

Abstract
Key points
Ageing‐induced endothelial dysfunction contributes to organ dysfunction and progression of cardiovascular disease. VE‐cadherin clustering at adherens junctions promotes protective endothelial functions, including endothelium‐dependent dilatation.
Ageing increased internalization and degradation of VE‐cadherin, resulting in impaired activity of adherens junctions.
Inhibition of VE‐cadherin clustering at adherens junctions (function‐blocking antibody; FBA) reduced endothelial dilatation in young arteries but did not affect the already impaired dilatation in old arteries. After junctional disruption with the FBA, dilatation was similar in young and old arteries.
Src tyrosine kinase activity and tyrosine phosphorylation of VE‐cadherin were increased in old arteries. Src inhibition increased VE‐cadherin at adherens junctions and increased endothelial dilatation in old, but not young, arteries. Src inhibition did not increase dilatation in old arteries treated with the VE‐cadherin FBA.
Ageing impairs the activity of adherens junctions, which contributes to endothelial dilator dysfunction. Restoring the activity of adherens junctions could be of therapeutic benefit in vascular ageing.
Abstract
Endothelial dilator dysfunction contributes to pathological vascular ageing. Experiments assessed whether altered activity of endothelial adherens junctions (AJs) might contribute to this dysfunction. Aortas and tail arteries were isolated from young (3–4 months) and old (22–24 months) F344 rats. VE‐cadherin immunofluorescent staining at endothelial AJs and AJ width were reduced in old compared to young arteries. A 140 kDa VE‐cadherin species was present on the cell surface and in TTX‐insoluble fractions, consistent with junctional localization. Levels of the 140 kDa VE‐cadherin were decreased, whereas levels of a TTX‐soluble 115 kDa VE‐cadherin species were increased in old compared to young arteries. Acetylcholine caused endothelium‐dependent dilatation that was decreased in old compared to young arteries. Disruption of VE‐cadherin clustering at AJs (function‐blocking antibody, FBA) inhibited dilatation to acetylcholine in young, but not old, arteries. After the FBA, there was no longer any difference in dilatation between old and young arteries. Src activity and tyrosine phosphorylation of VE‐cadherin were increased in old compared to young arteries. In old arteries, Src inhibition (saracatinib) increased: (i) 140 kDa VE‐cadherin in the TTX‐insoluble fraction, (ii) VE‐cadherin intensity at AJs, (iii) AJ width, and (iv) acetylcholine dilatation. In old arteries treated with the FBA, saracatinib no longer increased acetylcholine dilatation. Saracatinib did not affect dilatation in young arteries. Therefore, ageing impairs AJ activity, which appears to reflect Src‐induced phosphorylation, internalization and degradation of VE‐cadherin. Moreover, impaired AJ activity can account for the endothelial dilator dysfunction in old arteries. Restoring endothelial AJ activity may be a novel therapeutic approach to vascular ageing.
Keywords: adherens junctions, aging, endothelium, NO, VE‐cadherin
Key points
Ageing‐induced endothelial dysfunction contributes to organ dysfunction and progression of cardiovascular disease. VE‐cadherin clustering at adherens junctions promotes protective endothelial functions, including endothelium‐dependent dilatation.
Ageing increased internalization and degradation of VE‐cadherin, resulting in impaired activity of adherens junctions.
Inhibition of VE‐cadherin clustering at adherens junctions (function‐blocking antibody; FBA) reduced endothelial dilatation in young arteries but did not affect the already impaired dilatation in old arteries. After junctional disruption with the FBA, dilatation was similar in young and old arteries.
Src tyrosine kinase activity and tyrosine phosphorylation of VE‐cadherin were increased in old arteries. Src inhibition increased VE‐cadherin at adherens junctions and increased endothelial dilatation in old, but not young, arteries. Src inhibition did not increase dilatation in old arteries treated with the VE‐cadherin FBA.
Ageing impairs the activity of adherens junctions, which contributes to endothelial dilator dysfunction. Restoring the activity of adherens junctions could be of therapeutic benefit in vascular ageing.
Abbreviations
- AJ
adherens junctions
- FBA
function‐blocking antibody
- l‐NAME
N ω‐nitro‐l‐arginine methyl ester
- ROCK
Rho‐associated protein kinase
- TNFα
tumour necrosis factor‐α
Introduction
Ageing causes structural and functional deterioration of the arterial system that contributes to cardiovascular disease and organ dysfunction (Lakatta et al. 2009; Laurent, 2012). Endothelial dysfunction is a key contributor to this process of vascular ageing. Ageing arterial endothelial cells are compromised: their production of protective NO is reduced, their barrier function is impaired, they are highly susceptible to apoptosis, and they have a prominent pro‐inflammatory phenotype, including production of pathological mediators (Lakatta et al. 2009; Goel et al. 2010; Ungvari et al. 2010; Flavahan et al. 2016). Ageing endothelial cells are caught in a cycle of chronic stress, both producing and responding to pathological mediators, and their resulting frailty is considered a key instigator of vascular ageing (Ungvari et al. 2010; Wang et al. 2010a).
Maintenance of a normal protective endothelial cell layer is an active rather than a passive process (Murakami et al. 2008; Dejana et al. 2009; Flavahan et al. 2013; Flavahan, 2017). Protective features of arterial endothelial cells become fully engaged during the early postnatal period (Flavahan et al. 2013; Flavahan & Flavahan, 2014; Chang et al. 2016), and appear to be mediated by increased clustering of VE‐cadherin at adherens junctions, which promotes protective signalling including amplifying endothelial NO‐mediated dilatation (Flavahan et al. 2013). Indeed, endothelial adherens junctions promote endothelial and vascular stability, whereas their disruption compromises vascular integrity, increases vascular inflammation and fragility, and promotes the development of cardiovascular diseases (Murakami et al. 2008; Dejana et al. 2009; Giannotta et al. 2013; Flavahan, 2017). The present experiments were performed to determine whether ageing impairs the activity of endothelial adherens junctions and whether this could contribute to the ageing‐induced dysfunction in endothelial dilatation.
Methods
Ethical approval, animals
Young (3–4 months) and old (22–24 months) male F344 rats were obtained from Charles River and their National Institute on Aging colony, and killed by CO2 asphyxiation. Aortas and tail arteries were rapidly removed and placed in cold Krebs–Ringer bicarbonate solution (Flavahan et al. 2016). Animal use was approved by the Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals.
Functional responses
Tail arteries were cannulated with micropipettes, secured within a microvascular chamber (Living Systems, St Albans City, VT, USA), and maintained at a transmural pressure (P TM) of 60 mmHg in the absence of flow (Flavahan et al. 2016). The arteries were superfused with Krebs–Ringer bicarbonate solution (37°C, pH 7.4, 16% O2–5% CO2, balance N2), and the chamber was placed on the stage of an inverted microscope (Flavahan et al. 2016). Arterial diameter was continuously monitored and recorded (Biopac, Santa Barbara, CA, USA) (Flavahan et al. 2016). Concentration–effect curves to vasodilators were obtained during constriction to phenylephrine, and acetylcholine responses were determined in paired arteries with one segment serving as control. When analysing the effects of N ω‐nitro‐l‐arginine methyl ester (l‐NAME; 100 μm) to inhibit NO synthase or saracatinib (5 μm) to inhibit Src kinase (refers to the Src family of tyrosine kinases), arteries were incubated with these drugs for 60–90 min before and during exposure to acetylcholine. To determine the role of VE‐cadherin clustering at adherens junctions on vasodilator responses, the arteries were exposed to intraluminal administration of a function blocking antibody against VE‐cadherin (BV13, 50 μg ml−1; eBioscience, San Diego, CA, USA) or a control antibody (50 μg ml−1; eBioscience) for 2.5–3 h before analysing vasodilator responses (Flavahan et al. 2013). This antibody binds to an N‐terminal sequence in the most proximal extracellular domain (EC1) of VE‐cadherin (Corada et al. 2001; May et al. 2005).
Immunofluorescence
Tail arteries were mounted in specialized ‘flipper’ chambers (Living Systems) that enabled the blood vessel assembly to be rapidly (∼1 s) transferred from control solution to paraformaldehyde (3%, 4°C, 30 min) (Flavahan et al. 2013). Arteries were cut open longitudinally during fixation. Arteries were rinsed in phosphate‐buffered saline (PBS; 3 × 10 min), permeabilized (Triton X‐100, 0.5%, 15 min), rinsed again (PBS, 3 times) and then incubated in donkey serum (1.5%, 15 min) to reduce non‐specific binding. Arteries were incubated overnight with a goat polyclonal antibody to VE‐cadherin (C‐19, 1:500 dilution, Santa Cruz Biotechnology, Dallas, TX, USA). Arteries were rinsed (PBS, 3 × 15 min), then incubated (120 min) with Rhodamine or Alexa Fluor 488‐labelled donkey anti‐goat antibody (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA, USA), and DRAQ5 (5 μm, nuclear stain, Biostatus, Loughborough, UK). Samples were viewed using a (Leica, Buffalo Grove, IL, USA), AOBS‐equipped SP5 laser scanning microscope. Images (1024 × 1024 pixels) were obtained using sequential acquisition, a pinhole of 1 Airy unit, scan speed of 400 Hz, six‐line averaging, an optical zoom of 3.0, and excitation/emission settings for Alexa Fluor 488 (488 nm/492–541 nm), Rhodamine (543 nm/555–620 nm) and DRAQ (633 nm/650–750 nm). For quantitative comparison of fluorescence, young and old arteries or control and treated arteries were processed at the same time using the same instrument settings. Z‐Stacks were obtained for the complete endothelial layer (0.25 micron steps). The width and intensity of adherens junctions was assessed using line profiles of VE‐cadherin fluorescent staining, as previously described (Flavahan et al. 2013). Maximal Z‐stack projections of line profiles were generated by selecting pixels with the maximal intensity from the z‐sections. Peak fluorescence intensities are expressed as absolute detector units (Zhao et al. 2015). The width of adherens junctions was determined as the distance in micrometres that VE‐cadherin intensity was above the immediate background level (Flavahan et al. 2013). This approach involved a non‐selective survey of endothelial junctions (Flavahan et al. 2013), although taking care to avoid gaps in the junctions. This is in contrast to a previous study that purposefully measured the widest part of VE‐cadherin junctional staining with the apparent goal of assessing gaps in the junctions (Huynh et al. 2011).
Arterial lysis
For sequential lysis, rat arteries were first lysed with TTX lysis buffer (50 mm Tris pH 7.6, 150 mm NaCl, 0.5% Triton X‐100, 0.5% deoxycholate, 2 mm EDTA, 20 μg ml−1 leupeptin, 20 μg ml−1 aprotinin, 10 mm NaF, 0.5 mm 4‐benzenesulfonyl fluoride hydrochloride (AEBSF)–HCl, 1 mm sodium orthovanadate), followed by centrifugation at 15 000 g (4°C, 10 min), to obtain the TTX‐soluble lysate (supernatant). The TTX‐insoluble pellets were subsequently lysed with sodium dodecyl sulfate (SDS) lysis buffer (TTX lysis buffer with 2% SDS), boiled for 5 min, and then clarified by centrifugation (15 000 g, room temperature, 10 min) to obtain the TTX‐insoluble SDS‐soluble lysates. Rat arteries were also lysed using a one‐step lysis method, whereby rat arteries were directly lysed with the SDS lysis buffer, boiled for 5 min before centrifugation at 15 000 g at room temperature to obtain the total lysates. Total protein concentrations were measured using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
Cell surface labelling
A Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific) was used to label VE‐cadherin on the endothelial cell surface. Briefly, aortas, which had been opened longitudinally, were incubated with Sulfo‐NHS‐SS‐biotin in cold Krebs–Ringer bicarbonate solution at 4°C for 30 min, according to the manufacturer's protocol. The reaction was stopped by the addition of quenching solution. Arterial lysates were then obtained using the one‐step lysis method. The biotinylated surface proteins were purified with immobilized NeutrAvidin gel followed by elution with sample buffer containing DTT, according to the manufacturer's protocol.
Immunoprecipitation and immunoblotting
The reliability of commercially available antibodies to tyrosine‐phosphorylated VE‐cadherin has been questioned (Orsenigo et al. 2012; Wessel et al. 2014). Therefore, immunoprecipitation was used to assess tyrosine phosphorylation of VE‐cadherin. Briefly, 100–300 μg of total protein in immunoprecipitation buffer (25 mm Tris‐Cl, pH 7.2, 150 mm NaCl, 2 mm EDTA, 20 μg ml−1 leupeptin, 20 μg ml−1 aprotinin, 10 mm NaF, 0.5 mm AEBSF–HCl, 1 mm sodium orthovanadate) was incubated with mouse monoclonal phosphotyrosine antibody (clone 4G10, Millipore, Billerica, MA, USA) (3 h, 4°C), followed by protein A/G agarose beads (Thermo Fisher Scientific) (18 h, 4°C). When using SDS‐solubilized proteins, the lysates was diluted with immunoprecipitation buffer to reduce the SDS concentration to 0.1% (this concentration was confirmed not to affect the immunoprecipitation procedure). Immunoprecipitated proteins were eluted by adding Laemmli sample buffer (Bio‐Rad Laboratories, Hercules, CA, USA) to the beads.
Protein samples were boiled in Laemmli sample buffer for 5 min. For improved visualization of different VE‐cadherin species, agarose (1%)‐acrylamide (4%) composite gels were employed, using a slight modification of a previously published approach (Tatsumi & Hattori, 1995). Briefly, agarose (0.3 g) was completely dissolved in 15 ml of deionized water by heating at 100°C before being cooled to 50°C in a water bath; 4 ml of acrylamide solution (30% acrylamide/Bis solution, 19:1, Bio‐Rad) was added to 7.5 ml of 1.5 m Tris buffer (pH 8.8) and 3.5 ml deionized water with 0.2% SDS, and warmed to 37°C in a water bath. The agarose and acrylamide solutions were then mixed, followed by addition of ammonium persulfate and tetramethylethylenediamine, then poured into gel casting cassettes and allowed to solidify at room temperature for 2 h. Proteins were transferred to nitrocellulose membranes (Bio‐Rad), and incubated with the primary antibodies (18 h, 4°C): rat monoclonal VE‐cadherin antibody (BV13, 1:400, eBioscience), mouse monoclonal claudin‐5 (1:2,000, Thermo Fisher Scientific), rabbit monoclonal Src (1:2000) or rabbit monoclonal phospho‐Src (Tyr416, 1:2000, Cell Signaling Technology, Danvers, MA, USA). After extensive washing, the membranes were incubated with peroxidase‐conjugated secondary antibody (goat‐anti‐rabbit IgG, goat‐anti‐rat IgG, goat‐anti‐mouse IgG) (Jackson ImmunoResearch Laboratories) for 1 h, room temperature. Blots were then developed using enhanced chemiluminescence and quantified using ImageJ.
Drugs
Acetylcholine, l‐NAME and phenylephrine were from Sigma‐Aldrich (St Louis, MO, USA), DEA‐NONOate was from Enzo Life Sciences (Farmingdale, NY, USA), and saracatinib was from Cayman Chemical Co. (Ann Arbor, MI, USA)
Data analysis
Vasomotor responses were expressed as percentage change in baseline diameter. Agonist concentrations causing 50% dilatation of the phenylephrine constriction (EC50) were calculated by regression analysis and compared as log EC50 (Flavahan et al. 2016). Maximum responses were determined as the maximal observed dilatation of the constriction to phenylephrine (Flavahan et al. 2016). For functional and biochemical analyses, data are expressed as means ± SEM, for n animals from which arteries were studied. For image analysis of adherens junctions, data are expressed as means ± SEM for N junctions, assessed using blood vessels from at least three different animals. Statistical evaluation of the data was performed by Student's t test for paired or unpaired observations. When more than two means were compared, analysis of variance was used (Instat 3 software; GraphPad Software, La Jolla, CA, USA). If a significant F value was found, then the Tukey–Kramer test for multiple comparisons was employed to identify differences among groups. Values were considered to be statistically different when P < 0.05.
Results
VE‐cadherin, endothelial adherens junctions and ageing arteries
Biochemical analysis
Endothelial adherens junctions are formed by the clustering and engagement of VE‐cadherin on adjacent endothelial cells. The pattern of VE‐cadherin expression was different between young and old arteries. In young aortas, there was expression of a 140 kDa VE‐cadherin species and a lower molecular mass 115 kDa species (Fig. 1). These species were identified using an antibody directed to the most proximal extracellular domain (EC1) of VE‐cadherin (Fig. 1) (Corada et al. 2001; May et al. 2005) and confirmed using an antibody to EC3/EC4 domains (data not shown). When assessed using a single lysis approach, expression of the 140 kDa VE‐cadherin species was reduced in old compared to young aortas (to 13.4 ± 2.1% of levels in young aortas, n = 5, P < 0.001) and in old compared to young tail arteries (to 18.4 ± 2.2% of levels in young arteries, n = 3, P < 0.001) (Fig. 1 A). In contrast, expression of the 115 kDa VE‐cadherin species was reduced in young compared to old aortas (25.9 ± 9.2% of levels in old aortas, n = 5, P < 0.01) (Fig. 1 A). Expression of the 115 kDa species was reduced in tail arteries compared to aortas, and was generally below detectable levels (Fig. 1 A). Expression of claudin‐5, a transmembrane component of endothelial tight junctions (Harris & Nelson, 2010), was decreased in young compared to old arteries (to 55.1 ± 6.4% of levels in old aortas, n = 3, P < 0.05; to 36.5 ± 8.3% of levels in old tail arteries, n = 3, P < 0.05) (Fig. 1 A).
Figure 1. Immunoblot analysis of VE‐cadherin expression in young (yng) and old arteries.
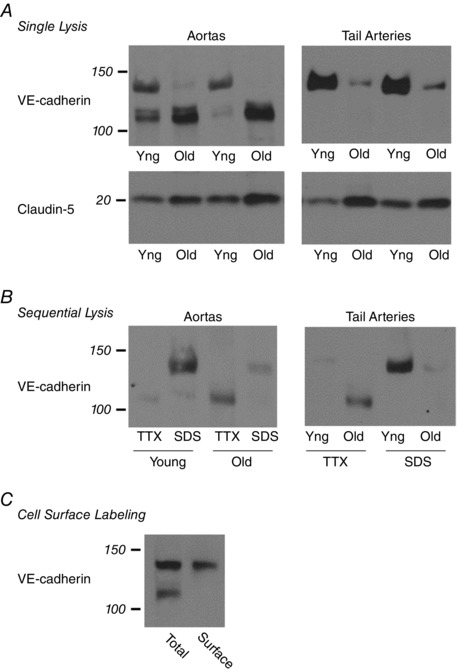
A, aortas and tail arteries were processed using the single lysis approach with SDS‐containing lysis buffer and representative blots are presented. Upper panel, two prominent bands for VE‐cadherin were observed at 140 and 115 kDa: levels of the 140 kDa species were highest in young compared to old aortas and tail arteries, whereas levels of the 115 kDa species were highest in old compared to young aortas. Lower panel, expression of claudin‐5, a transmembrane protein involved in endothelial tight junctions, was higher in old compared to young arteries. B, aortas and tail arteries were processed using sequential lysis in TTX‐containing buffer (TTX‐soluble fraction) followed by SDS‐containing buffer (TTX‐insoluble fraction). Representative blots are presented. The influence of ageing on levels of the 140 and 115 kDa VE‐cadherin species were the same as described in A. In young and old arteries, the 140 kDa species was captured in the TTX‐insoluble fraction (SDS), whereas the 115 kDa species was present in the TTX‐soluble pool (TTX). C, cell surface proteins in young aortas were labelled using Sulfo‐NHS‐SS‐Biotin and captured with immobilized NeutrAvidin gel. A representative blot from three experiments is presented. The 140 kDa but not the 115 kDa species was localized to the cell surface.
In cultured cells, the clustering of VE‐cadherin at adherens junctions is associated with attachment of the junctional complex to the actin cytoskeleton and translocation of VE‐cadherin from the TTX‐soluble to the TTX‐insoluble pool (Lee et al. 1999). When arteries were processed using sequential lysis, the 140 kDa VE‐cadherin species was present predominantly in the TTX‐insoluble fraction, whereas the 115 kDa species was present predominantly in the TTX‐soluble fraction (Fig. 1 B). In young arteries, which had the highest levels of the 140 kDa VE‐cadherin protein, the level of the 140 kDa protein in TTX‐insoluble fractions was 26.8 ± 4.4‐fold higher (aortas, n = 6, P < 0.001) and 30.9 ± 7.6‐fold higher (tail arteries, n = 6, P < 0.001) than levels in TTX‐soluble fractions (Fig. 1 B). In contrast, in old aortas, which had the highest level of the 115 kDa protein, the level of the protein in the TTX‐insoluble fraction was 3.3 ± 0.8‐fold lower compared to the TTX‐soluble fraction (n = 6, P ≤ 0.01) (Fig. 1 B). As observed with the single lysis approach, expression of the 140 kDa VE‐cadherin species was reduced in old compared to young arteries (to 24.1 ± 4.3% of levels in young aortas, n = 6, P < 0.001; to 28.5 ± 4.8% of levels in young tail arteries, n = 6, P < 0.001) (Fig. 1 B)
When cell‐surface proteins were labelled with impermeant and cleavable Sulfo‐NHS‐SS‐Biotin, followed by NeutrAvidin purification and immunoblot analysis of VE‐cadherin, the 140 kDa but not the 115 kDa species was found to be present on the endothelial cell surface (Fig. 1 C).
Immunofluorescence analysis
In young tail arteries, VE‐cadherin immunofluorescence identified continuous adherens junctions delineating the borders of endothelial cells with a width of 1.40 ± 0.02 μm (N = 349) and peak intensity of 3298.7 ± 30.8 (N = 349) (Fig. 2). In ageing tail arteries, the width of adherens junctions was significantly decreased (1.24 ± 0.02 μm, P < 0.001, N = 419) as was the peak intensity (2607.7 ± 29.1, P < 0.001, N = 419), and there were numerous areas of severe thinning and disrupted junctions (Fig. 2).
Figure 2. Endothelial adherens junctions in young (left) and old (right) tail arteries, assessed by immunofluorescence staining of VE‐cadherin (red; DRAQ5 nuclear staining: light blue).
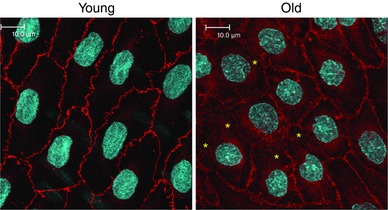
Images are maximal projections of Z‐stacks comprising the entire endothelial layer. The intensity of VE‐cadherin staining at adherens junctions and the width of the junctions were decreased in old compared to young arteries. Indeed, there are numerous areas in old endothelium with severe thinning or disruption of the junctions (*). Fluorescence images are presented in their original unprocessed state. White scale bar is 10 μm.
Role of impaired junctional activity in ageing‐induced endothelial dilator dysfunction
Dilatation to acetylcholine was reduced in old compared to young tail arteries, which was characterized by a decrease in the maximal response and a rightward shift in the concentration–effect curve to the agonist (Fig. 3 and Table 1). Inhibition of NO synthase with l‐NAME (100 μm) suppressed responses to acetylcholine in young and old tail arteries (Fig. 3 and Table 1), and after l‐NAME, the residual dilatation to acetylcholine was greater in old arteries (Fig. 3 and Table 1). Dilatation to the NO donor NONOate was not significantly different between young and old tail arteries (Fig. 3 and Table 1).
Figure 3. Dilatation of young and old rat isolated tail arteries to the endothelial agonist acetylcholine (A) or the NO donor DEA NONOate (B).
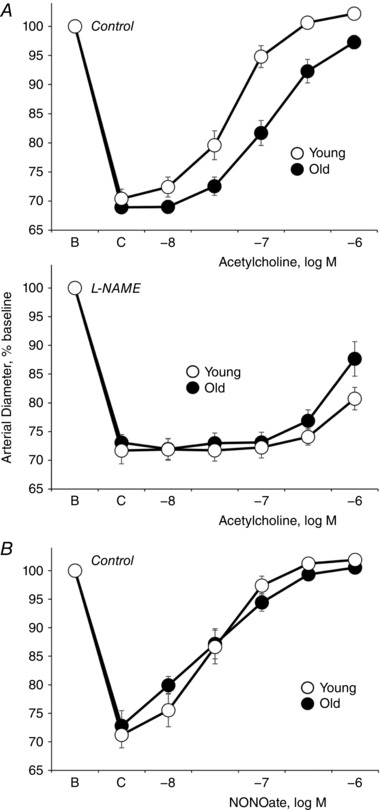
Responses to acetylcholine (A) were assessed under control conditions (upper) and after inhibition of NO synthase with l‐NAME (lower). Dilator responses were analysed in pressurized arteries (P TM of 60 mmHg) during stable constriction to phenylephrine (C). Data are expressed relative to baseline diameter (B) and presented as means ± SEM for A, n = 15 (control old), n = 15 (control young), or n = 8–9 (l‐NAME‐treated arteries); B, n = 6. Ageing decreased dilatation to acetylcholine under control conditions. After l‐NAME, the residual dilatation to acetylcholine (1 μm) was greater in old compared to young arteries. Ageing did not significantly affect dilatation to NONOate. See Table 1 for analyses.
Table 1.
Vasodilatation in young and old rat tail arteries to acetylcholine or DEA‐NONOate
| Treatment | Maximal effect | Log EC50 |
|---|---|---|
| Acetylcholine | ||
| Old, control (15) | 91.6 ± 3.6 ††† | −6.86 ± 0.09††† |
| Young, control (15) | 108.1 ± 2.1 | −7.33 ± 0.06 |
| Old, l‐NAME (9) | 56.1±10.1† | — |
| Young, l‐NAME (8) | 29.3 ± 7.3 | — |
| Old, control (6) | 82.6 ± 7.6† | −6.63 ± 0.14†† |
| Old, saracatinib (6) | 100.7 ± 1.5* | −7.22 ± 0.11*** |
| Young, control (6) | 104.2 ± 2.9 | −7.18 ± 0.08 |
| Young, saracatinib (6) | 102.4 ± 2.7 | −7.11 ± 0.07 |
| Old, control (6) | 98.1 ± 1.0††† | −6.92 ± 0.10†† |
| Old, VE‐cadherin FBA (6) | 92.1 ± 5.1 | −6.86 ± 0.10 |
| Young, control (6) | 111.1 ± 2.1 | −7.28 ± 0.06 |
| Young, VE‐cadherin FBA (6) | 102.4 ± 3.1** | −6.92 ± 0.07*** |
| Old, VE‐cadherin FBA (6) | 90.1 ± 3.0 | −6.73 ± 0.13 |
| Old, VE‐cadherin FBA plus saracatanib (6) | 82.1 ± 7.4 | −6.63 ± 0.17 |
| DEA‐NONOate | ||
| Old (6) | 103.2 ± 2.9 | −7.49 ± 0.15 |
| Young (6) | 106.8 ± 2.4 | −7.54 ± 0.08 |
Data presented as means ± SEM, with number of replicates indicated in parentheses. †Statistically significant difference compared to the corresponding group of young arteries: † P < 0.05, †† P < 0.01, ††† P < 0.001. *Statistically significant difference compared to the matching ‘control’ group: * P < 0.05, ** P < 0.01, *** P < 0.001. For ‘Old VE‐cadherin FBA plus saracatinib’, the matching control group is ‘Old VE‐cadherin FBA’.
To evaluate the role of VE‐cadherin clustering at adherens junctions in the endothelium‐dependent dilatation to acetylcholine, arterial endothelial cells were exposed to a function‐blocking antibody (FBA) to VE‐cadherin (BV13, 50 μg ml−1) or a control antibody (50 μg ml−1) (Flavahan et al. 2013). The FBA inhibits VE‐cadherin clustering and disrupts endothelial adherens junctions (Corada et al. 2001; Flavahan et al. 2013). FBA‐mediated disruption of VE‐cadherin clustering inhibited the dilatation to acetylcholine in young arteries, causing a decrease in the maximal response and a rightward shift in the concentration–effect curve to the agonist, but had no significant effect on old arteries (Fig. 4 and Table 1). After inhibition of VE‐cadherin clustering, there was no longer any significant difference between the dilatation to acetylcholine in young and old arteries (Fig. 4 and Table 1).
Figure 4. Effects of a function blocking antibody to VE‐cadherin (VEC FBA) compared to a control antibody (Control) on dilatation to acetylcholine in young and old rat isolated tail arteries.
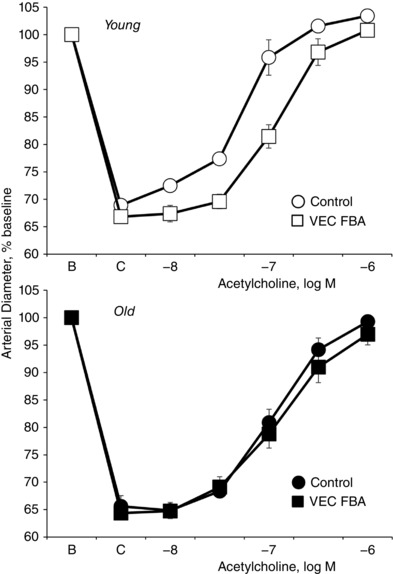
Dilatation was analysed in paired pressurized arteries (P TM of 60 mmHg) during stable constriction to phenylephrine (C). One artery of each pair was treated with a VE‐cadherin function blocking antibody (BV13, 50 μg ml−1) and the other with a control antibody (50 μg ml−1). Data are expressed relative to baseline diameter (B) and presented as means ± SEM for n = 6. The VE‐cadherin FBA inhibited dilatation to acetylcholine in young but not old arteries. Indeed, after the FBA, there was no longer any significant difference in dilatation to acetylcholine between young and old arteries. See Table 1 for analyses.
Role of elevated Src activity in ageing‐induced impairment of endothelial junctions and endothelial dilatation
VE‐cadherin is continually internalized and degraded in cultured endothelial cells, and this process is markedly increased following exposure to inflammatory mediators resulting in impaired activity of adherens junctions (Orsenigo et al. 2012). Internalization of VE‐cadherin is initiated following tyrosine phosphorylation of the protein by Src kinases (Komarova & Malik, 2010; Orsenigo et al. 2012).
Src activity, measured as phosphorylation of Tyr416 relative to total Src, was significantly increased by 2.2 ± 0.5‐fold in old compared to young arteries (n = 10, P < 0.05) (Fig. 5). This was associated with increased levels of tyrosine‐phosphorylated 115 kDa VE‐cadherin in old compared to young arteries (47.0 ± 9.1% and 5.7 ± 2.1% for old and young arteries, respectively, n = 3, P < 0.01, expressed relative to total 115 kDa in old lysates) (Fig. 5). In contrast, there was minimal to no tyrosine phosphorylation of the 140 kDa VE‐cadherin species detected in either old or young arteries (0.2 ± 0.2% and 2.1 ± 1.0% for old and young arteries, respectively, n = 3, NS, expressed relative to total 140 kDa in young lysates) (Fig. 5).
Figure 5. Tyrosine phosphorylation and ageing arteries.
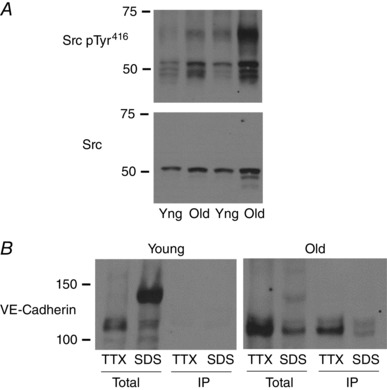
A, Src kinase activity, assessed by immunoblot analysis of total compared to Tyr416 phosphorylation, was increased in old compared to young (yng) tail arteries. Arteries were processed using the single lysis approach and a representative blot is presented. B, tyrosine phosphorylation of VE‐cadherin in old and young aortas was assessed by immunoprecipitation of tyrosine‐phosphorylated proteins followed by immunoblots for VE‐cadherin. This approach was performed on TTX‐soluble (TTX) and TTX‐insoluble (SDS) fractions, obtained using the sequential lysis approach. Representative blots for old and young arteries are presented. Tyrosine phosphorylation was detected in the 115 kDa but not 140 kDa VE‐cadherin species, and levels of tyrosine‐phosphorylated 115 kDa protein were increased in old compared to young aortas.
The selective Src inhibitor saracatinib (5 μm, 60 min) reduced Src activity in old rat tail arteries, measured as phosphorylation of Tyr416 relative to total Src (to 9.6 ± 3.3% of control untreated arteries, n = 4, P < 0.001) (Fig. 6). Saracatinib (5 μm) concomitantly increased the level of the 140 kDa VE‐cadherin species by 61.6 ± 8.4% (n = 4, P < 0.001), increased the intensity of VE‐cadherin staining at adherens junctions (from 2586.3 ± 34.9 to 3255.0 ± 36.4 (P < 0.001), and increased junctional width (from 1.19 ± 0.02 μm to 1.56 ± 0.03 μm, P < 0.001) (n = 351 and 325 junctions in untreated and saracatinib‐treated arteries, respectively) (Figs 6 and 7).
Figure 6. Effect of the Src inhibitor saracatinib on levels of VE‐cadherin (upper panel) and on Src activity (lower panels) in old tail arteries.
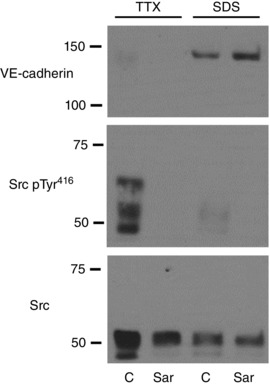
Paired old tail arteries were incubated under control conditions (C) or in the presence of saracatinib (Sar, 5 μm, 90 min, at a P TM of 60 mmHg), and then processed using the sequential lysis approach to obtain TTX‐soluble (TTX) and TTX‐insoluble (SDS) fractions. Representative blots are presented. Saracatinib reduced Src activity, as assessed by immunoblot analysis of Tyr416‐phosphorylated compared to total Src. The Src inhibitor also increased levels of the 140 kDa VE‐cadherin species, which was associated with the TTX‐insoluble fraction.
Figure 7. Effect of the Src inhibitor saractinib on endothelial adherens junctions in old tail arteries, assessed by immunofluorescence staining of VE‐cadherin (green; DRAQ5 nuclear staining: light blue).
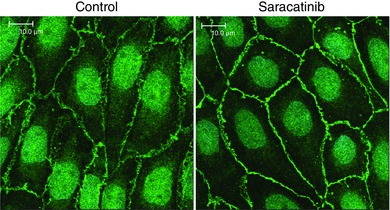
Paired old tail arteries were incubated under control conditions or in the presence of saracatinib (5 μm, 90 min, at a P TM of 60 mmHg) before being processed for staining. Images are maximal projections of Z‐stacks comprising the entire endothelial layer. Saracatinib increased the intensity of VE‐cadherin staining at adherens junctions and the width of the junctions. Fluorescent images are presented in their original unprocessed state. White scale bar is 10 μm.
Saracatinib (5 μm) also increased dilatation to acetylcholine in old arteries, causing an increase in the maximal response and a leftward shift in the concentration–effect curve to the agonist (Fig. 8 and Table 1). However, saracatinib (5 μm) did not significantly affect the response to acetylcholine in young arteries (Fig. 8 and Table 1). Indeed, after saracatinib (5 μm), there was no longer any significant difference between the dilatation to acetylcholine in young and old arteries (Fig. 8 and Table 1). In old arteries treated with the function‐blocking antibody to VE‐cadherin, saracatinib (5 μm) no longer significantly affected the dilatation to acetylcholine (Fig. 8 and Table 1).
Figure 8. Effects of the Src inhibitor saracatinib on dilatation to acetylcholine in young (bottom) and old (top, middle) rat isolated tail arteries.
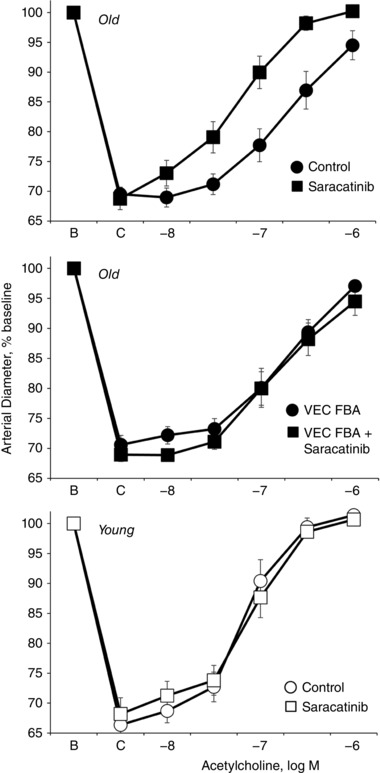
Dilatation was analysed in pressurized arteries (P TM of 60 mmHg) during stable constriction to phenylephrine (C). Data are expressed relative to baseline diameter (B) and presented as means ± SEM for n = 6. Saracatinib (5 μm) increased dilatation to acetylcholine in old (top) but not young (bottom) arteries. After saracatinib, there was no longer any significant difference in dilatation to acetylcholine between young and old arteries. When old arteries were treated with VE‐cadherin FBA (BV13, 50 μg ml−1), which did not affect dilatation in old arteries (see Fig. 4), the Src inhibitor no longer had any significant effect on dilatation to acetylcholine in old arteries (middle). See Table 1 for analyses.
Discussion
Endothelial dysfunction, including impaired endothelial dilatation, is considered a key contributor to the pathological process of vascular ageing (Ungvari et al. 2010; Hasegawa et al. 2012; Laurent, 2012). The present study demonstrates that ageing caused thinning and disruption of arterial endothelial adherens junctions, which was associated with increased phosphorylation of VE‐cadherin, loss of VE‐cadherin from adherens junctions and degradation of the protein in the endothelium of old arteries. Ageing was also associated with increased Src activity, and Src inhibition increased junctional levels of VE‐cadherin and restored the morphology of endothelial adherens junctions in old arteries. These molecular changes in adherens junctions were closely paralleled by changes in the contribution of endothelial adherens junctions to endothelium‐dependent dilatation, which was reduced in old compared to young arteries and was restored by Src inhibition. Impaired activity of arterial endothelial adherens junctions and interruption of VE‐cadherin‐dependent signalling is therefore an important mediator of endothelial dysfunction in ageing arteries, and likely to be a key mediator of vascular ageing.
Two prominent VE‐cadherin species were present in endothelium lining young arteries. The 140 kDa species was present on the cell surface and localized predominantly to the TTX‐insoluble fraction. The clustering of VE‐cadherin at adherens junctions in cultured cells is associated with attachment of the junctional complex to the actin cytoskeleton and movement of VE‐cadherin from the TTX‐soluble to the TTX‐insoluble fraction (Lee et al. 1999). The 140 kDa species is therefore the physiologically important VE‐cadherin species that is localized within adherens junctions of arterial endothelium. In contrast, the 115 kDa VE‐cadherin protein was not present on the cell surface and was localized predominantly to the TTX‐soluble fraction, which is consistent with this species being an internalized and degraded fragment. Although VE‐cadherin can be degraded by extracellular proteases that cleave the protein's extracellular domains (Hermant et al. 2003; Schulz et al. 2008; Sidibe et al. 2012), our immunoblot analyses employed an antibody that targets proximal amino acids of the first extracellular domain (EC1) of VE‐cadherin (Corada et al. 2001; May et al. 2005) and are proximal to cleavage sites of extracellular proteases (Hermant et al. 2003; Schulz et al. 2008; Sidibe et al. 2012). Therefore, the intracellular localization of a 115 kDa species containing this proximal N‐terminal sequence suggests that it was generated by intracellular and not extracellular cleavage of VE‐cadherin. In cultured endothelial cells, VE‐cadherin is internalized and degraded predominantly through a clathrin‐dependent endosomal–lysosomal pathway (Xiao et al. 2003b; Chiasson et al. 2009; Sawant et al. 2011; Orsenigo et al. 2012). Indeed, in cultured cells, the initial degradation of internalized VE‐cadherin generates an N‐terminal VE‐cadherin fragment that is ∼25 kDa smaller than the native protein (Su & Kowalczyk, 2016). This process and the subsequent degradation of VE‐cadherin into smaller fragments can occur rapidly (Xiao et al. 2003a; Orsenigo et al. 2012; Su & Kowalczyk, 2016). Therefore, the increased prominence of the 115 kDa VE‐cadherin species in aorta compared to tail arteries may reflect temporal differences in the degradative pathway of these endothelial cells. Because these VE‐cadherin species were observed in endothelial cells lining young arteries, it indicates that, as in cultured cells, the internalization and degradation of VE‐cadherin is a normal constitutive feature of native arterial endothelium (Xiao et al. 2003a; Orsenigo et al. 2012).
Ageing decreased the levels of the 140 kDa VE‐cadherin species in aortas and tail arteries, and concomitantly increased levels of the 115 kDa species. These results suggest that ageing increases the internalization and degradation of VE‐cadherin, resulting in loss of the physiologically important 140 kDa species from the cells and from adherens junctions. This was confirmed by immunofluorescence imaging of VE‐cadherin, demonstrating that ageing reduced the intensity of VE‐cadherin staining at adherens junctions and reduced the width of endothelial adherens junctions. A decreased junctional width is indicative of reduced clustering of VE‐cadherin and reduced stability and strength of the junctions (Sun et al. 2009; Liu et al. 2010; Flavahan et al. 2013; Breslin et al. 2015). Our results are consistent with previous studies demonstrating reduced endothelial junctional complexity and increased presence of thinning and open endothelial junctions (or gaps) in ageing aortas (Kao et al. 1994; Lee et al. 2001). The results are in apparent contrast to those of Hunyh et al. (2011) who proposed that the width of endothelial adherens junctions was increased in ageing mouse aortas. However, those authors purposefully assessed the widest part of endothelial junctions with the apparent goal of measuring open junctions or gaps in the endothelial monolayer (Huynh et al. 2011). Reduced proteostasis is considered one of the ‘hallmarks of ageing’, reflecting decreased activity of lysosomal and proteasomal pathways in ageing cells (Lopez‐Otin et al. 2013; De Meyer et al. 2015; Nussenzweig et al. 2015). Therefore, the increased internalization and degradation of VE‐cadherin in ageing endothelial cells is unlikely to reflect a generalized increase in proteolytic activity. Indeed, expression of claudin‐5, a transmembrane component of endothelial tight junctions (Harris & Nelson, 2010), was actually increased in ageing arteries, suggesting that VE‐cadherin may be specifically targeted by the ageing process.
The internalization and degradation of VE‐cadherin can be increased by inflammatory mediators and is preceded by a Src‐dependent increase in tyrosine phosphorylation of VE‐cadherin (Komarova & Malik, 2010; Orsenigo et al. 2012). Src phosphorylates VE‐cadherin at multiple sites including Tyr658 and Tyr731, which inhibits the binding of p120 catenin and β‐catenin, respectively, resulting in destabilization of the junctional complex and internalization of VE‐cadherin (Komarova & Malik, 2010; Orsenigo et al. 2012). Indeed, the binding of p120 catenin to VE‐cadherin masks an endocytic motif in VE‐cadherin, preventing internalization and degradation of the protein (Xiao et al. 2003a; Nanes et al. 2012). Src activity was increased in old compared to young arteries, and this was associated with increased levels of tyrosine‐phosphorylated VE‐cadherin (115 kDa) in old arteries. Because of the low levels of the 115 kDa species in tail arteries, the analysis of tyrosine phosphorylation of this species was restricted to aortas. We were unable to detect significant tyrosine phosphorylation of the 140 kDa VE‐cadherin species in aortas or tail arteries, which is likely to reflect rapid internalization and processing of phosphorylated VE‐cadherin (Xiao et al. 2003a; Orsenigo et al. 2012; Su & Kowalczyk, 2016). The Src inhibitor saracatinib reduced the ageing‐induced increase in Src activity and increased the level of the physiologically important 140 kDa VE‐cadherin species in old arteries. Moreover, the increased level of VE‐cadherin following Src inhibition was localized to the TTX‐insoluble fraction suggesting that it increased clustering of VE‐cadherin and strengthened adherens junctions in ageing endothelium. This was confirmed by immunofluorescence imaging of VE‐cadherin, which demonstrated increased intensity and increased width of endothelial adherens junctions following Src inhibition. These results suggest that internalization and degradation of VE‐cadherin, and the impairment of endothelial adherens junctions in old arteries are mediated, at least in part, by heightened Src activity, and that Src inhibition can block these pathological changes and restore endothelial junctional activity.
The clustering of VE‐cadherin at adherens junctions stimulates assembly of a macromolecular signalling complex that regulates endothelial cell signalling, morphology and function, including amplifying phosphoinositide 3‐kinase (PI3K)/Akt signalling (Lampugnani et al. 2002; Lampugnani & Dejana, 2007; Taddei et al. 2008; Komarova & Malik, 2010). Indeed, VE‐cadherin‐dependent signalling at adherens junctions amplifies endothelium‐dependent dilatation to acetylcholine, which was mediated by PI3K/Akt‐dependent activation of endothelial NO synthase (Flavahan et al. 2013; Flavahan & Flavahan, 2014). Ageing is well known to impair endothelium‐dependent, NO‐mediated dilatation (Csiszar et al. 2002; Kim et al. 2009; Trott et al. 2011), which was confirmed in the present study in rat tail arteries. Endothelium‐dependent, l‐NAME‐sensitive dilatation to acetylcholine was reduced in old compared to young arteries, whereas dilatation to an exogenous NO donor was not affected. These results are consistent with ageing‐induced impairment in endothelial NO activity. In tail arteries, an FBA to VE‐cadherin, which inhibits VE‐cadherin clustering and disrupts endothelial adherens junctions (Corada et al. 2001; Flavahan et al. 2013), decreased dilatation to acetylcholine in young arteries, but had no effect in ageing arteries. This confirms that VE‐cadherin signalling at adherens junctions can contribute to endothelial dilatation, but only in young arteries. Indeed, the lack of effect of the FBA in old arteries suggests that the functional role of VE‐cadherin and adherens junctions in endothelial dilatation is impaired by ageing, consistent with impaired endothelial junctions and increased degradation of VE‐cadherin in old arteries. Importantly, the decrease in acetylcholine‐induced dilatation following disruption of adherens junctions by the FBA in young arteries was of the same magnitude as the decreased dilatation associated with ageing‐induced impairment of the junctions in old arteries. Indeed, after disrupting endothelial adherens junctions with the FBA to VE‐cadherin, there was no longer any significant difference in dilatation to acetylcholine between young and old arteries. These data suggest that an ageing‐induced impairment of endothelial adherens junctions can account for the decreased endothelial dilatation in old arteries.
Concomitant with restoring the morphology of ageing endothelial adherens junctions and increasing VE‐cadherin levels in the TTX‐insoluble junctional fraction of old arteries, Src inhibition with saracatinib increased endothelium‐dependent dilatation to acetylcholine in old arteries. Saracatinib had no effect on acetylcholine‐induced dilatation in young arteries, and following Src inhibition there was no longer any difference in acetylcholine‐induced dilatation between young and old arteries. Therefore, Src inhibition completely reversed the ageing‐induced impairment in endothelium‐dependent dilatation. Importantly, treatment of old arteries with an FBA to VE‐cadherin, which itself had no effect in old arteries, prevented the ability of saracatinib to increase endothelial dilatation to acetylcholine in old arteries. Thus, the protective effect of Src inhibition on endothelial dilatation in old arteries was dependent on the clustering of VE‐cadherin at endothelial adherens junctions. The biochemical, morphological and functional data are therefore in agreement and indicate that the endothelial dilator dysfunction in old arteries is mediated by increased Src activity resulting in phosphorylation, internalization and degradation of VE‐cadherin, impairment of endothelial adherens junctions, and loss of the amplifying effect of VE‐cadherin junctional signalling on endothelial dilatation.
Endothelial permeability is increased in aortas and in the microcirculation of ageing animals (Ezekowitz et al. 1980; Belmin et al. 1993; Chen et al. 2003), which would be consistent with impaired function of endothelial adherens junctions (Hordijk et al. 1999; Dejana et al. 2008; Schulte et al. 2011). Interestingly, despite the prominent role of the endosomal–lysosomal pathway in degrading internalized VE‐cadherin, a proteasomal inhibitor prevented the increase in endothelial permeability and disruption of adherens junctions caused by a VE‐cadherin decoy protein, whereas a lysosomal inhibitor was without effect (Sawant et al. 2011). It is unclear if this reflects a role of the proteasome in VE‐cadherin degradation or an indirect effect to reduce VE‐cadherin ubiquitination, a key step in clathrin‐mediated internalization of the protein (Xiao et al. 2003b; Sawant et al. 2011). Although inhibiting VE‐cadherin internalization or degradation pathways may be of benefit in ageing endothelial cells, the approach was not investigated in the present study. The phosphorylation and internalization of VE‐cadherin reduces the strength and stability of adherens junctions, but additional signalling may be required to cause complete dissolution of adherens junctions, leading to the opening of intercellular gaps and increased permeability (Dejana et al. 2008; Orsenigo et al. 2012; Adam et al. 2016). For example, inflammatory mediators may contribute to junctional disruption by increasing endothelial actin–myosin contractility following activation of endothelial myosin light chain kinase by Src or following inhibition of myosin light chain phosphatase by Rho/Rho‐associated protein kinase (ROCK) (Garcia et al. 1999; Komarova & Malik, 2010). Indeed, increased deposition of stiff matrix components such as fibronectin in ageing arterial intima could contribute to junctional disruption by increasing endothelial Rho/ROCK activity (Fujiwara et al. 2004; Huynh et al. 2011; Krishnan et al. 2011). However, junctional dissolution and increased endothelial permeability, e.g. in response to tumour necrosis factor‐α (TNFα), can occur independently of Rho/ROCK signalling or of actin–myosin contractility (Marcos‐Ramiro et al. 2014; Adam et al. 2016). The status of endothelial contractility, stress fibres or Rho/ROCK signalling in the endothelium of ageing arteries is currently unknown. Although tight junctions are present in peripheral endothelial cells, their structure and functional role in regulating permeability is greatly diminished compared to cerebral endothelium and the blood–brain barrier (BBB) (Kniesel & Wolburg, 2000; Harris & Nelson, 2010; Lampugnani, 2012; Bauer et al. 2014). Because of this differential activity and regulation, the maintained expression of claudin‐5 in ageing peripheral endothelial cells, as observed in the present study, does not provide insight into the influence of ageing on cerebral endothelial cells and the BBB. Indeed, as with peripheral endothelial cells, BBB permeability is increased in ageing and may contribute to ageing‐induced declines in cerebral function (Farrall & Wardlaw, 2009).
Impaired activity of endothelial adherens junctions likely contributes to multiple aspects of endothelial dysfunction and arterial deterioration in the ageing vascular system. Ageing arterial endothelial cells are severely compromised: in addition to impaired endothelial dilatation and increased permeability, they are highly susceptible to apoptosis and have a prominent pro‐inflammatory phenotype, including production of pathological mediators such as angiotensin II, TNFα and endothelin‐1 (Belmin et al. 1995; Asai et al. 2000; Csiszar et al. 2004; Lakatta et al. 2009; Goel et al. 2010; Ungvari et al. 2010). These processes are regulated by VE‐cadherin‐dependent signalling within adherens junctions. Indeed, VE‐cadherin activity at adherens junctions is essential for maintaining the protective structure and function of the endothelium, such as promoting signalling that amplifies NO activity, reduces apoptosis, and inhibits inflammatory activity including reducing endothelial permeability and leukocyte extravasation (Dejana et al. 2009; Flavahan et al. 2013; Giannotta et al. 2013; Flavahan, 2017). Internalization and degradation of VE‐cadherin would not only reduce the protective activity of endothelial adherens junctions, but would also release β‐catenin (Valenta et al. 2012). Although free β‐catenin is targeted by a destruction complex, numerous mechanisms enable the protein to escape degradation and translocate to the nucleus (Lampugnani & Dejana, 2007; Beckers et al. 2008; Valenta et al. 2012). β‐Catenin transcriptional activity can increase expression of mediators that are known to play an important local role in vascular ageing, including the renin–angiotensin system, endothelin‐1, TNFα, interleukin 6, inducible nitric oxide synthase, fibronectin, cyclooxygenase 2, matrix metalloproteinase (MMP) 2 and MMP9 (Levy et al. 2002; Kim et al. 2005; Sun et al. 2006; Gelfand et al. 2011; Vikram et al. 2014; Zhou et al. 2015; Zhou & Liu, 2016). Indeed, arterial stiffening, a prominent feature of ageing arteries, reflects the activity of a local angiotensin II signalling system within the endothelium and intimal smooth muscle cells, which promotes inflammatory and fibrotic activity resulting in intimal deposition of poorly distensible proteins, such as fibronectin, and MMP‐mediated degradation of highly distensible elastin fibres (Wang et al. 2005, 2006, 2007, 2010a,b, 2014; Lakatta et al. 2009). Ageing‐induced arterial stiffness, which is also amplified by the ageing‐induced decrease in endothelial NO activity (Santhanam et al. 2010; Jung et al. 2013), disrupts organ blood flow and is a significant predictor of cardiovascular complications and death. The ageing arterial wall is also more susceptible to disease progression, displaying a lower threshold, increased severity and poorer prognosis for vascular disease, including atherosclerosis (Ungvari et al. 2010; Wang et al. 2010b). Impaired activity of endothelial adherens junctions also plays a key role in the initiation and progression of the atherosclerotic process, contributing to a breakdown in the endothelial barrier and enabling the subendothelial accumulation of atherogenic lipids, increased inflammatory activity and increased monocyte transmigration, and contributing to formation and expansion of the atherosclerotic plaque (Flavahan, 2017). Therefore, an ageing‐induced impairment of endothelial adherens junctions would be expected to amplify these pathological processes and contribute importantly to ageing‐induced deterioration of cardiovascular and organ function.
A limitation of the present study is that the mechanisms responsible for the ageing‐induced increase in Src activity were not identified. Ageing endothelial cells are caught in a chronic cycle of inflammatory stress, both producing and responding to pathological mediators (Flavahan et al. 2016). Indeed, the TNFα, angiotensin II and reactive oxygen species (ROS) locally produced by endothelial and intimal smooth muscle cells are considered key mediators of the endothelial pathology of ageing, including endothelial dilator dysfunction (Arenas et al. 2005; Csiszar et al. 2007; Wang et al. 2010a; Flavahan et al. 2016). As demonstrated in the present study, impaired activity of adherens junctions and loss of VE‐cadherin‐dependent signalling contributes to endothelial dilator dysfunction in old arteries. Therefore, these same mediators are likely to be involved in the ageing‐induced impairment of endothelial adherens junctions. Indeed, TNFα, angiotensin II and ROS are each capable of activating Src, increasing tyrosine phosphorylation of VE‐cadherin and disrupting adherens junctions in endothelial cells (Nwariaku et al. 2004; Angelini et al. 2006; Sano et al. 2006; Monaghan‐Benson & Burridge, 2009; Birukova et al. 2013; Dikalov et al. 2014).
The present study demonstrates using biochemical, morphological and functional analyses that endothelial adherens junctions are impaired in ageing arteries, reflecting phosphorylation, internalization and degradation of VE‐cadherin. This pathological process appears to be mediated by increased Src kinase activity, with Src inhibition increasing junctional expression of VE‐cadherin and restoring the morphology of adherens junctions. The impairment in endothelial NO‐mediated dilatation occurring in old arteries can be accounted for by the dysfunction of adherens junctions, reflecting a selective loss in the contribution of adherens junctions to endothelial dilatation that can be restored completely by Src inhibition. Impaired activity of endothelial adherens junctions is likely to contribute to multiple aspects of vascular ageing, including promoting the progression of vascular disease and contributing to organ dysfunction. Interventions designed to augment endothelial adherens junctions should be of therapeutic value in countering vascular ageing.
Additional information
Competing interests
None
Author contributions
The experiments were all performed in the laboratory of N.A.F. at Johns Hopkins University School of Medicine. The authors contributed in the following manner: conception or design of the work (N.A.F., S.F., F.C.); acquisition, analysis or interpretation of data for the work (N.A.F., S.F., F.C.); drafting the work or revising it critically for important intellectual content (N.A.F., S.F., F.C.). All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by an NIH award to N.A.F. (HD078639).
Acknowledgements
We are grateful to the Biological Resources Branch of the NIA for allowing us access to their Aged Rodent Colony.
References
- Adam AP, Lowery AM, Martino N, Alsaffar H & Vincent PA (2016). Src family kinases modulate the loss of endothelial barrier function in response to TNF‐α: crosstalk with p38 signaling. PLoS One 11, e0161975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini DJ, Hyun SW, Grigoryev DN, Garg P, Gong P, Singh IS, Passaniti A, Hasday JD & Goldblum SE (2006). TNF‐α increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol 291, L1232–L1245. [DOI] [PubMed] [Google Scholar]
- Arenas IA, Armstrong SJ, Xu Y & Davidge ST (2005). Chronic tumor necrosis factor‐α inhibition enhances NO modulation of vascular function in estrogen‐deficient rats. Hypertension 46, 76–81. [DOI] [PubMed] [Google Scholar]
- Asai K, Kudej RK, Shen YT, Yang GP, Takagi G, Kudej AB, Geng YJ, Sato N, Nazareno JB, Vatner DE, Natividad F, Bishop SP & Vatner SF (2000). Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler Thromb Vasc Biol 20, 1493–1499. [DOI] [PubMed] [Google Scholar]
- Bauer HC, Krizbai IA, Bauer H & Traweger A (2014). “You Shall Not Pass”—tight junctions of the blood brain barrier. Front Neurosci 8, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers CM, Garcia‐Vallejo JJ, van Hinsbergh VW & van Nieuw Amerongen GP (2008). Nuclear targeting of β‐catenin and p120ctn during thrombin‐induced endothelial barrier dysfunction. Cardiovasc Res 79, 679–688. [DOI] [PubMed] [Google Scholar]
- Belmin J, Bernard C, Corman B, Merval R, Esposito B & Tedgui A (1995). Increased production of tumor necrosis factor and interleukin‐6 by arterial wall of aged rats. Am J Physiol Heart Circ Physiol 268, H2288–H2293. [DOI] [PubMed] [Google Scholar]
- Belmin J, Corman B, Merval R & Tedgui A (1993). Age‐related changes in endothelial permeability and distribution volume of albumin in rat aorta. Am J Physiol Heart Circ Physiol 264, H679–H685. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Starosta V, Tian X, Higginbotham K, Koroniak L, Berliner JA & Birukov KG (2013). Fragmented oxidation products define barrier disruptive endothelial cell response to OxPAPC. Transl Res 161, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin JW, Zhang XE, Worthylake RA & Souza‐Smith FM (2015). Involvement of local lamellipodia in endothelial barrier function. PLoS One 10, e0117970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang F, Flavahan S & Flavahan NA (2016). Immature endothelial cells initiate endothelin‐mediated constriction of newborn arteries. J Physiol 594, 4933–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YM, Zhang JS & Duan XL (2003). Changes of microvascular architecture, ultrastructure and permeability of rat jejunal villi at different ages. World J Gastroenterol 9, 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasson CM, Wittich KB, Vincent PA, Faundez V & Kowalczyk AP (2009). p120‐catenin inhibits VE‐cadherin internalization through a Rho‐independent mechanism. Mol Biol Cell 20, 1970–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, Liao F, Lindgren M, Lampugnani MG, Breviario F, Frank R, Muller WA, Hicklin DJ, Bohlen P & Dejana E (2001). Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood 97, 1679–1684. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z & Ungvari Z (2007). Vasculoprotective effects of anti‐tumor necrosis factor‐alpha treatment in aging. Am J Pathol 170, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A & Kaley G (2002). Aging‐induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res 90, 1159–1166. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Koller A, Edwards JG & Kaley G (2004). Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics 17, 21–30. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F & Lampugnani MG (2008). The role of adherens junctions and VE‐cadherin in the control of vascular permeability. J Cell Sci 121, 2115–2122. [DOI] [PubMed] [Google Scholar]
- Dejana E, Tournier‐Lasserve E & Weinstein BM (2009). The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 16, 209–221. [DOI] [PubMed] [Google Scholar]
- De Meyer GR, Grootaert MO, Michiels CF, Kurdi A, Schrijvers DM & Martinet W (2015). Autophagy in vascular disease. Circ Res 116, 468–479. [DOI] [PubMed] [Google Scholar]
- Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassegue B, Griendling KK, Harrison DG & Dikalova AE (2014). Nox2‐induced production of mitochondrial superoxide in angiotensin II‐mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal 20, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezekowitz MD, Parker KH, Salpidoru N & Oxenham RC (1980). Effect of aging on permeability of cockerel aorta to 125I‐albumin. Am J Physiol Heart Circ Physiol 239, H642–H650. [DOI] [PubMed] [Google Scholar]
- Farrall AJ & Wardlaw JM (2009). Blood‐brain barrier: ageing and microvascular disease–systematic review and meta‐analysis. Neurobiol Aging 30, 337–352. [DOI] [PubMed] [Google Scholar]
- Flavahan NA (2017). In development … a new paradigm for understanding vascular disease. J Cardiovasc Pharmacol 69, 248–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan S, Chang F & Flavahan NA (2016). Local renin‐angiotensin system mediates endothelial dilator dysfunction in aging arteries. Am J Physiol Heart Circ Physiol 311, H849–H854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan S & Flavahan NA (2014). The atypical structure and function of newborn arterial endothelium is mediated by Rho/Rho kinase signaling. Am J Physiol Heart Circ Physiol 307, H628–H632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan S, Mozayan MM, Lindgren I & Flavahan NA (2013). Pressure‐induced maturation of endothelial cells on newborn mouse carotid arteries. Am J Physiol Heart Circ Physiol 305, H321–H329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Gu J & Sekiguchi K (2004). Rac regulates integrin‐mediated endothelial cell adhesion and migration on laminin‐8. Exp Cell Res 292, 67–77. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Verin AD, Schaphorst K, Siddiqui R, Patterson CE, Csortos C & Natarajan V (1999). Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60(src). Am J Physiol Lung Cell Mol Physiol 276, L989–L998. [DOI] [PubMed] [Google Scholar]
- Gelfand BD, Meller J, Pryor AW, Kahn M, Bortz PD, Wamhoff BR & Blackman BR (2011). Hemodynamic activation of β‐catenin and T‐cell‐specific transcription factor signaling in vascular endothelium regulates fibronectin expression. Arterioscler Thromb Vasc Biol 31, 1625–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotta M, Trani M & Dejana E (2013). VE‐cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 26, 441–454. [DOI] [PubMed] [Google Scholar]
- Goel A, Su B, Flavahan S, Lowenstein CJ, Berkowitz DE & Flavahan NA (2010). Increased endothelial exocytosis and generation of endothelin‐1 contributes to constriction of aged arteries. Circ Res 107, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ES & Nelson WJ (2010). VE‐cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol 22, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, Uno K, Gao J, Kaneko K, Shimosawa T, Asano T, Fujita T, Oka Y & Katagiri H (2012). Blockade of the nuclear factor‐κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125, 1122–1133. [DOI] [PubMed] [Google Scholar]
- Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T & Gulino‐Debrac D (2003). Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem 278, 14002–14012. [DOI] [PubMed] [Google Scholar]
- Hordijk PL, Anthony E, Mul FP, Rientsma R, Oomen LC & Roos D (1999). Vascular‐endothelial‐cadherin modulates endothelial monolayer permeability. J Cell Sci 112, 1915–1923. [DOI] [PubMed] [Google Scholar]
- Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB & Reinhart‐King CA (2011). Age‐related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med 3, 112ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SM, Jandu S, Steppan J, Belkin A, An SS, Pak A, Choi EY, Nyhan D, Butlin M, Viegas K, Avolio A, Berkowitz DE & Santhanam L (2013). Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am J Physiol Heart Circ Physiol 305, H803–H810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CH, Chen JK & Yang VC (1994). Ultrastructure and permeability of endothelial cells in branched regions of rat arteries. Atherosclerosis 105, 97–114. [DOI] [PubMed] [Google Scholar]
- Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC & Berkowitz DE (2009). Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol 107, 1249–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Xiong H, Zhang Z & Ren B (2005). β‐Catenin activates the growth factor endothelin‐1 in colon cancer cells. Oncogene 24, 597–604. [DOI] [PubMed] [Google Scholar]
- Kniesel U & Wolburg H (2000). Tight junctions of the blood‐brain barrier. Cell Mol Neurobiol 20, 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y & Malik AB (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72, 463–493. [DOI] [PubMed] [Google Scholar]
- Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VW, Carman CV, Brain JD, Fredberg JJ, Butler JP & van Nieuw Amerongen GP (2011). Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol Cell Physiol 300, C146–C154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG, Wang M & Najjar SS (2009). Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am 93, 583–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani MG (2012). Endothelial cell‐to‐cell junctions: adhesion and signaling in physiology and pathology. Cold Spring Harb Perspect Med 2, a006528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani MG & Dejana E (2007). Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb Res 120 Suppl 2, S1–S6. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Zanetti A, Breviario F, Balconi G, Orsenigo F, Corada M, Spagnuolo R, Betson M, Braga V & Dejana E (2002). VE‐cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell 13, 1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent S (2012). Defining vascular aging and cardiovascular risk. J Hypertens 30 Suppl, S3–S8. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI & Hla T (1999). Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine‐1‐phosphate. Cell 99, 301–312. [DOI] [PubMed] [Google Scholar]
- Lee WC, Chao WT & Yang VC (2001). Effects of high‐cholesterol diet on the interendothelial clefts and the associated junctional complexes in rat aorta. Atherosclerosis 155, 307–312. [DOI] [PubMed] [Google Scholar]
- Levy L, Neuveut C, Renard CA, Charneau P, Branchereau S, Gauthier F, Van Nhieu JT, Cherqui D, Petit‐Bertron AF, Mathieu D & Buendia MA (2002). Transcriptional activation of interleukin‐8 by β‐catenin‐Tcf4. J Biol Chem 277, 42386–42393. [DOI] [PubMed] [Google Scholar]
- Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM & Chen CS (2010). Mechanical tugging force regulates the size of cell‐cell junctions. Proc Natl Acad Sci USA 107, 9944–9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos‐Ramiro B, Garcia‐Weber D & Millan J (2014). TNF‐induced endothelial barrier disruption: beyond actin and Rho. Thromb Haemost 112, 1088–1102. [DOI] [PubMed] [Google Scholar]
- May C, Doody JF, Abdullah R, Balderes P, Xu X, Chen CP, Zhu Z, Shapiro L, Kussie P, Hicklin DJ, Liao F & Bohlen P (2005). Identification of a transiently exposed VE‐cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood 105, 4337–4344. [DOI] [PubMed] [Google Scholar]
- Monaghan‐Benson E & Burridge K (2009). The regulation of vascular endothelial growth factor‐induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem 284, 25602–25611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV & Simons M (2008). The FGF system has a key role in regulating vascular integrity. J Clin Invest 118, 3355–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes BA, Chiasson‐MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA & Kowalczyk AP (2012). p120‐catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol 199, 365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig SC, Verma S & Finkel T (2015). The role of autophagy in vascular biology. Circ Res 116, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwariaku FE, Liu Z, Zhu X, Nahari D, Ingle C, Wu RF, Gu Y, Sarosi G & Terada LS (2004). NADPH oxidase mediates vascular endothelial cadherin phosphorylation and endothelial dysfunction. Blood 104, 3214–3220. [DOI] [PubMed] [Google Scholar]
- Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M & Dejana E (2012). Phosphorylation of VE‐cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 3, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Hosokawa K, Kidoya H & Takakura N (2006). Negative regulation of VEGF‐induced vascular leakage by blockade of angiotensin II type 1 receptor. Arterioscler Thromb Vasc Biol 26, 2673–2680. [DOI] [PubMed] [Google Scholar]
- Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM & Berkowitz DE (2010). Decreased S‐nitrosylation of tissue transglutaminase contributes to age‐related increases in vascular stiffness. Circ Res 107, 117–125. [DOI] [PubMed] [Google Scholar]
- Sawant DA, Tharakan B, Adekanbi A, Hunter FA, Smythe WR & Childs EW (2011). Inhibition of VE‐cadherin proteasomal degradation attenuates microvascular hyperpermeability. Microcirculation 18, 46–55. [DOI] [PubMed] [Google Scholar]
- Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S & Vestweber D (2011). Stabilizing the VE‐cadherin‐catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J 30, 4157–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P & Reiss K (2008). ADAM10 regulates endothelial permeability and T‐Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res 102, 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidibe A, Mannic T, Arboleas M, Subileau M, Gulino‐Debrac D, Bouillet L, Jan M, Vandhuick T, Le Loet X, Vittecoq O & Vilgrain I (2012). Soluble VE‐cadherin in rheumatoid arthritis patients correlates with disease activity: evidence for tumor necrosis factor α‐induced VE‐cadherin cleavage. Arthritis Rheum 64, 77–87. [DOI] [PubMed] [Google Scholar]
- Su W & Kowalczyk AP (2016). The VE‐cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol Biol Cell 28, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Xiong H, Kim TH, Ren B & Zhang Z (2006). Positive inter‐regulation between β‐catenin/T cell factor‐4 signaling and endothelin‐1 signaling potentiates proliferation and survival of prostate cancer cells. Mol Pharmacol 69, 520–531. [DOI] [PubMed] [Google Scholar]
- Sun X, Shikata Y, Wang L, Ohmori K, Watanabe N, Wada J, Shikata K, Birukov KG, Makino H, Jacobson JR, Dudek SM & Garcia JG (2009). Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1‐phosphate‐induced endothelial barrier enhancement. Microvasc Res 77, 304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S & Dejana E (2008). Endothelial adherens junctions control tight junctions by VE‐cadherin‐mediated upregulation of claudin‐5. Nat Cell Biol 10, 923–934. [DOI] [PubMed] [Google Scholar]
- Tatsumi R & Hattori A (1995). Detection of giant myofibrillar proteins connectin and nebulin by electrophoresis in 2% polyacrylamide slab gels strengthened with agarose. Anal Biochem 224, 28–31. [DOI] [PubMed] [Google Scholar]
- Trott DW, Seawright JW, Luttrell MJ & Woodman CR (2011). NAD(P)H oxidase‐derived reactive oxygen species contribute to age‐related impairments of endothelium‐dependent dilation in rat soleus feed arteries. J Appl Physiol 110, 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungvari Z, Kaley G, de Cabo R, Sonntag WE & Csiszar A (2010). Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 65, 1028–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta T, Hausmann G & Basler K (2012). The many faces and functions of β‐catenin. EMBO J 31, 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikram A, Kim YR, Kumar S, Naqvi A, Hoffman TA, Kumar A, Miller FJ Jr, Kim CS & Irani K (2014). Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc‐regulated reactive oxygen species. Arterioscler Thromb Vasc Biol 34, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Jiang L, Monticone RE & Lakatta EG (2014). Proinflammation: the key to arterial aging. Trends Endocrinol Metab 25, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Khazan B & Lakatta EG (2010a). Central arterial aging and angiotensin II signaling. Curr Hypertens Rev 6, 266–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Monticone RE & Lakatta EG (2010b). Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens 19, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhang J, Jiang LQ, Spinetti G, Pintus G, Monticone R, Kolodgie FD, Virmani R & Lakatta EG (2007). Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 50, 219–227. [DOI] [PubMed] [Google Scholar]
- Wang M, Zhang J, Spinetti G, Jiang LQ, Monticone R, Zhao D, Cheng L, Krawczyk M, Talan M, Pintus G & Lakatta EG (2005). Angiotensin II activates matrix metalloproteinase type II and mimics age‐associated carotid arterial remodeling in young rats. Am J Pathol 167, 1429–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, Monticone R & Lakatta EG (2006). Matrix metalloproteinase 2 activation of transforming growth factor‐β1 (TGF‐β1) and TGF‐β1‐type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol 26, 1503–1509. [DOI] [PubMed] [Google Scholar]
- Wessel F, Winderlich M, Holm M, Frye M, Rivera‐Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, Nottebaum AF & Vestweber D (2014). Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE‐cadherin. Nat Immunol 15, 223–230. [DOI] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V & Kowalczyk AP (2003a). Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol 163, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V & Kowalczyk AP (2003b). Mechanisms of VE‐cadherin processing and degradation in microvascular endothelial cells. J Biol Chem 278, 19199–19208. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Flavahan S, Leung SW, Xu A, Vanhoutte PM & Flavahan NA (2015). Elevated pressure causes endothelial dysfunction in mouse carotid arteries by increasing local angiotensin signaling. Am J Physiol Heart Circ Physiol 308, H358–H363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, Hou FF, Kahn M & Liu Y (2015). Multiple genes of the renin‐angiotensin system are novel targets of Wnt/β‐catenin signaling. J Am Soc Nephrol 26, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L & Liu Y (2016). Wnt/β‐catenin signaling and renin‐angiotensin system in chronic kidney disease. Curr Opin Nephrol Hypertens 25, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]