

Abstract
Amino acid-derived cross-conjugated trienes were used as a starting point for the synthesis of a discovery library of over 200 polycyclic 5-iminooxazolidin-2-ones, hydantoins, and acylureas. The main feature of this library synthesis is a triple branching strategy which provides efficient access to five skeletally diverse scaffolds. In addition, four sets of building blocks were applied in both a front end and a back end diversification strategy. Multiple fused rings were obtained by cyclization of diamides with phosgene and stereoselective Diels-Alder reactions with maleimides. The 5-iminooxazolidin-2-one scaffold was rearranged into the isomeric hydantoin scaffold through a sequence of ring opening and ring closing reactions.
Keywords: Library Design, Skeletal Diversity, Appendage Diversity, Polycycles, Diels Alder Reaction
Graphical Abstract

Introduction
The search for new biological probes capable of modulating biological pathways has led to the rapid development of diversity oriented synthesis (DOS), a strategy used to access larger numbers of structurally unique compounds.1,2,3 In addition to occupying new chemical space, these molecules need to bind to proteins, be of a defined molecular complexity,4 be structurally rigid, and possess three-dimensionality.5 Many DOS libraries are inspired by natural products because of their richness in stereochemical and three-dimensional structural diversity.6,7 Although it is not easy to measure the diversity of a certain collection of compounds,8,9 four diversity elements have been described: building block, stereochemical, appendage, and skeletal.10 Recently, skeletal diversity has evolved as the most powerful element in the design of small molecule discovery libraries.11 It can be generated through branching strategies12 or through rearrangement and fragmentation processes13 in the library design. Polymorphic scaffolds14 and sigma-elements that contain appendages with pre-encoded skeletal information as defined by Schreiber15 have been described as alternative strategies to obtain skeletal diversity.
Distributing compounds into chemical descriptor space can be achieved best by combining several diversity elements. For the discovery library of polycyclic nonaromatic 5-iminooxazolidin-2-ones, hydantoins, and acylureas presented here, a triple branching strategy (see Scheme 1) allowing for the combination of appendage and skeletal diversity elements was employed (Figure 1).
Scheme 1.

Synthesis of polycyclic 5-iminooxazolidin-2-ones, hydantoins, and acylureas.
Figure 1.

δ-Lactams A as key intermediates for a structurally diverse discovery library.
The amino acid-derived δ-lactam A containing an electron deficient cross-conjugated triene system and diamide functionalities provides a densely functionalized key intermediate, which serves as a starting point in the skeletal diversity strategy for the generation of multiple scaffolds. The amide functionalities are cyclized with phosgene to form a sterically biased and electronically tuned 5-iminooxazolidin-2-one triene system. The electron deficient triene A undergoes a single cycloaddition reaction with dienophiles followed by the newly formed diene moiety reacting with another dienophile in a second Diels-Alder reaction or it can undergo a hetero-Diels-Alder reaction involving the carbonyl group.16
Further diversification of the 5-iminooxazolidin-2-one moiety can potentially be achieved by ring-opening reactions with amines and rearrangement into the isomeric hydantoin scaffold, an important pharmacophore.17 Although 4-iminooxazolidin-2-ones are of interest as fungicides,18 little is known about the isomeric 5-iminooxazolidin-2-ones.19 Therefore it was appealing to synthesize a discovery library based on this new motif. Furthermore, targeted scaffolds B–F are all nonaromatic and polycyclic in nature, a class of compounds of interest because of their natural product like character.20
Results and Discussion
Library Design
In addition to five highly functionalized scaffolds differing significantly in volume, flexibility, number of fused rings and their ability to donate and accept hydrogen bonds, the library design involved four sets of building blocks. Two triene containing methyl esters 1{1–2}16 (Figure 2) and a set of six amines 2{1–6} (Figure 3) were used for the essential conversion of 1{1–2} to 5{1–2,1–6} (Scheme 1). Cyclopropylmethylamine 2{1} was chosen as a small lipophilic building block, methoxyethylamine 2{2} and glycyl methyl ester 2{3} as more polar substituents, benzylamine 2{4} and 2-aminomethylpyridine 2{6} to include an aromatic and heteroaromatic system and tryptamine 2{5} because of its hydrogen bond donor abilities. Five maleimides 3{1–5} were applied as symmetric dienophiles in a Diels-Alder reaction in order to avoid the formation of regioisomers (Figure 4). Maleimide itself 3{1} was selected to include a second hydrogen bond donor, N-methyl and N-ethyl maleimide 3{2} and 3{3} because of their relatively small substituents, N-phenyl maleimide 3{4} as an aromatic representative and methyl N-acetate maleimide 3{5} because of its more polar character. The last set of building blocks consisted of six amines 4{1–6} that were used to open the 5-iminooxazolidin-2-one moiety (Figure 5). Again, the selection included small polar (4{1}, 4{6}) and lipophilic (4{4}) aliphatic substituents as well as a polar (4{2}) and lipophilic (4{5}) aromatic representatives. N-(2-Aminoethyl)-morpholine 4{3} was chosen because of its basic nitrogen and ethanolamine (4{1}) because of its ability to act as hydrogen bond donor.
Figure 2.
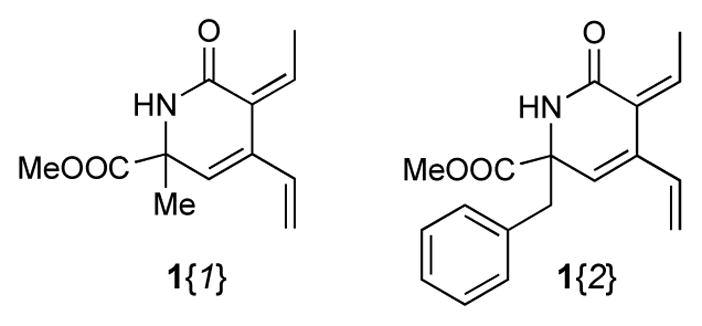
Building Blocks 1{1–2} used as starting points in the library synthesis.
Figure 3.
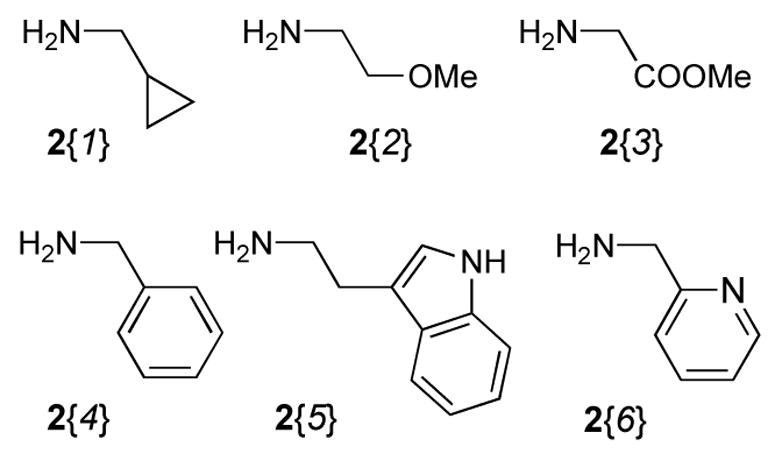
Building Blocks 2{1–6} used in the library synthesis.
Figure 4.
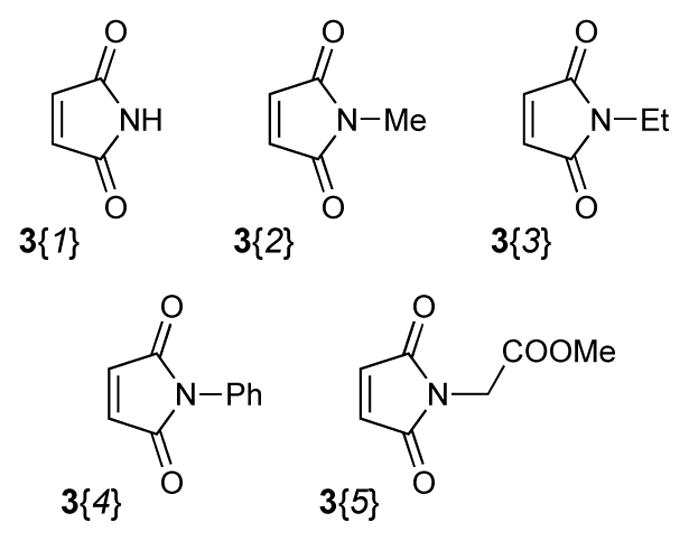
Building Blocks 3{1–5} used in the library synthesis.
Figure 5.
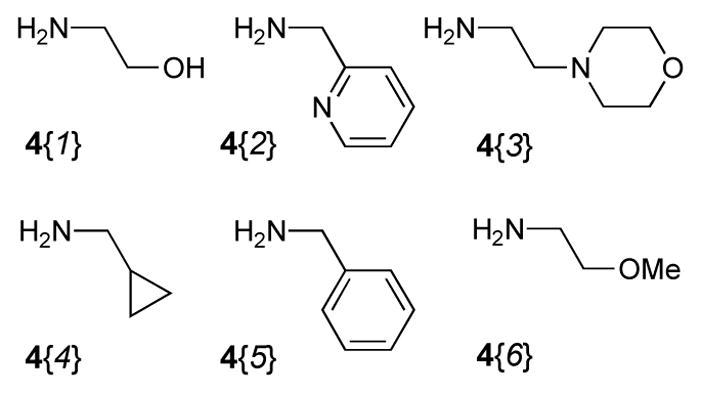
Building Blocks 4{1–6} used in the library synthesis.
Library Synthesis
The cross-conjugated trienes 1{1–2} were obtained via a Rh(I)-catalyzed Alder-ene reaction of propiolamides.16,21 Saponification of the methyl esters 1{1–2} with lithium hydroxide, followed by HOBt/DMAP/EDCI mediated amidation with amines 2{1–6} provided amides 5{1–2,1–6} in yields ranging from 18–93% (Scheme 1, Table 1) for the two steps.
Table 1.
Yields in % for the synthesis of amides 5{1–2,1–6} from methyl esters 1{1–2} and amines 2{1–6}
| 5{1–2,1–6} | 2{1} | 2{2} | 2{3} | 2{4} | 2{5} | 2{6} |
|---|---|---|---|---|---|---|
| 1{1} | 68 | 18 | 65 | 75 | 93 | -a |
| 1{2} | 55 | 51 | 83 | 30 | 79 | 55 |
not attempted as 5{2,6} did not undergo a selective reaction with phosgene in the next step.
The reaction conditions for the cyclization reaction with phosgene were optimized on small scale (0.1 mmol) with diamide 5{1,4} prior to applying it on large scale to all diamides 5{1–2,1–6}. Diamide 5{1,4} was treated with three equivalents of phosgene and triethylamine in dichloromethane at −10 ºC for 30 min. The reaction mixture was then warmed to 0 ºC and quenched by adding a saturated ammonium chloride/brine (1/10) solution. 5-Iminooxazolidin-2-one 7{1,4} was the only observed product. In contrast, when the reaction was carried out on larger scale under otherwise identical conditions the 5-iminooxazolidin-2-one 7{1,4} was immediately hydrolyzed back to 5{1,4} while quenching with saturated ammonium chloride/brine (1/10) because of the large amount of HCl generated. This could be prevented by using six equivalents triethylamine and quenching the reaction mixture with a saturated sodium bicarbonate solution. All 5-iminooxazolidin-2-ones 7{1–2,1–5} were very acid sensitive and converted back into the starting material if a purification by chromatography on silica was attempted. Thus, they were not purified and instead used crude in the following Diels-Alder reaction. The pyridine containing diamide 5{2,6} was the only diamide that did not undergo a cyclization reaction with phosgene. Upon addition of phosgene, the reaction mixture turned black and no product could be detected.
On large scale, it proved to be necessary to warm the reaction mixture from −10 ºC to room temperature prior to quenching it with saturated sodium bicarbonate to obtain complete conversion. However, carbamate 6{1–2,1–5} was now formed as a side product. Quenching the reaction at −10 ºC or 0 ºC led to incomplete reactions but prevented the carbamate formation (Table 2, footnotes b und c). Although a rather unusual reaction, the formation of N,N-diethyl carbamates from phosgene and triethylamine has been described.22
Table 2.
Yields in % for the synthesis of 5-iminooxazolidin-2-ones 8{1–2,1–5,1–5}, 9{1–2,1–5,1–5} and 10{1–2,1–5,1–5} from amides 5{1–2,1–5} and maleimides 3{1–5}a
| 8{1–2,1–5,1–5}/9{1–2,1–5,1–5}/10{1–2,1–5,1–5} | 3{1} | 3{2} | 3{3} | 3{4} | 3{5} |
|---|---|---|---|---|---|
| 5{1,1} | 14/-/13 | 22/–/58 | 20/5/38 | 24/–/37 | 21/–/37 |
| 5{1,2} | 13/-/13 | 12/3/17 | 13/–/18 | 10/–/17 | 17/-/13 |
| 5{1,3} | 20/-/7 | 22/–/35 | 24/–/46 | 32/-/31 | 27/–/30 |
| 5{1,4} | 27/–/33 | 28/8/42 | 28/–/47 | 25/–/53 | 27/–/32 |
| 5{1,5} | 11/–/13 | 15/–/18 | 16/–/60 | 22/–/39 | 20/–/39 |
| 5{2,1} | 18/-/5 | 25/–/43 | 27/–/53 | 28/–/42 | 22/–/37 |
| 5{2,2} | 18/–/29 | 17/–/39 | 13/–/51 | 33/–/61 | 12/7/29 |
| -/35/36b | |||||
| 5{2,3} | 20/-/12 | 25/–/58 | 27/–/40 | 18/3/50 | 24/–/39 |
| 5{2,4} | -/-/32c | -/-/61c | -/-/74c | -/4/19c | -/-/68c |
| 5{2,5} | 17/11/42 | 24/–/57 | 19/–/72 | 13/2/61 | 9/–/70 |
Yield in % for carbamate 8{1–2,1–5,1–5}/double Diels-Alder adduct 9{1–2,1–5,1–5}/Diels-Alder adduct 10{1–2,1–5,1–5}.
Phosgene reaction quenched at −10 ºC.
Phosgene reaction quenched at 0 ºC.
Assuming quantitative conversion in the cyclization step 5-iminooxazolidin-2-ones 6{1–2,1–5} and 7{1–2,1–5} were taken on to the next reaction. Heating each in a Radley’s Carousel for 2 h at 90 ºC with 1.3 equivalents maleimide 3{1–5} in toluene gave the Diels-Alder cycloadducts which could be easily purified on an ISCO Optix 10 chromatography station. The carbamates 8{1–2,1–5,1–5} were obtained in 0–33 % while the adducts 10{1-2–1-5,1–5} were isolated in 5–74 % yield (Table 2). In several cases a second Diels-Alder reaction occurred providing adduct 9{1–2,1–5,1–5} in 0–11 % yield which could also be separated by chromatography (Table 2–3). It is thought that in those cases the cyclization reaction did not go to completion and thus the maleimide was present in excess. This hypothesis is supported by the cyclization reaction of 5{2,2} which was quenched at −10 ºC, preventing completion of the reaction, and the crude reaction mixture was applied to the Diels-Alder reaction with 3{4}. In this case, carbamate 6{2,2,4} was not observed, but the double Diels-Alder adduct 9{2,2,4} was obtained in 35 % yield (Table 2, footnote b). An isomerization of the exocyclic double bond in 10{1–2,1–5,1–5} was noticed 23 but the E/Z-isomers were not separated because the diene system was reduced in the following step.
Table 3.
Purities in % for the double Diels-Alder adducts 9{1–2,1–5,1–5}, purity detection by ELS.
| Compound | Purity |
|---|---|
| 9{1,1,3} | >99 |
| 9{1,2,2} | 64 |
| 9{1,4,2} | 71 |
| 9{2,2,4} | 90a |
| 9{2,2,5} | >99 |
| 9{2,3,4} | 99 |
| 9{2,5,4} | >99 |
Purity estimated by 1H NMR.
Because of concern for the reactivity24 of the exocyclic double bond of the lactam, the diene in 8{1–2,1–5,1–5} and 10{1–2,1–5,1–5} was reduced by hydrogenation with Pd/C to give carbamates 11{1–2,1–5,1–5} in 23–99 % yield (Table 4) and lactams 12{1–2,1–5,1–5} in 17–85 % yield (Table 5). Most reactions were complete after 4 h, but the indole containing substrates required reaction times of 24 h.
Table 4.
Yields (Purities) in % for the hydrogenation of carbamates 11{1–2,1–5,1–5} from 8{1–2,1–5,1–5}, purity detection by ELS
| 11{1–2,1–5,1–5} | 3{1} | 3{2} | 3{3} | 3{4} | 3{5} |
|---|---|---|---|---|---|
| 5{1,1} | 92 (>99) | 99 (>99) | 85 (99) | 70 (>99) | 90 (96) |
| 5{1,2} | 85 (>99) | 51 (97) | 65 (93) | 75 (70) | 62 (>99) |
| 5{1,3} | 73 (98) | 59 (95) | 82 (93) | 70 (87) | 99 (90) |
| 5{1,4} | 70 (>99) | - | 86 (99) | 89 (>99) | 78 (97) |
| 5{1,5} | 87 (>99) | 70 (>99) | 61 (96) | 27 (86) | 25 (>99) |
| 5{2,1} | 96 (98) | 96 (99) | 96 (98) | 69 (>99) | 78 (90) |
| 5{2,2} | 80 (>99) | 81 (98) | 86 (98) | 86 (70) | 67 (98) |
| 5{2,3} | 96 (97) | 83 (96) | 86 (86) | 64 (98) | 79 (96) |
| 5{2,4} | - | - | - | - | - |
| 5{2,5} | 63 (94) | 23 (>99) | 38 (>99) | 39 (90)a | 41 (>99) |
Purity estimation by 1H NMR.
Table 5.
Yields (Purities) in % for the hydrogenation of Diels-Alder adducts 12{1–2,1–5,1–5} from 10{1–2,1–5,1–5}, purity detection by ELS
| 12{1–2,1–5,1–5} | 3{1} | 3{2} | 3{3} | 3{4} | 3{5} |
|---|---|---|---|---|---|
| 5{1,1} | 51 (93) | 48 (<50) | 35 (82) | 57 (>95) | 50 (<50) |
| 5{1,2} | 23 (>99) | 35 (57) | 52 (<50) | 43 (<50) | 44 (<50) |
| 5{1,3} | 18 (91) | 74 (97) | 36 (93) | 53 (94) | 60 (98) |
| 5{1,4} | 25 (88) | 58 (95) | 32 (75) | 36 (91) | 46 (87) |
| 5{1,5} | 24 (93) | 50 (77) | 23 (90)b | 17 (84) | 43 (93) |
| 5{2,1} | 61 (99) | 79 (94) | 44 (>99) | 46 (98) | 73 (80) |
| 5{2,2} | 76 (78) | 85 (97) | 63 (99) | 45 (95) | 32 (95) |
| 5{2,3} | 25 (98) | 83 (94) | 83 (80)a | 24 (80) | 46 (84)a |
| 5{2,4} | 46 (90) | 61 (77)a | 37 (94) | 34 (94) | 53 (76) |
| 5{2,5} | 17 (<50) | 42 (95)b | 53 (85) | 33 (98) | 29 (94) |
Purity detection by UV 254 nm.
Purity estimation by 1H NMR.
The structure of the carbamates was confirmed by an X-ray structure analysis of 11{1,1,4} (Figure 6a). Both Diels-Alder reactions took place in a diastereoselective fashion. The initial Diels-Alder cycloaddition reaction occurred with endo selectivity from the opposite face of the R1 substituent as seen in the X-ray structure analysis of 12{2,1,4} (Figure 6b). Based on comparison studies with a previously described double Diels-Alder adduct16 the stereochemistry for 9{1–2,1–5,1–5} has been tentatively assigned as resulting from an addition of the second dienophile from the less hindered convex face in endo mode.
Figure 6.

X-Ray structures of 11{1,1,4}, 12{2,1,4} and 13{1,1,4}.34
Although the 5-iminooxazolidin-2-ones 12{1–2,1–5,1–5} are very stable compounds, e.g. stirring in 1M HCl/MeOH for 24 h did not result in any reaction or decomposition, the 5-iminooxazolidin-2-one ring system can be opened by reaction with an excess primary amine in chloroform at room temperature. This reaction tolerated a wide range of amines and eighty-seven acylureas 14{1–2,1–5,1–5,1–6} were prepared (Table 6). The structure of those tricycles was confirmed by an X-ray structure of 14{1,1,4,1},25 clearly showing the amide as well as the acylurea functionality. In several cases a less polar side product was observed which could be easily separated by chromatography on silica and was identified as hydantoin 13{1–2,1–5,1–5} based on a characteristic chemical shift change of a 13C NMR resonance from 147 to 173 ppm. The structure was later confirmed by the X-ray structure analysis of 13{1,1,4} (Figure 6c). Alternatively, stirring the acylureas 14{1–2,1–5,1–5,1–6} in wet methanol for 24 h resulted in a selective conversion into the corresponding hydantoins 13{1–2,1–5,1–5} in 85–89 % yield. In all cases, the substituent R2 on the amide was preserved in the molecule while the substituent R4 on the acylurea was cleaved off. The formation of hydantoins by an intramolecular nucleophilic attack of an amide hydrogen on an ureido carbonyl,26 carbamoyl carbonyl27 or benzotriazole carbonyl28 has been reported previously but in all cases a base was required. The intramolecular cyclization between the amide hydrogen and the acylurea carbonyl of 14{1–2,1–5,1–5,1–6} took place under very mild conditions in wet methanol. As all library members will be stored in the UPCMLD compound collection as solution in DMSO at −78 ºC, it was important to investigate the stability of the acylurea 14{1–2,1–5,1–5,1–6} in DMSO-d6 under those conditions. While 14{2,5,2,1} had a half life time in DMSO-d6 at room temperature of only 21 days and completely rearranged into 13{2,5,2,1} after two months, no rearrangement was observed if stored at −78 ºC after three months.
Table 6.
Yields (purities) in % for the formation of the hydantoins 13{1–2,1–5,1–5} and acylureas 14{1–2,1–5,1–5,1–6} from the Diels-Alder adducts 12{1–2,1–5,1–5} and amines 4{1–6}, purity detection by ELSa
| 13{1–2,1–5,1–5}/14{1–2,1–5,1–5,1–6} | 4{1} | 4{2} | 4{3} | 4{4} | 4{5} | 4{6} |
|---|---|---|---|---|---|---|
| 12{1,1,1} | 43/22 (>99/>99) | - | - | - | - | - |
| 12{1,1,2} | -/87 (-/83) | -/81 (-/84) | - | -/85 (-/82) | - | - |
| 12{1,1,3} | -/94 (-/>99) | -/99 (-/>99) | - | - | - | - |
| 12{1,1,4} | -/92 (-/>99) | -/94 (-/>99) | 17/47 (>99/90b) | - | - | - |
| 12{1,1,5} | -c/58 (-c/98) | -/82 (-/95) | - | - | - | |
| 12{1,3,2} | 16/56 (>99/>99) | -/73 (-/97) | - | - | - | - |
| 12{1,3,3} | -/85 (-/97) | -/93 (-/98) | 19/46 (93/90b) | - | - | - |
| 12{1,3,4} | 6/38 (>99/71) | -/64 (-/96) | - | - | - | - |
| 12{1,3,5} | -/67 (-/90b) | -/65 (-/95b) | - | - | - | - |
| 12{1,4,1} | -/64 (-/>99) | -/80 (-/>99) | - | - | - | - |
| 12{1,4,2} | -/96 (-/>99) | -/90 (-/>99) | -/67 (-/98) | -/80 (-/>99) | -/40 (-/>99) | -/42 (-/>99) |
| 12{1,4,4} | -/52 (-/60) | -/52 (-/>99) | 8/54 (91/95b) | -/72 (-/>99) | - | - |
| 12{1,4,5} | 59/- (94/-) | -/70 (-/98) | -/7 (-/95b) | - | - | - |
| 12{1,5,2} | -/76 (-/>99) | -/88 (-/98) | - | - | - | - |
| 12{1,5,3} | -/95 (-/88) | -/95 (-/>99) | -/56 (-/90b) | -/79 (-/>99) | - | - |
| 12{1,5,4} | -/63 (-/98) | - | - | - | - | - |
| 12{1,5,5} | -/59 (-/90b) | -/41 (-/90b) | - | - | - | - |
| 12{2,1,1} | -/63 (-/>99) | - | - | - | - | - |
| 12{2,1,2} | -/61 (-/87) | -/52 (-/>99) | 28/37 (95b/99) | -/61 (-/97) | - | - |
| 12{2,1,3} | -d/70 (-d/>99) | -/66 (-/>99) | -/50 (-/95b) | -/62 (-/99) | - | - |
| 12{2,1,4} | -/73 (-/>99) | -/36 (-/>99) | -/16 (-/90b) | - | - | - |
| 12{2,1,5} | -/53 (-/99) | -/10 (-/>99) | - | -/37 (-/90) | - | - |
| 12{2,2,3} | 33/- (98/-) | -/54 (-/98) | 9/- (99/-) | -/60 (-/99) | - | - |
| 12{2,2,4} | -/51 (-/99) | -/86 (-/99) | - | - | - | - |
| 12{2,3,4} | -/5 (-/>99) | 16/62 (95/94) | - | - | - | - |
| 12{2,4,1} | - | 22/55 (95/90b) | - | - | - | - |
| 12{2,4,2} | - | -/45 (-/>99) | 41/50 (<50/90b) | -/54 (-/67e) | - | - |
| 12{2,4,3} | 41/35 (95/95) | - | - | - | - | - |
| 12{2,4,4} | 44/36 (86/>99) | -/19 (-/>99) | - | - | - | - |
| 12{2,4,5} | 16/36 (81/97) | -/51 (-/92) | -/42 (-/90) | - | - | - |
| 12{2,5,2} | -/89 (-/>99) | -/98 (-/99) | -/85 (-/95b) | -/97 (-/>99) | - | - |
| 12{2,5,3} | -/53 (-/90b) | -/46 (-/77e) | - | -/66 (-/93) | -/73 (-/>99) | - |
| 12{2,5,4} | - | -/65 (-/>99) | -/63 (-/90b) | - | - | - |
| 12{2,5,5} | -/50 (-/99) | -/74 (-/79e) | -/71 (-/95b) | - | - | - |
Yield (purity) in % for hydantoin 13{1–2,1–5,1–5}/acylurea 14{1–2,1–5,1–5,1–6}.
Purity estimated by 1H NMR.
Hydantoin 13{1,1,5} was synthesized from pure acylurea 14{1,1,5,1} by stirring in wet MeOH in 85% yield (purity >99%).
Hydantoin 13{2,1,3} was synthesized from pure acylurea 14{2,1,3,1} by stirring in wet MeOH in 89% yield (purity 95%).
Purity detected by UV 254 nm.
In contrast to 12{1–2,1–5,1–5}, carbamates 11{1–2,1–5,1–5} did not undergo ring opening with amines. Carbamate 11{2,1,4} was irradiated in the microwave in chlorobenzene with benzylamine 4{5} and after irradiation for 15 min at 180 ºC no reaction was observed.
Purity Analysis
Altogether seven double Diels-Alder adducts 9{1–2,1–5,1–5}, fourty-four carbamates 11{1–2,1–5,1–5}, fifty Diels-Alder adducts 12{1–2,1–5,1–5}, eighteen hydantoins 13{1–2,1–5,1–5} and eighty-seven acylureas 14{1–2,1–5,1–5,1–6} were synthesized. All compounds were analyzed by LC/MS-ELSD (Tables 2–6). One hundred sixty-seven compounds (81% of all compounds) passed the purity criteria of 90% by ELSD for the UPCMLD compound collection. The average purity by ELSD of the accepted library members was 96.6% (Table 7). The Diels-Alder adducts 12{1–2,1–5,1–5} contained the highest number of rejected compounds and the accepted compounds from this group had a slightly lower overall purity (94.6%) because in several cases those compounds were contaminated with small amounts of the corresponding double Diels-Alder adducts.
Table 7.
Number of compounds and purities in % for the five polycyclic scaffolds.
Physicochemical Profiling
3-D structures of all library members were built and minimized using the MM2 force field in Macro Model 8.6. The physicochemical profiling of the two hundred and six library members was analyzed computationally using QikProp 2.1.29 Besides slightly elevated molecular weights in case of the double Diels-Alder adducts 9{1–2,1–5,1–5}, carbamates 11{1–2,1–5,1–5} and acylureas 14{1–2,1–5,1–5,1–6} all molecular descriptors show values well within the range that is considered tool-30 and drug-like31 (Table 8). Because of the combination of skeletal and appendage diversity, distinct differences in the physicochemical properties exist not only within the five compound classes but also among the scaffolds. A difference in the average molecular weight of around 160 Daltons can be observed between the double Diels-Alder adducts 9{1–2,1–5,1–5} and the Diels-Alder adducts 12{1–2,1–5} and hydantoins 13{1–2,1–5,1–5}, which also have a clearly smaller solvent accessible volume. While the acylureas 14{1–2,1–5,1–5,1–6} attract attention because of their increased ability to act as hydrogen bond donors, the double Diels-Alder adducts 9{1–2,1–5,1–5} are especially rich in the number of hydrogen bond acceptors. The average logP and logS values differ by almost an order of magnitude between the carbamates 11{1–2,1–5,1–5) and the Diels-Alder adducts 12{1–2,1–5,1–5}. The distinction is even more dramatic regarding the number of rotatable bonds with an average of 4.7 for the relatively rigid hydantoins 13{1–2,1–5,1–5} and 9.0 for the more flexible carbamates 11{1–2,1–5,1–5}. As expected due to their size, the double Diels-Alder adducts 9{1–2,1–5,1–5} have a lower predicted permeability than all other scaffolds.
Table 8.
Physicochemical Profiling for the five scaffolds (average ± standard deviation), calculated with QikProp 2.1.29
| Scaffolds | 9{1–2, 1–5,1–5} | 11{1–2, 1–5,1–5} | 12{1–2, 1–5,1–5} | 13{1–2, 1–5,1–5} | 14{1–2,1–5, 1–5,1–6} |
|---|---|---|---|---|---|
| MW [g/mol] | 648 ± 114 | 586 ± 60 | 489 ± 60 | 490 ± 52 | 591 ± 68 |
| Volume [Å3] | 1724±302 | 1676±156 | 1371±160 | 1371±138 | 1699±180 |
| SASA [Å2] | 837 ± 138 | 826 ± 66 | 687 ± 71 | 682 ± 60 | 847 ± 79 |
| Fused Rings | 6 | 4 | 4 | 4 | 3 |
| HBD | 0.1 ± 0.4 | 0.4 ± 0.6 | 0.4 ± 0.6 | 0.1 ± 0.3 | 1.5 ± 0.8 |
| HBA | 12.1 ± 2.0 | 11.3 ± 1.2 | 8.6 ± 1.2 | 8.0 ± 1.0 | 10.2 ± 1.4 |
| logP | 3.9 ± 2.4 | 4.4 ±1.4 | 3.5 ± 1.5 | 3.9 ± 1.4 | 3.8 ± 1.4 |
| logS | −5.2 ± 2.4 | −5.1 ± 1.5 | −4.2 ± 1.4 | −4.4 ± 1.1 | −4.0 ± 1.4 |
| Rotatable Bonds | 5.6 ± 2.2 | 9.0 ± 1.4 | 6.0 ± 1.4 | 4.7 ± 1.4 | 8.4 ± 1.5 |
| Caco-Perm [nm/sec] | 131 ± 113 | 729 ± 544 | 388 ± 299 | 503 ± 317 | 564 ± 477 |
Conclusions
Two amino-acid derived cross-conjugated trienes were applied to the synthesis of a discovery library of nonaromatic polycyclic small molecules. Those key building blocks were converted into a sterically biased and electronically tuned 5-iminooxazolidin-2-one scaffold which allowed excellent stereocontrol in the following Diels-Alder reactions. Although the 5-iminooxazolidin-2-ones are very stable compounds, a conversion into acylureas and hydantoins was accomplished. The combination of skeletal and appendage diversity led to a library of 206 tri- to hexacyclic 5-iminooxazolidin-2-ones, acylureas and hydantoins. The physicochemical profiling of all compounds demonstrated a high degree of diversity within each compound class and among the five scaffolds. All compounds were analyzed by LC/MS and are currently being evaluated in several high-throughput-screening programs. Results on their biological activities can be accessed in PubChem (http://pubchem.ncbi.nlm.nih.gov).
Experimental Section
General
All solvents or reagents were used without further purification. Reactions were monitored by TLC analysis (EM Science pre-coated silica gel 60 F254 plates, 250 μm layer thickness) and visualization was accomplished with a 254 nm UV light and by staining with Vaughn’s reagent (4.8 g (NH4)6Mo7O24·4H2O, 0.2 g Ce(SO4)2·4H2O in 10 mL conc. H2SO4 and 90 mL H2O), KMnO4 (1.0 g KMnO4, 1.0 g K2CO3, 2 mL 5% aqueous NaOH, 100 mL H2O) and p-anisaldehyde (2.5 mL p-anisaldehyde, 2 mL acetic acid, 3.5 mL conc. H2SO4, 92 mL EtOH). NMR spectra were recorded in CDCl3 or DMSO-d6 (298 K) at 300.1 MHz (1H) or 75.5 MHz (13C) using a Bruker Avance 300 with XWIN-NMR software. Chemical Shifts (δ) are reported in parts per million (ppm). Tetramethylsilane (1H), chloroform-d (13C), or DMSO-d6 (13C) were used as internal standards. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = dublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, app = apparent), integration and coupling constants. IR spectra were obtained on a Nicolet AVATAR 360 FTIR E.S.P. Spectrometer. Mass spectra were obtained on a Micromass Autospec double focusing instrument (EI) or a Waters Q-Tof mass spectrometer (ESI). Melting points were obtained using a heating rate of 2 ºC/min on a MelTemp melting point apparatus with digital temperature reading and are reported uncorrected.
Compounds were analyzed by reverse-phase HPLC (Alltech Prevail C-18, 100 × 4.6 mm, 1 mL/min, 50–70% MeCN, 50–30% H2O) with UV (210 and 254 nm), ELS (Nebulizer 45ºC, Evaporator 45ºC, N2 flow 1.25 SLM) and MS detection using a Thermo Finnigan Surveyor LC and LCQ Advantage MS system (ESI positive mode).
General procedure for the formation of trienes 1{1–2} (Protocol A)
A 1L-three-necked round bottom flask equipped with a stir-bar and condenser was charged with allenyne (1.00 eq, 10.90 mmol) and toluene (400 mL). Argon was bubbled through the solution for 5 min and [Rh(CO)2Cl]2 (0.05 eq, 0.55 mmol) was added. The reaction was heated at 90 °C for 1 h, then a second portion of catalyst (0.025 eq, 0.27 mmol) was added. After an additional 2.5 h of heating at this temperature the reaction was complete by tlc. The reaction was allowed to cool to room temperature and the solution was loaded directly onto a silica gel column, eluting first with hexanes (200 mL) then 40% ethyl acetate/hexanes. The product-containing fractions were combined and concentrated to afford triene 1{1} in 74% yield. The spectral data matched that previously reported.21
General protocol for the saponification and amidation of esters 1{1–2} (Protocol B)
The methyl ester 1{1–2} (1.00 eq, 5.0–10 mmol) was dissolved in THF (100–200 mL) and water (100–200 mL) was added slowly to maintain a homogeneous solution. Lithium hydroxide hydrate (2.00 eq) was carefully added and the solution was stirred for 5 min. After complete hydrolysis a saturated solution of ammonium chloride was added (100 mL), the reaction mixture was acidified to pH = 1 with 1 M-HCl, extracted with ethyl acetate (3 × 50 mL), and dried over sodium sulfate. Removal of all volatile components in vacuo gave the crude acid in quantitative yield which was used in the following step without further purification.
The crude acid was dissolved in dry dichloromethane (150–300 mL) and treated with 1-hydroxybenzotriazole (HOBt, 1.00 eq), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI, 1.00 eq) and 4-dimethylaminopyridine (DMAP, 1.05 eq). The selected amine 2{1–6} (3.00 eq of volatile 2{1} or 1.50 eq of non-volatile 2{2–6}) was added over 10 min while stirring at room temperature. After 4 h the reaction mixture was extracted with water (100 mL) and saturated ammonium chloride (2 × 100 mL). The combined aqueous fractions were re-extracted with dichloromethane (50 mL) and all organic fractions were combined and dried over magnesium sulfate. Removal of all volatile components in vacuo provided crude diamide 5{1–2,1–5} which was purified on an ISCO Companion chromatography station (40 g silica gel, hexane/ethyl acetate). Compounds 5{1–2,1–5} were obtained in yields of 18–93% (Table 1).
(Z)-N-(Cyclopropylmethyl)-5-ethylidene-2-methyl-6-oxo-4-vinyl-1,2,5,6-tetrahydro-pyridine-2-carboxamide (5{1,1})
IR (film) 2929, 1673, 1635, 1530, 1403, 1273 cm−1; 1H NMR (CDCl3) δ 6.45-6.25 (m, 4 H), 5.85 (s, 1 H), 5.49 (dd, 1 H, J = 17.2, 1.5 Hz), 5.26 (dd, 1 H, J = 10.8, 1.5 Hz), 3.18-3.00 (m, 2 H), 2.31 (d, 3 H, J = 7.5 Hz), 1.61 (s, 3 H), 0.98-0.80 (m, 1 H), 0.50-0.40 (m, 2 H), 0.20-0.10 (m, 2 H); 13C NMR (CDCl3) δ 171.8, 165.7, 137.5, 135.3, 133.3, 125.2, 123.4, 117.4, 61.1, 43.7, 26.6, 15.5, 10.1, 2.7, 2.7; MS (ESI) m/z (rel. intensity) 543 ([2M + Na]+, 15), 283 ([M + Na]+, 100); HRMS (EI) m/z calculated for C15H20N2O2Na 283.1422, found 283.1419.
General protocol for 5-iminooxazolidin-2-one formation and Diels-Alder reaction of diamides 5{1–2,1–5} (Protocol C)
Diamide 5{1–2,1–5} (1.00 eq, 2.5–7.5 mmol) was dissolved in dichloromethane (100–200 mL), cooled to −10 °C and treated with triethylamine (6.00 eq). Phosgene (20% in toluene, 3.00 eq) was slowly added while keeping the temperature at −10 °C. After stirring for another 30 min at −10 °C, the reaction mixture was allowed to warm up to room temperature and stirred for an additional 30 min. The reaction mixture was extracted with sodium bicarbonate (100 mL), water (100 mL), and brine (100 ml). The combined aqueous fractions were reextracted with dichloromethane, and all organic fractions were combined and dried over sodium sulfate. Removal of all volatile components in vacuo provided a mixture of crude 5-iminooxazolidin-2-ones 6{1–2,1–5} and 7{1–2,1–5} in toluene which was used immediately in the next step without further purification. Acidic workup or chromatography over silica gel led to the hydrolysis of the desired product to starting material 5{1–2,1–5}. Quenching the reaction at −10 °C led to an incomplete reaction, but prevented the formation of the carbamate side product 6{1–2,1–5}.
A mixture of crude 5-iminooxazolidin-2-ones 6{1–2,1–5} and 7{1–2,1–5} (1.00 eq, 5.0–10 mmol assuming a quantitative conversion in the previous step) was dissolved in toluene (50–100 mL) and divided into five batches of equal volume. Using Radley’s Carousel Reaction Station (6 Place or 12 Place) maleimide 3{1}, N-methylmaleimide 3{2}, N-ethylmaleimide 3{3}, N-phenylmaleimide 3{4} and methyl 2-(N-maleimide)acetate 3{5}32 (1.30 eq) were added at once to the five batches of 5-iminooxazolidin-2-one and the reaction mixture was heated to 90 ºC for 1.5 h. After cooling to room temperature all volatile components were removed in vacuo and the residues were purified on an ISCO Optix 10 parallel chromatography station (12 g silica gel, hexane/ethyl acetate). In general, carbamates 8{1–2,1–5,1–5} (first eluting), double Diels-Alder adducts 9{1–2,1–5,1–5} (second eluting), and Diels-Alder adducts 10{1–2,1–5,1–5} (third eluting, in most cases a mixture of E/Z isomers33) could be separated (Table 2). In some cases, a second chromatography on an ISCO Companion chromatography station (12 g silica gel, hexane/ethyl acetate) was necessary to obtain pure material. If the reaction mixture was quenched at −10 ºC during the formation of 5-iminooxazolidin-2-one 7{1–2,1–5} (Table 2), no carbamate 6{1–2,1–5} was obtained. The double Diels-Alder adduct 9{1–2,1–5,1–5} (Table 3) was especially observed if the formation of 5-iminooxazolidin-2-one 7{1–2,1–5} was incomplete and the maleimides 3{1–5} were therefore present in greater excess.
rac-(8aR,11aS,11bR,11cS,Z)-1-(Cyclopropylmethylimino)-10-ethyl-11c-methyl-3,9,11-trioxo-6-vinyl-1,3,8,8a,9,10,11,11a,11b,11c-decahydrooxazolo[4,3-a]pyrrolo-[3,4-h]isoquinoline-5-yl diethylcarbamate (8{1,1,3})
IR (film) 2979, 2936, 1811, 1735, 1697, 1379, 1346, 1254, 1145, 995 cm−1; 1H NMR (CDCl3) δ 6.18 (dd, 1 H, J = 17.7, 11.4 Hz), 6.00 (ddd, 1 H, 6.9, 4.2, 1.9 Hz), 5.42 (dd, 1 H, J = 17.6, 1.7 Hz), 5.37 (dd, 1 H, J = 11.4, 1.8 Hz), 3.83 (dd, 1 H, J = 8.7, 4.6 Hz), 3.68-3.51 (m, 1 H), 3.49-3.24 (m, 7 H), 3.19 (app td, 1 H, J = 7.5, 0.9 Hz), 2.94-2.79 (m, 2 H), 2.22 (dddd, 1 H, J = 15.2, 7.4, 4.3, 0.9 Hz), 1.78 (s, 3 H), 1.23 (app t, 3 H, J = 7.1 Hz), 1.16 (app t, 3 H, J = 7.1 Hz), 1.38-1.08 (m, 1 H), 1.00 (app. t, 3 H, J = 7.2 Hz), 0.60-0.40 (m, 2 H), 0.30-0.18 (m, 2 H); 13C NMR (CDCl3) δ 178.8, 176.6, 153.1, 151.6, 148.0, 133.2, 132.8, 126.7, 122.5, 120.0, 110.6, 62.4, 52.5, 44.5, 42.3, 42.0, 41.3, 41.1, 33.7, 27.8, 25.7, 13.9, 13.0, 12.9, 11.5, 3.4, 3.2; MS (EI) m/z (rel. intensity) 510 (15), 466 (7), 385 (61), 286 (20), 160 (23), 100 (100), 72 (84); HRMS (EI) m/z calculated for C27H34N4O6 510.2478, found 510.2485.
rac-(3aS,3bR,3c1R,8R,8aS,11aR,11bS,12aR,E)-4-(Cyclopropylmethylimino)-2,10-diethyl-3c1,8-dimethyl-4,5,8,8a,11a,11b,12,12a-octahydro-5-oxa-2,6a,10-triazatri-cyclopenta[a,e,j]phenalene-1,3,6,7,9,11(2H,3aH,3bH,3c1H,6aH,10H)-hexaone (9{1,1,3})
IR (film) 2981, 2940, 1833, 1742, 1696, 1443, 1403, 1350, 1316, 1294, 1277, 1229 cm−1; 1H NMR (CDCl3) δ 3.95 (dd, 1 H, J = 9.7, 5.5 Hz), 3.50-3.30 (m, 7 H), 3.26 (dd, 1 H, J = 8.7, 6.6 Hz), 3.06 (dd, 1 H, J = 8.7, 5.7 Hz), 2.95 (app. t, 1 H, J = 3.9 Hz), 2.71 (ddd, 1 H, J = 14.5, 13.0, 5.5 Hz), 2.63-2.57 (m, 1 H), 2.53 (ddd, 1 H, J = 14.5, 4.9, 2.4 Hz), 2.28-2.20 (m, 1 H), 1.89 (d, 3 H, J = 7.4 Hz), 1.56 (s, 3 H), 1.10 (app t, 3 H, J = 7.2 Hz), 1.05-0.95 (m, 1 H), 0.98 (app t, 3 H, J = 7.2 Hz), 0.53-0.42 (m, 2 H), 0.28-0.15 (m, 2 H); 13C NMR (CDCl3) δ 177.9, 176.1, 175.7, 175.6, 155.9, 150.6, 150.0, 147.1, 131.9, 60.0, 52.6, 46.1, 42.6, 41.4, 39.4, 39.0, 35.4, 33.9, 33.9, 33.2, 29.8, 22.8, 15.4, 13.1, 12.9, 11.4, 3.4, 3.2; MS (ESI) m/z (rel. intensity) 1095 ([2M + Na]+, 50), 1073 ([2M + 1]+, 25), 975 (25), 559 ([M + Na]+, 25), 537 ([M + 1]+, 100), 483 (55), 412 (35); HRMS (ESI) m/z calculated for C28H33N4O7 537.2349, found 537.2341.
rac-Methyl 2-((1Z,6Z,8aR,11aS,11bR,11cS)-11c-benzyl-1-(benzylimino)-6-ethyli-dene-3,5,9,11-tetraoxo-1,5,6,8a,9,11,11a,11c-octahydrooxazolo[4,3-a]pyrrolo[3,4-h]-isoquinolin-10(3H,8H,11bH)-yl)acetate (10{2,4,5})
IR (film) 2928, 1838, 1742, 1712, 1421, 1365, 1300, 1223 cm−1; 1H NMR (CDCl3) δ 7.40-7.18 (m, 8 H), 6.84-6.80 (m, 2 H), 6.61 (q, 1 H, J = 7.4 Hz), 6.17-6.12 (m, 1 H), 4.68 (d, 1 H, J = 14.1 Hz), 4.58 (d, 1 H, J = 14.1 Hz), 4.15 (d, 1H, J = 17.2 Hz), 4.08 (d, 1 H, J = 17.2 Hz), 3.92 (dd, 1 H, J = 8.9, 5.3 Hz), 3.67 (s, 3 H), 3.32-3.29 (m, 1 H), 3.29 (d, 1 H, J = 13.5 Hz), 3.15 (d, 1 H, J = 13.5 Hz), 3.00-2.93 (m, 2 H), 2.29 (d, 3 H, J = 7.5 Hz), 2.30-2.20 (m, 1 H); 13C NMR (CDCl3) δ 177.5, 175.8, 166.0, 159.3, 150.2, 146.7, 140.6, 137.8, 134.8, 132.1, 129.8, 129.5, 128.5, 128.3, 128.0, 127.8, 127.0, 123.5, 65.6, 52.3, 51.6, 47.9, 41.0, 40.5, 39.3, 39.2, 24.9, 15.5; MS (ESI) m/z (rel. intensity) 1157 ([2M + Na]+, 10), 590 ([M + Na]+, 100), 568 ([M + 1]+, 10), 413 (40); HRMS (ESI) m/z calculated for C32H29N3O7Na 590.1903 found 590.1877.
General protocol for the hydrogenation of Diels-Alder adducts 8{1–2,1–5, 1–5} and 10{1–2,1–5,1–5} (Protocol D)
The Diels-Alder adducts 8{1–2,1–5,1–5} or 10{1–2,1–5 1–5} (1.00 eq, 0.050–0.50 mmol), respectively, were dissolved in dry THF (10 mL) and treated with 20 wt% Pd/C (10%). Using a Radley’s Carousel reaction station (12 Place) a single balloon filled with hydrogen was attached and the system was flushed five times with hydrogen. The reaction mixtures were stirred in parallel for 4 h, after which the Pd/C was removed through filtration over a plug of Celite, followed by washing with dichloromethane (3 × 20 mL). In the case of indole containing substrates 8{1–2,5,1–5} or 10{1–2,5,1–5} another 20 wt% Pd/C (10%) was added after 4 h, the system was flushed again five times with hydrogen and stirred for an additional 20 h. All volatile components were removed in vacuo and the residues were purified on an ISCO Optix 10 parallel chromatography station (12 g silica gel, hexane/ethyl acetate) to afford the reduced Diels-Alder adducts 11{1–2,1–5,1–5} or 12{1–2,1–5,1–5}, respectively (Tables 4–5).
rac-(8aR,11aS,11bR,11cS,Z)-1-(Cyclopropylmethylimino)-6-ethyl-10,11c-dimethyl-3,9,11-trioxo-1,3,8,8a,9,10,11,11a,11b,11c-decahydrooxazolo[4,3-a]pyrrolo[3,4-h]iso-quinolin-5-yl diethylcarbamate (11{1,1,2})
IR (film) 2973, 2935, 1810, 1734, 1699, 1431, 1383, 1255, 1140, 977 cm−1; 1H NMR (CDCl3) δ 5.85 (ddd, 1 H, J = 7.0, 4.1, 2.2 Hz), 3.83 (dd, 1 H, J = 8.7, 4.5 Hz), 3.70-3.59 (m, 1 H), 3.43-3.18 (m, 6 H), 2.87 (ddd, 1 H, J = 15.4, 8.0, 1.3 Hz), 2.87 (s, 3 H), 2.83-2.77 (m, 1 H), 2.30-2.10 (m, 3 H), 1.76 (s, 3 H), 1.24 (app t, 3 H, J = 7.1 Hz), 1.17 (app t, 3 H, J = 7.1 Hz), 1.15-1.05 (m, 1 H), 0.97 (app. t, 3 H, J = 7.4 Hz), 0.53-0.40 (m, 2 H), 0.27-0.20 (m, 2 H); 13C NMR (CDCl3) δ 179.1, 176.9, 153.3, 152.0, 147.9, 132.9, 132.3, 120.0, 113.0, 62.2, 52.4, 43.9, 42.3, 42.0, 41.4, 41.3, 27.7, 25.5, 24.9, 17.4, 13.9, 13.0, 11.4, 3.4, 3.2; MS (ESI) m/z (rel. intensity) 1019 ([2M + Na]+, 25), 521 ([M + Na]+, 100), 499 ([M + 1]+, 45); HRMS (ESI) m/z calculated for C26H34N4O6Na 521.2376, found 521.2356.
rac-Methyl 2-((8aR,11aS,11bR,11cS,Z)-1-(benzylimino)-6-ethyl-11c-methyl-3,5,9,11-tetraoxo-1,7,8,8a,9,11,11a,11c-octahydrooxazolo[4,3-a]pyrrolo[3,4-h]isoquinolin-10(3H,5H,11bH)-yl)acetate (12{1,4,5})
IR (film) 2953, 1833, 1741, 1708, 1420, 1326, 1281, 1220, 975 cm−1; 1H NMR (CDCl3) δ 7.34-7.24 (m, 5 H), 4.73 (d, 1 H, J = 14.6 Hz), 4.63 (d, 1 H, J = 14.6 Hz), 4.15 (d, 1 H J = 17.0 Hz), 4.05 (d, 1 H, J = 17.0 Hz), 3.89 (dd, 1 H, J= 9.5, 5.5 Hz), 3.69 (s, 3 H), 3.17 (ddd, 1 H, J = 9.6, 5.3, 2.4 Hz), 3.09 (d, 1 H, J = 5.4 Hz), 2.67-2.59 (m, 2 H), 2.52-2.40 (m, 1 H), 2.37-2.22 (m, 2 H), 2.16-2.02 (m, 1 H), 1.74 (s, 3 H), 0.95 (app. t, 3 H, J = 7.4 Hz); 13C NMR (CDCl3) δ 177.3, 175.7, 166.3, 158.3, 152.0, 146.9, 145.1, 138.3, 133.5, 128.5, 127.8, 127.2, 60.4, 52.6, 51.8, 40.9, 40.7, 39.9, 39.3, 29.7, 25.9, 19.9, 19.0, 12.2; MS (ESI) m/z (rel. intensity) 516 ([M + Na]+, 20), 494 ([M + 1]+, 100), 333 (50); HRMS (ESI) m/z calculated for C26H28N3O7 494.1927, found 494.1944.
General protocol for the formation of acylureas 14{1–2,1–5,1–5,1–6} (Protocol E)
Using 16 mm test tubes, the reduced Diels-Alder adducts 12{1–2,1–5,1–5} (1.0 eq, 0.030–0.060 mmol) were dissolved in chloroform (1 mL), treated with amines 4{1–6} (5.0 eq) and stirred in parallel for 2 h at room temperature. All volatile components were removed in vacuo and the residues were purified on an ISCO Optix 10 parallel chromatography station (4 g silica gel, hexane/ethyl acetate) to afford amides 14{1–2,1–5,1–51–6}. In several cases, hydantoins 13{1–2,1–5,1–5} were observed as side product and could be easily separated as the first eluting component (Table 6).
rac-(3aR,9S,9aR,9bS)-9-Benzyl-N8,N9-bis(cyclopropylmethyl)-6-ethyl-2-methyl-1,3,7-trioxo-3,3a,4,5,7,9,9a,9b-octahydro-1H-pyrrolo[3,4-h]isoquinoline-8,9(2H)-dicarboxamide (14{2,1,2,4})
IR (film) 3324, 2960, 1699, 1542, 1437, 1385, 1319, 1287, 1170 cm−1; 1H NMR (CDCl3) δ 9.18 (app t, 1 H, J = 5.3 Hz), 7.93 (app t, 1 H, J = 5.1 Hz), 7.18-7.14 (m, 3 H), 7.04-7.01 (m, 2 H), 4.02 (d, 1 H, J = 14.4 Hz), 3.52 (d, 1 H, J = 14.3 Hz), 3.52 (dd, 1 H, J = 9.3, 3.6 Hz), 3.38-3.20 (m, 3 H), 3.14-3.00 (m, 3 H), 2.86 (s, 3 H), 2.23-1.96 (m, 4 H), 1.72-1.58 (m, 1 H), 1.51-1.39 (m, 1 H), 1.20-1.08 (m, 1 H), 1.02-0.87 (m, 1 H), 0.57 (app t, 3 H, J = 7.4 Hz), 0.60-0.51 (m, 2 H), 0.48-0.42 (m, 2 H), 0.34-0.28 (m, 2 H), 0.21-0.16 (m, 2 H); 13C NMR (CDCl3) δ 178.5, 178.3, 170.3, 166.5, 154.6, 142.8, 138.4, 132.7, 129.8, 128.3, 126.8, 66.9, 45.5, 44.9, 44.7, 44.6, 42.2, 41.0, 25.0, 24.7, 20.5, 19.4, 12.5, 10.9, 10.2, 3.7, 3.6, 3.6, 3.2; MS (ESI) m/z (rel. intensity) 569 ([M + Na]+, 25), 547 ([M + 1]+, 25), 518 (15), 476 (100), 472 (60); HRMS (ESI) m/z calculated for C31H39N4O5 547.2920, found 547.2883.
General protocol for the formation of hydantoins 13{1–2,1–5,1–5} (Protocol F)
Acylurea 14{1–2,1–5,1–5,1–6} (1.0 eq, 0.030 mmol) was dissolved in methanol (1 mL) and stirred for 16 h. All volatile components were removed in vacuo and the residue was purified on an ISCO Companion chromatography station (4 g silica gel, hexane/ethyl acetate) to afford hydantoin 13{1–2,1–5,1–5} in 85–89% yield (Table 6, footnote c and d).
rac-Methyl 2-((8aR,11aS,11bR,11cS)-2-(cyclopropylmethyl)-6-ethyl-11c-methyl-1,3,5,9,11-pentaoxo-2,3,7,8,8a,9,11,11a-octahydro-1H-imidazo[5,1-a]pyrrolo[3,4-h]isoquinolin-10(5H,11bH,11cH)-yl)acetate (13{1,1,5})
IR (film) 2953, 1796, 1727, 1416, 1368, 1326, 1214 cm−1; 1H NMR (CDCl3) δ 4.15 (d, 1 H, J = 17.1 Hz), 4.05 (d, 1 H, J = 17.0 Hz), 3.69 (s, 3 H), 3.66 (dd, 1 H, J = 9.5, 5.1 Hz), 3.53 (dd, 1 H, J = 14.0, 6.8 Hz), 3.45 (dd, 1 H, J = 14.0, 7.4 Hz), 3.24 (ddd, 1 H, J = 9.5, 5.9, 3.1 Hz), 3.04 (d, 1 H, J = 5.1 Hz), 2.70-2.59 (m, 2 H), 2.51-2.23 (m, 3 H), 2.16-2.08 (m, 1 H), 1,67 (s, 3 H), 1.28-1.20 (m, 1 H), 0.96 (app t, 3 H, J = 7.5 Hz), 0.52-0.46 (m, 2 H), 0.39-0.35 (m, 2 H); 13C NMR (CDCl3) δ 177.5, 175.1, 173.5, 166.4, 158.3, 151.6, 143.4, 134.3, 60.6, 52.7, 43.8, 41.2, 40.2, 39.6, 39.4, 28.0, 25.7, 20.1, 19.2, 12.5, 9.5, 3.9, 3.7; MS (ESI) m/z (rel. intensity) 480 ([M + Na]+, 100), 458 ([M + 1]+, 10).
Stability study of acylureas 14{1–2,1–5,1–5,1–6} in DMSO
Acylurea 14{2,5,2,1} (0.0050 mmol) was dissolved in DMSO-d6 (0.7 mL, Cambridge Isotope Laboratories, Inc, D 99.9%) and stored in an NMR tube at room temperature for several weeks. The composition was determined regularly by 1H NMR studies. Complete and selective conversion of acylurea 14{2,5,2,1} to hydantoin 13{2,5,2} was observed after two months. An identical sample was stored in a −78ºC freezer and no rearrangement was observed over three months.
Acknowledgments
We gratefully acknowledge financial support provided by NIGMS (P50-GM067082). The authors also thank Ms. Leslie A. Twining for LC-MS/UV/ELSD analyses, Dr. Steven Geib for performing several X-ray analyses, and Dr. Branko Mitasev and Prof. Peter Wipf for stimulating discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Webb TR. Curr Opin Drug Disc Dev. 2005;8:303–308. [PubMed] [Google Scholar]
- 2.(a) Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; (b) Burke MD, Lalic G. Chem Biol. 2002;9:535–541. doi: 10.1016/s1074-5521(02)00143-6. [DOI] [PubMed] [Google Scholar]; (c) Spring DR. Org Biomol Chem. 2003;1:3867–3870. doi: 10.1039/b310752n. [DOI] [PubMed] [Google Scholar]; (d) Burke MD, Schreiber SL. Angew Chem Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; (e) Taylor SJ, Taylor AM, Schreiber SL. Angew Chem Int Ed. 2004;43:1681–1685. doi: 10.1002/anie.200353466. [DOI] [PubMed] [Google Scholar]
- 3.Tan DS. Nat Chem Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 4.Selzer P, Roth HJ, Ertl P, Schuffenhauer A. Curr Opin Chem Biol. 2005;9:310–316. doi: 10.1016/j.cbpa.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Liao Y, Hu Y, Wu J, Zhu Q, Donovan M, Fathi R, Yang Z. Curr Med Chem. 2003;10:2285–2316. doi: 10.2174/0929867033456738. [DOI] [PubMed] [Google Scholar]
- 6.(a) Arya P, Joseph R, Chou DTH. Chem Biol. 2002;9:145–156. doi: 10.1016/s1074-5521(02)00105-9. [DOI] [PubMed] [Google Scholar]; (b) Arya P, Joseph R, Gan Z, Rakic B. Chem Biol. 2005;12:163–180. doi: 10.1016/j.chembiol.2005.01.011. [DOI] [PubMed] [Google Scholar]; (c) Reayi A, Arya P. Curr Opin Chem Biol. 2005;9:240–247. doi: 10.1016/j.cbpa.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 7.(a) Abel U, Koch C, Speitling M, Hansske FG. Curr Opin Chem Biol. 2002;6:453–458. doi: 10.1016/s1367-5931(02)00338-1. [DOI] [PubMed] [Google Scholar]; (b) Ortholand JY, Ganesan A. Curr Opin Chem Biol. 2004;8:271–280. doi: 10.1016/j.cbpa.2004.04.011. [DOI] [PubMed] [Google Scholar]; (c) Boldi A. Curr Opin Chem Biol. 2004;8:281–286. doi: 10.1016/j.cbpa.2004.04.010. [DOI] [PubMed] [Google Scholar]; (d) Tan DS. Comb Chem High Throughput Screening. 2004;7:631–643. doi: 10.2174/1386207043328418. [DOI] [PubMed] [Google Scholar]
- 8.Fergus S, Bender A, Spring DR. Curr Opin Chem Biol. 2005;9:304–309. doi: 10.1016/j.cbpa.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 9.(a) Haggarty SJ, Clemons PA, Wong JC, Schreiber SL. Comb Chem High Throughput Screening. 2004;7:669–676. doi: 10.2174/1386207043328319. [DOI] [PubMed] [Google Scholar]; (b) Kim YK, Arai MA, Arai T, Lamenzo JO, Dean EF, Patterson N, Clemons PA, Schreiber SL. J Am Chem Soc. 2004;126:14740–14745. doi: 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]
- 10.Thomas GL, Wyatt EE, Spring DR. Curr Opin Drug Discovery Dev. 2006;9:700–712. [PubMed] [Google Scholar]
- 11.Burke MD, Berger EM, Schreiber SL. Science. 2003;302:613–618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]
- 12.(a) Kwon O, Park SB, Schreiber SL. J Am Chem Soc. 2002;124:13402–13404. doi: 10.1021/ja028086e. [DOI] [PubMed] [Google Scholar]; (b) Wyatt EE, Fergus S, Galloway WRJD, Bender A, Fox DJ, Plowright AT, Jessiman AS, Welch M, Spring DR. Chem Commun. 2006:3296–3298. doi: 10.1039/b607710b. [DOI] [PubMed] [Google Scholar]
- 13.Yeager AR, Min GK, Porco JA, Jr, Schaus SE. Org Lett. 2006;8:5065–5068. doi: 10.1021/ol0618252. [DOI] [PubMed] [Google Scholar]
- 14.Couladouros E, Strongilos AT. Angew Chem Int Ed. 2002;41:3677–3680. doi: 10.1002/1521-3773(20021004)41:19<3677::AID-ANIE3677>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.(a) Sello JK, Andreana PR, Lee D, Schreiber SL. Org Lett. 2003;5:4125–4127. doi: 10.1021/ol035773h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burke MD, Berger EM, Schreiber SL. J Am Chem Soc. 2004;126:14095–14104. doi: 10.1021/ja0457415. [DOI] [PubMed] [Google Scholar]
- 16.Mitasev B, Yan B, Brummond KM. Heterocycles. 2006;70:367–388. [Google Scholar]
- 17.(a) Edmunds JJ, Klutchko S, Hamby JM, Bunker AM, Connolly CJC, Winters RT, Quin J, III, Sircar I, Hodges JC, Panek RL, Keiser JA, Doherty AM. J Med Chem. 1995;38:3759–3771. doi: 10.1021/jm00019a005. [DOI] [PubMed] [Google Scholar]; (b) Hudkins RL, DeHaven-Hudknis DL, Doukas P. Bioorg Med Chem Lett. 1997;7:979–984. [Google Scholar]; (c) Scholl S, Koch A, Henning D, Kempter G, Kleinpeter E. Struct Chem. 1999;10:355–366. [Google Scholar]; (d) Stilz HU, Guba W, Jablonka B, Just M, Klingler O, König W, Wehner V, Zoller G. J Med Chem. 2001;44:1158–1176. doi: 10.1021/jm001068s. [DOI] [PubMed] [Google Scholar]; (e) Ooms F, Wouters J, Oscari O, Happaerts T, Bouchard G, Carrupt PA, Testa B, Lambert DM. J Med Chem. 2002;45:1748–1756. doi: 10.1021/jm010896y. [DOI] [PubMed] [Google Scholar]; (f) Jansen M, Potschka H, Brandt C, Löscher W, Dannhardt G. J Med Chem. 2003;46:64–73. doi: 10.1021/jm020955n. [DOI] [PubMed] [Google Scholar]; (g) Zha C, Brown GB, Brouillette WJ. J Med Chem. 2004;47:6519–6528. doi: 10.1021/jm040077o. [DOI] [PubMed] [Google Scholar]
- 18.(a) Fujinami A, Ozaki T, Yamamoto S. Agric Biol Chem. 1971;35:1707. [Google Scholar]; (b) Fujinami A, Ozaki T, Yamamoto S, Akiba K, Ooba S, Tanaka K, Kameda N, Nodera K, Ohishi T. Ger Offen. 1970 2002410. [Google Scholar]; Chem Abstr. 1970;73:87912c. [Google Scholar]
- 19.(a) Boger DL, Weng JH, Miyazaki S, McAtee JJ, Castle SL, Kim SH, Mori Y, Rogel O, Strittmatter H, Jin Q. J Am Chem Soc. 2000;122:10047–10055. [Google Scholar]; (b) Breitenmoser R, Linden A, Heimgartner H. Helv Chim Acta. 2002;85:990–1018. [Google Scholar]
- 20.(a) Messer R, Fuhrer CA, Häner R. Curr Opin Chem Biol. 2005;9:259–265. doi: 10.1016/j.cbpa.2005.04.002. [DOI] [PubMed] [Google Scholar]; (b) Arya P, Quevillon S, Joseph R, Wei CQ, Gan Z, Parisien M, Sesmilo E, Reddy PT, Chen ZX, Durieux P, Laforce D, Campeau LC, Khadem S, Couve-Bonnaire S, Kumar R, Sharma U, Leek DM, Daroszewska M, Barnes ML. Pure Appl Chem. 2005;77:163–178. [Google Scholar]
- 21.(a) Brummond KM, Chen H, Sill P, You L. J Am Chem Soc. 2002;124:15186–15187. doi: 10.1021/ja027588p. [DOI] [PubMed] [Google Scholar]; (b) Brummond M, Mitasev B. Org Lett. 2004;6:2245–2248. doi: 10.1021/ol0492391. [DOI] [PubMed] [Google Scholar]; (c) Brummond KM, Painter TO, Probst D, Mitasev B. Org Lett. 2007;9:347. doi: 10.1021/ol062842u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brimble MA, Duckworth MS, Lee CKY. Aust J Chem. 1998;51:907–913. [Google Scholar]
- 23.E/Z-Isomerization at elevated temperatures has been observed previousely in case of related scaffolds with methyl and phenyl substituents at the exocyclic double bond, unpublished results.
- 24.(a) Rishton GM. Drug Discov Today. 1997;2:382–384. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]; (b) Rishton GM. Drug Discov Today. 2002;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 25.The X-ray structure analysis of 14{1,1,4,1} showed significant disorder in the outer ligands and was therefore not published here.
- 26.Nagase T, Mase T, Fukami T, Hayama T, Fujita K, Niiyama K, Takahashi H, Kumagai U, Urakawa Y, Nagasawa Y, Ihara M, Nishikibe M, Ishikawa K. Bioorg Med Chem Lett. 1995;5:1395–1400. [Google Scholar]
- 27.(a) Savrda J, Chertanova L, Wakselman M. Tetrahedron. 1994;50:5309–5322. [Google Scholar]; (b) Hulme C, Ma L, Romano JJ, Morton G, Tang SY, Cherrier MP, Choi S, Salvino J, Labaudiniere R. Tetrahedron Lett. 2000;41:1889–1893. [Google Scholar]; (c) Vázquez J, Royo M, Albericio F. Lett Org Chem. 2004;1:224–226. [Google Scholar]
- 28.(a) Opacic N, Barbaric M, Zorc B, Cetina M, Nagl A, Frkovic D, Kralj M, Pavelic K, Balzarini J, Andrei G, Snoeck R, De Clercq E, Raic-Malic S, Mintas M. J Med Chem. 2005;48:475–482. doi: 10.1021/jm040869i. [DOI] [PubMed] [Google Scholar]; (b) Opacic N, Zorc B, Cetina M, Mrvos-Sermek D, Raic-Malic S, Mintas M. J Peptide Res. 2005;66:85–93. doi: 10.1111/j.1399-3011.2005.00276.x. [DOI] [PubMed] [Google Scholar]
- 29.QikProp 2.1. Schroedinger Inc; New York: 2003. MW is the Molecular weight of the molecule, Volume is the total solvent-accessible volume in cubic angstroms using a probe with a 1.4 Å radius, SASA is the total solvent accessible surface area (SASA) in square angstroms using a probe with a 1.4 Å radius, HBD is the estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution, HBA is the estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution, logP is the predicted octanol/water partition coefficient, logS is the predicted aqueous solubility (S in moles/liter is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid), rotatable bonds is the number of non-trivial (not CX3), non-hindered (not alkene, amide, small ring) rotatable bonds, Caco-Perm. is the predicted apparent Caco-2 cell permeability in nm/sec Duffy EM, Jorgensen WL. J Am Chem Soc. 2000;122:2878–2888.Jorgensen WL, Duffy EM. Adv Drug Delivery Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x.Jorgensen WL. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361.
- 30.(a) Lipinski C. Drug Discov Today Technologies. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]; (b) Lipinski C, Hopkins A. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]; (c) Werner S, Iyer PS, Fodor MF, Coleman CMC, Twining LA, Mitasev B, Brummond KM. J Comb Chem. 2006;8:368–380. doi: 10.1021/cc050160c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; (b) Egan WJ, Merz KM, Jr, Baldwin JJ. J Med Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]; (c) Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 32.Compound 3{5} was synthesized in analogy to the following protocols: Reddy PY, Kondo S, Toru T, Ueno YJ. Org Chem. 1997;62:2652–2654. doi: 10.1021/jo962202c.Werner S, Curran DP. Org Lett. 2003;5:3293–3296. doi: 10.1021/ol035214a.
- 33.The assignment of the E/Z isomers was made by a characteristic chemical shift difference for the exocyclic methylene protons in the 1H NMR spectra, which showed a resonance around 7.0 ppm for the E-isomers and around 6.6 ppm for the Z-isomers. For reference data on a similar system see: Yates P, Helferty PH. Can J Chem. 1983;61:936–945.
- 34.Crystal structures have been deposited at the Cambridge Crystallographic Data Centre: CCDC 675555 (11{1,1,4}), CCDC 675554 (12{2,1,4}) and CCDC 675556 (13{1,1,4}).