Although T3 affected neuronal maturation, the vast similarities between MCT8-deficient and control neural cells suggest that the TH-linked phenotypes in AHDS patients may instead be due to lowered neural exposure to T3. As such, it was critical to assess the possible dysregulation of T3 transport across the BBB, and, hence, this study developed a novel iPSC-based model of diseased human BMECs. The capability to model the MCT8-deficient BBB in human disease in the appropriate genetic background can only be done using the iPSC-based model. The model possesses many BBB-relevant properties (Lippmann et al., 2012), including physiologically relevant TEER values and efflux transporter activity, and expresses a comparable set of BBB genes (TJ proteins, ATP-binding cassette [ABC], SLC transporters, etc.) to those expressed in cultured primary human BMECs and to BMECs purified from human brain tissue. Finally, the iBMECs generated from the control and MCT8-deficient lines were very similar on a transcriptome-wide basis. Combined, these data suggest that this novel in vitro BBB model can uniquely provide a reasonable facsimile of the in vivo human BBB in a genetic background relevant to human MCT8 deficiency.
Human iBMECs expressed LAT1, LAT2, MCT10, DIO3, and lower levels of DIO2, similar to purified human BMECs. However, as previously described (Lippmann et al., 2012), iBMECs did not express OATP1C1, which, importantly, contrasts with TH transporter expression in rodents and zebrafish (Mayerl et al., 2012; Roberts et al., 2008; Vatine et al., 2013). As a result, MCT8-deficient iBMECs transported significantly lower levels of T3 in the blood-to-brain direction compared to controls. This reduced transport was unlikely due to changes in paracellular permeability, since control and MCT8-deficient iBMECs had no significant differences in TEERs and fluorescein permeability. Strikingly, the net transport of T3 across the MCT8-deficient BBB was completely abolished when corrected for the paracellular route. Moreover, when measuring the bidirectional transport of T3, MCT8 clearly mediated the polarized blood-to-brain transport of T3. Finally, the T3 transport effects also manifested in isogenic lines in which the MCT8 deficiency was either gene corrected in a patient line or mutated in a control line, clearly demonstrating that MCT8 deficiency alone can severely impact T3 transport across the BBB. Taken together, these results suggest that, in the human iBMEC model and possibly the human BBB, MCT8 is the key TH transporter that is capable of transporting T3 across the BBB into the brain and is, therefore, crucial for delivering T3 to the developing brain, where it is required for neuronal maturation (Figure 7).
Figure 7. MCT8 Deficiency Leads to Reduced Transport of T3 from the Blood-stream into the Brain.

To reach the human CNS, T3 crosses from the bloodstream through the blood-brain barrier (BBB) via MCT8-dependent transport by brain endothelial cells. T3 is then transported through MCT8 (black) and other TH transporters (gray) expressed on astrocyte and neuronal cell membranes, where it is thought to promote neuronal maturation. In the MCT8-deficent brain, T3 is unable to cross the BBB, resulting in T3 starvation of the brain, which in turn results in reduced neuronal maturation.
Intriguingly, despite the fact that MCT8 played a role in transporting T4 in and out of iBMECs, T4 transport across MCT8-deficient iBMECs was not significantly affected, suggesting that the role of MCT8 in T3 and T4 transport is different at the human BBB. In the mouse brain, T4 may cross the BBB via other non-Mct8 transporters and can be converted into T3 by deiodination in astrocytes (Heuer and Visser, 2013). Our results suggest that, like in the mouse, T4 can be transported through alternative transporters. While we confirm that, unlike in rodents, OATP1C1 is absent in the human BBB, human iBMECs do express LAT1, LAT2, and MCT10, which may transport T4 (Zevenbergen et al., 2015). Thus, since MCT8-deficient patients manifest lower free T4 serum concentrations, it is likely that the lower T4 concentrations observed in the post-mortem MCT8-deficient brain (López-Espíndola et al., 2014) reflect the serum concentrations rather than abrogated MCT8-mediated transport.
TH research commonly measures TH permeability using radiolabels; however, this is considered an indirect method. Therefore, this study developed a novel platform combining iBMECs with an MS assay to measure the transport of THs across the diseased human BBB. LC-MS/MS is highly accurate and allows higher content screening since different molecules can be distinguished within the same sample. As such, T3, T4, and free iodine can be distinguished to avoid any possible radiolabeled assay artifacts. The MS approach showed that blood-to-brain T3 transport is significantly decreased in the MCT8-deficient BBB, corroborating the radiolabeled T3 transport assays. In addition to the transport of natural thyroid hormones, the iBMEC-MS platform can be used to measure the comparative transport of T3 analogs that have been explored to treat MCT8-deficient patients or to screen for drugs that could restore T3 transport across the BBB. Since our data suggest that MCT8-deficient neural cells can mature normally in the presence of T3, one therapeutic approach may be to activate T3 signaling using BBB-permeable analogs that can enter the CNS in an MCT8-independent fashion. As an example, DITPA is a TH analog that can activate the T3 signaling cascade and has been tested in animal models and MCT8-deficient patients (Ferrara et al., 2015; Verge et al., 2012). However, species differences in TH transport mechanisms preclude addressing whether this TH analog would be transported across the human MCT8-deficient BBB. The proof-of-concept iBMEC-MS approach showed that, compared with T3 transport, DITPA is inefficiently transported across the human BBB in a manner that is MCT8 independent. Moreover, DITPA had a lower iBMEC permeability than that reported for BBB-impermeant sucrose using the same iBMEC model (Lippmann et al., 2012). This therefore predicts that DITPA might have a limited effect on the neurological condition of MCT8 deficiency. However, the lack of neurological improvement in response to DITPA treatment, which was at doses 1,000-fold higher than replacement doses of T3, may also be due to the late treatment initiation of MCT8-deficient patients (at ages of 9–21 months) (Verge et al., 2012). Development of this MS approach is ongoing in order to test additional TH analog candidates, such as 3,5,3′-triiodothyroacetic acid (TRIAC). More generally, the iBMEC platform holds great potential for high-throughput screening of molecules that can be delivered into the human CNS or impact the diseased BBB.
In conclusion, this study describes the first iPSC-based model of a BBB-related disease in the form of MCT8 deficiency. Disease-in-a-dish modeling with various CNS cell types provided a broader picture of the disease that ultimately suggests a new multicellular mechanism. For the first time, it is shown that human BMECs have perturbed MCT8 transport of T3 and that this is a likely cause of neurological symptoms in MCT8 deficiency. Specifically, the results predict that a lack of a functional MCT8 in the BBB results in the T3 starvation of the brain, which in turn results in deficient neuronal maturation (Figure 7). This concept of BBB disease modeling and drug screening can be further applied to other BBB-related diseases with genetic transporter alterations, such as GLUT1 deficiency (Larsen et al., 2015). Moreover, several neurodegenerative diseases, such as Alzheimer’s and Huntington’s diseases (Di Marco et al., 2015), and amyotrophic lateral sclerosis (Bataveljic et al., 2014) have been suggested to involve BBB dysfunction. In a parallel paper (Lim et al., 2017), the authors use a complimentary approach to interrogate abnormal signaling at the Huntington’s disease BBB. Together, these reports support the use of iPSCs to generate clinically relevant models of diseased human BBB. As such, this iPSC-based model could have far-reaching implications to advance the understanding and treatment of other neurological diseases.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Clive Svendsen, at clive.svendsen@cshs.org.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary cultures were obtained under consent and privacy guidelines of the University of Chicago. All procedures were performed in accordance with the IRB guidelines at the Cedars-Sinai Medical Center under IRB-SCRO Protocols Pro00021505 and Pro00032834.
METHOD DETAILS
Reprograming and characterization of MCT8-deficient iPSCs
Fibroblasts obtained from skin biopsies of two severely affected male patients with MCT8-deficiency and one parental control were reprogrammed into iPSCs by transfecting with non-integrating episomal plasmids expressing OCT4, SOX2, C-MYC and KLF4. Patient 2558 has a single base pair deletion in the 3rd exon of the MCT8 gene, predicted to produce a truncated and non-functional MCT8 protein that fails to migrate to the plasma membrane (Dumitrescu et al., 2004) and patient 3601 has a C to T transition that results in replacement of Proline to Leucine (P321L) causing the mutant MCT8 to not transport T3 across cell membranes (Kersseboom et al., 2013). Fibroblasts from the 2558 and 3601 patients in addition to fibroblasts from the father of the 3601 patient were reprogrammed to generate iPSCs (CS58iMCT8, CS01iMCT8 and CS03iCTR respectively). Three clones were selected from each cell line for further evaluation. Additional control lines, CS02iCTR, CS00iCTR and CS14iCTR, were also included (see Table S1 for cell line specific details).
iPSC characterization
iPSCs were characterized at the Cedars-Sinai iPSC Core Facility as previously described (Mattis et al., 2015). Successful reprograming of each iPSC clone was verified by immunocytochemistry (ICC) for the pluripotency markers NANOG, TRA160, OCT4, SSEA4, TRA181 and SOX2 as well as the activity of the pluripotency marker alkaline phosphatase (AP) (Figures S1A–S1C). In addition, cells were subjected to the array-based PluriTest (Muller el., 2011, Illumina), which confirmed that the iPSCs were pluripotent and novel (Figure S1D). Chromosomal abnormalities can occur during the reprograming process and overtime during cell passaging (Hamada et al., 2012). G-band karyotype analysis confirmed that the iPSC lines showed no chromosomal abnormalities (Figures S1A–S1C). Finally, the persistence of the MCT8 gene mutations in the iPSC lines was verified by DNA sequencing (Figure S1E).
Generation of isogenic control lines
In order to exclude clonal variation, the CRISPR/Cas9 system was used to generate isogenic lines with either induced or corrected MCT8 mutations. The gRNA 5′-CACCGCTTCCTCATCAGAATGCTGG-3′ designed to target the 3rd exon of the MCT8 gene was cloned into the pX330-mCherry expression construct (Bakondi et al., 2016) (Life Technologies, Carlsbad, CA) using manufacturer instructions. Electroporation of the gRNA in healthy background cells (CS03iCTR) (Figure S2A) efficiently generated a variety of indel mutations from which the CS03iCTRmut was selected. This isogenic line incorporated an 8-base pair (bp) deletion (Figures S2A and S2B), which caused a frameshift that introduced an early stop codon and thereby resulted in a truncated protein, mimicking the truncated non-functional MCT8 protein in the CS58iMCT8 iPSCs (Figure S2C).
Specific correction of the P321L mutation in the CS01iMCT8 cells was obtained by CRISPR-mediated homology directed repair (HDR) achieved by electroporation of the same gRNA (Figure S2A) in combination with a 100 bp long single-stranded DNA oligonucleotide (SSODN, 5′-GTGGTGTCTGCTGGGAGTAGCATTTTCTCCATGTCCTTCCCCTTCCTCATCAGAATGCTGGGGGATAAGAT CAAGCTGGCCCAAACCTTCCAGGTGCTGA-3′). In addition to the T to C correction, the SSODN included a silent mutation (C to G) at the location of the protospacer adjacent motif (PAM) site to prevent retargeting by the Cas9-gRNA complex. The 100 bp oligonucleotide was purchased from IDT and PAGE-Purified. The generated CS01iMCT8cor isogenic line (Figure S2D) demonstrated a corrected MCT8 sequence (Figure S2E). In both isogenic lines, PCR confirmed the absence of off-site mutations (Table S2) and karyotyping verified the lack of chromosomal abnormalities possibly evoked by the gene editing process (Figures S2F and S2G).
iPSCs were maintained on Matrigel (BD Biosciences) in mTeSR1 medium (StemCell Technologies, Inc.), cultured in 6-well plates and prepared for transfection by passaging at a high density using an EZ passage tool (Invitrogen). At 80% confluency, cells were treated with 10 μM Y-27632 dyhydrochloride, Rho-associated kinase inhibitor (ROCKi, Tocris Biosciences) for 24 hr. A Neon Transfection System (Thermo Fisher) was used for DNA delivery by electroporation. iPSCs were washed with cold phosphate buffered saline (PBS) and treated with 1mL Accutase (Millipore) at 37°C for 3–5 min. The cells were then washed with PBS and resuspended in buffer Rat approximately 1×106 cells/100 μl with 5 μg of Px330-MCT8ex3-mCherry vector (per 1×106 cells) with or without the 5 μg of ssODN (per 1×106 cells) in a Neon pipette tip and electroporated with a single pulse (1100 Voltage, 30 Width). Following electroporation, cells were plated at 1×106 cells per well into matrigel coated 6-well plates in conditioned mTeSR1 supplemented with 10 μM ROCKi. After 48 hr, cells were sorted by fluorescence-activated cell sorting (FACS, BD FACS ARIA III, Cedars-Sinai Flow Cytometry Core) for positive mCherry expression. Sorted cells were replated at 3000 cells/cm2 on Matrigel in conditioned mTeSR1 supplemented with ROCKi for 24 hr, then gradually changed to fresh mTeSR1 without ROCKi and expanded for 7–14 days. Individual colonies were manually selected and further expanded in 24-well plates on Matrigel in mTeSR for 5–10 days. Colonies were collected and genomic DNA was isolated (Quick-gRNA MiniPrep, Zymo Research) and MCT8 was analyzed by DNA sequencing (Genewiz). The MCT8I2E3F: 5′-CTGAGGGTCAGTGGCAAGTG-3′ and MCT8E3R: 5′-CTAGGCAGGTTGAGGGTAGC-3′ primers were used for PCR (Takara Ex Taq, DNA polymerase). Potential off-target sites were predicted by an online tool (crispr.mit.edu). The top ten putative exonic off-target sites were selected and analyzed by PCR amplification followed by DNA sequencing (Table S1).
Generations EZ-spheres from iPSCs
EZ-spheres were generated from iPSC colonies as previously described (Ebert et al., 2013). Briefly, iPSC colonies grown on Matrigel-coated plates (0.5 mg/plate) in mTESR1 (StemCell Technologies, Inc.) media were removed by gentle scraping and placed directly into DMEM:F12 7:3 supplemented with 100 ng/ml basic fibroblast growth factor (bFGF, Chemicon), 100 ng/ml epidermal growth factor (Chemicon), and 5 μg/ml heparin (Sigma) in ultra-low attachment flasks and were passaged weekly using a chopping technique (Svendsen et al., 1998).
Differentiation of EZ-spheres into neural cultures
To induce neural differentiation, spheres were dissociated with Accutase (EMD Millipore) and plated onto poly-ornithine/laminincoated (Sigma) coverslips or laminin-coated culture or MEA plates in DMEM:F12 with 2% B27, 1% N2 Supplement, 20 ng/ml brain-derived neurotrophic factor (BDNF) for 2–6 weeks. For T3-induced gene expression, cells were deprived of T3 for 48 hr, using B27 without T3. Next, T3 was added to the media as needed at 0.1–100 nM for 24–48 hr. For T3 deprivation differentiation assays, cells were grown with B27 without T3, and then supplemented with 100 nM of T3 as needed.
Radiolabelled TH accumulation assays
Neural differentiated EZ-spheres or iBMECs were plated at confluence on a 24-well plate and were washed with Transport Buffer (Dulbecco’s PBS containing 0.1% D-glucose and 0.1% BSA). TH uptake was tested by incubation of the cells for 5 to 60 min at 37°C with 1 nM (2 × 105 cpm) [125I] T3 or [125I] T4 (Perkin Elmer) in 0.5 mL of Transport Buffer. After incubation, cells were washed with the medium, lysed with 0.1M NaOH, and counted in a gamma counter. For measurement of neural TH efflux, cells were loaded for 1 hr with Transport Buffer containing 1 nM (2 × 105 cpm) [125I] T3 or [125I] T4. After removal of the medium, cells were washed and incubated for 5 to 60 min with Transport Buffer. Finally, medium was removed and cells were washed with Transport Buffer and lysed with 0.1M NaOH. The lysate was analyzed in a gamma counter and the resulting slope was plotted. Values were corrected for protein concentrations by a Bradford assay (Bio-Rad).
MEA recording
EZ-spheres were dissociated into single cells and plated on laminin-coated M768-GLx 12-well plates (Axion BioSystems) at typical densities of 100,000 cells/well. Cells were then differentiated as described above. Each monolayer was monitored before recordings to confirm the presence of a full monolayer of cells. Simultaneous recordings from 64 extracellular electrodes per well were made using the Maestro (Axion BioSystems) micro electrod array (MEA) system at a constant temperature of 37°C. Data were sampled at 12.5 kHz, digitized and analyzed using Axion integrated Studio software (Axion BioSystems) with a 200 Hz high pass and 2.5 kHz low pass filter and an adaptive spike detection threshold set at 6 times the SD for each electrode at 1 s binning.
RNA-seq
Libraries for RNA-Seq were prepared with KAPA Stranded RNA-Seq Kit. The workflow consists of mRNA enrichment, cDNA generation, and end repair to generate blunt ends, A-tailing, adaptor ligation and PCR amplification. Different adaptors were used for multiplexing samples in one lane. Sequencing was performed on Illumina NextSeq 500 for a single read 75 run. Data quality check was done on Illumina SAV. Demultiplexing was performed with Illumina Bcl2fastq2 v 2.17 program.
Differentiation of iPSCs into iBMECs
iPSCs were maintained between passages 15–40 on Matrigel (BD Biosciences) in mTeSR1 medium (StemCell Technologies, Inc.). For differentiation, cells were passaged onto Matrigel in mTeSR1medium for 2–3 days of expansion until reaching a density of 2–3×105 cells/well and then switched to unconditioned medium lacking bFGF for 6 days. Human endothelial serum-free medium (hESFM; Life Technologies) supplemented with 20 ng/mL bFGF, 10 μM All-trans retinoic acid (RA) (Sigma) and 1% platelet-poor plasma derived bovine serum (Biomedical Technologies, Inc.) was then added for 2 days. Cells were then gently dissociated with Accutase for 35 min and plated onto 1.12 cm2 Transwell-Clear permeable inserts (0.4 μm pore size) coated with a mixture of collagen IV (400 mg/mL; Sigma) and fibronectin (100 mg/mL; Sigma) in H2O. Resultant purified human iPSC-derived BMECs were then grown in endothelial cell medium (ECM) for 24 hr (without bFGF and RA). At 24 hr, iBMEC-plated Transwell inserts were placed on top of 14–28 days EZ-sphere differentiated neural cultures plated on a 12-well plate.
Paracellular permeability, efflux transporters and TH transport assays across iBMECs
iBMECs plated on a Transwell were washed with Transport Buffer. For paracellular permeability assays, 1 μM sodium fluorescein was added to the upper chamber. Aliquots were then extracted from the bottom chamber every 15 min and replaced with fresh buffer and fluorescence (485 nm excitation and 530 nm emission) was quantified at the end of the experiment on a plate reader. For efflux transporters experiments, cells were pre-incubated with 10 mM cyclosporin A (Sigma), 1 mM Ko143 (Sigma), or 10 mM MK571 (Sigma), depending on the experiment, for 1 hr at 37°C on a rotating platform, then co-incubated with the inhibitor and compound of interest (rhodamine 123 (R123), carboxymethyl-2′,7’-dichlorofluorescein diacetate (DCFDA) or FL-prazosin) and was quantified using a plate reader. For radiolabelled TH transport assays, 1 nM (2 × 105 cpm) [125I] T3 or [125I] T4 (Perkin Elmer) was added to the upper chamber and measured in the bottom chamber using a gamma counter. The rate of accumulation was used to calculate Pe values. Monolayer fidelity was confirmed at the beginning and at the end of each experiment by measuring TEER by an EVOM2 system and a Chopstick Electrode (World Precision Instruments).
Immunocytochemistry
Immunocytochemistry was conducted as previously described (Lippmann et al., 2012) with minor modifications. Cells were blocked in PBS containing 5%–10% donkey serum (Sigma) at 4°C over night. Triton X-100 at 0.1% was included if membrane permeabilization was required. Primary antibodies were MCT8 1:100 (SLC16A2, Atlas Antibodies, Cat# HPA003353), βIII-Tubulin 1:1000 (Tuj1α, Sigma-Aldrich, Cat#T8660), GFAP 1:1000 (DAKO), Glut-1 1:100 (Thermo Fisher, Cat# MA5–11315), PECAM1, OCCLUDIN, CLAUDIN5 (according to Lippmann et al., 2012). Appropriate fluorescent secondary antibodies (Life Technologies) at 1:1000 were incubated for 1 hr at room temperature. Cells were then counterstained with Dapi. Images were captured with a Leica AF3500 microscope.
Immunohistochemistry
The adult human brain post-mortem tissue was provided by the Harvard Brain Tissue Resource Center, McLean Hospital (Belmont, MA). Research was conducted in compliance with the policies and principles contained in the Federal Policy for the Protection of Human Subjects. The formalin fixed paraffin embedded human brain was sectioned at 8 μm on a rotary microtome. Tissue was dewaxed through Xylene treatment followed by 100% ethanol and 95% ethanol and rehydrated in distilled water. Sections were then quenched in 0.3% H2O2 for 30 min and washed 3 times in PBS. Antigen retrieval was performed with 0.1 M sodium citrate pH 7.2 at 75°C for 40 min and cooled to room temperature. Sections were blocked in 3% normal donkey serum in 0.25% Triton X-100 for 1 hr, then incubated with primary antibodies (MCT8 1:100, Atlas Antibodies, Cat# HPA003353 and Glut-1 1:100, Thermo Fisher, Cat# MA5–11315) over night at room temperature and washed 3 times. Secondary antibodies (Life Technologies) were incubated at 1:1000 for 1 hr at room temperature and washed 3 times with PBS. Sections were than incubated in 0.1% Sudan Black B in 70% ethanol for 30 min, rinsed in PBS, stained with DAPI and mounted with aqueous mounting media.
Western blot
MCT8 western blot with iPSCs, iBMECs and iPSC-derived neural cells was performed as previously described (Mattis et al., 2015). Collected cells were washed in PBS, pelleted at 1000 rpm, and lysed using LB1 buffer containing protease inhibitor (50 mM HEPES-KOH, pH 7.5), 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40 and 0.25% Triton X-100, 1 mM PMSF Protease Inhibitor (Thermo Fisher), incubated on ice for 30 min, sonicated 3 times at 20 Amps for 10–15 s on/off. The lysate was pelleted at 16,000 g at 4°C for 30 min. Remaining pellets were resuspended in NENT buffer containing Protease Inhibitor (20 mM Tris-Cl, pH 8.0), 100 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 0.1 mM PMSF Protease Inhibitor, sonicated 3 times at 20 Amps for 10–15 s on/ off and centrifuged for 20 min at 16,000 g. The supernatants were combined. Protein concentrations were determined using a Bio-Rad Protein assay according to manufacturer’s instructions. 35 μg of protein was loaded into a 10% Mini-PROTEAN TGX precast gels (Bio-Rad), separated with 95V for 1–1.5 hr, and electro-transferred onto PVDF membrane (BioRad Turbo transfer) in a semi-dry transfer system for 7 min at 1.3 A. The membrane was blocked in 2% dry non-fat milk in Tris-buffered saline plus 0.1% Tween 20 (Sigma-Aldrich) for 1 hr at RT and then exposed to primary antibody that recognizes the amino-terminal domain of the MCT8 protein (1:1000, rabbit, Sigma-Aldrich) (Wirth et al., 2009) or β-actin (1:1000, rabbit, Sigma-Aldrich) to ensure equal protein loading in block overnight at 4°C. Anti-rabbit secondary antibodies conjugated to peroxidase (1:5000, Peroxidase AffiniPure Goat Anti-Rabbit IgG, Jackson ImmunoResearch Laboratories Inc.) was applied in block for 45 min at RT, followed by chemiluminescence imaging using the Clarity Western ECL Blotting Substrate kit (Bio-Rad).
qRT-PCR
Total RNA was isolated using the RNeasy Mini Kit (QIAGEN) and digested by DNase I (RQ1 DNase, Promega). Complementary DNA was generated from 0.5 mg total RNA using a Reverse Transcript System (Promega A3500). Primer sequences are included in Table S6. qPCR was performed using a 384-well format (BioRad). GAPDH was included as an endogenous control. Data were analyzed using the 2−ΔΔCT method and normalized to the maximum readout in the run.
Measurements of iodothyronines by LC/MS/MS
T4 and T3 were simultaneously measured by liquid chromatography (dual LC Shimadzu Prominence system, Shimadzu, Columbia, MD) followed by tandem mass spectrometry (Ferrara et al., 2013). (Q-Trap 6500, Sciex) with a TurboVion source. After addition of the internal standard 13C6 - T3 and 13C6- T4 (Iso Sciences) to 100 μL sample of Transport Buffer collected from the top or bottom chamber, the iodothyronines were extracted with 1:4 (v/v) and with 100 μL of EtOH:NH4OH (98:2). The combined supernatants were evaporated using a speed vacuum (Thermo Scientific). The residue was then reconstituted in 100 μL of 0.1% Formic acid in water and 40 μL of reconstituted extract was injected into Kinetex C18–100A (2.6 μ, 30 × 3 mm, Phenomenex, CA) column, protected by a Phenomenex C18-RP guard cartridge in 40°C column oven. lodothyronines were chromatographed with 0.1% formic acid in deionized water (aqueous mobile phase A) and 0.1% formic acid in methanol (organic mobile phase B). The gradient was 5 to 90% B in 2 min with flow rate of 0.4 ml/min. The positive ion multiple reaction monitoring (MRM) mode was used for detection. The MRM transition monitored was: m/z 777.5 > 731.5 for T4; m/z 651.5 > 605.5 for T3; m/z 651.5 > 605.5 for 13C6 - T3, and m/z 783.5 > 737.3 for 13C6- T4. All the MRM data was processed with Multiquant (Sciex).
Measurements of DITPA by LC/MS/MS
DITPA was measured with the same LC-MS/MS instruments as the iodothyronines but with different chromatographic protocols and negative mode in MS (Verge et al., 2012). The internal standard 3,3,5-triiodothyropropionic acid (TITPA) was first added to the Transport Buffer. Then, DITPA and TITPA were extracted using conditioned methanol and water through a 30 mg, 1 mL Oasis HLB extraction cartridge (Waters, Milford, MA). Finally, DITPA and TITPA were eluted from the cartridge with acetonitril and methanol and evaporated to dryness using speedvac. The residues were reconstituted in acetonitril: 2 mM ammonium acetate (1:1). 40 μl were injected into a 3.3 cm × 3 mm, 3 μm SUPELCOSIL LC-18 DB analytical column (SUPELCO). DITPA and TITPA were chromatographed using a gradient (25 to 85% of B in 3 min) elution with 2 mM ammonium acetate (pH 3.7, Buffer A) and acetonitrile (buffer B) and detected by monitoring ions at a mass to charge ratio m/z 508.8 > 126.8 for DITPA and 634.2 > 126.6 (TITPA), respectively. DITPA was obtained from Sigma-Aldrich and TITPA from Toronto Research Chemicals. All the MRM data was processed with Multiquant (Sciex).
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE97324.
QUANTITATION AND STATISTICAL ANALYSIS
Statistical Analysis
All experiments were performed in duplicates or triplicates with a minimum of three independent neural or iBMEC differentiations, including a single clone for each cell line. When comparing two cell lines, statistical analyses were performed using an unpaired Student’s t test. For dose-dependent experiments, linear relationship was applied to test if the R-values are significantly different than 0. For analysis across multiple MCT8-deficent and control cell lines, data was analyzed by ANOVA with nested design to account for cell line groupings, followed by Tukey test to adjust for multiple comparisons. Residuals were inspected to confirm data met assumptions required for parametric testing and to confirm the absence of influential outliers. Any data that failed to meet assumptions necessary for parametric testing was log-transformed prior to analysis. For all testing the level of significance was set at a two-sided p < 0.05 and error bars represent standard deviation (SD).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SLC16A2 (MCT8) | Atlas Antibodies | HPA003353; RRID: AB_611613 |
| Tuj1a (BIII Tubulin) | Sigma-Aldrich | T8660; RRID: AB_477590 |
| GFAP | Dako | Z0334; RRID: AB_10013382 |
| Glut-1 | Thermo Fisher | MA5–11315; RRID: AB_10979643 |
| Biological Samples | ||
| Postmortem human cerebellum from a healthy adult | Harvard Brains Tissue Resource Center, McLean Hospital | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| matrigel | BD Bioscienses | 356234 |
| mTeSRI | StemCell Technologies | 85851 |
| Rocki Y-27632 | Tocris Biosciences | 1254 |
| Accutase | EMD Millipore | SCR005 |
| bFGF | EMD Millipore | 01–106 |
| EGF | EMD Millipore | GF144 |
| Heparin | Sigma-Aldrich | 84020 |
| TRIIODOTHYRONINE, ([125I]-T3) | Perkin Elmer | NEX110 |
| Thyroxine, ([125I]-T4) | Perkin Elmer | NEX111 |
| Human endothelial serum-free medium | Life Technologies | 11111–044 |
| All-trans-Retinoic acid | Sigma-Aldrich | R2625 |
| Platelet-poor Plasma Derived Serum, Bovine | Biomedical Technologies | BT-214 |
| Ko143 hydrate | Sigma-Aldrich | K2144 |
| MK571, Sodium Salt | Sigma-Aldrich | 475874-M |
| Cyclosporin A | Sigma-Aldrich | C3662 |
| 2′, 7’ -Dichlorofluorescin diacetate | Sigma-Aldrich | D6883 |
| Rhodamine 123 | Sigma-Aldrich | 83702 |
| FL Prazosin | Fischer Scientific | B7433 |
| Diiodothyropropionic acid (DITPA) | Sigma-Aldrich | Not available |
| 3,3,5-triiodothyropropionic acid (TITPA) | Toronto Research Chemicals | Not available |
| Critical Commercial Assays | ||
| PluriTest | Muller | Illumina HT12v4 |
| Oasis HLB 96-well Plate 30 mg | Waters | WAT058951 |
| Kinetex C18–100A (2.6 μ, 30 × 3 mm) | Phenomenex | 00A-4462-Y0 |
| LiChrospher RP-18 | Phenomenex | RP-18 |
| SUPELCOSIL LC-18-DB HPLC Column | Sigma | 58978C30 SUPELCO |
| Experimental Models: Cell Lines | ||
| CS03iCTR | iPS-core, Cedars-Sinai Medical Center | |
| CS01iMCT8 | iPS-core, Cedars-Sinai Medical Center | |
| CS58iMCT8 | iPS-core, Cedars-Sinai Medical Center | |
| Oligonucleotides | ||
| See Table S6 for details | ||
| Recombinant DNA | ||
| pX330-mCherry | Bakondi et al., 2016 | Addgene (64324) |
| pX330-mCherry-MCT8 | This manuscript | |
| Software and Algorithms | ||
| Axion integrated Studio software | Axion BioSustems |
Highlights.
MCT8-deficient iPSCs generate neurons with normal TH-dependent neuronal maturation
Differentiation to brain endothelial cells models the blood-brain barrier
MCT8-deficient brain endothelial cells show defects in thyroid hormone transport
A platform to test candidate drug transport across the diseased BBB was established
ACKNOWLEDGMENTS
The authors would like to thank Catherine Bresee, MS, for the statistical analysis. This work was supported by the Sherman Family Foundation, the Board of Governors Regenerative Medicine Institute at Cedars-Sinai Medical Center (CNS), and the NIH (NS083688 and AA020476, E.V.S. and S.P.P.; NS085351, S.P.P. and E.V.S.; and R37DK15070, S.R.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2017.04.002.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Bauer et al., 2014; Geier et al., 2013; Huntley et al., 2014; Liebner et al., 2011; Obermeier et al., 2013.
REFERENCES
- Allan W, Herndon CN, and Dudley FC (1944). Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am. J. Ment. Defic 48, 325–334. [Google Scholar]
- Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, Levy R, Akhtar AA, Breunig JJ, Svendsen CN, and Wang S (2016). In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther 24, 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataveljic D, Milosevic M, Radenovic L, and Andjus P (2014). Novel molecular biomarkers at the blood-brain barrier in ALS. BioMed Res. Int 2014, 907545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer HC, Krizbai IA, Bauer H, and Traweger A (2014). “You Shall Not Pass”-tight junctions of the blood brain barrier. Front. Neurosci 8, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal J (2000). Thyroid Hormones in Brain Development and Function In Endotext, De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, and Rebar R, et al. , eds. (South Dartmouth, MA: MDText.com). [Google Scholar]
- Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, Morte B, and Bernal J (2009). Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3′-triiodo-L-thyronine. Endocrinology 150, 2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A, and Frangi AF (2015). Vascular dysfunction in the pathogenesis of Alzheimer’s disease-A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis 82, 593–606. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Ibrahim A, and Barres BA (2012). The T3-induced gene KLF9 regulates oligodendrocyte differentiation and myelin regeneration. Mol. Cell. Neurosci 50, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, and Refetoff S (2004). A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am. J. Hum. Genet 74, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Weiss RE, Millen K, and Refetoff S (2006). Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147, 4036–4043. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF Jr., Mattis VB, Lorson CL, Thomson JA, and Svendsen CN (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Shelley BC, Hurley AM, Onorati M, Castiglioni V, Patitucci TN, Svendsen SP, Mattis VB, McGivern JV, Schwab AJ, et al. (2013). EZ spheres: a stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res. (Amst.) 10, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara AM, Liao XH, Gil-Ibáñez P, Marcinkowski T, Bernal J, Weiss RE, Dumitrescu AM, and Refetoff S (2013). Changes in thyroid status during perinatal development of MCT8-deficient male mice. Endocrinology 154, 2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara AM, Liao XH, Ye H, Weiss RE, Dumitrescu AM, and Refetoff S (2015). The Thyroid Hormone Analog DITPA Ameliorates Metabolic Parameters of Male Mice With Mct8 Deficiency. Endocrinology 156, 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, and Visser TJ (2003). Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem 278, 40128–40135. [DOI] [PubMed] [Google Scholar]
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, et al. (2004). Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364, 1435–1437. [DOI] [PubMed] [Google Scholar]
- Geier EG, Chen EC, Webb A, Papp AC, Yee SW, Sadee W, and Giacomini KM (2013). Profiling solute carrier transporters in the human blood-brain barrier. Clin. Pharmacol. Ther 94, 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M, Malureanu LA, Wijshake T, Zhou W, and van Deursen JM (2012). Reprogramming to pluripotency can conceal somatic cell chromosomal instability. PLoS Genet. 8, e1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer H, and Mason CA (2003). Thyroid hormone induces cerebellar Purkinje cell dendritic development via the thyroid hormone receptor alpha1. J. Neurosci 23, 10604–10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer H, and Visser TJ (2013). The pathophysiological consequences of thyroid hormone transporter deficiencies: Insights from mouse models. Biochim. Biophys. Acta 1830, 3974–3978. [DOI] [PubMed] [Google Scholar]
- Ho R, Sances S, Gowing G, Amoroso MW, O’Rourke JG, Sahabian A, Wichterle H, Baloh RH, Sareen D, and Svendsen CN (2016). ALS disrupts spinal motor neuron maturation and aging pathways within gene coexpression networks. Nat. Neurosci 19, 1256–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley MA, Bien-Ly N, Daneman R, and Watts RJ (2014). Dissecting gene expression at the blood-brain barrier. Front. Neurosci 8, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MM (1984). The role of thyroid hormone deiodination in the regulation of hypothalamo-pituitary function. Neuroendocrinology 38, 254–260. [DOI] [PubMed] [Google Scholar]
- Kersseboom S, Kremers GJ, Friesema EC, Visser WE, Klootwijk W, Peeters RP, and Visser TJ (2013). Mutations in MCT8 in patients with Allan-Herndon-Dudley-syndrome affecting its cellular distribution. Mol. Endocrinol 27, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J, Johannesen KM, Ek J, Tang S, Marini C, Blichfeldt S, Kibaek M, von Spiczak S, Weckhuysen S, Frangu M, et al. ; MAE working group of EuroEPINOMICS RES Consortium (2015). The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia 56, e203–e208. [DOI] [PubMed] [Google Scholar]
- Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, and Sheen J (2013). Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol 31, 688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Czupalla CJ, and Wolburg H (2011). Current concepts of blood-brain barrier development. Int. J. Dev. Biol 55, 467–476. [DOI] [PubMed] [Google Scholar]
- Lim RG, Quan C, Reyes-Ortiz AM, Lutz SE, Kedaigle AJ, Gipson TA, Wu J, Vatine GD, Stocksdale J, Casale MS, et al. (2017). Huntington’s Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 19, 1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, and Shusta EV (2012). Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol 30, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, and Shusta EV (2014). A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep 4, 4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Espíndola D, Morales-Bastos C, Grijota-Martínez C, Liao XH, Lev D, Sugo E, Verge CF, Refetoff S, Bernal J, and Guadaño-Ferraz A (2014). Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J. Clin. Endocrinol. Metab 99, E2799–E2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis VB, and Svendsen CN (2011). Induced pluripotent stem cells: a new revolution for clinical neurology? Lancet Neurol. 10, 383–394. [DOI] [PubMed] [Google Scholar]
- Mattis VB, Tom C, Akimov S, Saeedian J, Østergaard ME, Southwell AL, Doty CN, Ornelas L, Sahabian A, Lenaeus L, et al. (2015). HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet 24, 3257–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerl S, Visser TJ, Darras VM, Horn S, and Heuer H (2012). Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology 153, 1528–1537. [DOI] [PubMed] [Google Scholar]
- Mayerl S, Müller J, Bauer R, Richert S, Kassmann CM, Darras VM, Buder K, Boelen A, Visser TJ, and Heuer H (2014). Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J. Clin. Invest 124, 1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Mayerl S, Visser TJ, Darras VM, Boelen A, Frappart L, Mariotta L, Verrey F, and Heuer H (2014). Tissue-specific alterations in thyroid hormone homeostasis in combined Mct10 and Mct8 deficiency. Endocrinology 155, 315–325. [DOI] [PubMed] [Google Scholar]
- Núñez B, Martínez de Mena R, Obregon MJ, Font-Llitjós M, Nunes V, Palacín M, Dumitrescu AM, Morte B, and Bernal J (2014). Cerebral cortex hyperthyroidism of newborn mct8-deficient mice transiently suppressed by lat2 inactivation. PLoS ONE 9, e96915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier B, Daneman R, and Ransohoff RM (2013). Development, maintenance and disruption of the blood-brain barrier. Nat. Med 19, 1584–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM (2012). Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab 32, 1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, and Zerangue N (2008). Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 149, 6251–6261. [DOI] [PubMed] [Google Scholar]
- Shelley BC, Gowing G, and Svendsen CN (2014). A cGMP-applicable expansion method for aggregates of human neural stem and progenitor cells derived from pluripotent stem cells or fetal brain tissue. J. Vis. Exp (88), 51219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, and Caldwell MA (1998). A new method for the rapid and long term growth of human neural precursor cells. J. Neurosci. Methods 85, 141–152. [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, and Heuer H (2007). Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J. Clin. Invest 117, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, and Terasaki T (2011). Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem 117, 333–345. [DOI] [PubMed] [Google Scholar]
- Vatine GD, Zada D, Lerer-Goldshtein T, Tovin A, Malkinson G, Yaniv K, and Appelbaum L (2013). Zebrafish as a model for monocarboxyl transporter 8-deficiency. J. Biol. Chem 288, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge CF, Konrad D, Cohen M, Di Cosmo C, Dumitrescu AM, Marcinkowski T, Hameed S, Hamilton J, Weiss RE, and Refetoff S (2012). Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. J. Clin. Endocrinol. Metab 97, 4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser WE, Jansen J, Friesema EC, Kester MH, Mancilla E, Lundgren J, van der Knaap MS, Lunsing RJ, Brouwer OF, and Visser TJ (2009). Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum. Mutat 30, 29–38. [DOI] [PubMed] [Google Scholar]
- Visser WE, Swagemakers SM, Ozgur Z, Schot R, Verheijen FW, van Ijcken WF, van der Spek PJ, and Visser TJ (2010). Transcriptional profiling of fibroblasts from patients with mutations in MCT8 and comparative analysis with the human brain transcriptome. Hum. Mol. Genet 19, 4189–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GR (2008). Neurodevelopmental and neurophysiological actions of thyroid hormone. J. Neuroendocrinol 20, 784–794. [DOI] [PubMed] [Google Scholar]
- Wirth EK, Roth S, Blechschmidt C, Hölter SM, Becker L, Racz I, Zimmer A, Klopstock T, Gailus-Durner V, Fuchs H, et al. (2009). Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan- Herndon-Dudley syndrome. J. Neurosci 29, 9439–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann G, Mohácsik P, Balkhi MY, Gereben B, and Lechan RM (2015). Endotoxin-induced inflammation down-regulates L-type amino acid transporter 1 (LAT1) expression at the blood-brain barrier of male rats and mice. Fluids Barriers CNS 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, and Koenig RJ (2000). Gene regulation by thyroid hormone. Trends Endocrinol. Metab 11, 207–211. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Zada D, Tovin A, Lerer-Goldshtein T, Vatine GD, and Appelbaum L (2014). Altered behavioral performance and live imaging of circuit-specific neural deficiencies in a zebrafish model for psychomotor retardation. PLoS Genet. 10, e1004615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zevenbergen C, Meima ME, Lima de Souza EC, Peeters RP, Kinne A, Krause G, Visser WE, and Visser TJ (2015). Transport of Iodothyronines by Human L-Type Amino Acid Transporters. Endocrinology 156, 4345–4355. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE97324.