

Abstract
Many cellular stresses activate senescence, a persistent hyporeplicative state characterized in part by expression of the p16INK4a cell cycle inhibitor. Senescent cell production occurs throughout life and plays beneficial roles in a variety of physiological and pathological processes including embryogenesis, wound healing, host immunity and tumor suppression. Meanwhile, the steady accumulation of senescent cells with age also has adverse consequences. These non-proliferating cells occupy key cellular niches and elaborate pro-inflammatory cytokines, contributing to aging-related diseases and morbidity. This model suggests that the abundance of senescent cells in vivo predicts ‘molecular’, as opposed to chronologic, age, and that senescent cell clearance may mitigate aging-associated pathology.
I. INTRODUCTION
‘Cellular senescence’ is a special form of durable cell cycle arrest that serves to prevent cancer in mammals. While cellular senescence has become critical to the scientific underpinning of cancer biology and aging research, the concept has been consistently undervalued since its original description. The discoverer of senescence, Dr. Leonard Hayflick, had early difficulty with peer-review. The 1961 letter rejecting his first manuscript on the topic was signed by no less than Peyton Rous, winner of the 1966 Nobel Prize for his discovery of tumor viruses. Despite the widespread replication of Hayflick’s findings, skeptics would argue for decades that senescence was an in vitro artifact, of little relevance to in vivo biology. However, recent findings have established a clear role for cellular senescence in certain physiological and pathological states. Admittedly, issues related to the definition and heterogeneity of senescent cells in vivo remain (Sharpless and Sherr, 2015), but the confirmed predictions regarding health and disease afforded by our modern understanding of senescence have become too important for even committed skeptics to discount.
Cellular senescence (or merely ‘senescence’) is a stress-induced, durable cell cycle arrest of previously replication-competent cells. While the term initially described the finite proliferative capacity of cultured normal human fibroblasts (Hayflick and Moorhead, 1961), cells with features of senescence were later discovered in vivo, with the number of such cells increasing with age in mammalian species including humans (Dimri et al., 1995; Krishnamurthy et al., 2004; Liu et al., 2009a; Melk et al., 2004). The hallmarks of senescent cells have been reviewed (Campisi, 2013; Childs et al., 2015; Sharpless and Sherr, 2015), but to summarize, senescent cells are characterized by a set of core features including durable growth arrest, expression of anti-proliferative molecules (e.g. p16INK4a), and activation of damage sensing signaling pathways (e.g. p38MAPK and NF-kB) resulting in expression of a number of senescence-associated transcripts (Figure 1). This growth arrest of formerly replication competent cells is often triggered by persistent DNA damage response (DDR) or stress signaling, and executed by constitutive activation of the p16INK4a-RB and/or p53 pathways (Campisi, 2013; Childs et al., 2015; Munoz-Espin and Serrano, 2014). While senescent cells are hyporeplicative, they are metabolically active (Dorr et al., 2013), and often capable of performing functions of the replication-competent cells from which they derive.
Figure 1. Characteristics of cellular senescence.
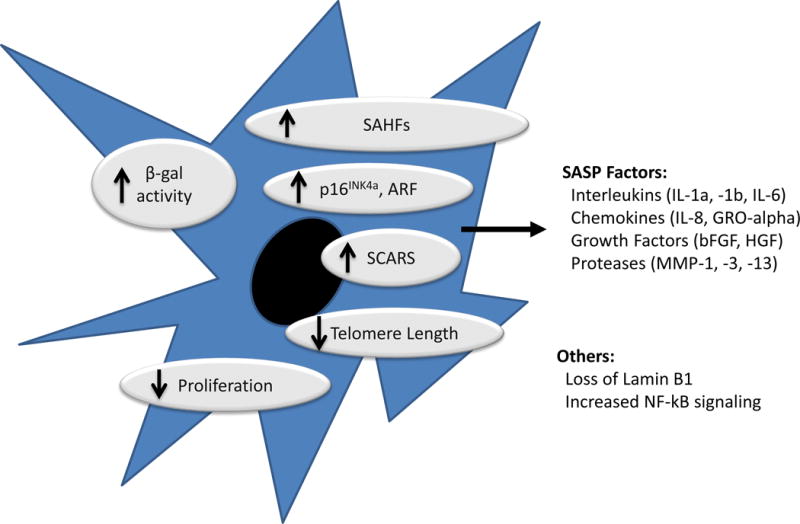
Senescent cells exhibit durable growth arrest, increased expression of the products of the CDKN2a locus (p16INK4a and to a lesser extent ARF) and characteristic changes in cellular structures and protein expressions (e.g. elaboration of SASP factors). Senescent cells in vitro exhibit changes in cellular morphology (e.g. increased cell spreading) and increased SA-β-galactosidase activity, but these markers have been less useful for in vivo recognition. Several other markers (e.g. short telomeres; SCARS; activated NF-kB and DNA damage response; SAHFs) are often associated with cellular senescence, but these markers are neither sensitive nor specific for the senescent state. Loss of the Lamin B1 is an interesting, new marker of senescence that is under investigation.
Activation of the CDKN2a locus, producing p16INK4a and ARF, is observed in most senescent cells, and may play a causal role in their growth arrest. The CDKN2A locus is repressed in most normal tissues, becoming activated at times of tissue damage or cellular stress. In young healthy organisms, therefore, expression of p16INK4a is low or undetectable, but its expression increases exponentially in most tissues with aging (Krishnamurthy et al., 2004; Zindy et al., 1997). This extreme dynamic range and strong association with senescence has made activation of the p16INK4a promoter the preferred genetic tool for in vivo studies (Baker et al., 2011; Burd et al., 2013; Demaria et al., 2014; Yamakoshi et al., 2009).
Senescent cells often exhibit increased lysosomal β-galactosidase activity that is responsible for the characteristic senescence associated β-gal (SA-β-gal) staining near neutral pH, and secrete cytokines with potent effects. This suite of pro-inflammatory cytokines is referred to as the senescence-associated secretory phenotype (SASP) (Coppe et al., 2008). While SASP cytokines may differ from cell type to cell type, their production largely depends on stress-induced NF-kB and p38MAPK signaling (Chien et al., 2011; Freund et al., 2011; Kang et al., 2015), and is regulated by mTOR-dependent protein translation (Herranz et al., 2015; Laberge et al., 2015). Several other features of cellular senescence have been reported (Figure 1), but while of great biological interest, none are reliable for the in vivo recognition of senescent cells. A significant hindrance to the field has been this lack of a ‘magic marker’ of senescence for in vivo identification, but likely no single such marker exists (Sharpless and Sherr, 2015).
Senescence is a distinct form of growth arrest
It is important to differentiate senescence from other forms of cell cycle arrest. While all types of growth arrest are characterized by the presence of active, hypophosphorylated RB-family members, these arrests differ significantly in their durability, associated features, and mechanisms of initiation and/or maintenance (Table 1).
Table 1.
Senescence versus other forms of growth arrest
| Senescence | Quiescence | Exhaustion | Terminal Differentiation | |
|---|---|---|---|---|
| Cell Type | Many if not most replication competent cell (adipose progenitors, lymphocytes, pancreatic beta cells, renal epithelia, keratinocytes, somatic tissue progenitors, etc) | Many if not all dividing cells (somatic stem and progenitor cells, lymphocytes, hepatocytes, renal/pulmonary epithelia, chondrocytes, glia, etc) | T lymphocytes Most somatic SCs |
Many cell types (neurons, cardiomyocytes, osteocytes, etc) |
| Growth Arrest | Near permanent | Reversible | Defective in response to antigenic challenge (T cells) Limited proliferative potential (Somatic SCs) |
Varied by cell type |
| DNA content | 2N or 4N | 2N | 2N | Usually 2N (with some exceptions, e.g. megakaryocytes, striated muscle) |
| Effectors | p16INK4a, p21CIP1, ARF, p53 and RB Short, dysfunctional telomeres p16INK4a expression |
p18INK4c, p21CIP1, p27KIP1, p107, p130, repressive E2Fs | ? | p18INK4c, p21CIP1, p27KIP1, p107, p130, repressive E2Fs |
| Markers | Persistent DNA damage response SASP SAHF SA-β-gal staining Loss of Lamin B1 |
None | Expression of PD-1, TIM-3, LAG-3 (T cells) Varied (Somatic SCs) |
None |
SASP, senescence associated secretory phenotype; SAHF, senescence associated heterochromatin foci; SC, stem cell;
Quiescence
Quiescence is the process where a replication-competent cell undergoes reversible cell cycle arrest in response to mitogen deprivation, contact inhibition, etc. Like senescent cells, quiescent cells are non-dividing and maintain the identity of the previously cycling cell, but the key distinction is that quiescent cells can re-enter the cell cycle when growth conditions are restored.
Terminal differentiation
Terminal differentiation describes the developmentally programmed process through which undifferentiated precursors generate specialized effector cells. While terminal differentiation is usually accompanied by permanent withdrawal from the cell cycle, unlike senescence, cells undergoing terminal differentiation often exhibit functional and morphological changes, losing their original cellular identity. Terminal differentiation is most often developmentally specified, whereas senescence has been thought of as a stochastic response to cellular damage. This latter distinction has become less well demarcated with the appreciation that senescence plays a role in embryogenesis (Munoz-Espin et al., 2013; Storer et al., 2013) whereas exogenous stress can drive somatic stem cells to undergo terminal differentiation (Inomata et al., 2009; Matatall et al., 2016; Wang et al., 2012).
Exhaustion
Exhaustion is a term applied to two distinct types of cellular hypofunction. In T lymphocytes, it refers to a form of growth arrest observed after prolonged antigenic stimulation. This hyporeplicative state differs from senescence by demonstrating increased expression of cell surface markers such as PD1, TIM3 and LAG3 (Akbar and Henson, 2011; Crespo et al., 2013) and by being reversible, for example by blocking PD1 activation. Exhaustion can also be used in the context of somatic stem cells, generally occurring after excessive proliferation as a result of physiological aging or stress (Ruzankina and Brown, 2007). Unlike senescence, exhausted stem cells remain capable of proliferation and self-renewal, but exhibit defective lineage specification and/or reduced ability to generated progeny.
Cellular senescence is a regulated response to stress
Although senescence can occur in many different settings, it generally involves a replication-competent cell receiving a pro-mitogenic signal with simultaneous, persistent anti-proliferative signals (Demidenko and Blagosklonny, 2008; Takahashi et al., 2006). For example, high-level RAS signaling is thought to induce senescence by providing a concomitant MAPK-dependent proliferation signal while also activating the CDKN2a locus via “replication stress” in the same cell. Other signals thought to induce senescence include telomere dysfunction, oxidative stress, changes in chromatin architecture, mitochondria dysfunction, mitotic stress and proteotoxicity (Childs et al., 2015; Munoz-Espin and Serrano, 2014; van Deursen, 2014). It is unclear if these different stresses induce senescence through a common biochemical structure, or via distinct mechanisms.
Senescence is initiated as a response to a cellular insult, leading initially to a transient cell cycle arrest, usually via p53 activation and expression of its downstream mediators (e.g. p21CIP1). If the precipitating stress is transient, senescence usually does not occur, and the cell can resume cycling upon resolution of the stress. In cultured cells, the onset of irreversible senescence requires a prolonged period (>4 days) of stimuli driving the cell both to divide and not to divide (Chen et al., 2002; Dai and Enders, 2000). With stressors that persist, activation of the CDKN2A locus occurs, enforcing a durable growth arrest. Interestingly, in this latter phase, p53 signaling may decline despite persistent DNA damage, combined with decreased p21CIP1 (Stein et al., 1999). Therefore, while p21CIP1 may participate in the initiation of senescence, it is not required for the most important aspect of senescence: the durable nature. This persistence can be further enforced by heterochromatization of E2F targets (Narita et al., 2003), effects of SASP cytokines (Acosta et al., 2013; Acosta et al., 2008; Coppe et al., 2008), self-inflicted DNA damage signaling (Passos et al., 2010), and/or degradation of nuclear lamina and chromatin (Dou et al., 2015; Ivanov et al., 2013). It is worth noting, however, that the absolute durability of this growth arrest in vivo remains controversial. Both genetic and pharmacological approaches have demonstrated cells with a “senescence-like” phenotype that can be reversed by further oncogenic events (Souroullas and Sharpless, 2015).
Recently, it has also become clear that senescent cells actively suppress apoptosis (Childs et al., 2014). For example, navitoclax, an inhibitor of the anti-apoptotic BCL2, BCL-W and BCL-XL proteins, specifically induces apoptosis of senescent cells, and decreases the burden of senescent cells in mice (Chang et al., 2016; Zhu et al., 2015a). This feature of senescent cells has proven key to their targeting for therapeutic intent in vivo.
Kinetics of Senescence Accumulation in Vivo
The rate of senescent cell accumulation is not linear throughout the organismal lifespan, but accelerates with aging, as evidenced by an exponential increase in the expression of some markers of senescence (i.e. telomeric foci, p16INK4a) (Herbig et al., 2006; Liu et al., 2009a). It is thought this does not reflect a global increase in the per cell expression of these markers with aging, but rather an increase in the total number of senescent cells in aging tissues (Helman et al., 2016; Herbig et al., 2006; Wang et al., 2009). The exponential accumulation of senescent cells with age can result from an increase in senescent cell production and/or a decrease in their clearance (Figure 2).
Figure 2. Mechanisms of senescent cell accumulation with aging.
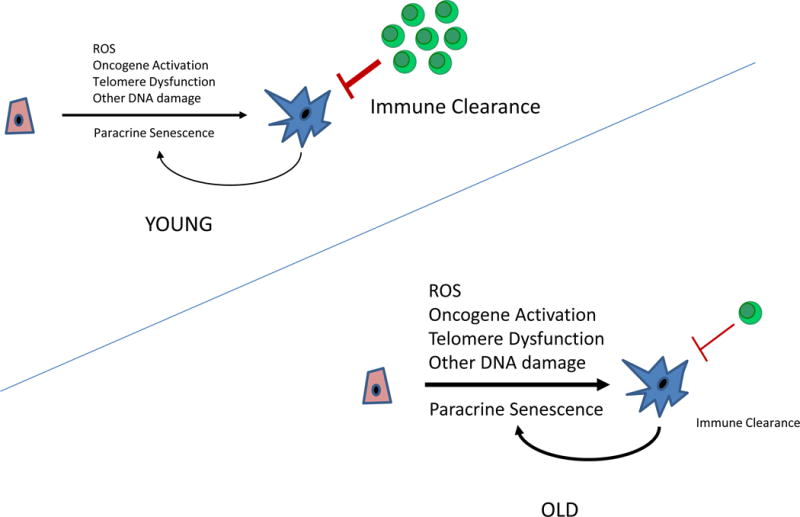
The rate at which senescent cells accumulate increases with aging. This may reflect an increased rate of senescent cell production due to changes in DNA repair, telomere dysfunction and/or decreased senescent cell clearance by immune system. It has also been suggested that senescent cells can induce the formation of other senescent cells in a paracrine manner.
There is evidence that senescent cells can be cleared under physiological conditions. Lowe and colleagues showed that senescent hepatic cells can be efficiently cleared through an immune response (Kang et al., 2011; Xue et al., 2007). Likewise, cells with senescent features that are generated at the site of a UV-induced skin burn (unpublished observation) or a cutaneous wound (Demaria et al., 2014) rapidly disappear after injury, which may reflect immunologic clearance. One hypothesis, therefore, suggests that a reason for the sharp increase in senescent cells with aging reflects a precipitous decline in the clearance of senescent cells in aged organisms, likely reflecting an age-related decline in immune function (Munoz-Espin and Serrano, 2014). While this model has appeal, there are examples of very long-lived senescent cells in vivo. For example, DNA damaging agents increase the burden of senescent cells in young mammals (Chang et al., 2016; Demaria et al., 2017), and the long-term accumulation of radiation-induced senescent cells in the mouse appears independent of immune status (Le et al., 2010). Likewise, young adult women cured of breast cancer through treatment with cytotoxic chemotherapy exhibit increased expression of markers of cellular senescence for decades after chemotherapy (Sanoff et al., 2014). Lastly, cutaneous nevi, where would-be cancer cells undergo oncogene-induced senescence, can persist for an entire adult lifespan. Therefore, while there is evidence for clearance of senescent cells under some settings, there are also examples of senescent cells persisting for decades.
II. ROLES OF SENESCENCE IN HEALTH AND DISEASE
It is important to recognize that the organismal effects of senescence in specific contexts are pleiotropic. Although an imperfect classification, the effects of senescent cells can be thought of as beneficial (Figure 3) or detrimental (Figure 4) with regard to host physiology and disease, although in some contexts, senescent cells affect a disease state in a complex manner: both promoting and opposing certain conditions.
Figure 3. Beneficial roles of cellular senescence.
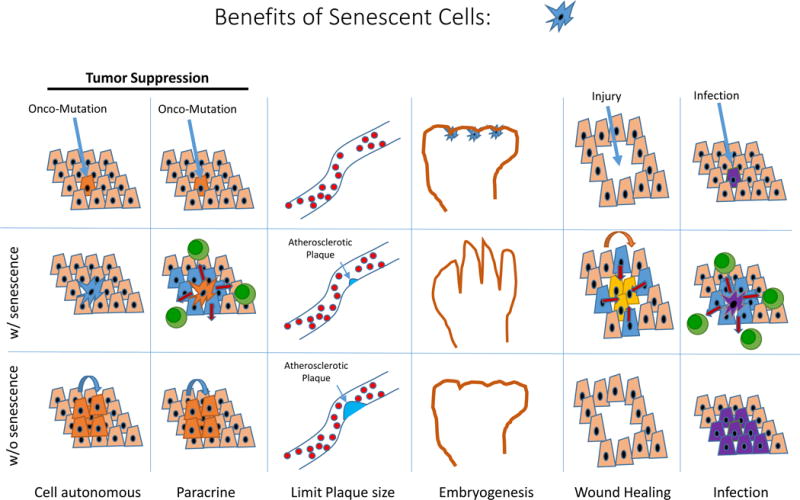
Senescence affords tumor suppression in a cell intrinsic manner, and perhaps also by augmenting local anti-tumor immunity. The activation of the CDKN2a locus appears to limit the size of atherosclerotic plaques, thereby reducing anatomic obstruction. Senescence resulting from p21CIP expression during embryogenesis may be required for certain aspects of fetal development. Senescence contributes to wound healing and host immunity.
Figure 4. Detrimental effects of senescent cells.
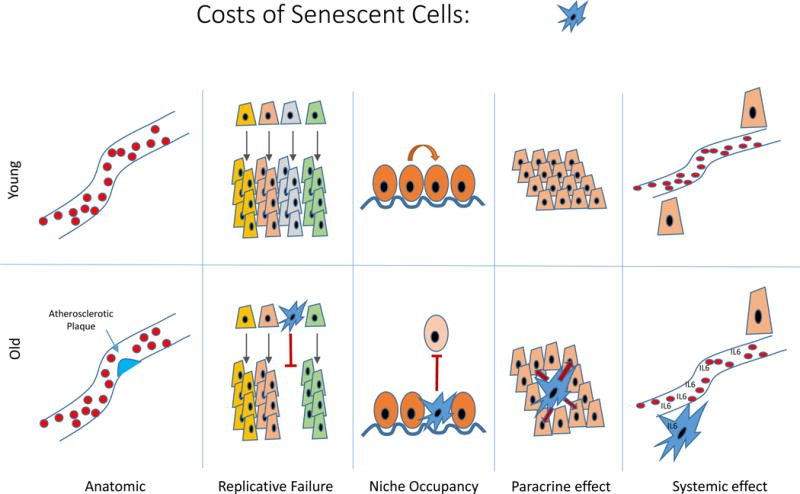
The accumulation of senescent cells can lead to anatomic lesions (e.g. as in an atherosclerotic plaque). The loss of replicative capacity of certain senescent cells (e.g. T cells, pancreatic β-cells) may lead to defects in tissue regeneration. The occupancy of prized physiological niches by senescent cells may impair tissue homeostasis. The paracrine and endocrine elaboration of pro-inflammatory hormones and enzymes promotes tissue dysfunction locally and at the organismal level.
Developmental Senescence
Cells with features of senescence have been identified in a number of transient anatomical structures in the developing embryo, and appear to play a role in shaping organogenesis (Munoz-Espin et al., 2013; Storer et al., 2013). Such cells are hyporeplicative and express SA-β-gal, but are not associated with DNA damage, do not depend on p53 and p16INK4a expression for proliferative arrest and do not secrete the typical range of SASP cytokines. Instead, embryonic senescence is developmentally programmed, induced by paracrine signaling, and mediated by the expression of the cell cycle inhibitor p21CIP1 (Munoz-Espin et al., 2013; Storer et al., 2013).
Wound Healing
Rapid and potent activation of p16INK4a has been observed in various models of wounding in adult animals (Demaria et al., 2014; Jun and Lau, 2010). Using p16INK4a–reporter mice, activation of the p16INK4a promoter is seen within 2–3 days of tissue wounding, peaks between 4 to 7 days, and then resolves over 2–3 weeks (Demaria et al., 2014; Sorrentino et al., 2014a). In addition to p16INK4a expression, cells with other features of senescence -NF-kB activation and expression of SASP cytokines - are observed at sites of wounding, and appear important for optimal healing, as clearance of p16INK4a-expressing cells delays healing (Baker et al., 2016; Demaria et al., 2014). As p16INK4a-deficeint animals do not exhibit defects in wound healing, these observations suggest some feature of p16INK4a-expressing cells, but not p16INK4a itself, aids in tissue remodeling at a wound. The likely candidates for this effect are SASP components, with a particular role identified for PDGF-AA elicited by senescent fibroblasts and endothelial cells (Demaria et al., 2014).
Gudkov and colleagues have recently suggested that some p16INK4a–expressing cells localizing to tissue injury are specialized macrophages recruited by inflammatory cues. Beads that elaborate SASP-components attract macrophages, which show some features of senescence (e.g. SA-β-gal and p16INK4a expression) (Hall et al., 2016). It is likely that some bone marrow-derived cells found in nascent neoplasms or after UV injury are also p16INK4a–expressing macrophages (Burd et al., 2013; Sorrentino et al., 2014a), and these cells may accumulate with aging. It is unclear if these cells are recruited pre-existing senescent macrophages or are induced to senesce by the SASP cytokines; it is also unclear if these cells are indeed “senescent” (i.e. SASP-expressing, long-lived and persistently hyporeplicative) or they merely represent a transient response to injury. It is clear, however, that many non-macrophage cell types (e.g. melanocytes, pancreatic beta-cell, adipocytes, T cells) do undergo senescence as a means to prevent malignant conversion and accumulate with aging.
Tumor suppression
Cellular senescence is best known as a cell-intrinsic mechanism to prevent neoplastic transformation. As the role of senescence in cancer prevention is familiar (Childs et al., 2015; Munoz-Espin and Serrano, 2014; Perez-Mancera et al., 2014; Sherr, 2004), we won’t discuss this function of senescence other than to make one relevant point: cellular senescence is more important than forms of cell death for tumor suppression in mammals. For example, mice with major defects in apoptosis are not markedly predisposed to neoplasia (Lindsten et al., 2000), whereas even subtle perturbations of the senescence machinery dramatically influence cancer susceptibility. Mice with loss of a single copy of Trp53 or p16INK4a are tumor-prone (Donehower et al., 1992; Sharpless et al., 2001); while mice bearing an extra copy of either gene are cancer resistant (Garcia-Cao et al., 2002; Matheu et al., 2004). Genome-wide analyses have demonstrated that loss of p16INK4A and/or p53 function are the most common genetic events in human cancers, with perhaps all cancers harboring events that disrupt senescence (Beroukhim et al., 2010; Kandoth et al., 2013). Even in the case of p53, a protein capable of initiating apoptosis or senescence, its ability to prevent cancer predominantly depends on the induction of senescence (Brady et al., 2011; Liu et al., 2004; Sluss et al., 2004).
Since apoptosis is older in evolutionarily terms, the rise of senescence as the major mammalian tumor suppressor mechanism suggests it has evolutionary advantages over apoptosis, for example in the handling of damaged cells. Unlike apoptosis, which irrevocably removes cells, senescence arrests these cells in a functional but non-dividing state. Apoptosis reduces clonal diversity, which is undesirable in situations where population diversity is essential for proper physiological function. For example, senescent memory T lymphocytes carry essential information of past infections and apoptotic death of these cells results in the loss of that memory. Consistent with this notion, senescent T cells exclusively exhibit a differentiated effector memory phenotype, showing elevated cytotoxicity and enhanced cytokine secretion compared to naïve T cells (Akbar and Henson, 2011). Also, cells censored through apoptosis may need to be compensated by hypertrophy of remaining cells, or replaced through increased proliferation of somatic stem cells. Hypertrophy has been associated with tissue dysfunction in many organs (e.g. heart), while increased stem cell proliferation can deplete their limited replicative potential, leading to stem cell exhaustion (Ruzankina and Brown, 2007). Finally, senescent cells may provide an important local but persistent signal of oncogenic stress, and thereby facilitate cancer cell immune surveillance. This evolutionary preference of senescence over cell death for tumor suppression merits careful consideration given therapeutic efforts to destroy senescent cells (“senolysis”), which in effect seek to convert senescent cells to dead ones.
Host immunity
Senescence plays complex roles on both cellular and innate immunity. T cells in old organisms express high levels of p16INK4a as well as a subset of SASP factors, and T cell senescence with aging or chronic HIV infection has been suggested to lead to a loss of diversity in the T cell repertoire and immune aging (Chou and Effros, 2013). In contrast, given that certain viruses depend on host cell proliferation for viral replication, it has also been postulated that cellular senescence may have evolved as a host anti-viral defense (Reddel, 2010). Viral infection can induce cellular senescence directly via inducing cell fusion (Chuprin et al., 2013) or DNA damage (Martinez et al., 2016), and perhaps indirectly via prolonged cytokine signaling (e.g. interferons and TNFa) with the induction of nearby ‘paracrine’ senescence (Acosta et al., 2013; Coppe et al., 2008). It has been shown that the replication of certain viruses is impaired in senescent cells (Baz-Martinez et al., 2016), and senescent cells can recruit innate immune cells which might prevent the further spreading of a viral infection. To defeat senescence mechanisms, certain viruses produce oncoproteins (e.g. E6/7 (human papilloma virus), E1a/b (adenovirus) and large T antigen (polyomavirus)) to inhibit the RB and/or p53 pathways. Therefore, senescence has been argued to provide a direct barrier to certain types of infection (e.g. by limiting host cell proliferation) and as a means to activate innate immunity, but may compromise cellular immunity with aging or chronic infection.
Atherosclerosis
In addition to the suppression of neoplastic proliferation, senescence may play a role in the suppression of other types of detrimental proliferation. For example, increased expression of the CDKN2a/b (encoding p16INK4a, ARF and p15INK4b) locus has been associated with reduced susceptibility to atherosclerotic vascular disease (Harismendy et al., 2011; Liu et al., 2009b). In fact, regulatory polymorphisms of the CDKN2a/b locus, discovered by genome-wide association studies (GWAS), appear to be the strongest common determinant of atherosclerotic disease in the human genome (Holdt and Teupser, 2012). How increased CDKN2a/b expression might prevent atherosclerosis is not completely understood, although an emerging view is that expression of the locus in intimal macrophages of atherosclerotic plaques limits their proliferation and accumulation, thereby limiting plaque growth (Gizard et al., 2005; Kuo et al., 2011). While CDKN2a activation appears to prevent atherosclerotic plaque formation, it also appears that the accumulation of senescent cells contributes to atherogenesis, as the clearance of p16INK4a-expressing macrophages from atherosclerotic lesions in murine models decreases plaque size (Childs et al., 2016). These results suggest a complex model in which the anti-proliferative component of senescence (e.g. CDKN2a activation) is beneficial whereas other aspects of senescence (e.g. SASP production) are detrimental with regard to atherogenesis.
Type 2 Diabetes
Like atherosclerosis, single-nucleotide polymorphisms (SNPs) near the CDKN2a/b locus have also been associated with type 2 diabetes in large and replicated GWAS (see (Jeck et al., 2012)). Unlike atherosclerosis, a functional effect of the diabetes SNPs on CDKN2a/b expression has not been demonstrated. Given that p16INK4a is highly expressed in the insulin-producing β-cells of the pancreas, and loss of p16INK4a has been associated with enhanced β-cell replication in aging mice (Krishnamurthy et al., 2006), one interpretation of this result has been that individuals with a congenital susceptibility to express increased levels of p16INK4a in β-cells have decreased β-cell replication and an increased risk of type 2 diabetes. Correspondingly, humans harboring hypomorphic alleles of p16INK4a that confer increased risk to melanoma are also associated with an enhanced capacity for pancreatic insulin secretion (Pal et al., 2016). Indeed, recent work has suggested that a very high frequency (>50%) of β-cells from aged humans or mice exhibit features of senescence (Helman et al., 2016). Further complexity to this model, however, has been suggested by the finding that on a per-cell basis, senescent β-cells secrete considerably more insulin than non-senescent β-cells, and islets from old mice have increased glucose-stimulated insulin secretion compared to young islets. To make matters even more confusing, it has also been suggested that increased Cdkn2a expression might increase peripheral sensitivity to insulin through an effect on muscle or liver (Gonzalez-Navarro et al., 2013). Therefore, senescence may influence diabetes predisposition by an effect on β-cells, as well as effects on tissue insulin resistance.
Fatigue
A recent development relates to chemotherapy-induced fatigue (asthenia), an important and often durable side effect of treatment for cancer. Campisi and colleagues studied the role of senescent cells in the toxicities of cytotoxic chemotherapy by inducing senescence in murine tissues using a variety of DNA damaging chemotherapy agents. When these senescent cells were cleared, several side effects of chemotherapy were ameliorated. In particular, animals that had undergone senolysis were less fatigued, recovering activity levels to that of untreated mice (Demaria et al., 2017). In accord with these murine findings, women with breast cancer and higher levels of a peripheral blood marker of senescence prior to chemotherapy were 9 times more likely to experience chemotherapy-induced asthenia than women demonstrating low levels of this senescence marker (Demaria et al., 2017). Although older patients are at greater risk for asthenia, the predictive effect of the senescence biomarker in this cohort was independent of patient age. These results suggest that an increased total body burden of senescent cells may cause fatigue.
Other Age-Related Disease
Beyond the aforementioned, senescence has been argued to contribute to other age-related phenotypes. This includes disparate associations with hair graying, sarcopenia, adiposity, neurogenesis, fibrosis and glaucoma (reviewed in (Campisi, 2013; Childs et al., 2015; Jeck et al., 2012; Munoz-Espin and Serrano, 2014; van Deursen, 2014). The evidence linking senescence to these diverse pathologies generally consists of the following:
Senescence markers accumulate in relevant tissues with aging. Expression of several senescence markers (e.g. p16INK4a, dysfunctional telomeres, SASP cytokines) increase markedly with aging in cells linked to these pathologies. For example, loss of neural or muscle stem cell function with aging is thought to contribute to reduced neurogenesis and sarcopenia respectively, and such cells strongly express p16INK4a with aging (Molofsky et al., 2006; Sousa-Victor et al., 2014).
Expression of certain senescence regulators cause some aspects of aging. It has been shown that age-related increase of p16INK4a expression in murine T cells, hematopoietic progenitor cells, neural stem cell, pancreatic β-cells and muscle stem cells causes functional declines of these cells with aging (Janzen et al., 2006; Krishnamurthy et al., 2006; Liu et al., 2011; Molofsky et al., 2006; Signer et al., 2008; Sousa-Victor et al., 2014). Likewise, excess activation of p53 has been suggested to contribute to a variety of progeroid phenotypes in murine models (Maier et al., 2004; Tyner et al., 2002).
Maneuvers to modulate the abundance of senescent cells affect age-related phenotypes. A few means to accelerate or retard the production of senescent cells have been described in vivo. For example, ‘gerontogenic’ exposures such as tobacco use or cytotoxic chemotherapy appear to augment the production of senescent cells in humans or mice (Liu et al., 2009a; Sanoff et al., 2014; Sorrentino et al., 2014a). Likewise, a few groups have shown that certain growth factors or signaling molecules can delay the expression of senescence markers with aging, and thereby provide a functional resistance to chronological aging (Berent-Maoz et al., 2012; Chen et al., 2011). Perhaps some of the strongest evidence linking senescent to aging pathology comes from experiments demonstrating that the “senolytic” clearance of senescent cells can reduce age-related phenotypes.
These data notwithstanding, there has been skepticism as to whether senescent cells could really wreak such havoc in an old organism. Specifically, senescent cells are challenging to identify in vivo (Sharpless and Sherr, 2015), and disparate studies have suggested that at most only a small percentage of replication-competent cells are senescent in a given tissue of an elderly person. Given that the vast majority of cells in tissue from an aged host would remain functional, could such a small fraction of cells undergoing senescence cause disease? We believe it could given the mechanisms by which senescent cells can effect pathology (Figure 4). While not all tissues exhibit high frequencies of senescent cells, even if senescence occurred in only 1-5% of cells with advanced age in the tissues that do demonstrate substantial senescence (e.g. T cells, β-cells, adipose, kidney, etc.), this would be a massive number of dysfunctional cells. Consider that even very small tumors can produce dramatic paraneoplastic syndromes via the production of specific cytokines, and it seems clear that a much larger number of senescent cells could exert a substantial effect on host physiology. Moreover, if the small number of cells that undergo senescence are of a particularly important fraction of cells, then the loss of function of these cells could have an outsized effect on tissue integrity and function. For example, early lymphoid progenitors comprise a rare minority of hematopoietic precursors, and appear to lose replicative function with aging in a CDKN2a-dependent manner (Berent-Maoz et al., 2012; Signer et al., 2008), but the loss of this miniscule subset of cells may have prodigious effects on the entire adaptive immune system.
III. TRANSLATING SENESCENCE
The association of senescence with disease states suggests several means to translate these findings for therapeutic benefit. First, since humans are genetically heterogeneous with regard to the regulation of senescence, humans with different ‘set-points’ to activate senescence should differ in their susceptibility to diseases of aging. Second, if senescent cells cause aging-related pathology, then the measurement of senescent cells in an individual should provide information independent of chronological age related to that individual’s risk for age-related disease. Finally, if senescent cells are harmful, than the clearance of senescent cells should produce benefits.
Set points
It is non-controversial that key mediators of senescence (e.g. telomerase, p16INK4a, Rb, p53) play a critical role in human susceptibility to a common disease of aging: cancer. While null alleles of these senescence mediators in the heterozygous state yield high-penetrance cancer syndromes, it is now clear that even subtle perturbations of the senescence machinery influences human cancer risk. For example, GWAS and candidate analysis have identified non-coding regulatory polymorphisms near the CDKN2a locus that affect the lifetime risk of melanoma, pancreatic cancer, leukemia and glioma. Therefore, humans with differing set-points to activate the senescence machinery differ in their susceptibility to neoplastic disease.
Beyond cancer, however, there are also strong unbiased genome-wide data linking senescence to a wide range of other common age-associated human diseases. This topic has been reviewed (Childs et al., 2015; Jeck et al., 2012; Munoz-Espin and Serrano, 2014), but in brief, CDKN2a/b has been associated through GWAS with various types of atherosclerotic disease (myocardial infarction, stroke, aortic aneurysm), type 2 diabetes, glaucoma and endometriosis. Likewise, TERT, the regulatory subunit of telomerase, has been associated with several cancers and pulmonary fibrosis, a common feature of pulmonary aging. These results are supported by examinations of non-malignant disease in individuals with congenital alterations in senescence regulation. For example, telomerase-deficient humans demonstrate an excess incidence of some age-related phenotypes: pulmonary fibrosis, emphysema, graying, bone marrow hypoplasia and cirrhosis (Armanios and Blackburn, 2012); suggesting the premature activation of senescence in such individuals causes some aspects of aging.
Estimating Risk of Age-Associated Disease
A robust and precise theory of aging should provide a means to measure age, and the notion that aspects of aging are caused by the accumulation of senescent cells has provided hope in this regard. Specifically, we believe an organism’s ‘molecular age’ can be estimated by measuring the total-body burden of senescent cells. This notion of molecular age should in turn prove ‘useful’ in some clinical sense; that is, knowing an organism’s molecular age should predict its susceptibility to certain disease or likelihood of recovering from a given insult.
While work in model organisms suggests this association is valid, the measurement of senescence in humans has proven challenging. There have been long-standing efforts related to two potential markers of senescence—the elaboration of cytokines (e.g. of IL6, a canonical SASP component) and telomere dysfunction—and the merits and faults of these approaches are reviewed elsewhere (Matjusaitis et al., 2016; Sharpless and Sherr, 2015; Sorrentino et al., 2014b). More recent work suggests that measurement of expression of the CDKN2a locus may be useful in this regard. One area of exploration has been renal transplantation, where an important problem is that organs from older donors generally function less well when transplanted. Since there is a shortage of donor kidneys, there is interest in using kidneys from older individuals, and therefore markers of physiological age have been of keen interest. A few groups have studied the utility of p16INK4a expression in this setting, looking at p16INK4a mRNA level in ‘time zero’ biopsies of human kidneys at the time of their harvest for transplantation. Increased expression of p16INK4a in renal biopsies was found to be a strong predictor of long-term graft function, providing greater predictive capacity than the chronological age of the donor or other molecular biomarkers (Sturmlechner et al., 2017).
While p16INK4a expression is a marker of senescence in many human tissues, most p16INK4a–expressing tissues are difficult to sample for routine purposes. In the peripheral blood, only lymphocytes, especially T cells, express significant levels of p16INK4a with aging (Liu et al., 2009a). Therefore, in order to determine senescence in a non-invasive manner on large numbers of individuals at low cost, we have relied on p16INK4a determination in peripheral blood T lymphocytes (PBTL) (Liu et al., 2009a; Rosko et al., 2015; Sanoff et al., 2014). Age-promoting behaviors (tobacco use, physical inactivity) accelerate the rate at which p16INK4a–expressing T cells accumulate with aging. Likewise, exposure to cytotoxic chemotherapy, bone marrow transplantation or chronic HIV infection all appear to markedly and durably increase p16INK4a expression. This effect can be strong; for example, a few months of adjuvant chemotherapy for breast cancer on average increases T cell p16INK4a to an equivalent amount as 15 years of chronological aging (Sanoff et al., 2014). As mentioned, PBTL p16INK4a expression prior to therapy appears to predict the risk of asthenia in breast cancer patients treated with chemotherapy (Demaria et al., 2017) and ongoing large clinical trials are exploring the utility of this marker of molecular age in other clinical settings. In addition to markers of senescence, other promising biomarkers of molecular age have been proposed such as changes in DNA methylation (Hannum et al., 2013; Horvath, 2013), which strongly correlate with chronological age. It is currently unclear if alterations in DNA methylation carry related or independent information to markers of senescence with regard to physiological age.
Clearing senescent cells to treat aging-related pathologies
In 2011, Van Deursen and colleagues employed a genetic approach to study the benefits of eliminating senescent cells from progeroid mice (Baker et al., 2011), followed by similar experiments in normally aged mice (Baker et al., 2016). These experiments employed a transgenic strain that expressed an inducible dimerizable caspase 8 under the control of a fragment of the p16INK4a promoter. In this strain, p16INK4a-expressing cells were selectively killed by the systemic administration of a small molecule. Using this model, the authors showed strong effects of senolysis in old animals, which have been confirmed employing similar genetic strains (Chang et al., 2016), as well as through pharmacological approaches (Chang et al., 2016; Roos et al., 2016; Zhu et al., 2015a; Zhu et al., 2015b).
To date, the clearance of senescent cells has been suggested to benefit several areas of pathology. For example, glomerulosclerosis and decline in renal function in aged mice are rescued by clearance of p16INK4a -expressing senescent tubular brush-border epithelial cells, whereas senolysis of ciliated epithelial cells and fibroblasts in the pericardium reduces age-related cardiomyocyte hypertrophy and improves cardiac stress tolerance (Baker et al., 2016). Likewise, navitoclax-mediated senolysis ameliorates the aging-related functional decline of hematopoietic and skeletal muscle stem cells (Chang et al., 2016), and clearance of senescent intimal foam cells attenuates the pathologies of atherosclerosis at all stages of pathogenesis (Childs et al., 2016). Senescent cell clearance also delays the onset of multiple types of cancer and extends the murine life span (Baker et al., 2016).
These observations suggest several questions. First, if there is physiological clearance of senescent cells in healthy organisms, can this natural senolysis be beneficially augmented? It is thought the clearance of senescent cells can result from the activity of both adaptive and innate immunity; the latter resulting from the increased expression of ligands (e.g. NKG2D) on senescent cells that target them for selective killing by natural killer (NK) lymphocytes (Sagiv et al., 2016). Therefore, mechanisms to augment cellular immunity or NK function might be therapeutically beneficial in some settings.
Additionally, can senolysis be accomplished for therapeutic purposes in humans? In some ways, this question is reminiscent of a fundamental issue in cancer research: can one identify a target that can be inhibited in an undesirable target cell without toxicity to healthy cells. A key difference between cancer therapy and senolysis, however, is that senescent cells are not genetically unstable, and therefore resistance to senolytics may not develop with the same rapidity seen with many cancer therapeutics. The finding of small molecules that accomplish clearance of senescent cells with therapeutic benefit in mice suggests such human agents may be within reach.
A Path to Senolysis in Humans
To date, the most promising targets of would-be senolytics seem to be inhibitors of pro-survival BCL-proteins, given the need of senescent cells to suppress apoptosis to survive (Chang et al., 2016; Yosef et al., 2016). This class of agents had undergone extensive testing in humans with chronic leukemia, leading to the FDA approval of venetoclax, a selective BCL2 inhibitor. Venetoclax is not a potent senolytic in vitro, whereas the related molecule, navitoclax, is one of the strongest senolytics described. Navitoclax inhibits, BCL-2, BCL-XL and BCL-W, suggesting senolysis requires inhibition of a broader spectrum of anti-apoptotic effectors than BCL-2 alone. Lamentably, navitoclax use in humans has been limited by on-target thrombocytopenia (Vogler et al., 2011), and therefore the use of this agent for systemic senolysis would not be straightforward. It is possible, however, that broad-spectrum BCL-proteins inhibitor could be adapted for senolysis in humans with acceptable toxicity through new approaches in formulation, delivery (e.g. intra-articular, inhalation) or administration schedule.
It is worth discussing how a senolytic might be developed clinically. Even if one were to stipulate that a good agent to effect senolysis can be developed, the clinical development path of such a compound is not simple. First, any such agent would require a marker of in vivo effect (i.e. a pharmacodynamic assay), and given the challenges of defining senescent cells in vivo, this is not a trivial concern. Additionally, the initial indication for a senolytic is unclear. Since such agents might induce toxicity, their use for conditions of minimal morbidity would be challenging. The ideal condition would be highly symptomatic, common and where the use of senolytics would pose the least risk. Finally, what might be the toxicity of a successful senolytic? Even if such an agent had few off-target effects, the on-target toxicity could be formidable. The cancer experience has taught us that the acute toxicity of simultaneously clearing a burden of dysfunctional cells can be severe. For example, ‘tumor lysis syndrome’ is a feared complication of effective cancer therapies, where the rapid, simultaneous death of cancer cells causes renal injury and metabolic abnormalities. Additionally, since senescent cells appear to play a role in wound healing, it is possible that acute senolysis could impair wound healing in some tissues. This type of toxicity is already observed in humans treated with anti-angiogenic agents or high-dose corticosteroids, and that experience has suggested that impaired wound healing, especially in older patients, can be significant. Furthermore, a theoretical concern relates to the evolutionary rationale for senescence. If a key function of senescent cells is to carry on functions of the normal, non-senescent tissue, their sudden clearance could adversely affect organ function. For example, Ben Porath and colleagues have shown that a high fraction (>50%) of pancreatic β-cells express p16INK4a and other markers of senescence, while remaining functional (i.e. secreting insulin) (Helman et al., 2016). If a senolytic led to the death of these cells, such a rapid loss of β-cell mass could lead to significant defects in glucose handling. Lastly, if a function of senescent cells is to occupy certain niches thereby decreasing the need for somatic stem cell proliferation, then the clearance of senescent cells might impose a proliferative burden on somatic stem cells to fill unoccupied niches post-senolysis. Excess stem cell proliferation, however, can lead to stem cell exhaustion, and is clearly oncogenic. Despite these concerns, we believe therapeutic trials of senolytics are possible, especially if the initial focus is on conditions that are highly morbid, and by prioritizing local as opposed to systemic administration. It is further reassuring that transgene-mediated senolysis in mice affords benefits without significantly affecting parenchymal tissue function, suggesting senescent cells may not be key for maintaining essential tissue function (Baker et al., 2016)
IV. CONCLUSIONS AND FUTURE DIRECTIONS
Cellular senescence provides clear benefits to the host manifesting as a reduced incidence of cancer and certain other diseases characterized by excess proliferation, as well as enhanced wound healing. However, the accumulation of senescent cells with aging causes host toxicity, manifesting as a decreased ability to recover from insult and increased susceptibility to certain age-associated diseases.
This model makes important predictions. In particular, it suggests an explanation for the linkage, through GWAS, of the CDKN2a/b and TERT loci to a variety of important human diseases, particularly those associated with aging. Additionally, the ability to measure the burden of senescent cells in vivo could provide a measure of ‘molecular age’, making it possible to predict which organisms of a given chronologic age are most likely develop certain types of pathology. Also, it would provide a means to understand if environmental exposures are age-promoting (i.e. “gerontogenic”, like inhaled tobacco (Sorrentino et al., 2014a)) or anti-aging (like exercise). Finally, the hope of addressing certain common disease states through the controlled clearance of senescent cells holds particular translational appeal. Senolysis provides an alternative to the traditional prevention paradigm of ‘anti-aging therapy’ - life-style or pharmacological maneuvers to slow the onset of age-induced decrepitude. The prevention paradigm, however, poses substantial problems from a clinical investigation point of view (e.g. long trial times and intolerance of toxicity). Therefore, it is exciting that this evolving notion of the role of senescent cells in human disease suggests a therapeutic approach to age-related pathologies: pharmacologic or immunological means to clear accumulated senescent cells. Such therapeutic trials are simpler from a clinical trials point of view, yet provide hope for a means to revert the toxicities that result from the accumulation of an overwhelming burden of senescent cells during a human lifespan.
Acknowledgments
This work has been supported by grants from the Glenn Foundation, the Ellison Medical Foundation, the Burroughs Wellcome Foundation, the NCI and the NIA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COI: NES is an inventor on patents related to this work. He is a co-founder and board member of HealthSpan Diagnostics, in which he holds equity. He is a consultant to Unity Biotechnology, in which he holds equity.
References
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nature cell biology. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11:289–295. doi: 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH. The telomere syndromes. Nature reviews Genetics. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, R AS, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring P16-positive cells shorten healthy lifespan. Nature. 2016 doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baz-Martinez M, Da Silva-Alvarez S, Rodriguez E, Guerra J, El Motiam A, Vidal A, Garcia-Caballero T, Gonzalez-Barcia M, Sanchez L, Munoz-Fontela C, et al. Cell senescence is an antiviral defense mechanism. Scientific reports. 2016;6:37007. doi: 10.1038/srep37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berent-Maoz B, Montecino-Rodriguez E, Signer RA, Dorshkind K. Fibroblast growth factor-7 partially reverses murine thymocyte progenitor aging by repression of Ink4a. Blood. 2012;119:5715–5721. doi: 10.1182/blood-2011-12-400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–351. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annual review of physiology. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature medicine. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang W, Gao YF, Su XQ, Zhai ZH. Senescence-like changes induced by expression of p21(waf1/Cip1) in NIH3T3 cell line. Cell research. 2002;12:229–233. doi: 10.1038/sj.cr.7290129. [DOI] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes & development. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO reports. 2014;15:1139–1153. doi: 10.15252/embr.201439245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nature medicine. 2015;21:1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JP, Effros RB. T cell replicative senescence in human aging. Current pharmaceutical design. 2013;19:1680–1698. doi: 10.2174/138161213805219711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes & development. 2013;27:2356–2366. doi: 10.1101/gad.227512.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS biology. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current opinion in immunology. 2013;25:214–221. doi: 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CY, Enders GH. p16 INK4a can initiate an autonomous senescence program. Oncogene. 2000;19:1613–1622. doi: 10.1038/sj.onc.1203438. [DOI] [PubMed] [Google Scholar]
- Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer discovery. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Developmental cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell cycle. 2008;7:3355–3361. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee XH, Basile G, Acosta M, Scott C, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereirasmith O, et al. A Biomarker That Identifies Senescent Human-Cells in Culture and in Aging Skin in-Vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501:421–425. doi: 10.1038/nature12437. [DOI] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–109. doi: 10.1038/nature15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. The EMBO journal. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, Serrano M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. The EMBO journal. 2002;21:6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, Sevestre H, Krimpenfort P, Corsini A, Rochette J, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. The Journal of clinical investigation. 2005;115:3228–3238. doi: 10.1172/JCI22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navarro H, Vinue A, Sanz MJ, Delgado M, Pozo MA, Serrano M, Burks DJ, Andres V. Increased dosage of Ink4/Arf protects against glucose intolerance and insulin resistance associated with aging. Aging cell. 2013;12:102–111. doi: 10.1111/acel.12023. [DOI] [PubMed] [Google Scholar]
- Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, et al. Aging of mice is associated with P16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging. 2016 doi: 10.18632/aging.100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Experimental cell research. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, et al. P16-induced senescence of pancreatic beta cells enhances insulin secretion. Nature medicine. 2016 doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nature cell biology. 2015;17:1205–1217. doi: 10.1038/ncb3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:196–206. doi: 10.1161/ATVBAHA.111.232678. [DOI] [PubMed] [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome biology. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inomata K, Aoto T, Binh NT, Okamoto N, Tanimura S, Wakayama T, Iseki S, Hara E, Masunaga T, Shimizu H, et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 2009;137:1088–1099. doi: 10.1016/j.cell.2009.03.037. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. Lysosome-mediated processing of chromatin in senescence. The Journal of cell biology. 2013;202:129–143. doi: 10.1083/jcb.201212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging cell. 2012;11:727–731. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nature cell biology. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. The Journal of clinical investigation. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CL, Murphy AJ, Sayers S, Li R, Yvan-Charvet L, Davis JZ, Krishnamurthy J, Liu Y, Puig O, Sharpless NE, et al. Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2483–2492. doi: 10.1161/ATVBAHA.111.234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nature cell biology. 2015;17:1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le ON, Rodier F, Fontaine F, Coppe JP, Campisi J, DeGregori J, Laverdiere C, Kokta V, Haddad E, Beausejour CM. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging cell. 2010;9:398–409. doi: 10.1111/j.1474-9726.2010.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Molecular cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nature genetics. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, Sharpless NE. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood. 2011;117:3257–3267. doi: 10.1182/blood-2010-09-304402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, Thomas NE, Sharpless NE. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging cell. 2009a;8:439–448. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PloS one. 2009b;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes & development. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez I, Garcia-Carpizo V, Guijarro T, Garcia-Gomez A, Navarro D, Aranda A, Zambrano A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence. 2016;7:427–442. doi: 10.1080/21505594.2016.1144001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, King KY. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell reports. 2016;17:2584–2595. doi: 10.1016/j.celrep.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheu A, Pantoja C, Efeyan A, Criado LM, Martin-Caballero J, Flores JM, Klatt P, Serrano M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes & development. 2004;18:2736–2746. doi: 10.1101/gad.310304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing research reviews. 2016;29:1–12. doi: 10.1016/j.arr.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Melk A, Schmidt BM, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney international. 2004;65:510–520. doi: 10.1111/j.1523-1755.2004.00438.x. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nature reviews Molecular cell biology. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Pal A, Potjer TP, Thomsen SK, Ng HJ, Barrett A, Scharfmann R, James TJ, Bishop DT, Karpe F, Godsland IF, et al. Loss-of-Function Mutations in the Cell-Cycle Control Gene CDKN2A Impact on Glucose Homeostasis in Humans. Diabetes. 2016;65:527–533. doi: 10.2337/db15-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Molecular systems biology. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nature reviews Cancer. 2014;14:547–558. doi: 10.1038/nrc3773. [DOI] [PubMed] [Google Scholar]
- Reddel RR. Senescence: an antiviral defense that is tumor suppressive? Carcinogenesis. 2010;31:19–26. doi: 10.1093/carcin/bgp274. [DOI] [PubMed] [Google Scholar]
- Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging cell. 2016 doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosko A, Hofmeister C, Benson D, Efebera Y, Huang Y, Gillahan J, Byrd JC, Burd CE. Autologous hematopoietic stem cell transplant induces the molecular aging of T-cells in multiple myeloma. Bone marrow transplantation. 2015;50:1379–1381. doi: 10.1038/bmt.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Brown EJ. Relationships between stem cell exhaustion, tumour suppression and ageing. British journal of cancer. 2007;97:1189–1193. doi: 10.1038/sj.bjc.6604029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging. 2016;8:328–344. doi: 10.18632/aging.100897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanoff HK, Deal AM, Krishnamurthy J, Torrice C, Dillon P, Sorrentino J, Ibrahim JG, Jolly TA, Williams G, Carey LA, et al. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. Journal of the National Cancer Institute. 2014;106:dju057. doi: 10.1093/jnci/dju057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nature reviews Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–246. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- Signer RA, Montecino-Rodriguez E, Witte ON, Dorshkind K. Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16Ink4a and Arf. Genes & development. 2008;22:3115–3120. doi: 10.1101/gad.1715808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Armata H, Gallant J, Jones SN. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Molecular and cellular biology. 2004;24:976–984. doi: 10.1128/MCB.24.3.976-984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino JA, Krishnamurthy J, Tilley S, Alb JG, Jr, Burd CE, Sharpless NE. p16INK4a reporter mice reveal age-promoting effects of environmental toxicants. The Journal of clinical investigation. 2014a;124:169–173. doi: 10.1172/JCI70960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino JA, Sanoff HK, Sharpless NE. Defining the toxicology of aging. Trends in molecular medicine. 2014b;20:375–384. doi: 10.1016/j.molmed.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souroullas GP, Sharpless NE. mTOR signaling in melanoma: oncogene-induced pseudo-senescence? Cancer Cell. 2015;27:3–5. doi: 10.1016/j.ccell.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–+. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Molecular and cellular biology. 1999;19:2109–2117. doi: 10.1128/mcb.19.3.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal ageing and disease. Nature reviews Nephrology. 2017;13:77–89. doi: 10.1038/nrneph.2016.183. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nature cell biology. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler M, Hamali HA, Sun XM, Bampton ET, Dinsdale D, Snowden RT, Dyer MJ, Goodall AH, Cohen GM. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood. 2011;117:7145–7154. doi: 10.1182/blood-2011-03-344812. [DOI] [PubMed] [Google Scholar]
- Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging cell. 2009;8:311–323. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148:1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, Mann DJ, Ohmura M, Hirao A, Saya H, et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. The Journal of cell biology. 2009;186:393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nature communications. 2016;7:11190. doi: 10.1038/ncomms11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging cell. 2015a doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging cell. 2015b;14:644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]