

Abstract
Genome instability is a hallmark of cancer cells and dysregulation or defects in DNA repair pathways cause genome instability and are linked to inherited cancer predisposition syndromes. Ionizing radiation can cause immediate effects such as mutation or cell death, observed within hours or a few days after irradiation. Ionizing radiation also induces delayed effects many cell generations after irradiation. Delayed effects include hypermutation, hyper-homologous recombination, chromosome instability and reduced clonogenic survival (delayed death). Delayed hyperrecombination (DHR) is mechanistically distinct from delayed chromosomal instability and delayed death. Using a green fluorescent protein (GFP) direct repeat homologous recombination system, time-lapse microscopy and colony-based assays, we demonstrate that DHR increases several-fold in response to low-LET X rays and high-LET carbon-ion radiation. Time-lapse analyses of DHR revealed two classes of recombinants not detected in colony-based assays, including cells that recombined and then senesced or died. With both low- and high-LETradiation, DHR was evident during the first two weeks postirradiation, but resolved to background levels during the third week. The results indicate that the risk of radiation-induced genome destabilization via DHR is time limited, and suggest that there is little or no additional risk of radiation-induced genome instability mediated by DHR with high-LET radiation compared to low-LET radiation.
INTRODUCTION
Ionizing radiation is an effective tool in the fight against cancer, with over a million patients receiving radiotherapy annually in the U.S. The goal of radiotherapy is to deliver lethal doses to the tumor volume while minimizing doses to normal tissue. Normal tissue tolerance is generally dose limiting because excessive radiation doses to normal tissue cause early and late complications. Early effects include skin reactions, local hair loss, intestinal problems, local pain and swelling. Late effects are observed six months or more after therapy, depend on the site of irradiation, and include neurological problems, infertility, joint pain, lymphedema, cardiovascular problems, bone loss and secondary tumors (1–3). Genome instability is a hallmark of cancer cells (4), reflecting genetic changes ranging from point mutations to chromosome rearrangements and whole chromosome gain or loss (5). Recently, it has become clear that genome instability can precede tumor initiation, thus genome instability is not just a consequence of the cancer state, but rather an active driver of tumorigenesis, accelerating the acquisition of mutations in proteins that regulate key cellular processes required for tumor growth and spread (4, 6–10).
Cells can repair DNA damage arising spontaneously or induced by genotoxic chemicals or radiation, restoring the chemical structure of DNA, but DNA repair processes sometimes fail to restore the genetic information at or near DNA lesions. Such “misrepair” results in small-to-large-scale genetic changes detected shortly after genotoxin exposure (11–13). For example, base damage can be bypassed by error-prone translesion DNA polymerases producing point mutations; non-homologous end joining (NHEJ) can result in loss or gain of nucleotides at repair junctions; and even so-called “error-free” homologous recombination (HR) repair can result in small- or large-scale loss of heterozygosity, inversions, deletions, amplifications and chromosomal translocations (14). Some of these changes occur during DNA repair (e.g., NHEJ), while others, especially those mediated by HR, can arise during DNA replication of damaged templates, either from error-prone translesion synthesis, or from reciprocal recombination (crossover) events associated with replication fork restart (15, 16). Ionizing and nonionizing radiation also induce late effects seen many cell generations after exposure, including mutations, chromosome translocations, chromosome aberrations, micronuclei, microsatellite instability, giant cells and cell death (17–21). We defined delayed hyperrecombination (DHR) as a novel delayed event induced by ionizing and nonionizing radiation (22–24). Although both delayed chromosomal instability (DCI) and DHR are induced by low to moderate radiation doses at high frequencies (indicative of nontargeted effects), DCI and DHR arise through distinct mechanisms, since delayed death is associated with DCI, but not DHR (24). HR levels are tightly regulated, and both increased or decreased levels of key HR proteins can suppress repair, trigger genome instability and predispose to cancer (25–31). Thus, the high viability and increased potential for large-scale genome rearrangement of radiation-induced DHR may pose significant risk for secondary tumor induction and/or tumor progression.
Typically, the benefits of radiotherapy far outweigh the low risk of secondary tumor induction. Nevertheless, questions remain about the relative risks of treatments with low- versus high-LET radiation (32). To help model these risks and to gain further insight into DHR mechanism(s) and persistence, we investigated DHR induction by low- and high-LET radiation (X-ray and carbon-ion radiations) in the well-characterized RKO36 cell system in which DHR is monitored with a single-copy, integrated GFP direct repeat HR substrate (24). In past studies DHR was detected by fluorescence microscopy of individual colonies with both GFP+ cells and GFP− cells (mixed GFP+/− colonies) that arose ~1 week after irradiation. In the current study, we developed a new method to measure DHR over longer time periods, and by using time-lapse microscopy we identified two novel classes of DHR products that are not detected in colony-based assays, namely individual cells that show delayed recombination but die or senesce. We further demonstrate that low-dose X-ray or carbon-ion radiations induce DHR that persists for two weeks, however, by the third week, HR returns to background levels. These results indicate that radiation-induced DHR is a transient phenotype with limited potential for long-term genome destabilizing potential, and that this risk is no greater for high-LET carbon-ion radiation than low-LET X rays.
MATERIALS AND METHODS
Cell Culture and Irradiation
RKO36 is a derivative of human colorectal carcinoma cell line RKO (33) bearing a single integrated copy of a EGFP direct repeat HR substrate (Fig. 1A) (24). Cells were grown in DMEM with glutamine (Sigma-Aldrich, Tokyo, Japan) supplemented with 1 mM sodium pyruvate, 1X antibiotic-antimycotic (Life Technologies, Grand Island, NY) and 10% fetal bovine serum (Nichirei Bioscience, Tokyo, Japan) at 37°C in 5% CO2 in air. Cells were X-ray irradiated using a Pantak HF320 X-ray machine (250 kV peak, 13 mA; half-value layer, 1.65-mm copper filter), or with monoenergetic carbon ions (290 MeV/nucleon, LET 70 keV/μm) generated by the Heavy Ion Medical Accelerator facility at the Japan National Institute of Radiological Sciences (Chiba, Japan). Limited experiments were performed with iron ions (500 MeV/nucleon, LET 200 keV/μm) due to beam time restrictions.
FIG. 1.

RKO36 cell fates. Panel A: The RKO36 HR substrate consists of 1 kbp direct repeats, comprising EGFP coding (GFP) and poly(A) sequences, and a neo resistance marker. One EGFP is driven by the CMV promoter but inactivated by an XhoI linker frameshift (FS) mutation, and the second lacks a promoter. GFP+ recombinants can arise by GC, which eliminates the XhoI mutation, or by SSA or crossovers that delete one GFP and the intervening sequence. These outcomes were distinguished by PCR with primers 1, 2 and 3. Panel B: Schematic of time-lapse and end-point DHR assays. Pure GFP− populations produced by FACS were irradiated (or mock irradiated) and surviving cells were seeded to dishes for visualization for seven days by time-lapse microscopy; resultant colonies were also scored in the end-point assay. For each condition, separate dishes were incubated in parallel, and after seven days these cells were harvested, and GFP− cells were generated by FACS and analyzed by time-lapse and end-point assays. This procedure was then repeated for a third week. Colonies arising one week postirradiation were expanded and genomic DNA was prepared for PCR analysis to distinguish GC and SSA products. The sorted GFP+ cells can be used to study delayed mutation (to GFP−) but this was not performed in this study. Panel C: Individual frames of time-lapse movies show examples of immediate HR (all GFP+) and DHR (mixed GFP+/−) colonies at indicated times; yellow arrows indicate GFP+ cells arising in the middle frame. Immediate HR and DHR cells that subsequently senesced or died are shown in the lower three rows.
Cell Survival and RBE Calculation
Cells were irradiated at ~50% confluence in T12.5 or T25 flasks, harvested, counted and dispersed to 35-mm dishes at appropriate concentrations to yield 100–150 surviving colonies per dish. Cells were incubated for seven days, and colonies were fixed with 100% ethanol, stained with crystal violet and counted after drying. Cloning efficiency was determined using nonirradiated cells and normalized survival curves were generated using Prism software version 5.0 (GraphPad Software Inc., La Jolla, CA) as follows. Exponential one phase decay nonlinear regressions were generated for 2–3 replicate determinations per data point. Goodness of fit (R2) was determined with 16 and 9 degrees of freedom for X rays and carbon ions, respectively. RBE was calculated from the quotient of the doses that yielded 10% survival for X rays and carbon ions.
Flow Cytometric Analysis of GFP Fluorescence and Autofluorescence
RKO36 GFP− and GFP+ cells from irradiated cultures or nonirradiated control cultures were harvested and washed once in phosphate buffered saline (PBS). Samples were diluted to ~106 cells/ml in PBS, and 30,000 cells per sample were analyzed with a FACSCalibur™ flow cytometer (BD Biosciences, San Jose, CA) using 488-nm excitation and GFP fluorescent emissions with filters at 530/30 nm, using CellQuest™ software for data collection (BD Biosciences). FlowJo® software (Tree Star) was used for data analysis.
Cell Sorting
Cultures of RKO36 cells are largely GFP−, with rare GFP+ cells arising by spontaneous HR during culture expansion. RKO36 cells were sorted into GFP− and GFP+ populations as follows. Cells were harvested and washed once in PBS, diluted to ~107 cells/ml and sorted using a FACSAria™ cell sorter (BD Biosciences) with excitation laser and emission filters, as noted above. Since radiation induces transient green autofluorescence (see Results), nonirradiated GFP− and GFP+ cells were used to establish sorting gates. FACSDiva™ software version 4.1.2 was used for data collection and sorting.
Time-Lapse and Colony (End Point) Analysis of Radiation-Induced DHR
DHR was scored by time-lapse microscopy in irradiated or control cells by seeding appropriate numbers of cells into 3.5 cm dishes to yield ~500 viable colonies per dish. Cells were incubated overnight, plastic lids were replaced with optically transparent glass lids, and dishes were incubated in a time-lapse microscopy system (Olympus LCV-110; Olympus, Tokyo, Japan) (34). Images were acquired each hour for seven days with MetaMorph software version 7.5.6.0 (MDS Analytical Technologies, Winnersh, UK) using the following conditions: 10× magnification, 2 × 2 camera binning, 30 ms (transillumination) and 500 ms (GFP) exposure times, and 6.0 × 6.4-mm image acquisition frames in the center of each tissue culture dish. Individual images were assembled into movies with Meta-Morph® Offline software version 7.5.6.0 (Molecular Devices, Sunnyvale, CA) using AVI2JPG version 6.10 (Novo, Tokyo, Japan) compression software. Four movies were generated per dish, each capturing 25% of the 6.0 × 6.4-mm acquisition frame.
The number of adhered cells was determined at early time points for each movie. Individual cells were classified into specific phenotypes as they developed into colonies, failed to divide (senescent) or died (dislodged from dish) during week-long incubation periods (see Results). Still images were extracted from movies by using an AVI to JPEG converter and processed using ImageJ software (National Institutes of Health, Bethesda, MD) (35). Percentages of each phenotype were calculated based on the number of initial adherent cells in each movie.
After acquisition of each week-long time-lapse movie, whole-dish, live-colony composite images were generated using a BIOREVO BZ-9000 imaging system and software (Keyence Corp., Osaka, Japan) and used to score each colony as GFP−, GFP+ or GFP+/− (end-point assay). Image files were processed using Photoshop (Adobe Systems Inc., San Jose, CA) to determine GFP status and ImageJ (35) to count colonies. In some cases, GFP status was confirmed in fixed colonies by immunofluorescence microscopy (see below). Percentage DHR was calculated as the ratio (×100) of mixed GFP+/− colonies to total initial cells (time lapse) or total colonies (end point).
Molecular Analysis of HR Products
DHR clonal isolates for PCR analysis were generated by sorting single RKO36 GFP+ recombinant cells into individual wells of 96-well tissue culture dishes seven days postirradiation. After colony formation, cells from wells with a single colony were transferred into six-well plates and expanded, and genomic DNA was prepared using purification columns (Promega Inc., Madison, WI). PCR distinguishes gene conversion, which retains the parental GFP direct repeat structure, from GFP deletions via single-strand annealing (SSA or, less likely, crossover) repair events. PCR amplification was performed with a T3 forward primer (5′-CTCGAAATTAACCCTCACTAAAGG; Fig. 1A, primer 1) paired with a reverse primer between the two GFP genes (5′-TTCCAAACTGGAACAACACTCAAC) or downstream of the distal GFP gene (5′-GATTTAGTGCTTTACGGCACCTC) (Fig. 1A, primers 2 and 3, respectively). Primers 1 + 2 yield a 1,512 bp fragment from the parent and GC products; primers 1 + 3 produce a 1,422 bp fragment from deletion products.
Immunofluorescence Detection of 53BP1 Foci and GFP
Cells were seeded onto Nunc chamber glass slides 24 h prior to X-ray or carbon-ion irradiations. After irradiation, cells were washed in cold PBS and fixed for 10 min in 4% w/v paraformaldehyde in PBS. Cells were permeabilized for 2 min with 0.2% v/v Triton™ X-100 (Sigma-Aldrich, Tokyo, Japan) in PBS and washed twice in PBS. Antibodies were diluted with 4% w/v BSA in PBS. The slides were incubated with rabbit anti-53BP1 (Bethyl Laboratories Inc., Montgomery, TX) or mouse anti-GFP monoclonal antibodies (Life Technologies) and incubated for 1 h in a humidified incubator at 37°C, washed three times in PBS and then processed in a similar fashion with Alexa Fluor® 555-conjugated anti-rabbit IgG or Cy3-conjugated anti-mouse IgG antibodies (Sigma-Aldrich). Slides were incubated in PBS containing 4′,6-diamidino-2-phenylindole (DAPI) for 5 min to stain DNA and mounted using Vectashield® antifade medium (Vector® Laboratories, Burlingame, CA). Immunofluorescent images were acquired using a BX51 fluorescent microscope controlled by DP Controller software version 2.2.1.227 (Olympus) and processed using DP Manager software version 2.2.1.195 (Olympus).
RESULTS
Time-Lapse Microscopy Reveals DHR and Previously Undetected Cell Fates
In prior studies DHR was detected as mixed GFP+/− colonies arising 7–10 days, after derivatives of human RKO or hamster–human hybrid GM10115 cells were exposed to low-to-moderate doses of ionizing radiation or ultraviolet (UV) radiation (22–24). In the current study, we modified the assay to detect DHR over a longer time period and to expand the range of detectable outcomes. Studies were performed in RKO36 cells, which carry a single, integrated copy of a GFP direct repeat HR reporter (Fig. 1A). The upstream GFP gene is inactivated by a frameshift mutation and the downstream copy has a complete coding sequence but is inactive because it lacks a promoter. HR produces GFP+ recombinants by gene conversion, which preserves the gross structure of the reporter, or by SSA or crossovers that delete one GFP copy and the intervening sequence; HR outcomes were distinguished by PCR (Fig. 1A). Here we use the term “HR” as inclusive of both conservative gene conversion (RAD51-dependent) and SSA (RAD51-independent) events, since both involve recombination between homologous sequences.
DHR was analyzed over three-week periods after low- or high-LET irradiation (Fig. 1B). Parental RKO36 cells were first sorted into GFP− and GFP+ populations. Although not part of the current study, GFP+ cells can be used to study delayed mutation to GFP−. GFP− cells were irradiated and split into two cultures. One culture was analyzed for seven days by time-lapse microscopy, followed by end-point analysis of resultant colonies, classified as GFP− (nonrecombinant), GFP+ (recombinant induced immediately by radiation) or mixed GFP+/− (DHR). Clonal isolates of GFP+ DHR products were expanded for PCR analysis of HR outcomes. The second culture of GFP− cells was sorted seven days after plating to obtain pure GFP− populations, which were analyzed as above for time-lapse and end-point assays 8–14 days postirradiation; this procedure was repeated for a third week (days 15–21 postirradiation). In the initial week of analysis, both DHR and immediate HR products were detectable as mixed GFP+/− and full GFP+ colonies, respectfully. In the second and third weeks, all GFP+ colonies (pure or mixed GFP+/−) were classified as DHR because these periods began with pure (sorted) GFP− cultures. Time-lapse microscopy revealed immediate HR and DHR products, as in the end-point assay, and as expected there was good agreement between the two assays (see below). Time-lapse microscopy also revealed several other events not detectable in the end-point assay, including cells that were GFP+ within hours of irradiation and senesced or died and those that became GFP+ at later times and senesced or died (Fig. 1C). In this study, we analyzed cell fates after exposure to equitoxic doses of low-LET X rays or high-LET carbon ions using the end-point assay; time-lapse microscopy was performed on the carbon-ion-irradiated cells.
RKO36 Displays Typical Survival and DNA Damage Responses to Low- and High-LET Radiation
We performed colony-based survival assays in response to acute doses of low-LET X rays and high-LET (70 keV/μm) carbon ions and determined the relative biological effect (RBE) at 10% survival (D10). As is typical for mammalian cells, carbon ions were more cytotoxic to RKO cells per unit dose than X rays, yielding an RBE of ~3 (Fig. 2). This value was used in subsequent experiments to compare effects of equitoxic doses of X rays and carbon ions. The higher RBE of carbon ions is thought to reflect complex, clustered DNA damage (36–38). 53BP1 is a DNA damage response protein that is rapidly recruited to double-strand breaks (DSBs) detected as subnuclear foci with cognate antibodies. X rays produced small 53BP1 foci, but the clustered damage created by carbon ions yielded larger foci (Supplementary Fig. S1; http://dx.doi.org/10.1667/RR14748.1.S1), characteristic of those observed with various types of high-LET radiation (39, 40). These large foci may reflect 53BP1 recruited to many lesions along an individual, high-LET radiation track. RKO36 cells display normal p53 and p21 responses to radiation (24), and equitoxic doses of X rays and carbon ions induced apoptosis with similar frequencies and kinetics (Supplementary Fig. S2; http://dx.doi.org/10.1667/RR14748.1.S1).
FIG. 2.
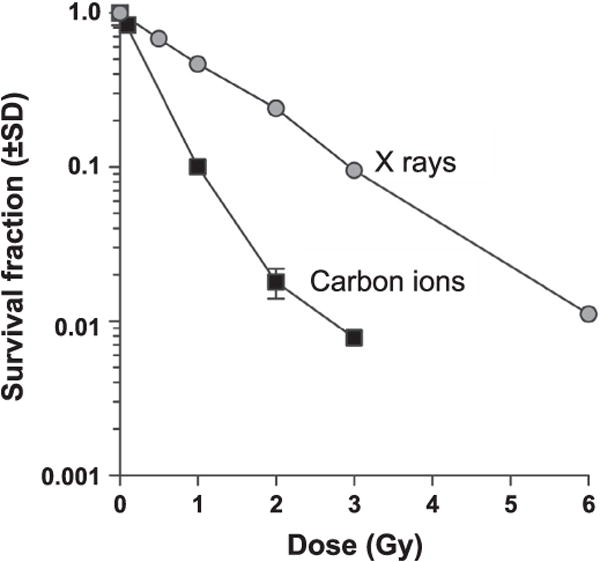
Exposure dose-response curves. RKO36 cells were irradiated with X rays, carbon ions and survival was measured by clonogenic assay. Data are averages (±SD) for three replicates per determination.
Radiation Induces Transient Autofluorescence That Can Confound Early Detection of GFP+ Cells
To facilitate HR product analysis, GFP-based HR reporters are typically employed as single integrated copies, as in RKO36 cells. The level of GFP expression for a particular reporter, after a productive HR event, varies with integration site, reflecting position effects (41) and with copy number. The single copy GFP reporter in RKO36 cells produces a moderately bright GFP signal that is several-fold above the level of background autofluorescence. While developing the time-lapse microscopy assay to analyze DHR, we performed control experiments using flow-cytometry and observed that ~50% of cells irradiated with 6 Gy of X rays or 2 Gy of carbon ions showed green fluorescence at a level comparable to nonirradiated GFP+ cells (Supplementary Fig. S3A; http://dx.doi.org/10.1667/RR14748.1.S1). Because it was highly unlikely that radiation induced HR at such high frequencies, we considered the possibility that the radiation was instead inducing autofluorescence in RKO36 cells, as reported for other cell types (42). Radiation increased green fluorescence in both GFP− and GFP+ cells (Supplementary Fig. S3B), indicating that the effect is independent of GFP expression. Green fluorescence also increased with dose, but this is a transient effect, since it was not detectable 7–8 days postirradiation (Supplementary Fig. S3C; and data not shown). Consistent with this effect occurring independently of GFP, it was observed in cells carrying a GFP reporter (GFP− and GFP+ RKO36 cells) and in cells lacking a reporter (parent RKO and HeLa cells) (Supplementary Fig. S3D). The magnitude of the effect was similar among RKO cell lines with or without GFP and in HeLa cells: 6 Gy X-ray doses increased autofluorescence by ~2.5 fold in each case (Supplementary Fig. S3E). Despite the transient increase in autofluorescence, GFP+ cells were distinguishable from fainter GFP− cells; during the first week postirradiation when autofluorescence was maximal, GFP+ cells were confirmed by staining with anti-GFP antibodies (Supplementary Fig. S3F).
Carbon-Ion Radiation Slightly Induces Immediate HR
DSBs created within or near HR reporter loci stimulate HR at high frequency. Radiation is relatively inefficient at stimulating HR at a reporter locus because DSBs induced by radiation at low to moderate doses are generally random and thus rarely occur near reporter loci. Immediate HR (IHR) was assayed by time-lapse microscopy, with 3–4 determinations per condition (Fig. 3). We observed apparent increases in IHR after 0.6–2 Gy carbon-ion irradiations, but absolute IHR frequencies were quite low, and only at the 1 Gy dose was there a significant difference in IHR between irradiated cells and nonirradiated controls (Fig. 3, left-side panel). Sometimes IHR events were followed by senescence or death. These too were rare, and were even less frequent after irradiation (Fig. 3, right-side panel), perhaps because carbon ions create damage that is highly lethal, as this would reduce the chance for cells to attach and senesce or die at later times. This idea is consistent with the reduction in senescent cells (regardless of GFP status) with increasing carbon-ion irradiations (see below).
FIG. 3.
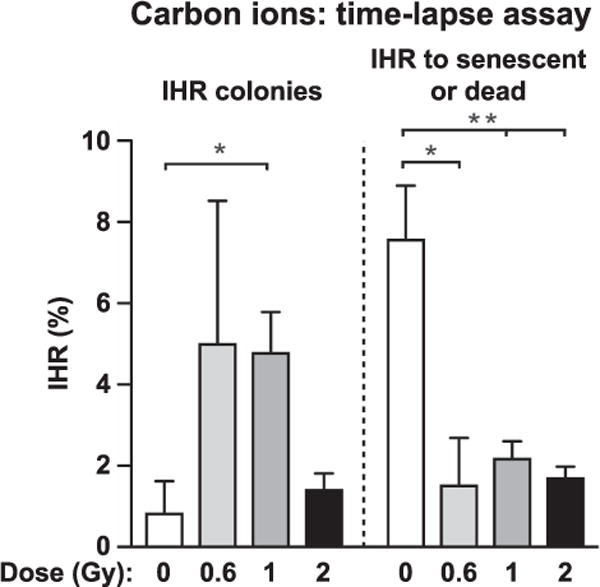
Frequencies of carbon-ion irradiation induced immediate HR (IHR) and IHR to senescence or death. Phenotypes were determined by time-lapse microscopy with 97–1,659 initial cells scored per condition (average number of cells scored = 730). Values are averages (+SEM) for 3–4 determinations and statistics were calculated by t tests. *P < 0.05; **P < 0.01.
Low- and High-LET Radiation Induces DHR for Two Weeks and Then Resolves to Background Levels
DHR is induced 7–10 days after UV and low-LET irradiations, detected with an end-point assay (22–24). We measured DHR after exposure to equitoxic doses of X rays or carbon ions to determine whether DHR is differentially induced by low- versus high-LET radiation. We also extended the assay period to three weeks to define DHR persistence, and scored events by time-lapse microscopy and end-point assays. In this and subsequent time-lapse and end-point assays, values were fairly consistent among determinations, and data were pooled. Each cell (or colony) was categorized as, for example, DHR or not DHR, and statistics were determined by Fisher exact tests. Thus, absolute values rather than averages/error bars are presented. During the first and second weeks postirradiation, X rays induced statistically significant increases in DHR, but levels returned to background by the third week (Fig. 4). DHR was also induced by carbon ions, and similar to X rays, it persisted for two weeks before resolving in the third week postirradiation (Fig. 5). At equitoxic doses, DHR appeared to be induced approximately twofold more efficiently by carbon ions than X rays during the first two weeks (scored by end-point assay), but with statistically significant differences in only two cases: week 1 for 6 Gy X rays vs. 2 Gy carbon ions (P < 0.0001); and week 2 for 3 Gy X rays vs. 1 Gy carbon ions (P = 0.0084). A larger study testing a range of LET radiations may confirm a trend of increasing DHR with increasing LET. Together, the results indicate that equitoxic doses of high-LET radiation induce DHR with similar or slightly higher efficiency than low-LET radiation, and that regardless of dose or LET, DHR persists for two weeks before resolving to background levels.
FIG. 4.

X-ray-induced DHR. DHR was scored by end-point assay, with 2–4 dishes monitored and 201–2,606 colonies scored per condition (average number of colonies scored = 1,080). Statistics calculated by Fisher exact tests. *P < 0.05; **P < 0.01; ***P < 0.001.
FIG. 5.
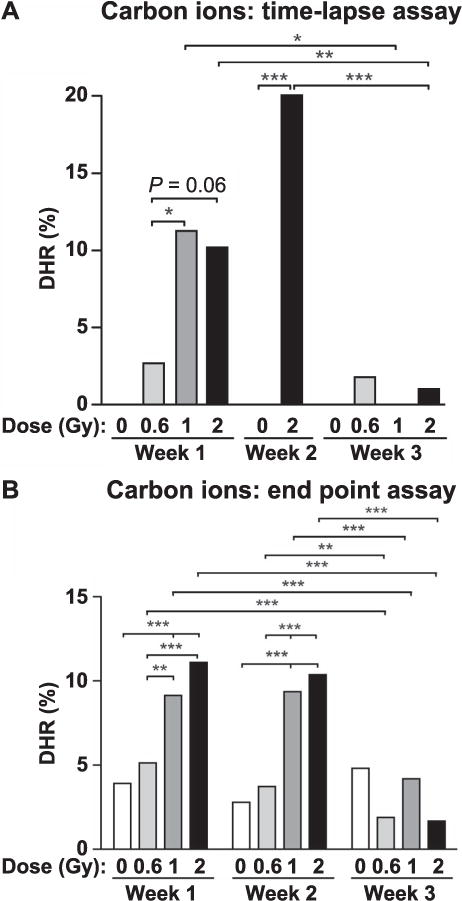
Carbon-ion-induced DHR measured by time-lapse and endpoint assays. Time-lapse values were determined as described in Fig. 3 legend, with 74–1,659 initial cells scored per condition (average number of cells scored = 353). End-point values and statistics were determined as described in Fig. 4 legend, with 488–1,814 colonies scored per condition (average number of cells scored = 1,165).
Carbon-Ion-Induced DHR is Frequently Associated with Cell Senescence or Death
As noted above, the time-lapse assay revealed cell fates not detected in traditional colony-based assays, that is, cells that displayed DHR that subsequently senesced or died (Fig. 1C). We quantitated these events and found that senescence or death subsequent to DHR was not induced by carbon ions in the first week postirradiation, but with a moderate (1 Gy) dose, these cell fates appeared to increase during the second and third week postirradiation (Fig. 6A). At the highest dose (2 Gy), there was little or no induction of DHR to senescence/death over the three-week period (Fig. 6A). Consistent with a ~50% plating efficiency, approximately half of cells visualized by time-lapse microscopy either senesced or died, regardless of GFP status (Fig. 6B). However, total senescence and death did increase with dose, particularly during the initial two weeks of analysis, although at the highest carbon ion dose (2 Gy), senescent and dead cells were less frequent in weeks 2 and 3 (Fig. 6B), probably because severely damaged cells were cleared from the population by these later times.
FIG. 6.
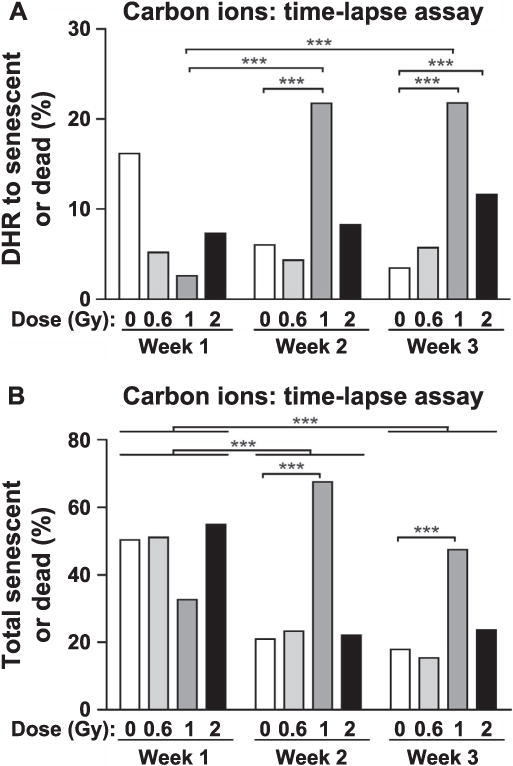
Carbon-ion-induced DHR to senescence or death (panel A) and total senescent or dead cells (panel B). These events were determined by time-lapse assays using the same samples and statistical analyses described in Fig. 5 legend.
Because the time-lapse assay revealed DHR cells that senesced, we examined lone cells (those not belonging to a colony) in end-point assay dishes in the first and second weeks after carbon-ion irradiation, and scored GFP status by fluorescence microscopy. This revealed significant fractions of DHR to senescent cells, even in nonirradiated controls, and these events increased among populations exposed to 1–2 Gy radiation (Supplementary Fig. S4; http://dx.doi.org/10.1667/RR14748.1.S1). The time-lapse assay showed less frequent senescent/dead cells in the second and third weeks postirradiation (Fig. 6B), and this trend was also apparent for DHR to senescent cells identified in the end-point assay (Supplementary Fig. S4).
Repeat Deletion is the Dominant HR Outcome after Exposure to Low- or High-LET Radiation
The GFP direct repeat in RKO36 cells can produce a functional GFP protein by gene conversion without a crossover, which maintains the gross repeat structure, or by SSA or crossovers, which delete one repeat and the 217 bp sequence between repeats (Fig 1A). Genomic DNA was prepared from GFP+ clones isolated one week after equitoxic exposures to X rays (3 Gy), carbon ions (1 Gy) or iron ions (1 Gy). In all cases, the majority of GFP+ products arose by deletion (Supplementary Fig. S5; http://dx.doi.org/10.1667/RR14748.1.S1), most likely via SSA. There was no statistically significant difference between X-ray versus carbon-ion products, nor X-ray versus iron-ion products, but the higher fraction of deletions after iron-ion irradiation (100%) was significantly different from that of carbon ions (82%) (Supplementary Fig. S5). Larger sample sets might reveal a statistically significant difference between X rays and iron ions.
DISCUSSION
Homologous recombination has critical roles in maintaining genome stability and tumor suppression, and it is tightly regulated in tissues and during the cell cycle (43–47). HR is particularly important during DNA replication, since it can accurately repair and restart stalled or collapsed replication forks; when HR is defective, broken forks may be repaired by alternative NHEJ, producing translocations and other gross genome rearrangements (48–50). HR dysregulation includes defects in HR proteins that reduce HR efficiency (51–58), as well as defects in HR suppressors, such as the Bloom syndrome gene (BLM), which confer a hyper-HR phenotype (59, 60). Both hypo-and hyper-HR phenotypes are associated with cancer predisposition, emphasizing the importance of proper regulation of HR (29, 43). Increased HR levels may also reflect increased levels of DNA damage as a result of metabolic defects that increase reactive oxygen species (ROS) levels, or defects in other repair pathways that increase DNA damage load and replication stress (15, 61, 62). The precise integration site of the GFP HR substrate in RKO36 cells is unknown. As with expression of transgenes, HR can vary with integration site (“position effects”). In a previously published study, we showed that X-ray-induced DHR varied several-fold among RKO36 and several other cell lines carrying the same GFP HR substrate (24). A key question concerns the potential association between radiation-induced DHR and mutagenesis or carcinogenesis. In a study of UV-induced DHR, increased mutagenesis (at HPRT) was observed among DHR colonies, but there was no evidence that DHR specifically caused mutagenesis, or vice versa, since mutations arising in DHR colonies were point mutations (22), as opposed to deletions that might have arisen via SSA (e.g., between intronic Alu repeats). However, DHR and mutagenesis may be triggered by the same underlying cause, such as increased ROS, as discussed further below. To date, no studies have correlated radiation-induced DHR with mutagenesis or carcinogenesis, so these remain important open questions for the future that may be addressed with advanced mutation detection systems, and transgenic mice such as RaDR-GFP mice developed in the Engelward laboratory (63).
Because genome stability requires proper HR regulation, radiation-induced DHR raises significant concerns, especially given that DHR is efficiently induced by radiation doses that approximate those delivered to patients for CT scans (23, 64). Unlike DCI, DHR is associated with high cell viability, indicating that the genome instability associated with DHR does not typically cause lethal chromosome rearrangements, as appears to be the case with DCI (24). The high viability of DHR cells would tend to increase risk of genome instability and cancer, since such cells would not be cleared from tissues as readily as DCI cells. On the other hand, DCI can persist for years (19), whereas the risks associated with persistence of genome instability driven by DHR appear to be limited to approximately a two-week period postirradiation (Figs. 4 and 5). Although unlikely, we cannot rule out that DHR rises and falls in a cyclic fashion; addressing this question would require DHR monitoring over longer time periods. Earlier studies indicated that although DHR and DCI are both radiation induced with high efficiency, they are nonetheless mechanistically distinct, since cells that express DCI do not express DHR and vice versa (24). In addition to differences in viability, the current study revealed the vast difference in the persistence of DHR and DCI, highlighting yet another mechanistic difference between these phenotypes.
We show that DHR is stimulated by both low- and high-LET radiation, and that under both conditions it persists for two weeks before resolving in the third week after an acute radiation dose. The trigger(s) for DHR remain a mystery, although it is clear that, similar to DCI (19, 65, 66), the target for radiation-induced DHR is as large as the nucleus and perhaps the entire cell (23). DHR does not appear to be induced in nonirradiated cells, i.e., as a bystander effect (23). It is instructive to consider the potential triggers and persistence mechanisms of DCI, as more studies have addressed this question than for DHR. Changes in gene expression are not implicated in DCI, nor are DSBs alone sufficient to trigger DCI (67, 68), although DNA is at least part of the DCI target (69). Epigenetic changes are potential drivers of bystander effects, and may contribute to, or regulate, radiation-induced DCI. DCI may be regulated by DDR factors, but given the large target size, it is unlikely that DCI reflects specific alterations to DDR machinery. There is evidence that DCI is stimulated by persistent high levels of ROS as a result of mitochondrial dysfunction (70–73). This idea is supported by evidence that suppressing ROS with radical scavengers suppresses radiation-induced DCI (74). Interestingly, increased ROS levels were not associated with increased oxidative DNA damage (75), raising the possibility that oxidative damage to other cellular components may trigger DCI, or perhaps that oxidative DNA damage repair itself triggers DCI. Another feature of DCI that may drive persistence is the fact that large-scale chromosome rearrangements can themselves be destabilizing (and therefore self-propagating), as evidenced by bridge-breakage-fusion cycles (76, 77).
Although DCI and DHR are both induced by radiation, the mutually exclusive nature of these phenotypes argue that they are mechanistically distinct. The vast difference in persistence (weeks vs. years) is further evidence that DCI and DHR are distinct, and suggests that the proposed persistence mechanisms for DCI (elevated ROS, mitochondrial dysfunction) do not underlie the more transient DHR phenotype. One potential clue that may help explain the induction and transient persistence of DHR is the observation that radiation induces transient autofluorescence. We confirmed the findings of Schaue et al. (42), that X rays cause a dose-dependent increase in autofluorescence. In that study, autofluorescence was measured with a 488-nm excitation wavelength, the effect was proportional to dose, and it was observed with many different cell lines (42), similar to our results (Supplementary Fig. S3; http://dx.doi.org/10.1667/RR14748.1.S1). Radiation-induced autofluorescence correlated with mitochondrial changes in NAD/NADH/FAD concentrations, calcium homeostasis and cellular morphology, and effects were observed 1–3 days postirradiation (42). We extended these findings by showing that autofluorescence is induced by low- and high-LET radiation (Fig. S3A) and that it persists for ~7 days (Supplementary Fig. S3C). Although radiation-induced autofluorescence subsides before DHR (Figs. 4 and 5), it is still possible that these phenomena are related. Autofluorescence is only detected with relatively high doses of radiation (≥2 Gy) whereas DHR is induced by doses of a few cGy (23), thus autofluorescence and DHR might share the same trigger, but DHR may have a lower threshold, or there may be other changes associated with altered NAD/NADH/FAD/calcium/morphology that induce DHR for longer periods than autofluorescence. If DHR is indeed linked to the transient effects of radiation on NAD/NADH/FAD levels, calcium levels and/or cellular morphology, one intriguing possibility is that these effects may be mediated by NAD-regulation of Sirtuins, which regulate chromatin structure and function. It is well known that chromatin regulates many DNA dynamic processes, including HR (78–86). Schaue et al. (42) demonstrated that radiation induction of autofluorescence could be attenuated by treating cells with the calcium ionophore A23187, or low-nutrient conditions that prevent radiation-induced cell swelling. It will be interesting to determine if DHR is similarly attenuated by calcium ionophores and low-nutrient conditions, and if so, such treatments may mitigate radiation-induced DHR and thereby reduce risks associated with DHR-mediated genome instability.
Among the DHR products analyzed, we observed a strong bias toward deletion events (Supplementary Fig. S5; http://dx.doi.org/10.1667/RR14748.1.S1). These deletions likely arise by SSA, given that deletions increase with decreasing length of the sequence between repeats (87), and the HR substrate in RKO36 cells has a short, 217 bp sequence between GFP repeats. There was no difference in the deletion:GC ratios between X-ray and carbon-ion products, but there was a striking absence of GC products after iron-ion exposure. High-LET DNA damage is refractory to repair by NHEJ (37, 88), and it promotes end resection, even in G1 cells in which resection is normally suppressed (89). Both GC and SSA require end resection, so this alone cannot account for the lack of GC after iron irradiation. SSA generally requires more end resection than RAD51-dependent HR, as complementary strands in repeats must be exposed to allow RAD52 to mediate annealing (90). The lack of GC products after iron-ion irradiation could result from more extensive end resection (89), which would promote SSA, and/or from complex damage suppressing RAD51-mediated strand invasion or other HR steps subsequent to end resection. Both SSA and GC events are stimulated by DSBs, but it is unlikely that the events observed here result directly from radiation-induced DSBs, since this would probably require DSBs in or between the GFP repeats, or within several kbp of the repeats, and these would be exceedingly rare with radiation doses compatible with cell survival. A more likely explanation is that SSA and GC events result from replication stress associated with DNA damage (91, 92), induced by ROS or other factors during ~8 cell divisions occurring over the course of the one-week time-lapse assays, or ~16 cell divisions occurring over the course of the two-week end-point assays. Although RAD51-mediated HR and SSA both involve interactions between segments sharing extensive homology, SSA is nonconservative, leading to large deletions when it occurs between linked repeats, and translocations when repeats reside on different chromosomes (93). Thus, SSA is inherently genome-destablizing, and it has been implicated in cancer predisposition associated with defects in RAD51-dependent HR, such as BRCA2 mutations (28).
In summary, this study demonstrates that the risks of genome instability associated with DHR are limited to a two-week period postirradiation, that DHR is similarly induced by X rays and carbon ions, and that carbon ions appear to pose a similar, or perhaps slightly greater risk of DHR-mediated genome destabilization via SSA than X rays. SSA-associated risk does appear to increase for extremely high-LET ions like iron, but it is important to note that the carcinogenic properties of such ions may be mitigated in part by their highly lethal nature. Thus, high-LET radiation induced delayed genomic instability is only relevant when cells receive sublethal doses. These and other considerations contribute to the substantial complexity of risk assessment for radiation exposures from terrestrial and space environments, and from diagnostic and therapeutic clinical procedures (94, 95).
Supplementary Material
Acknowledgments
We thank Mr. T. Maeda and the FACS support team of the National Institute of Radiological Sciences (NIRS), as well as M. Akaishizawa, Y-Q. Fang, F. Maruyama, K. Dozono and M. King for technical assistance and S. Bailey and H. Liber for helpful discussions. We appreciate the support provided by the Chang Yung-Fa Fund to the NIRS International Open Laboratory. This research was supported by the National Institutes of Health (NIH grant no. GM084020 to JAN). This report is dedicated to the memory of William F. Morgan, an important pioneer in the field of radiation-induced delayed genome instability.
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR14748.1) contains supplementary information that is available to all authorized users.
References
- 1.Hawley L. Management of acute toxicity associated with radiotherapy. Br J Hosp Med (Lond) 2013;74:C173–6. doi: 10.12968/hmed.2013.74.sup11.c173. [DOI] [PubMed] [Google Scholar]
- 2.Oeffinger KC, Baxi SS, Novetsky Friedman D, Moskowitz CS. Solid tumor second primary neoplasms: who is at risk, what can we do? Semin Oncol. 2013;40:676–89. doi: 10.1053/j.seminoncol.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall EJ, Giaccia AJ. Radiobiology for the radiobiologist. 7th. Philadelphia: J. B. Lippincott; 2011. [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Allen CP, Fujimori A, Okayasu R, Nickoloff JA. Radiation-induced delayed genome instability and hypermutation in mammalian cells. In: Mittelman D, editor. Stress-induced mutagenesis. New York Heidelberg Dordrecht London: Springer; 2013. [Google Scholar]
- 6.Hanks S, Coleman K, Reid S, Plaja A, Firth H, FitzPatrick D, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–61. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 7.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 8.Pikor L, Thu K, Vucic E, Lam W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013;32:341–52. doi: 10.1007/s10555-013-9429-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61:818–22. [PubMed] [Google Scholar]
- 10.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Benjamin MB, Little JB. X rays induce interallelic homologous recombination at the human thymidine kinase gene. Mol Cell Biol. 1992;12:2730–38. doi: 10.1128/mcb.12.6.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao J, DePrimo SE, Stringer JR. Cell cycle dependence of radiation-induced homologous recombination in cultured monkey cells. Mutat Res. 1997;374:233–43. doi: 10.1016/s0027-5107(96)00237-0. [DOI] [PubMed] [Google Scholar]
- 13.Liber HL, Idate R, Warner C, Bailey SM. Radiation quality and mutagenesis in human lymphoblastoid cells. Radiat Res. 2014;182:390–5. doi: 10.1667/RR13817.1. [DOI] [PubMed] [Google Scholar]
- 14.Shen Z, Nickoloff JA. Mammalian homologous recombination repair and cancer intervention. In: Wei Q, Li L, Chen DJ, editors. DNA repair, genetic instability, and cancer. Singapore: World Scientific Publishing Co; 2007. [Google Scholar]
- 15.Allen C, Ashley AK, Hromas R, Nickoloff JA. More forks on the road to replication stress recovery. J Mol Cell Biol. 2011;3:4–12. doi: 10.1093/jmcb/mjq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budzowska M, Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem Biophys. 2009;53:17–31. doi: 10.1007/s12013-008-9039-y. [DOI] [PubMed] [Google Scholar]
- 17.Dahle J, Kvam E. Induction of delayed mutations and chromosomal instability in fibroblasts after UVA-, UVB-, and X-radiation. Cancer Res. 2003;63:1464–9. [PubMed] [Google Scholar]
- 18.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res. 2003;159:581–96. doi: 10.1667/0033-7587(2003)159[0581:nadeoe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 19.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat Res. 2003;159:567–80. doi: 10.1667/0033-7587(2003)159[0567:nadeoe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 20.Roy K, Kodama S, Suzuki K, Watanabe M. Delayed cell death, giant cell formation and chromosome instability induced by X-irradiation in human embryo cells. J Radiat Res. 1999;40:311–22. doi: 10.1269/jrr.40.311. [DOI] [PubMed] [Google Scholar]
- 21.Chang WP, Little JB. Delayed reproductive death as a dominant phenotype in cell clones surviving X-irradiation. Carcinogenesis. 1992;13:923–8. doi: 10.1093/carcin/13.6.923. [DOI] [PubMed] [Google Scholar]
- 22.Durant ST, Paffett KS, Shrivastav M, Timmins GS, Morgan WF, Nickoloff JA. UV radiation induces delayed hyperrecombination associated with hypermutation in human cells. Mol Cell Biol. 2006;26:6047–55. doi: 10.1128/MCB.00444-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang L, Kim PM, Nickoloff JA, Morgan WF. Targeted and non-targeted effects of low-dose ionizing radiation on delayed genomic instability in human cells. Cancer Res. 2007;67:1099–104. doi: 10.1158/0008-5472.CAN-06-3697. [DOI] [PubMed] [Google Scholar]
- 24.Huang L, Grimm S, Smith LE, Kim PM, Nickoloff JA, Goloubeva OG, et al. Ionizing radiation induces delayed hyperrecombination in mammalian cells. Mol Cell Biol. 2004;24:5060–68. doi: 10.1128/MCB.24.11.5060-5068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–78. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 26.Myung K, Datta A, Chen C, Kolodner RD. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet. 2001;27:113–16. doi: 10.1038/83673. [DOI] [PubMed] [Google Scholar]
- 27.Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, et al. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging. 2011;3:515–32. doi: 10.18632/aging.100325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–16. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–3. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- 30.Kim PM, Allen C, Wagener BM, Shen Z, Nickoloff JA. Overexpression of human RAD51 and RAD52 reduces double-strand break-induced homologous recombination in mammalian cells. Nucleic Acids Res. 2001;29:4352–60. doi: 10.1093/nar/29.21.4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertrand P, Lambert S, Joubert C, Lopez BS. Overexpression of mammalian Rad51 does not stimulate tumorigenesis while a dominant-negative Rad51 affects centrosome fragmentation, ploidy and stimulates tumorigenesis, in p53-defective CHO cells. Oncogene. 2003;22:7587–92. doi: 10.1038/sj.onc.1206998. [DOI] [PubMed] [Google Scholar]
- 32.Newhauser WD, Durante M. Assessing the risk of second malignancies after modern radiotherapy. Nat Rev Cancer. 2011;11:438–48. doi: 10.1038/nrc3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brattain MG, Brattain DE, Fine WD, Khaled FM, Marks ME, Kimball PM, et al. Initiation and characterization of cultures of human colonic carcinoma with different biological characteristics utilizing feeder layers of confluent fibroblasts. Oncodev Biol Med. 1981;2:355–66. [PubMed] [Google Scholar]
- 34.Araki R, Jincho Y, Hoki Y, Nakamura M, Tamura C, Ando S, et al. Conversion of ancestral fibroblasts to induced pluripotent stem cells. Stem Cells. 2010;28:213–20. doi: 10.1002/stem.282. [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–75. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okayasu R. Repair of DNA damage induced by accelerated heavy ions–a mini review. Int J Cancer. 2012;130:991–1000. doi: 10.1002/ijc.26445. [DOI] [PubMed] [Google Scholar]
- 37.Okayasu R, Okada M, Okabe A, Noguchi M, Takakura K, Takahashi S. Repair of DNA damage induced by accelerated heavy ions in mammalian cells proficient and deficient in the non-homologous end-joining pathway. Radiat Res. 2006;165:59–67. doi: 10.1667/rr3489.1. [DOI] [PubMed] [Google Scholar]
- 38.Sutherland BM, Bennett PV, Schenk H, Sidorkina O, Laval J, Trunk J, et al. Clustered DNA damages induced by high and low LET radiation, including heavy ions. Phys Med. 2001;17:202–4. [PubMed] [Google Scholar]
- 39.Jakob B, Scholz M, Taucher-Scholz G. Biological imaging of heavy charged-particle tracks. Radiat Res. 2003;159:676–84. doi: 10.1667/0033-7587(2003)159[0676:biohct]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Jakob B, Splinter J, Taucher-Scholz G. Positional stability of damaged chromatin domains along radiation tracks in mammalian cells. Radiat Res. 2009;171:405–18. doi: 10.1667/RR1520.1. [DOI] [PubMed] [Google Scholar]
- 41.Fukushige S, Sauer B. Genomic targeting with a positive-selection lox integration vector allows highly reproducible gene expression in mammalian cells. Proc Natl Acad Sci U S A. 1992;89:7905–09. doi: 10.1073/pnas.89.17.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaue D, Ratikan JA, Iwamoto KS. Cellular autofluorescence following ionizing radiation. PLoS ONE. 2012;7:e32062. doi: 10.1371/journal.pone.0032062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35:909–23. doi: 10.15252/embj.201693860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durant ST, Nickoloff JA. Good timing in the cell cycle for precise DNA repair by BRCA1. Cell Cycle. 2005;4:1216–22. doi: 10.4161/cc.4.9.2027. [DOI] [PubMed] [Google Scholar]
- 46.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–47. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 47.Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin Cancer Biol. 2016:37–38. 51–64. doi: 10.1016/j.semcancer.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:7720–5. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res. 2014;329:85–93. doi: 10.1016/j.yexcr.2014.09.030. [DOI] [PubMed] [Google Scholar]
- 50.Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen. 2015;793:166–75. doi: 10.1016/j.mrgentox.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Brenneman MA, Weiss AE, Nickoloff JA, Chen DJ. XRCC3 is required for efficient repair of chromosome breaks by homologous recombination. Mutat Res. 2000;459:89–97. doi: 10.1016/s0921-8777(00)00002-1. [DOI] [PubMed] [Google Scholar]
- 52.Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Mol Cell. 2002;10:387–95. doi: 10.1016/s1097-2765(02)00595-6. [DOI] [PubMed] [Google Scholar]
- 53.Cui X, Brenneman MA, Meyne J, Oshimura M, Goodwin EH, Chen DJ. The XRCC2 and XRCC3 genes are required for chromosome stability in mammalian cells. Mutat Res. 1999;434:75–88. doi: 10.1016/s0921-8777(99)00010-5. [DOI] [PubMed] [Google Scholar]
- 54.Lio YC, Schild D, Brenneman MA, Redpath JL, Chen DJ. Human Rad51C deficiency destabilizes XRCC3, impairs recombination, and radiosensitizes S/G2-phase cells. J Biol Chem. 2004;279:42313–20. doi: 10.1074/jbc.M405212200. [DOI] [PubMed] [Google Scholar]
- 55.Schoenmakers EFPM, Huysmans C, Van de Ven WJM. Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) uterine leiomyomas. Cancer Res. 1999;59:19–23. [PubMed] [Google Scholar]
- 56.Sigurdsson S, Van Komen S, Bussen W, Schild D, Albala JS, Sung P. Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev. 2001;15:3308–18. doi: 10.1101/gad.935501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takata M, Sasaki MS, Sonoda E, Fukushima T, Morrison C, Abala JS, et al. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol Cell Biol. 2000;20:6476–82. doi: 10.1128/mcb.20.17.6476-6482.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiese C, Hinz JM, Tebbs RS, Nham PB, Urbin SS, Collins DW, et al. Disparate requirements for the Walker A and B ATPase motifs of human RAD51D in homologous recombination. Nucleic Acids Res. 2006;34:2833–43. doi: 10.1093/nar/gkl366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheok CF, Bachrati CZ, Chan KL, Ralf C, Wu L, Hickson ID. Roles of the Bloom’s syndrome helicase in the maintenance of genome stability. Biochem Soc Trans. 2005;33:1456–9. doi: 10.1042/BST0331456. [DOI] [PubMed] [Google Scholar]
- 60.Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol. 2007;14:677–9. doi: 10.1038/nsmb1267. [DOI] [PubMed] [Google Scholar]
- 61.Carr AM, Lambert S. Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol. 2013;425:4733–44. doi: 10.1016/j.jmb.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 62.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sukup-Jackson MR, Kiraly O, Kay JE, Na L, Rowland EA, Winther KE, et al. Rosa26-GFP direct repeat (RaDR-GFP) mice reveal tissue- and age-dependence of homologous recombination in mammals in vivo. PLoS Genet. 2014;10:e1004299. doi: 10.1371/journal.pgen.1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCollough CH, Bushberg JT, Fletcher JG, Eckel LJ. Answers to common questions about the use and safety of CT scans. Mayo Clin Proc. 2015;90:1380–92. doi: 10.1016/j.mayocp.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 65.Baverstock K. Radiation-induced genomic instability: a paradigm-breaking phenomenon and its relevance to environmentally induced cancer. Mutat Res. 2000;454:89–109. doi: 10.1016/s0027-5107(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 66.Kaplan MI, Morgan WF. The nucleus is the target for radiation-induced chromosomal instability. Radiat Res. 1998;150:382–90. [PubMed] [Google Scholar]
- 67.Limoli CL, Kaplan MI, Phillips JW, Adair GM, Morgan WF. Differential induction of chromosomal instability by DNA strand-breaking agents. Cancer Res. 1997;57:4048–56. [PubMed] [Google Scholar]
- 68.Morgan WF, Corcoran J, Hartmann A, Kaplan MI, Limoli CL, Ponnaiya B. DNA double-strand breaks, chromosomal rearrangements, and genomic instability. Mutat Res. 1998;404:125–8. doi: 10.1016/s0027-5107(98)00104-3. [DOI] [PubMed] [Google Scholar]
- 69.Limoli CL, Corcoran JJ, Milligan JR, Ward JF, Morgan WF. Critical target and dose and dose-rate responses for the induction of chromosomal instability by ionizing radiation. Radiat Res. 1999;151:677–85. [PubMed] [Google Scholar]
- 70.Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis. 1996;17:1633–9. doi: 10.1093/carcin/17.8.1633. [DOI] [PubMed] [Google Scholar]
- 71.Limoli CL, Hartmann A, Shephard L, Yang CR, Boothman DA, Bartholomew J, et al. Apoptosis, reproductive failure, and oxidative stress in Chinese hamster ovary cells with compromised genomic integrity. Cancer Res. 1998;58:3712–8. [PubMed] [Google Scholar]
- 72.Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res. 2006;66:10377–83. doi: 10.1158/0008-5472.CAN-05-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis. 2006;21:361–7. doi: 10.1093/mutage/gel048. [DOI] [PubMed] [Google Scholar]
- 74.Limoli CL, Kaplan MI, Giedzinski E, Morgan WF. Attenuation of radiation-induced genomic instability by free radical scavengers and cellular proliferation. Free Radic Biol Med. 2001;31:10–9. doi: 10.1016/s0891-5849(01)00542-1. [DOI] [PubMed] [Google Scholar]
- 75.Limoli CL, Giedzinski E, Morgan WF, Swarts SG, Jones GD, Hyun W. Persistent oxidative stress in chromosomally unstable cells. Cancer Res. 2003;63:3107–11. [PubMed] [Google Scholar]
- 76.Sabatier L, Ricoul M, Pottier G, Murnane JP. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol Cancer Res. 2005;3:139–50. doi: 10.1158/1541-7786.MCR-04-0194. [DOI] [PubMed] [Google Scholar]
- 77.Feijoo P, Dominguez D, Tusell L, Genesca A. Telomere-dependent genomic integrity: evolution of the fusion-bridge-breakage cycle concept. Curr Pharm Des. 2014;20:6375–85. doi: 10.2174/1381612820666140630085416. [DOI] [PubMed] [Google Scholar]
- 78.Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15:608–24. doi: 10.1038/nrc3985. [DOI] [PubMed] [Google Scholar]
- 79.Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A. 2012;109:11800–5. doi: 10.1073/pnas.1200583109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009;8:2662–3. doi: 10.4161/cc.8.16.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodeling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–83. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsukuda T, Lo YC, Krishna S, Sterk R, Osley MA, Nickoloff JA. INO80-dependent chromatin remodeling regulates early and late stages of mitotic homologous recombination. DNA Repair. 2009;8:360–9. doi: 10.1016/j.dnarep.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 83.Wu S, Shi Y, Mulligan P, Gay F, Landry J, Liu H, et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol. 2007;14:1165–72. doi: 10.1038/nsmb1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bao Y, Shen X. Chromatin remodeling in DNA double-strand break repair. Curr Opin Genet Dev. 2007;17:126–31. doi: 10.1016/j.gde.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 85.Sinha M, Peterson CL. Chromatin dynamics during repair of chromosomal DNA double-strand breaks. Epigenomics. 2009;1:371–85. doi: 10.2217/epi.09.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chambers AL, Downs JA. The RSC and INO80 chromatin-remodeling complexes in DNA double-strand break repair. Prog Mol Biol Transl Sci. 2012;110:229–61. doi: 10.1016/B978-0-12-387665-2.00009-2. [DOI] [PubMed] [Google Scholar]
- 87.Schildkraut E, Miller CA, Nickoloff JA. Gene conversion and deletion frequencies during double-strand break repair in human cells are controlled by the distance between direct repeats. Nucleic Acids Res. 2005;33:1574–80. doi: 10.1093/nar/gki295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang H, Wang X, Zhang P, Wang Y. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair. 2008;7:725–33. doi: 10.1016/j.dnarep.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 89.Yajima H, Fujisawa H, Nakajima NI, Hirakawa H, Jeggo PA, Okayasu R, et al. The complexity of DNA double strand breaks is a critical factor enhancing end-resection. DNA Repair. 2013;12:936–46. doi: 10.1016/j.dnarep.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 90.Nimonkar AV, Sica RA, Kowalczykowski SC. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc Natl Acad Sci U S A. 2009;106:3077–82. doi: 10.1073/pnas.0813247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–45. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 92.Groth P, Orta ML, Elvers I, Majumder MM, Lagerqvist A, Helleday T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 2012;40:6585–94. doi: 10.1093/nar/gks315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–94. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 94.Morgan WF, Bair WJ. Issues in low dose radiation biology: the controversy continues. A perspective Radiat Res. 2013;179:501–10. doi: 10.1667/RR3306.1. [DOI] [PubMed] [Google Scholar]
- 95.Morgan WF. Compelling issues compounding the understanding of low dose radiation effects: but do they matter? Health Phys. 2016;110:291–2. doi: 10.1097/HP.0000000000000469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.