

Abstract
Cardiovascular disease is a common co-morbidity found with obesity-linked type 2 diabetes. Current pharmaceuticals for these two diseases treat each of them separately. Yet, diabetes and cardiovascular disease share molecular signaling pathways that are increasingly being understood to contribute to disease pathophysiology, particularly in pre-clinical models. This review will focus on one such signaling pathway: that mediated by the G protein-coupled receptor, Prostaglandin E2 Receptor 3 (EP3), and its associated G protein in the insulin-secreting beta-cell and potentially the platelet, Gz. The EP3/Gz signaling axis may hold promise as a dual target for type 2 diabetes and cardiovascular disease.
Keywords: Diabetes, Type 2 Diabetes, Cardiovascular Disease, G proteins, GPCRs, Prostaglandins
Introduction
Diabetes is a chronic and life-threatening disease whose overall life risk has risen dramatically over the last two decades. In 2014, the Centers for Disease Control reported more than 29 million people in the US (9.3% of the population) with diagnosed or undiagnosed diabetes: primarily obesity-linked type 2 diabetes (T2D)(1). Further, in this same time period 86 million American adults were estimated to have pre-diabetes, and although it is traditionally considered adult onset, T2D affected the lives of thousands of children and adolescents (1). In addition to the indirect impact of T2D on productivity and healthy lifespan, the direct financial impact in the US is nearly $250 billion healthcare dollars per year, and rising (2).
T2D is tightly correlated with obesity and is a metabolic disease in which tissues normally sensitive to the peptide hormone, insulin, become resistant to its action. This, in conjunction with dysfunction and/or death of the insulin-secreting pancreatic β-cells, ultimately leads to a failure to maintain healthy blood glucose levels (3, 4). The loss of insulin secretion and function results in a host of metabolic defects, including hyperglycemia, and is associated with chronic comorbidities including hyperlipidemia, retinopathy, and cardiovascular disease (3).
Poorly managed T2D increases the risk for complications and comorbid conditions that adversely affect quality of life (3, 5). The vast increase in cardiovascular death rates and incidences of myocardial infarction and ischemic stroke by adults diagnosed with diabetes is alarming as compared to non-diabetics (6). The pathophysiology of cardiovascular disease and diabetes are tightly linked through many complex factors, including inflammation (6). Despite recent advancements in pharmaceutical therapies, treatments for T2D often fail, or are insufficient in managing blood glucose homeostasis (7) and preventing the associated disease risks.
There is an impending need for improved T2D therapies strategically targeting T2D pathology and its associated co-morbidities such as cardiovascular disease. Currently, a significant proportion of all pharmaceuticals target a specific membrane receptor family, the G protein-coupled receptors (GPCRs) (8). Many GPCRs are involved in pathways that could ultimately improve target tissue insulin sensitivity, β-cell function, and health (9). The most well-known GPCR as a T2D therapeutic target is the glucagon-like peptide 1 receptor (GLP-1R), a stimulatory Gs-coupled receptor that acts in multiple tissues to decrease the gastric emptying rate, improve insulin action, and improve insulin secretion (9, 10). In addition, recent studies have suggested a lower risk of cardiovascular events in individuals treated with certain GLP-1-based therapeutics (11–13). Yet, GLP-1-based therapeutics are not effective to reduce blood glucose in all individuals with diabetes (14). It is now becoming clear that inhibitory Gi-coupled receptors serve a counter-regulatory role in the beta-cell, and that these GPCR pathways can be up-regulated in T2D (15–19). In this review, we will give an overview of GPCRs as T2D therapeutic targets as well as discuss how a specific receptor, Prostaglandin E2 Receptor 3 (EP3), and its beta-cell-associated G-protein, Gz, have the potential to act as therapeutic targets for T2D and cardiovascular disease.
GPCRs as Therapeutic Targets for T2D
Traditional T2D therapies have focused on directly increasing peripheral insulin sensitivity and/or activating insulin secretory pathways in the β-cell. Metformin and other insulin sensitizers target peripheral and hepatic tissues to restore insulin sensitivity, thereby reducing blood glucose by reducing hepatic glucose output and improving peripheral glucose uptake. Metformin is the classical first-line therapeutic for T2D and has been in use since the 1950s, establishing itself with an excellent cardiovascular risk profile (20). Yet, many individuals progress to requiring additional pharmaceutical treatment in order to maintain healthy blood glucose levels. The thiazolidinedione, rosiglitazone, is a potent insulin sensitizer, but is associated with an increased risk of myocardial infarction, and is currently under selling restrictions in the US (20). Sulfonylureas and meglintides act on the ATP-sensitive potassium (KATP) channel to stimulate beta-cell membrane depolarization and exocytosis of insulin granules (21, 22), and are often combined with metformin as a dual therapy (23). Yet, the cardiovascular risk of sulfonylureas is questionable(20), and meglintides have not been studied in enough detail to determine their cardiovascular safety (20).
The GLP-1R was originally proposed as a T2D therapeutic due to its ability to promote insulin secretion from the beta-cell in a glucose-dependent manner (24), in contrast to sulfonylureas and meglintides, whose effects on insulin secretion are fully or partially glucose-independent, respectively. GLP-1 is now known to have effects on many other target tissues, acting in an autocrine, paracrine, or neuronal manner due to its rapid degradation in circulation by the activity of dipeptidyl peptidase 4 (DPP4) (25). Stable GLP-1 analogs such as exenatide and liraglutide and DPP4 inhibitors such as sitagliptin and vildagliptin have been in use clinically since the mid-late 2000s and are currently well-accepted therapeutics for T2D due to their positive effects on insulin secretion and whole-body glucose homeostasis. Further, while the cardiovascular risks of DPP-4 inhibitors is still unclear, the GLP-1 analogs have a very favorable cardiovascular risk profile (20). Yet, GLP-1-based therapeutics do not work to effectively reduce blood glucose in all individuals. For example, approximately 35–60% of T2D patients treated with sitagliptin fail to achieve their target of glycosylated hemoglobin levels (HbA1c) of less than 7 percent (26–28).
Most of what we know about GLP-1R signaling and therapeutic effects has been derived from rodent studies. In the beta-cell, ligand binding to the GLP-1R potentiates glucose stimulated insulin secretion through elevations in intracellular cyclic AMP (cAMP) production to induce the activity of protein kinase A (PKA) and EPAC2 (a guanine nucleotide exchange factor for Rap1 directly activated by cAMP), leading to increased glucose metabolism, membrane depolarization, Ca2+ influx, Ca2+-induced Ca2+ release (CICR) and direct activation of the exocytotic machinery on insulin secretory granules (Figure 1)(29–33). While activation of the Gs protein can stimulate cAMP production, in the beta-cell, effects on cAMP signaling and insulin secretion require beta-arrestin-1 (34). GLP-1R activation also induces β-cell hypertrophy and proliferative gene expression through the cAMP regulatory element binding protein (CREB) (Figure 1) (29, 30). Moreover, GLP-1 has effects in the hypothalamus that suppress appetite and increase energy expenditure, important aspects to controlling T2D pathophysiology. For further reading on the systemic effects of GLP-1, we direct readers to the comprehensive review by Baggio and Drucker (24).
Figure 1: Modulation of beta cell function and survival by prostaglandins.

Glucose enters beta cells via GLUT2 and then is metabolized, increasing the ATP/ADP ratio. KATP channels close due to the abundance of ATP, promoting membrane depolarization and the opening of voltage-dependent calcium channels (VDCCs) and influx of calcium. Calcium induces further intracellular calcium release and, along with ATP, stimulates exocytosis of insulin granules. Glucose-stimulated insulin secretion as well as proliferation and survival can be modulated by cyclic AMP (cAMP). Glucagon-like peptide-1 (GLP-1) enhances beta-cell function, proliferation and survival via activation of its G protein-coupled receptor (GPCR), GLP1R, which is coupled to a stimulatory G protein that activates adenylate cyclase to generate cAMP. Beta-cell cAMP signaling and effects on insulin secretion require the presence of beta-arrestin-1 (β-arr-1). Conversely, the arachidonic acid metabolite, prostaglandin E2 (PGE2) activates a different GPCR, EP3, to inhibit cAMP production and its downstream effects. Intracellular cAMP works by multiple mechanisms to regulate glucose-stimulated insulin secretion and proliferation and survival. Stimulation of EPAC2 by cAMP promotes release of GDP from Rap1 and binding of the more cellular abundant GTP resulting in Rap1’s active conformation and enhanced glucose stimulated insulin secretion. Rap1 activity is limited by Rap1GAP, which is a unique effector of Gz. PKA phosphorylates many substrates to enhance calcium mobilization and nuclear translocation of cAMP bound CREB to promote proliferation and survival.
Chronic inflammation, obesity and T2D pathogenesis
Metabolic tissues maintain immune homeostasis, and it is well established that obesity leads to chronic low-grade inflammation (35, 36). Immune infiltration in adipose, hepatic and pancreatic islet tissues has been well documented (37–39). Elevated macrophage infiltration into tissues leads to a pro-inflammatory cytokine profile, including changes in TNFα, IL-6, and IL-1β (40–42). Pro-inflammatory cytokine effects on metabolic tissues have been characterized mechanistically, and, in general, lead to alterations in the insulin receptor signaling pathway that induce insulin resistance. The induction of insulin resistance impairs glucose transport in insulin-sensitive tissues, leading to hyperglycemia (43–45). In this state, hepatic tissues are incapable of properly managing glucose homeostasis through insulin regulatory mechanisms that halt gluconeogenesis and induce glycogen storage, exacerbating hyperglycemia (46, 47).
Pro-inflammatory cytokines also induce prostaglandin (PG) synthesis, and disrupted PG profiles due to elevated pro-inflammatory signals have been documented in the obese, insulin resistant, and diabetic states (44, 48–54). PGs are transient lipid-derived molecules possessing a wide variety of effects on various tissues. Pro-inflammatory signal transduction activates the lipolytic enzyme, phospholipase A2, which functions to cleave AA from the membrane embedded phospholipids. Free AA is synthesized to prostaglandin G2 and prostaglandin H2 through a series of oxygenation and reduction reactions by the cyclooxygenase enzymes COX-1 and COX-2. PGH2 is the central precursor for all PGs. Its subsequent metabolism is tissue specific, but prostaglandin synthase enzymes result in the production of PGD2, PGF2α, PGE2, prostacyclin (PGI2), and thromboxane A2 (TXA2). Of primary concern in T2D is the negative effect of PGs on pancreatic islets as glucose homeostasis cannot be restored or managed without exogenous insulin once functional beta-cell mass has significantly declined.
Prostaglandin E2, the EP3 receptor and diabetes pathogenesis.
PGE2 and its receptor, Prostaglandin E2 receptor 3 (EP3), have been proposed as contributors to the progression of obesity, insulin resistance, and β-cell failure (48, 54, 55). PGE2 is the primary endogenous ligand for the E prostanoid GPCR family: EP1, EP2, EP3 and EP4. EP1 is a Gq-coupled GPCR whose activation induces phospholipase C beta (PLC-β) activity, triggering cytoplasmic Ca2+ influx. EP2 and EP4 are both Gs-coupled GPCRs responsible for elevated adenylate cyclase activity and cAMP production, as well as Ca2+ influx. Of the four receptors, only EP3 couples to Gi proteins, making it the only PGE2 receptor that reduces cAMP production upon ligand binding, while also inhibiting cytoplasmic Ca2+ flux via reduction in PKA, EPAC, and Gβγ activity.
The first elucidation of the G protein coupling and biological activity of EP3 in the β-cell was by the Robertson group using the hamster insulinoma cell line, HIT-T15. PGE2 binds specifically to HIT-T15 cell membranes and elicits a dose-dependent decrease in cAMP accumulation and insulin secretion (17). PGE2 binds in a similar manner to isolated guinea pig islets (17). In the presence of pertussis toxin (PTX)—which inactivates nearly all Gαi subfamily members by catalyzing the ADP ribosylation of a cysteine near their C termini—the maximal inhibitory effect of PGE2 on cumulative insulin secretion is reduced but not ablated, suggesting coupling of the HIT-T15 EP3 receptor to both PTX-sensitive and -insensitive Gi proteins (17). Perifusion assays to measure temporal insulin secretion in HIT-T15 cells demonstrate a single phase of insulin secretion when stimulated by glucose, with the addition of the inhibitor of cAMP degradation, isobutyl-1-methylxanthine (IBMX), providing a second phase of insulin secretion more consistent with that observed from isolated pancreatic islets (18). Addition of PGE2 to the perifused solution reduced insulin secretion by approximately 50% in both cases, while PTX pre-treatment reduced but did not completely block this inhibition (18). Interestingly, continuing the PTX treatment during the assay almost completely ablated the effects of PGE2 on inhibiting insulin secretion, although this was not necessarily explained by a lack of ADP ribosylation of Gi proteins, as both pre-treatment and continuous treatment of cells with PTX displayed the same completeness of ADP-ribosylation of Gα subunits (18). Further work by the Robertson group revealed the identity of PTX-sensitive Gi/o subfamily members in HIT-T15 cells: Gαi1, Gαi2, Gαi3, and Gαo subtypes including Gαo2 (56).
Because of the partial PTX insensitivity of the inhibitory response of PGE2 in HIT-T15 cells, we were intrigued with the possibility of EP3 coupling to the only PTX-insensitive Gi subfamily member, Gz (57). Gαz protein is readily detectable in isolated rat and mouse islets and insulinoma cell lines by Western blot or immunohistochemistry (58). Using the Ins-1(832/13) rat insulinoma cell line, the closely related eicosanoid, PGE1 (which has nearly the same affinity profile for the mouse EP receptors as PGE2 (59)) inhibited insulin secretion by approximately 50%, an effect that was unchanged by PTX pre-treatment (58). Overexpression of the RGS domain of the specific Gαz inhibitor, RGSZ1, or siRNA-mediated reduction in Gαz protein expression, completely or partially ablated the effect of PGE1 on insulin secretion, respectively, supporting the coupling of EP3 to Gz (58). The PTX-insensitivity of EP3 activity was confirmed in islets isolated from diabetic C57BL/6J LeptinOb/Ob mice, which show a 50% inhibition of insulin secretion by the selective EP3 agonist, sulprostone, an effect that is unchanged by PTX pre-treatment (60). In both the 2005 and 2012 publications cited above, the inhibition of insulin secretion by other inhibitory GPCR agonists, including those targeting the α2A adrenergic receptor and the somatostatin receptor, were completely ablated by PTX, demonstrating a full inhibition of PTX-sensitive Gi proteins (58, 60). Taken together, the studies by the Robertson and Kimple groups are not necessarily discordant, and differences in the ability of PGE1, PGE2, or sulprostone to activate Gi/o vs. Gz proteins; the concentration of PTX used; and/or species-specific differences in GPCR coupling or G protein expression could explain the partial vs. full PTX insensitivity of EP3 signaling found by the two groups. In sum, while EP3 may couple to other Gi proteins, it is certainly at least partially coupled to Gz in both insulinoma cell lines and isolated pancreatic islets (shown in Figure 1).
Elevated PGE2 acts in an autocrine or paracrine manner to bind to EP3. PGE2 is released into the media from cultured islet cells or insulinoma cell lines incubated in stimulatory glucose, effects that are completely ablated by addition of COX inhibitors (17, 61). Elevated PGE2 production is also observed from cultured mouse or human islets isolated from diabetic individuals (54). Coincidently, EP3 expression itself is also induced in islets isolated from diabetic mice and humans and corresponds with reduction in cAMP signaling and a blunted GLP-1 response in the β-cell (54).
The EP3/Gz signaling pathway has been proposed as a therapeutic target for beta-cell failure in T2D (54, 60). C57BL/6N mice lacking the catalytic alpha-subunit of Gz (Gαz-null) are resistant to glucose intolerance induced by high-fat feeding as a result of increased β-cell replication, leading to elevated insulin secretion even in the face of severe insulin resistance (60). This same mouse model, when administered a low daily dose of exenatide, is resistant to hyperglycemia induced by the beta-cell toxin streptozotocin due to improved beta-cell replication, survival, and function (62). These results lend strong support to the EP3/Gz signaling pathway as a therapeutic target for T2D. Yet, nearly all pharmaceuticals that target G-protein signaling pathways target the GPCR and not the G protein or other downstream signaling components, and EP3-null mice have been shown to have increased daytime food intake as compared to wild-type controls, which, over time, leads to increased adiposity and body weight (63). These mice exhibit fasting hyperglycemia, increased plasma insulin and leptin levels, and worsened insulin sensitivity and glucose tolerance as compared to wild-type controls at both 3 and 6 months of age (63). These metabolic findings were re-capitulated in a separate study, which also found leaving the EP3β variant intact was insufficient to protect from the spontaneous development of obesity, insulin resistance, and fasting hyperinsulinemia (55). Finally, a third study with whole-body EP3-null mice found that chow-fed animals have an essentially wild-type metabolic phenotype, while high-fat diet-fed EP3-null mice exhibit increased food intake, increased adiposity, more severe insulin resistance, and elevated serum triglycerides as compared to wild-type mice (48). This leads high-fat diet-fed EP3-null mice to have fasting hyperglycemia and fasting hyperinsulinemia with more severe glucose intolerance than wild-type controls, hallmarks of the diabetic condition (48).
Ceddia and colleagues determined there is no effect of EP3 loss on beta-cell function when pancreatic islets were assayed ex vivo (48). Therefore, the phenotype of the EP3-null mouse is likely not a beta-cell phenotype. EP3 is the E prostanoid receptor variant most highly expressed in white adipose tissue (64). The adipose tissue expression of total EP3 or any of its three splice variants is significantly down-regulated in mouse models of diabetes (55), and specific activation of adipose-tissue EP3 increases adiposity and decreases lipolysis (65). These results are consistent with the phenotype of EP3-null mice, which have increased adipogenesis and adipocyte size, contributors to the obesity phenotype, and augmented lipolysis (48, 55). Furthermore, the adipose tissue itself is more necrotic and inflamed in high-fat diet-fed EP3-null mice, leading to increased secretion of inflammation-associated adipokines (48). Finally, inhibition of adipocyte COX-2 augments obesity-associated adipose tissue inflammation and insulin resistance (50). Taken together, these results suggest systemic inhibition of EP3 may not be an appropriate T2D therapeutic strategy, but instead support a beta-cell-specific or biased antagonist therapy that would directly interfere with up-regulated EP3/Gz signaling in the beta-cell, leaving the beneficial effects of EP3 in tissues such as adipose intact.
Role of Gi proteins in cardiovascular disease
Platelets are key contributors to the development of cardiovascular disease, a primary co-morbid condition occurring in T2D. Like pancreatic β-cells, platelet function can be regulated by inhibitory G protein signaling. Platelets express four Gαi family members: Gαi1, Gαi2, Gαi3 and Gαz (66–68). The preferential coupling of Gαi family members to GPCRs was demonstrated using specific Gα subunit knock-out mice. Gαi2 preferentially couples to the ADP receptor P2Y12 (69) and Gαz prefers the α2A-adrenergic receptor for epinephrine (Figure 2) (70). Gαi2 and Gαz, but not Gαi3, regulate basal levels of cAMP in circulating platelets (71).
Figure 2: Regulation of platelet function by prostanoid receptors.
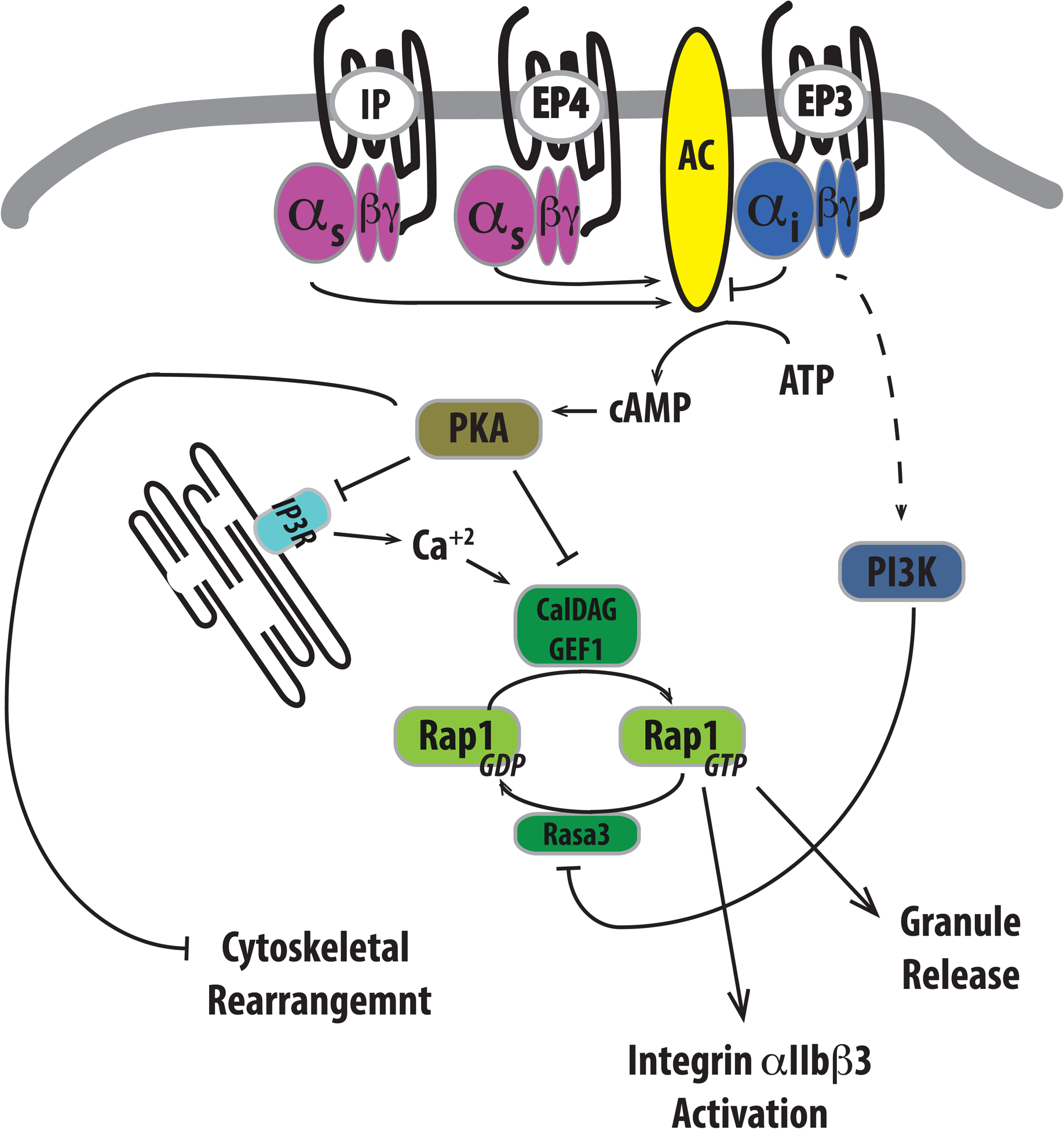
Prostaglandins, particularly prostaglandin E2 (PGE2), can have a profound impact on platelet function based on the prostanoid receptor that becomes activated. IP and EP4 couple to stimulatory heterotrimeric G proteins to activate adenylate cyclase and enhance cAMP production. In direct contrast, EP3 couples to an inhibitory G protein to prevent cAMP generation. Intracellular cAMP levels and activation of the PKA negatively influences platelet reactivity by phosphorylation and inactivation of specific proteins important to many aspects of platelet activation. Granule release and integrin αIIbβ3 activation can be prevented by PKA phosphorylation of the IP3 receptor (IP3R) on the ER, which prevents IP3-stimulated release of calcium and subsequent activation of the small GTPase Rap1 via CalDAG GEF1. PKA phosphorylation of CalDAG GEF1 can also impair its ability to activate Rap1. In contrast, βγ subunits previously associated with inhibitory G proteins have been reported to activate PI3K to inhibit RASA3 to promote prolonged activation of Rap1, thereby stimulating granule release and integrin activation.
In addition to the classical role of Gαi proteins in the inhibition of AC activity and reduction of cAMP, the Gβγ subunits of G proteins activate phosphoinositide 3-kinase (PI3K) to elicit signaling to promote platelet activation (66). Recent studies indicate that PI3K inhibits Rasa3, a Rapl GTPase activating protein, to promote platelet activation (Figure 2) (72).
It has been long appreciated that diabetics have increased sensitivity of platelets to ADP and epinephrine, two agents well-known to stimulate platelet activation (73). In both pre- clinical models and diabetic subjects, elevated plasma levels of the PGE2 metabolite, 15- keto-13,14-dihydroprostaglandin E2, can be found (60, 74–76). This is particularly evident during diabetic ketoacidosis (75, 76). Isolated whole blood or platelet-rich plasma from type 1 and/or type 2 diabetic subjects produce significantly more prostaglandin E-like material or PGE2 itself than isolates from control subjects (77, 78). In addition, platelets isolated from diabetic subjects generate increased amounts of prostaglandin E-like material or PGE2 itself in response to ADP, epinephrine, collagen and arachidonic acid (73, 79).
PGE2 can have profound effects on platelet function by binding to and activating EP receptors. The non-steroidal anti-inflammatory drugs (NSAIDs), such as acetylsalicylic acid (aspirin) target the COX enzymes to inhibit prostaglandin production, abolishing platelet hypersensitivity. Using receptor subtype specific or selective agonists and E prostanoid receptor-null mice, it was demonstrated that EP3, EP4 and IP receptors elicit functional responses in platelets. EP4 and IP receptors inhibited platelet function and enhanced VASP phosphorylation (80, 81), indicating that these two GPCRs are Gs coupled. Conversely, the EP3 receptor agonist promotes platelet aggregation, calcium signaling and P-selectin expression, which are associated with reduction of VASP phosphorylation (80, 81), suggesting coupling to a Gi family member. Antagonist-induced competitive inhibition of EP3 decreases thrombosis without impairing hemostasis (82).
EP3 antagonists have been suggested as anti-platelet activating therapeutics. Due to EP3 and Gz coupling in pancreatic β-cells (54), it is possible that EP3 also couples to Gz in platelets. Unfortunately, this functional relationship has not yet been confirmed, but is suggested in Figure 3: a summary of EP3 functions in various insulin-sensitive tissues and platelets and how these may be impacted in the diabetic environment.
Figure 3: PGE2 signaling contributes to the diabetic environment.

Chronic low-grade inflammation and hyperglycemia are hallmarks of diabetes mellitus with elevated circulating levels of pro-inflammatory cytokines, TNFα, IL-6 and IL-1β. These cytokines alter insulin receptor signaling leading to insulin resistance and also to enhanced synthesis of prostaglandins, including PGE2, in target tissues. PGE2 is rapidly degraded in the circulation, but elevated plasma levels of the PGE2 metabolite, 15-keto-13,14-dihydroprostaglandin E2 (dhPGE2), have been found in pre-clinical models of diabetes as well as individuals with diabetes mellitus. EP3-expressing, insulin-sensitive tissues such as adipose and liver become insulin resistant and contribute to prolonged hyperglycemia in different ways. Insulin resistance in hepatic tissues enhances gluconeogenesis and inhibits storage of glycogen, while adipocytes fail to uptake glucose for energy use. In addition, EP3 is pathologically up-regulated with diabetes onset in pancreatic islets. The coupling of EP3 to the inhibitory heterotrimeric G protein, Gz, in pancreatic islets results in reduced glucose stimulated insulin secretion and impaired maintenance of beta cell mass. Due to endothelial cell activation in the diabetic environment, the resting tone of platelets is perturbed leading to hypersensitivity. Independently and/or possibly together, platelet EP3 and Gz contribute to signaling pathways leading to platelet activation.
Targeting EP3 and Ga zln Diabetes
Prostaglandins are of great physiological importance and are required for proper immune function, among other physiological processes. Furthermore, while plasma levels of the PGE2 metabolite, 15-keto-13,14-dihydroprostaglandin E2, are elevated in the diabetic condition, 15-keto-13,14-dihydroprostaglandin E2 itself does not activate the EP3 receptor and its utility as a biomarker for either disease status or therapeutic response remains questionable. Finally, while EP3 receptor signaling is dyfunctionally up-regulated in the diabetic beta-cell, its expression in other tissues is required for normal processes such as the inhibition of lipolysis. Taken together, these findings suggest systemic targeting of prostaglandin production or direct inhibition of EP3 may not be successful pharmacological strategies for treating diabetes and its complications. However, the function of EP3 and its putative coupling to Gz, whose expression is quite limited, in both pancreatic islets and platelets provides a unique scenario in which development of biased- antagonist specifically targeting EP3 and Gz coupling could prove useful in the direct treatment of both hyperglycemia as well as associated cardiovascular disease.
References
- 1.Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services; 2014. [Google Scholar]
- 2.American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013. April;36(4):1033–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2011. January;34 Suppl 1:S62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liston A, Todd JA, Lagou V. Beta-Cell Fragility As a Common Underlying Risk Factor in Type 1 and Type 2 Diabetes. Trends Mol Med. 2017. January 20. [DOI] [PubMed] [Google Scholar]
- 5.Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. 2012. November;35(11):2402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selvin E, Marinopoulos S, Berkenblit G, Rami T, Brancati FL, Powe NR, et al. Meta-analysis: glycosylated hemoglobin and cardiovascular disease in diabetes mellitus. Annals of internal medicine. 2004. September 21;141(6):421–31. [DOI] [PubMed] [Google Scholar]
- 7.Triplitt C Improving treatment success rates for type 2 diabetes: recommendations for a changing environment. Am J Manag Care. 2010. August;16(7 Suppl):S195–200. [PubMed] [Google Scholar]
- 8.Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2016. December 02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reimann F, Gribble FM. G protein-coupled receptors as new therapeutic targets for type 2 diabetes. Diabetologia. 2016. February;59(2):229–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trujillo JM, Nuffer W. GLP-1 receptor agonists for type 2 diabetes mellitus: recent developments and emerging agents. Pharmacotherapy. 2014. November;34(11):1174–86. [DOI] [PubMed] [Google Scholar]
- 11.Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2016. November 10;375(19):1834–44. [DOI] [PubMed] [Google Scholar]
- 12.Kumarathurai P, Anholm C, Larsen BS, Olsen RH, Madsbad S, Kristiansen O, et al. Effects of Liraglutide on Heart Rate and Heart Rate Variability: A Randomized, Double-Blind, Placebo-Controlled Crossover Study. Diabetes Care. 2017. January;40(1):117–24. [DOI] [PubMed] [Google Scholar]
- 13.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2016. July 28;375(4):311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prasad-Reddy L, Isaacs D. A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs in context. 2015;4:212283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimple ME, Neuman JC, Linnemann AK, Casey PJ. Inhibitory G proteins and their receptors: emerging therapeutic targets for obesity and diabetes. Experimental & molecular medicine. 2014;46:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson RP, Gavareski DJ, Porte D, Jr., Bierman EL. Inhibition of in vivo insulin secretion by prostaglandin E1. J Clin Invest. 1974. August;54(2):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson RP, Tsai P, Little SA, Zhang HJ, Walseth TF. Receptor-mediated adenylate cyclase-coupled mechanism for PGE2 inhibition of insulin secretion in HIT cells. Diabetes. 1987. September;36(9):1047–53. [DOI] [PubMed] [Google Scholar]
- 18.Seaquist ER, Walseth TF, Nelson DM, Robertson RP. Pertussis toxin-sensitive G protein mediation of PGE2 inhibition of cAMP metabolism and phasic glucose-induced insulin secretion in HIT cells. Diabetes. 1989. November;38(11):1439–45. [DOI] [PubMed] [Google Scholar]
- 19.Tran PO, Gleason CE, Robertson RP. Inhibition of interleukin-1beta-induced COX-2 and EP3 gene expression by sodium salicylate enhances pancreatic islet beta-cell function. Diabetes. 2002. June;51(6):1772–8. [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Rajaratnam R. Cardiovascular safety of non-insulin pharmacotherapy for type 2 diabetes. Cardiovascular diabetology. 2017. February 02;16(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002. December;51 Suppl 3:S368–76. [DOI] [PubMed] [Google Scholar]
- 22.Principalli MA, Dupuis JP, Moreau CJ, Vivaudou M, Revilloud J. Kir6.2 activation by sulfonylurea receptors: a different mechanism of action for SUR1 and SUR2A subunits via the same residues. Physiological reports. 2015. September;3(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eriksson JW, Bodegard J, Nathanson D, Thuresson M, Nystrom T, Norhammar A. Sulphonylurea compared to DPP-4 inhibitors in combination with metformin carries increased risk of severe hypoglycemia, cardiovascular events, and all-cause mortality. Diabetes Res Clin Pract. 2016. July;117:39–47. [DOI] [PubMed] [Google Scholar]
- 24.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007. May;132(6):2131–57. [DOI] [PubMed] [Google Scholar]
- 25.D’Alessio D Is GLP-1 a hormone: Whether and When? Journal of diabetes investigation. 2016. April;7 Suppl 1:50–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia. 2006. November;49(11):2564–71. [DOI] [PubMed] [Google Scholar]
- 27.Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006. December;29(12):2632–7. [DOI] [PubMed] [Google Scholar]
- 28.Nonaka K, Kakikawa T, Sato A, Okuyama K, Fujimoto G, Kato N, et al. Efficacy and safety of sitagliptin monotherapy in Japanese patients with type 2 diabetes. Diabetes Res Clin Pract. 2008. February;79(2):291–8. [DOI] [PubMed] [Google Scholar]
- 29.Drucker DJ. Glucagon-like peptide-1 and the islet beta-cell: augmentation of cell proliferation and inhibition of apoptosis. Endocrinology. 2003. December;144(12):5145–8. [DOI] [PubMed] [Google Scholar]
- 30.Linnemann AK, Neuman JC, Battiola TJ, Wisinski JA, Kimple ME, Davis DB. Glucagon-Like Peptide-1 Regulates Cholecystokinin Production in beta-Cells to Protect From Apoptosis. Mol Endocrinol. 2015. July;29(7):978–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes. 2002. December;51 Suppl 3:S434–42. [DOI] [PubMed] [Google Scholar]
- 32.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005. October;85(4):1303–42. [DOI] [PubMed] [Google Scholar]
- 33.Seino S, Takahashi H, Fujimoto W, Shibasaki T. Roles of cAMP signalling in insulin granule exocytosis. Diabetes Obes Metab. 2009. November;11 Suppl 4:180–8. [DOI] [PubMed] [Google Scholar]
- 34.Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc Natl Acad Sci U S A. 2008. May 6;105(18):6614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apostolopoulos V, de Courten MP, Stojanovska L, Blatch GL, Tangalakis K, de Courten B. The complex immunological and inflammatory network of adipose tissue in obesity. Mol Nutr Food Res. 2015. September 1. [DOI] [PubMed] [Google Scholar]
- 36.Khodabandeloo H, Gorgani-Firuzjaee S, Panahi S, Meshkani R. Molecular and cellular mechanisms linking inflammation to insulin resistance and beta-cell dysfunction. Translational research: the journal of laboratory and clinical medicine. 2015. September 5. [DOI] [PubMed] [Google Scholar]
- 37.Divella R, De Luca R, Abbate I, Naglieri E, Daniele A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. Journal of Cancer. 2016;7(15):2346–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gunasekaran MK, Virama-Latchoumy AL, Girard AC, Planesse C, Guerin-Dubourg A, Ottosson L, et al. TLR4-dependant pro-inflammatory effects of HMGB1 on human adipocyte. Adipocyte. 2016. Oct-Dec;5(4):384–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh M, Iwabuchi K. Communication between natural killer T cells and adipocytes in obesity. Adipocyte. 2016. Oct-Dec;5(4):389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mito N, Hosoda T, Kato C, Sato K. Change of cytokine balance in diet-induced obese mice. Metabolism. 2000. October;49(10):1295–300. [DOI] [PubMed] [Google Scholar]
- 41.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003. December;112(12):1785–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mancuso P The role of adipokines in chronic inflammation. ImmunoTargets and therapy. 2016;5:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem. 2016. November 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bluher M Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clinical science. 2016. September 01;130(18):1603–14. [DOI] [PubMed] [Google Scholar]
- 45.Verma S, Hussain ME. Obesity and diabetes: An update. Diabetes & metabolic syndrome. 2016. June 17. [DOI] [PubMed] [Google Scholar]
- 46.Kim OK, Jun W, Lee J. Mechanism of ER Stress and Inflammation for Hepatic Insulin Resistance in Obesity. Annals of nutrition & metabolism. 2015;67(4):218–27. [DOI] [PubMed] [Google Scholar]
- 47.Duarte N, Coelho IC, Patarrao RS, Almeida JI, Penha-Goncalves C, Macedo MP. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. BioMed research international. 2015;2015:984578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceddia RP, Lee D, Maulis MF, Carboneau BA, Threadgill DW, Poffenberger G, et al. The PGE2 EP3 Receptor Regulates Diet-Induced Adiposity in Male Mice. Endocrinology. 2016. January;157(1):220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Alonso V, Titos E, Alcaraz-Quiles J, Rius B, Lopategi A, Lopez-Vicario C, et al. Prostaglandin E2 Exerts Multiple Regulatory Actions on Human Obese Adipose Tissue Remodeling, Inflammation, Adaptive Thermogenesis and Lipolysis. PloS one. 2016;11(4):e0153751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan PC, Hsiao FC, Chang HM, Wabitsch M, Hsieh PS. Importance of adipocyte cyclooxygenase-2 and prostaglandin E2-prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 2016. June;30(6):2282–97. [DOI] [PubMed] [Google Scholar]
- 51.Hu X, Cifarelli V, Sun S, Kuda O, Abumrad NA, Su X. Major role of adipocyte prostaglandin E2 in lipolysis-induced macrophage recruitment. J Lipid Res. 2016. April;57(4):663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015. December 22;112(51):15642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Alonso V, Claria J. Prostaglandin E2 signals white-to-brown adipogenic differentiation. Adipocyte. 2014. Oct-Dec;3(4):290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kimple ME, Keller MP, Rabaglia MR, Pasker RL, Neuman JC, Truchan NA, et al. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes. 2013. June;62(6):1904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu H, Fu JL, Miao YF, Wang CJ, Han QF, Li S, et al. Prostaglandin E2 receptor EP3 regulates both adipogenesis and lipolysis in mouse white adipose tissue. Journal of molecular cell biology. 2016. December;8(6):518–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seaquist ER, Neal AR, Shoger KD, Walseth TF, Robertson RP. G-proteins and hormonal inhibition of insulin secretion from HIT-T15 cells and isolated rat islets. Diabetes. 1992. November;41(11):1390–9. [DOI] [PubMed] [Google Scholar]
- 57.Casey PJ, Fong HK, Simon MI, Gilman AG. Gz, a guanine nucleotide-binding protein with unique biochemical properties. J Biol Chem. 1990. February 5;265(4):2383–90. [PubMed] [Google Scholar]
- 58.Kimple ME, Nixon AB, Kelly P, Bailey CL, Young KH, Fields TA, et al. A role for G(z) in pancreatic islet beta-cell biology. J Biol Chem. 2005. September 9;280(36):31708–13. [DOI] [PubMed] [Google Scholar]
- 59.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. British journal of pharmacology. 1997. September;122(2):217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kimple ME, Moss JB, Brar HK, Rosa TC, Truchan NA, Pasker RL, et al. Deletion of GalphaZ protein protects against diet-induced glucose intolerance via expansion of beta-cell mass. J Biol Chem. 2012. June 08;287(24):20344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metz SA, Robertson RP, Fujimoto WY. Inhibition of prostaglandin E synthesis augments glucose-induced insulin secretion is cultured pancreas. Diabetes. 1981. July;30(7):551–7. [DOI] [PubMed] [Google Scholar]
- 62.Brill AL, Wisinski JA, Cadena MT, Thompson MF, Fenske RJ, Brar HK, et al. Synergy Between Galphaz Deficiency and GLP-1 Analog Treatment in Preserving Functional beta-Cell Mass in Experimental Diabetes. Mol Endocrinol. 2016. May;30(5):543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanchez-Alavez M, Klein I, Brownell SE, Tabarean IV, Davis CN, Conti B, et al. Night eating and obesity in the EP3R-deficient mouse. Proceedings of the National Academy of Sciences of the United States of America. 2007. February 20;104(8):3009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Borglum JD, Pedersen SB, Ailhaud G, Negrel R, Richelsen B. Differential expression of prostaglandin receptor mRNAs during adipose cell differentiation. Prostaglandins Other Lipid Mediat. 1999. July;57(5–6):305–17. [DOI] [PubMed] [Google Scholar]
- 65.Wolf G Adipose-specific phospholipase as regulator of adiposity. Nutr Rev. 2009. Sep;67(9):551–4. [DOI] [PubMed] [Google Scholar]
- 66.Woulfe D, Jiang H, Mortensen R, Yang J, Brass LF. Activation of Rap1B by G(i) family members in platelets. J Biol Chem. 2002. June 28;277(26):23382–90. [DOI] [PubMed] [Google Scholar]
- 67.Gagnon AW, Manning DR, Catani L, Gewirtz A, Poncz M, Brass LF. Identification of Gz alpha as a pertussis toxin-insensitive G protein in human platelets and megakaryocytes. Blood. 1991. September 1;78(5):1247–53. [PubMed] [Google Scholar]
- 68.Williams AG, Woolkalis MJ, Poncz M, Manning DR, Gewirtz AM, Brass LF. Identification of the pertussis toxin-sensitive G proteins in platelets, megakaryocytes, and human erythroleukemia cells. Blood. 1990. August 15;76(4):721–30. [PubMed] [Google Scholar]
- 69.Yang J, Wu J, Kowalska MA, Dalvi A, Prevost N, O’Brien PJ, et al. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc Natl Acad Sci U S A. 2000. August 29;97(18):9984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jantzen HM, Milstone DS, Gousset L, Conley PB, Mortensen RM. Impaired activation of murine platelets lacking G alpha(i2). J Clin Invest. 2001. August;108(3):477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang J, Wu J, Jiang H, Mortensen R, Austin S, Manning DR, et al. Signaling through Gi family members in platelets. Redundancy and specificity in the regulation of adenylyl cyclase and other effectors. J Biol Chem. 2002. November 29;277(48):46035–42. [DOI] [PubMed] [Google Scholar]
- 72.Stefanini L, Paul DS, Robledo RF, Chan ER, Getz TM, Campbell RA, et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J Clin Invest. 2015. April;125(4):1419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Colwell JA, Halushka PV, Sarji K, Levine J, Sagel J, Nair RM. Altered platelet function in diabetes mellitus. Diabetes. 1976;25(2 SUPPL):826–31. [PubMed] [Google Scholar]
- 74.Axelrod L, Cornelius P, Kieffer JD. Plasma eicosanoid levels in rats with nonketotic diabetes mellitus: effect of severity. Metabolism: clinical and experimental. 1986. April;35(4):328–32. [DOI] [PubMed] [Google Scholar]
- 75.Axelrod L, Shulman GI, Blackshear PJ, Bornstein W, Roussell AM, Aoki TT. Plasma level of 13,14-dihydro-15-keto-PGE2 in patients with diabetic ketoacidosis and in normal fasting subjects. Diabetes. 1986. September;35(9):1004–10. [DOI] [PubMed] [Google Scholar]
- 76.McRae JR, Day RP, Metz SA, Halter JB, Ensinck JW, Robertson RP. Prostaglandin E2 metabolite levels during diabetic ketoacidosis. Diabetes. 1985. August;34(8):761–6. [DOI] [PubMed] [Google Scholar]
- 77.Chase HP, Williams RL, Dupont J. Increased prostaglandin synthesis in childhood diabetes mellitus. J Pediatr. 1979. February;94(2):185–9. [DOI] [PubMed] [Google Scholar]
- 78.Abbate R, Pinto S, Panetta A, Favilla S, Prisco D, Paniccia R, et al. Platelet synthesis of cyclooxygenase and lipoxygenase products in type I and type II diabetes. Prostaglandins Leukot Essent Fatty Acids. 1988. January;31(1):9–15. [DOI] [PubMed] [Google Scholar]
- 79.Halushka PV, Lurie D, Colwell JA. Increased synthesis of prostaglandin-E-like material by platelets from patients with diabetes mellitus. The New England journal of medicine. 1977. December 15;297(24):1306–10. [DOI] [PubMed] [Google Scholar]
- 80.Fabre JE, Nguyen M, Athirakul K, Coggins K, McNeish JD, Austin S, et al. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J Clin Invest. 2001. March;107(5):603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iyu D, Glenn JR, White AE, Johnson AJ, Fox SC, Heptinstall S. The role of prostanoid receptors in mediating the effects of PGE(2) on human platelet function. Platelets. 2010;21(5):329–42. [DOI] [PubMed] [Google Scholar]
- 82.Tilly P, Charles AL, Ludwig S, Slimani F, Gross S, Meilhac O, et al. Blocking the EP3 receptor for PGE2 with DG-041 decreases thrombosis without impairing haemostatic competence. Cardiovascular research. 2014. January 2. [DOI] [PubMed] [Google Scholar]