

Abstract
Tumors are dynamic pseudo-organs that contain numerous cell types interacting to create unique physiology. Within this network, the malignant cells encounter many challenges and rewire their metabolic properties accordingly. Such changes can be experienced and executed autonomously or through interaction with other cells in the tumor. The focus of this review is on the remodeling of the tumor microenvironment that leads to pathophysiologic interactions that are influenced and shaped by metabolism. They include symbiotic nutrient sharing, nutrient competition, and the role of metabolites as signaling molecules. Examples of such processes abound in normal organismal physiology, and such heterocelluar metabolic interactions are repurposed to support tumor metabolism and growth. The importance and ubiquity of these processes is just beginning to be realized, and insights into their role in tumor development and progression are being used to design new drug targets and cancer therapies.
Keywords: cancer-associated fibroblasts, crosstalk, hypoxia, immune, stroma
Features of the tumor microenvironment that affect metabolism
In normal organ physiology, parenchymal cells form the organ and define its function. These cells are supported by the connective tissue, which maintains the structural framework and integrates organ systems. Cells of the immune system function to protect against infection and to support tissue repair following injury.
Solid tumors are highly disorganized versions of normal organs populated with numerous cell types including endothelial cells (e.g. blood vessels), stromal fibroblasts, immune cells, and malignant cancer cells [1]. Transformation and growth of organ-resident parenchymal cells leads to the development of clonal malignant subtypes within a tumor. The diversity is achieved as transformed cells accumulate mutations. This leads to changes in signal transduction, the epigenome, and gene expression, which endow a spectrum of differentiation, metabolic, and proliferative states across the cancer cell populations in the tumor [2, 3]. These features encompass the intrinsic metabolic properties of the cancer cells [4–6]. Clonal heterogeneity is further diversified through interactions with other cells types in the tumor and properties of the tumor microenvironment (TME), collectively referred to herein as extrinsically influencing variables [7–10]. Interactions among the intrinsic metabolic networks and the extrinsic effecters leads to a spectrum of cancer cells within a tumor that exhibit varying metabolic requirements and properties (Fig. 1).
Figure 1.
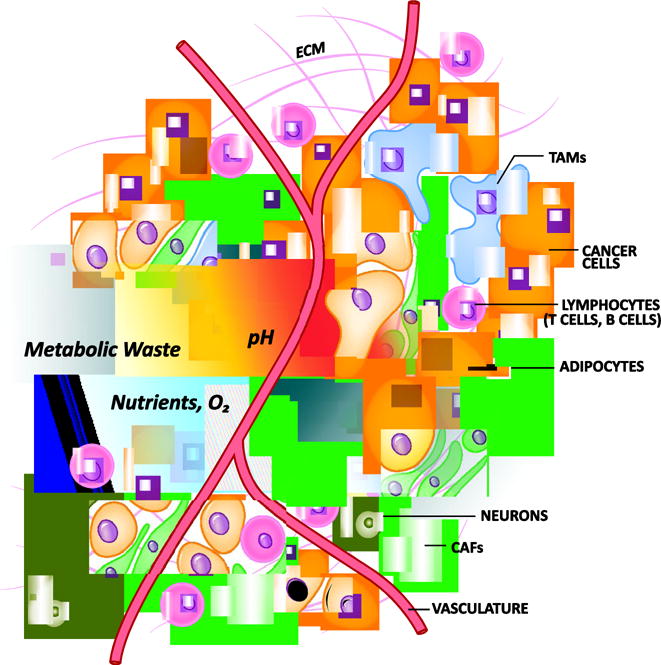
Features of the tumor microenvironment that contribute to metabolic heterogeneity. Intrinsic and extrinsic variables influence the metabolic properties of a tumor. Cell intrinsic variables include the genetic, epigenetic and metabolic programs active in each cell type. Extrinsic variables include interactions among the heterogenous cellular populations, nutrient availability, waste and pH gradients, oxygen (O2) tension, extracellular matrix (ECM) deposition, physical and oxidative pressures. CAF, cancer associated fibroblast; TAM, tumor associated macrophage.
The cadre of stromal, immune and malignant cells creates a TME that imposes many challenges for the cancer cells: physical pressure, oxidative stress, nutrient deprivation and competition, hypoxia and immune surveillance. For example, stromal fibroblasts, commonly referred to as cancer associated fibroblasts (CAF), can become activated and proliferate, release growth factors and cytokines, and deposit extracellular matrix proteins [11–13]. Cytokines recruit immune cells, which secrete extracellular matrix remodeling factors. Together these processes change the architecture of the organ creating a stiff fibrotic matrix, marked by increased interstitial pressure, which hinders the activity of the vascular system [14–16]. Deregulated proliferation and growth factor release further influences blood vessel development, which is characterized by leaky vessels that inefficiently deliver nutrients and remove waste products of cellular metabolism, like lactate [17, 18].
In addition to nutrients and waste, the insufficient tissue coverage and abnormal characteristics of tumor vasculature limits gas exchange and creates regions of hypoxia. The hypoxic response leads to enhanced glycolytic activity of the tumor and additional lactate deposition. The buildup of lactate acidifies the TME, which influences how the immune system recognizes and responds to the tumor [19–21]. Furthermore, nutrient limitation in the TME provides a context in which immune, stromal and cancer cells must compete for nutrients to carry out biosynthesis, bioenergetic and effector activities. Immune cells tend to be less adapted for nutrient competition, and this is a principle mechanism regulating anti-tumor immunity. Numerous additional mechanisms in the TME influence tumor immunity [22–24], including the activity of intracellular metabolic pathways in immune cells. These topics have been the subject of several recent reviews [25–27].
Intra-tumoral metabolic crosstalk
Recent findings suggest that intra-tumoral mechanisms of metabolite communication act symbiotically to support tumor metabolism, maintenance, and growth, or competitively to impair anti-tumor immunity. Heterocellular metabolic interactions in the TME are highly analogous to their normal physiological processes, repurposed to support tumor metabolism and growth. Examples of such processes abound in normal organismal physiology, including the glucose-lactate shuttle in the brain [28], a role for lactate as a signaling molecule in vasoconstriction [29], tryptophan-kynurenine metabolism in the brain and immune system [30], and the glucose-alanine cycle between muscle and liver [31]. Understanding the role of these metabolic interactions in tumor development and progression has the potential to inform the development of new drug targets and cancer therapies [32, 33].
Biosynthetic and bioenergetic metabolite cross-feeding and competition
Significant metabolic heterogeneity exists among the cells in a tumor [34–38]. This is in part established by lineage dependent gene expression programs, but proximity to the vasculature also strongly influences intratumoral metabolic heterogeneity. For example, while nutrient and oxygen access is proportional to vascular access, efficient waste removal is inversely proportional (Fig. 1). To counteract these metabolic challenges, heterocellular metabolite cross-feeding pathways are established that support bioenergetics, biosynthesis and the clearance/reuse of metabolic waste products. In a similar but opposing manner, the lack of degradation of a metabolite can also have pro-tumor growth properties. For example, metabolite catabolism by pro-tumorigenic cells can lead to a state of nutrient deficiency, and thus impaired activity of immune cells involved in tumor destruction. These processes have been described for many metabolites, proteins and cellular cargo and are discussed below.
Glycolysis and Lactate
Data supporting cooperative metabolic pathways that coordinate lactate and glucose metabolism among cells in a tumor have been reported in several types of cancer [9, 39–42]. For example, cancer cells in hypoxic tumor regions metabolize glucose through anaerobic glycolysis. This process is supported by well oxygenated cancer cells, which consume lactate discarded by the hypoxic cancer cells to fuel mitochondrial metabolism [43]. An important consideration for lactate sharing concerns the expression of appropriate intake/release transporters. Lactate is released from cells via monocarboxylate transporter 4 (MCT4), whose expression is increased in hypoxic tumor regions. In contrast, expression of the lactate importer MCT1 is increased in cancer cells in normoxic regions of tumors [43, 44]. These results provide a rationale for the existence of this pathway (Fig. 2A).
Figure 2.
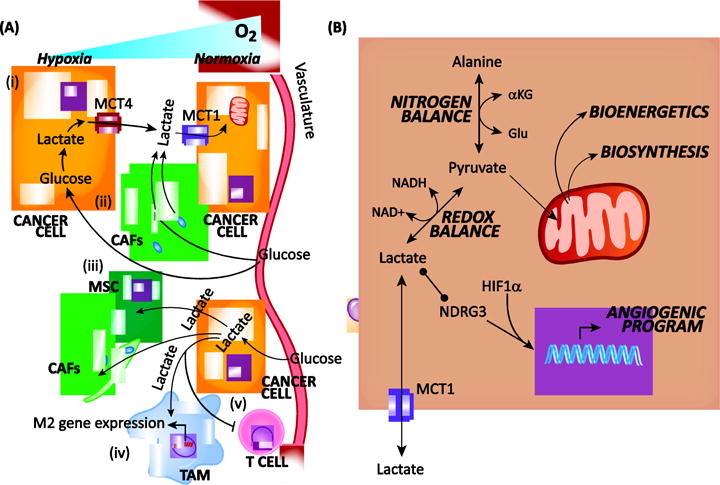
The multifaceted roles of lactate in tumor metabolism.
(A) Cross-feeding and crosstalk mechanisms. Glucose-derived lactate has been reported to (i) originate in hypoxic cancer cells and feed cancer cells within the same tumor that are in proximity to the vasculature; (ii) originate in cancer associated fibroblasts (CAF) and feed cancer cells; (iii) originate in cancer cells and feed mesenchymal stem cells (MSC) and CAFs; (iv) polarize macrophages toward an alternative polarized M2 / tumor associated macrophage (TAM) fate; and (v) inhibit anti-tumor T cells. Lactate is released through the MCT4 transporter and taken-in by the MCT1 transporter. (B) Intracellular metabolic and signaling functions. Metabolism of intracellular lactate regulates nitrogen and redox balance, bioenergetics, and biosynthesis. In addition, lactate can participate is signaling via direct binding to N-myc downstream-regulated gene 3 (NDRG3) and activation of a hypoxia inducible factor 1α (HIF1α)-dependent angiogenic program. aKG, alpha-ketoglutarate; Glu, glutamate; NAD+, nicotinamide adenine dinucleotide oxidized; NADH, nicotinamide adenine dinucleotide reduced.
To study the exchange of lactate among cancer cells more precisely, cell based pancreatic cancer models were used to illustrate that hypoxic cancer cells released lactate through MCT4, and that those grown in normoxia utilized MCT1 to directly import and metabolize lactate [44] (Fig. 2A). Similarly, acute hypoxia induced by pharmacological inhibition of angiogenesis drove pancreatic neuroendocrine cancer cells to reorganize into symbiotic networks that share glucose-derived carbon. In other words, glycolytic hypoxic cancer cells produced lactate, and this lactate was used by cancer cells in proximity to blood vessels, and thus oxygen [45]. Similar results were also observed following anti-angiogenic therapy in a breast cancer model [46].
In the studies described, this symbiotic intra-tumoral metabolism pathway is reported in pancreatic neuroendocrine, pancreatic ductal adenocarcinoma, lung, colon, and breast cancers, illustrating that it may represent a general phenomenon utilized across many cancer types (Fig. 2A). Perhaps most importantly, co-targeting this pathway with anti-angiogenesis agents and mTOR inhibitors revealed new, unique vulnerabilities in cancer metabolism, which occurred in part by activating glucose uptake in normoxic cancer cells [45].
This model of symbiotic metabolism is reminiscent of the glucose-lactate shuttle observed in the brain, in which glucose-derived lactate derived from astrocytes is shuttled to neurons [28]. The precise function of this pathway remains contentious and may serve several functions [47]. Similarly, the wiring and function of this symbiotic metabolic pathway amongst cells in a tumor remains to be fully elucidated. It is not clear why well oxygenated cancer cells with ready access to the blood stream utilize lactate in preference to glucose. This could potentially be explained by the profile and magnitude of glucose and lactate transporter expression, which are regulated by hypoxia. Further, the precise role(s) of lactate remain to be determined (Fig. 2B). It is suggested that lactate in this context is used for bioenergetics [43], but lactate has myriad functions. In the neuron-astrocyte model, lactate-glucose shuttling is thought to balance other fluxes, like recycling of the neurotransmitter glutamate. Lactate uptake also directly affects the redox state as oxidative metabolism by lactate dehydrogenase results in NAD+ consumption and NADH production [48]. Finally, lactate can act directly as a signaling metabolite to affect gene expression programs [20, 49–52].
Like oxygen tension-mediated alterations of glucose and lactate metabolism among cancer cells, similar changes are also observed through contact-mediated processes among prostate CAFs and cancer cells [53]. Prostate CAFs grown in the presence of cancer cells activate glucose transporter (i.e. GLUT1) expression, glucose uptake, MCT4 expression and lactate release. Concurrently, glycolytic activity is repressed in prostate cancer cells, and these cells begin to metabolize CAF-derived lactate. While oxygen availability does not regulate this pathway, it is similarly mediated by stabilized hypoxia-inducible factor 1α (HIF1α) in the CAFs, via reactive oxygen species. Consistent with these studies, TGF-β and PDGF signaling in colon CAFs leads to the activation of aerobic glycolysis, which is also mediated by HIF1α [54]. In this study, decreased intracellular alpha-ketoglutarate levels stabilized HIF1α by preventing the prolyl hydroxylase 1 (PHD1)-mediated degradation of HIF1α. The phenomenon, whereby CAFs take in glucose and release lactate to cancer cells, has been coined the “reverse Warburg effect” [40, 55]. However, this description does not reflect all CAF-cancer interactions, as numerous reports with opposing results exist. For example, breast and colon cancer cells consume glucose and release lactate to surrounding fibroblasts and mesenchymal stem cells [56, 57] (Fig. 2A). Similarly, pancreatic and ovarian CAFs have low glycolytic activity [58, 59] and consume lactate [59]. The observed discrepancies likely reflect dominant effects by the cell/tissue of origin on microenvironmental interactions.
Glucose availability in the TME is inversely proportional to local utilization, and this access has a strong impact on the activity of anti-tumor effector T cells [60, 61]. For example, glucose consumption by cancer cells in melanoma mouse models limited glucose available to T cells, thereby dampening their effector activity [61] (Fig. 3). This reduction occurred through a drop in the glycolytic intermediate phosphoenol-pyruvate, which limited T cell receptor-mediated calcium signaling. Similarly, glucose deprivation inhibited T cell responses in a sarcoma model. T cell responses were restored by blocking glucose uptake in cancer cells, because more glucose was then available for the tumor infiltrating T cells [60].
Figure 3.
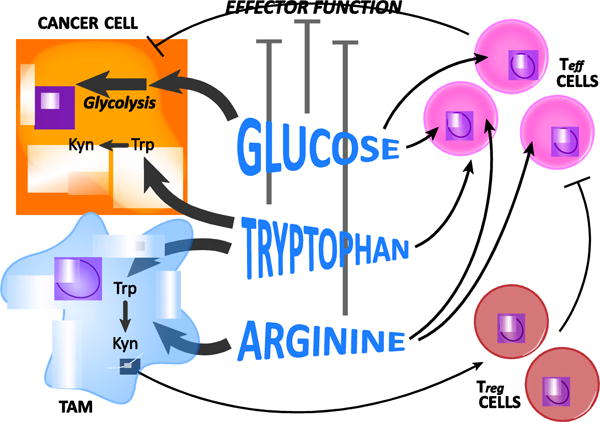
Heterocellular nutrient competition in the tumor microenvironment.
Glucose avid cancer cells impair anti-tumor T effector (Teff) cell effector function by limiting glucose availability. Tryptophan (Trp) avidity and metabolism by cancer cells and tumor associated macrophages (TAM) reduces tryptophan availability in the tumor microenvironment. This directly impairs anti-tumor Teff cells, and indirectly supports pro-tumor regulatory T cells (Treg) via release of the tryptophan metabolite kynurenine (Kyn). Similarly, arginine avidity and metabolism in TAMs depletes arginine in the tumor microenvironment and impairs anti-tumor T cell activity.
Amino Acids
Pancreatic CAFs (presumed to arise from activated pancreatic stellate cells; PSC) excrete the amino acid alanine in response to interaction with pancreatic cancer cells [58] (Fig. 4). This alanine is captured by the pancreatic cancer cells and used to fuel macromolecular biosynthesis. Of note, alanine-derived carbon can outcompete glucose and glutamine-derived carbon to fuel metabolism [58], two important biosynthetic substrates that are limiting in the nutrient austere pancreatic TME [62]. Furthermore, alanine secretion from PSCs requires the induction of autophagy, which is stimulated by cancer cells. And, genetic inhibition of autophagy in the PSCs blunts pancreatic tumor growth. Collectively, these results reveal a pathway of metabolic crosstalk between PSCs and cancer cells, where pancreatic cancer cells send signals to activate autophagy in PSCs, which stimulates access to alanine in the pancreatic TME to support tumor growth. Future studies will be required to determine how nutrients are obtained by PSCs, which also exist in the nutrient austere pancreatic TME. Utilization of metabolic waste products is likely an important contributor, as seen in the ovarian cancer-stroma crosstalk models described below [59, 63].
Figure 4.
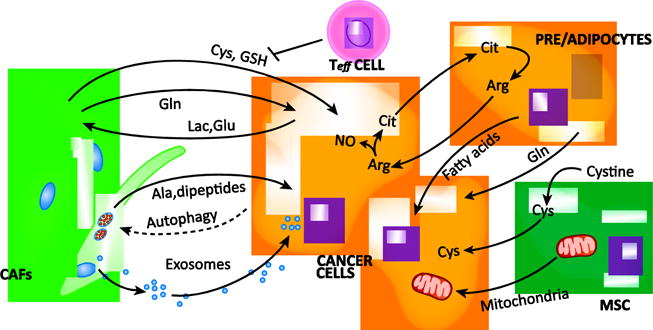
Heterocellular nutrient symbiosis in the tumor microenvironment.
Tumor-associated stromal cell populations engage in cooperative metabolic processes with cancer cells in the tumor microenvironment. Ovarian cancer associated fibroblasts (CAF) have been reported to provide cysteine (Cys) and reduced glutathione (GSH) to combat oxidative stress, a process blocked by T effector (Teff) cells. Ovarian CAFs also provide the cancer cells with glutamine (Gln) and utilize cancer cell-derived lactate (Lac) and glutamate (Glu) to regenerate Gln. In pancreatic cancer, CAFs provide the cancer cells with alanine (Ala) in an autophagy dependent manner. Similarly, breast CAFs provide the cancer cells with autophagy-derived dipeptides. CAF-derived exosomes are another means of nutrient transfer and provide nutrient cargo to prostate and pancreatic cancer cells. Cells of the adipocyte lineage have been reported to provide pancreatic cancer cells with Gln, ovarian and breast cancer cells with fatty acids, and to engage in an arginine (Arg) cycling pathway by way of citrulline (Cit) to activate nitric oxide (NO) production and subsequently glycolysis in the cancer cells. Mesenchymal stem cells have been reported to shuttle mitochondria and/or mitochondrial DNA in leukemia, lung and breast cancer, and to consume the cysteine (Cys) dimer cystine and provide leukemic cells with chemoprotective Cys.
Pancreatic tumors from older, obese patients are marked by the presence of adipocytes and these patients have worse outcomes [64–66]. Based on the observation that pancreatic cancers depend on glutamine metabolism for survival [67, 68], conditioned media from preadipocytes were also found to contain glutamine, which supported pancreatic cancer cell proliferation in glutamine-free growth media [69] (Fig. 4). Based on this, it is tempting to speculate that adipocyte-mediated glutamine release may be a causative factor in the worse survival outcome, though numerous other roles for obesity and inflammation also contribute [70, 71]. Detailed in vivo studies will be required to test this hypothesis.
Similarly, CAFs from ovarian tumors synthesize and release glutamine that is utilized by cancer cells [59] (Fig. 4). This metabolite sharing pathway, like that described for alanine above [58], is stimulated by conditioned media from the cancer cells. It has been hypothesized that this occurs through changes in intracellular fluxes that occur when CAFs consume cancer cell-excreted glutamate and lactate [72], both of which were shown to contribute to the CAF-derived glutamine using isotope tracers. Furthermore, blocking glutamine synthesis in CAFs, glutamine breakdown in ovarian cancer cells, or the combination, slowed tumor growth, with the combination having the greatest effect [59]. Similarly, cultured astrocytes can provide glutamine to brain cancer cells [73]. Glutamine sharing metabolic pathways in pancreatic [69], ovarian [59] and brain cancer [73] suggest that targeting metabolic crosstalk pathways specifically involving glutamine will provide new and additional therapeutic targets to treat cancer.
Adipose stromal cells in the ovarian TME have also been shown to engage in a symbiotic metabolism pathway with ovarian cancer cells, via arginine metabolism [63] (Fig. 4). Cancer cells consume and catabolize arginine to yield citrulline and nitric oxide through inducible nitric oxide synthase (iNOS). The nitric oxide inhibits oxidative phosphorylation and promotes glycolytic activity and proliferation. The citrulline generated in the cancer cells is released into the extracellular space, captured by stromal adipocytes, converted back into arginine, and excreted for cancer cells, thereby propagating this symbiotic metabolic shuttle.
The amino acid cysteine plays an important role in the maintenance of redox balance as one of the three component amino acids in glutathione. Given that cancer cells are often under intense redox stress, particularly when exposed to chemotherapies, there is increased demand for cysteine [74]. In blood, cysteine exists predominantly in the oxidized dimeric form cystine. However, chronic lymphoid leukemia (CLL) cells were found to be unable to readily take up cystine because they express very low levels of the transporter, system X (xCT) [75]. Accordingly, cancer associated bone marrow stromal cells consume cystine and release cysteine, which is then imported into the CLL cells through neutral amino acid transporters (Fig. 4). This cysteine is used to produce glutathione and protects the cancer cells from chemotherapy-induced cell death. In a similar study, ovarian CAFs promoted chemoresistance by releasing cysteine and glutathione to neighboring ovarian cancer cells [76]. The tumor-supporting function of ovarian CAFs is reversed by the presence of CD8+ T cells (Fig. 4). In this context, the T cells do not exhibit direct cytotoxicity against the cancer cells. Rather, they act on the CAFs through interferon-γ (IFNγ) release/signaling, which impairs cysteine and glutathione release from the CAFs, and thus sensitizes the ovarian cancer cells to chemotherapy.
Availability and metabolism of the amino acids tryptophan and arginine represent critical nodes of cell-cell interaction and immune regulation in normal physiology and across many cancer types [33, 77]. Under normal physiological conditions, tryptophan catabolism is important for the maintenance of placental immune privilege [78]. Like other immune regulatory metabolic pathways, tryptophan metabolism is coopted by malignant cells in tumors to repel the immune system. This occurs through two distinct and non-mutually exclusive mechanisms. First, local depletion of tryptophan by cancer cells and macrophages, through uptake and catabolism, suppresses antigen-specific T cell responses [79, 80] (Fig. 3). In addition, tryptophan metabolism produces immunosuppressive catabolites, most notably kynurenine [30], which is an endogenous aryl hydrocarbon receptor (AHR) ligand [81]. Activation of AHR signaling in CD4+ T cells primes their differentiation into the immunosuppressive T regulatory fate [82] and negatively regulates dendritic cell immunogenicity [83]. Collectively, these activities lead to the depletion of local tryptophan and its breakdown into immune-inhibitory AHR agonists, which impair the ability of the immune system to recognize and kill cancer cells. For these reasons, selective inhibitors of the first step in tryptophan metabolism are being tested in clinical trials to boost tumor immune responses [84].
Like tryptophan, the availability of extracellular arginine in the TME dictates the magnitude and duration of the T cell anti-tumor response [33]. Arginine within a tumor is rapidly catabolized by myeloid derived suppressor cells and macrophages creating a state of arginine deficiency. Cytotoxic T cells are sensitive to fluctuations in local amino acid availability, and their effector and thus anti-tumor activity is significantly diminished when arginine is depleted locally [85–87] (Fig. 3). The way in which T cell effector function is shaped by arginine limitation was thought to occur directly through amino acid deprivation, and thus protein biosynthesis mediated cellular exhaustion. However, T cells express recently identified arginine sensors that respond to arginine availability. The presence of arginine leads to arginine-sensor mediated activation of a gene expression program that enhances the bioenergetic profile of T cells, leading to a central memory like T cell state and improved anti-tumor activity [88].
Methods to exploit the arginine metabolism immunoregulatory axis in cancer have received significant clinical interest and are now being explored [33]. The principle strategies include methods to increase arginine in the TME through inhibition of arginine metabolizing enzymes like arginase. However, malignant cells also require arginine. Based on this, methods to deplete arginine in tumors are also being explored. As these studies progress, it will be interesting to see whether manipulating the arginine concentration in the TME has a greater effect on cancer or T cell metabolism, both of which benefit from increased local arginine [63, 85–87, 89, 90].
Fatty Acids
Ovarian cancer cells preferentially metastasize to the omentum, a tissue rich in adipocytes. Once established, the metastatic ovarian cancer microenvironment provides fatty acids to ovarian cancer cells to facilitate tumor growth [91] (Fig. 4). In this context, the fatty acids are used in beta-oxidation to support bioenergetics. Similar results were also found in breast cancer co-culture models, where the cancer cells promoted mobilization of fatty acids from adipocytes, which caused modest pro-proliferative and pro-migratory effects [92] (Fig. 4). In both of these diseases, the reasons for selective use of fatty acids over other bioenergetic nutrients (like glucose and amino acids) remain to be determined. It is tempting to speculate that the preference for fatty acids may owe to a tissue-specific feature in these cancers, which arise near adipose tissue. Accordingly, their metabolism has evolved to anticipate the availability of these nutrients.
Macromolecules and Organelles
In addition to basic molecular building blocks, there are also several reports of larger molecules released by non-cancer cells in the TME that support biosynthetic and bioenergetic needs of the cancer cells. For example, lung CAFs can be distinguished from normal fibroblasts by the release of dipeptides [93] (Fig. 4). This observation led to the finding that lung CAFs exhibit a high degree of basal autophagy, and together with the work on alanine release from pancreatic CAFs [58], suggest that autophagy is a major mechanism by which nutrients are transferred from CAFs to feed cancer cells in a tumor. Prostate and pancreatic CAF-derived exosomes have also been shown to contain nutrient cargo (including a spectrum of amino acids, fatty acids and TCA cycle metabolites) that are taken in and utilized by cancer cells [94, 95] (Fig. 4).
Similar symbiotic processes have also been reported for entire organelles in physiological and pathophysiological processes [96]. A pioneering study in cancer organelle transfer revealed that lung cancer cells depleted of mitochondrial DNA (called rho-zero cells; ρ0), and thus possessing an inactive electron transport chain (ETC), re-acquired ETC activity when co-cultured with bone marrow stromal cells. This occurred via the transfer of mitochondria and/or mitochondrial DNA from the stromal cells to the lung cancer cells [97] (Fig. 4). In a similar study employing two murine ρ0 cancer cell lines, the mitochondria-deficient cancer cell lines form tumors with an increased latency [98]. During the dormant phase, the cancer cells acquire mitochondria and/or mitochondrial DNA from cells of the host animal and form tumors with proficient mitochondria.
Recently, acute myeloid leukemia cells with intact mitochondria were found to acquire additional mitochondria from bone marrow stromal cells in co-culture models and in vivo [99] (Fig. 4). Furthermore, treatment with chemotherapy enhanced mitochondrial transfer. This study provides a more relevant physiological system than the pioneering work described above, illustrating that mitochondrial acquisition is advantageous even in cancers with fully functioning mitochondria. It now remains to be determined which of the various biological roles mediated by mitochondria is the most important.
Metabolites as Signaling Molecules
Beyond their utility in biosynthesis, metabolites also act as signaling molecules through autonomous and non-autonomous mechanisms [100]. In the latter case, metabolites exchanged within the TME can regulate signal transduction and gene expression in neighboring cells. For example, by binding to the protein NDRG3, lactate functions directly as a signaling molecule, which leads to the induction of the angiogenic response [29, 50] (Fig. 2B). This physiological mechanism acts in concert with the canonical HIF-1α response to induce blood vessel formation and is co-opted by tumors. Several additional growth-promoting signaling pathways have also been reported to be affected directly by lactate signaling in cancer [49, 51, 52]. Moreover, abundant lactate in the TME can directly affect immune cells. In melanoma, lactate suppresses natural killer and T cell anti-tumor immune responses [20, 101, 102]. Similarly, tumor-derived lactate drives macrophages toward an immunosuppressive fate [21, 103, 104]. In these contexts, lactate functions directly as a signaling molecule, though the contribution of lactate in this capacity relative to its role in metabolic pathway rewiring and microenvironment acidification have yet to be fully elucidated.
Purinergic signaling plays a role in numerous physiological and pathophysiological processes, not the least of which is tumor immunity [105–108]. Virtually all the cells in the TME express the machinery required to respond to and transduce signaling by adenosine and associated nucleotides (i.e. ATP, ADP, AMP). Generally, nucleotide phosphates are released into the TME either actively by transport or passively by dying cells. Nucleotide phosphates exhibit anti-tumor properties by facilitating antigen presentation on dendritic cells and cancer cell clearance by T cells. The exact cellular source(s) of the nucleotides in different cancer models have not been clearly discerned. In advanced cancers, tumor infiltrating immune cells overexpress nucleotide hydrolases, which covert adenosine nucleotides into the potent immunosuppressive molecule adenosine nucleoside [109, 110]. Of note, immunosuppressive T regulatory cells are the only immune cell that expresses the full complement of surface endonucleases (CD39 and CD73) to convert adenine nucleotides into adenosine [111], and adenosine levels are typically 1-2 orders of magnitude higher in tumors relative to normal tissues [112]. These observations suggest that adenosine nucleotides accumulate in tumor tissue and are converted into adenosine, which suppresses the anti-tumor immune response.
Metabolites derived from reactions in central carbon metabolism are substrates and products of enzymes that regulate the epigenome and thus their abundance regulates gene expression [113]. Notable examples include acetyl-coenzyme A (Ac-CoA) and S-adenosyl-methionine (SAM), which provide acetyl and methyl groups, respectively, that decorate histones and DNA. Pancreatic cancer cells grown in media conditioned by activated PSCs were found to exhibit profound epigenetic changes, most notably increased acetylation-mediated gene activation [114]. Gene signatures involved in anabolic glucose metabolism were significantly activated by acetylation [115]. These changes led to increased glycolytic flux and mitochondrial activity. It is tempting to speculate that these changes lead to increases in the Ac-CoA pools and thus act in a feed forward manner to potentiate an anabolic metabolic program [116]. Moreover, these results may overlap with the finding that activated PSCs provide pancreatic cancer cells with alanine-derived Ac-CoA units [58], suggesting that these metabolites similarly participate in the activation of metabolic programs in pancreatic cancer cells.
Concluding Remarks
Heterocellular interactions shape the metabolic nature of the TME to support tumor growth and evade immune destruction. The pathways and activities mediating these processes are gaining considerable attention from the basic science community and those interested in treating cancer alike. As we detail these alterations, it will be important to remember that cancer cells repurpose many, if not all, of these pathways from normal physiological processes [28–31, 78, 96, 117, 118]. Being mindful of this information will undoubtedly facilitate our understanding of pathophysiologic heterocellular communication pathways in cancer, while also reminding us to consider treatment-associated toxicities. Furthermore, an appreciation for the complex nature and importance of heterocellular communication within tumors warrants a reconsideration of the models being used to study tumor biology and metabolism (see Outstanding Questions). Such models should not operate in isolation and should include interactions between stromal, immune and malignant cell types. Indeed, even simple approaches utilizing these principles have led to important insights [58, 59, 76]. More elegant models, albeit limited in experimental flexibility, have been important in pushing the boundaries of our understanding of tumor metabolism in vivo, illustrating the importance of context [4, 5, 8–10]. Future studies integrating these concepts and techniques will be critical to more clearly elucidate the metabolic role of the TME and associated therapeutic vulnerabilities.
Outstanding Questions.
How do cell/tissue-of-origin features contribute to tumor-stroma and tumor-immune metabolic rewiring?
How should cancer biologists model heterotypic interactions in a facile and relevant experimental setting?
Will heterocellular metabolic pathways provide viable drug targets with a tractable therapeutic window?
Trends.
The tumor microenvironment is characterized by deregulated metabolic properties
Intrinsic features (e.g. genetic programs in cancer cells) and extrinsic characteristics (e.g. oxygen tension, nutrient availability, pH) contribute to the deregulated metabolic profile of a tumor
Malignant cells adapt through symbiotic metabolic interactions with other tumor cells
These processes in tumors are repurposed pathways of normal heterocellular metabolic crosstalk
Nutrient competition in the tumor microenvironment impairs effective anti-tumor immunity
Acknowledgments
We apologize for the omission of any primary citations. We would like to thank Drs. Deepak Nagrath and Christopher Halbrook for their feedback on the manuscript. C.A.L. and A.C.K. are inventors on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting GOT1 as a therapeutic approach. A.C.K. also holds a patent on the autophagic control of iron metabolism and is on the SAB and has ownership interests in Cornerstone Pharmaceuticals and Vescor Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Egeblad M, et al. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nature Reviews Cancer. 2016;16(9):553–565. doi: 10.1038/nrc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson SM, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23(3):517–28. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayers JR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353(6304):1161–5. doi: 10.1126/science.aaf5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuneva MO, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–70. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmona-Fontaine C, et al. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1700600114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christen S, et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016;17(3):837–848. doi: 10.1016/j.celrep.2016.09.042. [DOI] [PubMed] [Google Scholar]
- 9.Hensley CT, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164(4):681–94. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sellers K, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125(2):687–98. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moscat J, et al. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell. 2016;167(3):606–609. doi: 10.1016/j.cell.2016.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valencia T, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26(1):121–35. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 14.DuFort CC, et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys J. 2016;110(9):2106–19. doi: 10.1016/j.bpj.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain RK, et al. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321–46. doi: 10.1146/annurev-bioeng-071813-105259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DuFort CC, et al. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2016;150(7):1545–1557 e2. doi: 10.1053/j.gastro.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi SY, et al. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? J Pathol. 2013;230(4):350–5. doi: 10.1002/path.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brand A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24(5):657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gajewski TF, et al. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol. 2013;25(2):200–5. doi: 10.1016/j.coi.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabrilovich DI, et al. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson MO, et al. Nutrients and the microenvironment to feed a T cell army. Semin Immunol. 2016;28(5):505–513. doi: 10.1016/j.smim.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langston PK, et al. Metabolism Supports Macrophage Activation. Front Immunol. 2017;8:61. doi: 10.3389/fimmu.2017.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geeraerts X, et al. Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front Immunol. 2017;8:289. doi: 10.3389/fimmu.2017.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allaman I, et al. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34(2):76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Yamanishi S, et al. Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am J Physiol Heart Circ Physiol. 2006;290(3):H925–34. doi: 10.1152/ajpheart.01012.2005. [DOI] [PubMed] [Google Scholar]
- 30.Schwarcz R, et al. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13(7):465–77. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felig P. The glucose-alanine cycle. Metabolism. 1973;22(2):179–207. doi: 10.1016/0026-0495(73)90269-2. [DOI] [PubMed] [Google Scholar]
- 32.Anastasiou D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br J Cancer. 2017;116(3):277–286. doi: 10.1038/bjc.2016.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray PJ. Amino acid auxotrophy as a system of immunological control nodes. Nat Immunol. 2016;17(2):132–9. doi: 10.1038/ni.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birsoy K, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508(7494):108–12. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeBleu VS, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16(10):992–1003. 1–15. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sancho P, et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015;22(4):590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 37.Viale A, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–32. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boudreau A, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. 2016;12(10):779–86. doi: 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy KM, et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One. 2013;8(9):e75154. doi: 10.1371/journal.pone.0075154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sotgia F, et al. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 41.Draoui N, Feron O. Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments. Dis Model Mech. 2011;4(6):727–32. doi: 10.1242/dmm.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123(9):3685–92. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sonveaux P, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–42. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guillaumond F, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110(10):3919–24. doi: 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen E, et al. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016;15(6):1144–60. doi: 10.1016/j.celrep.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pisarsky L, et al. Targeting Metabolic Symbiosis to Overcome Resistance to Anti-angiogenic Therapy. Cell Rep. 2016;15(6):1161–74. doi: 10.1016/j.celrep.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dienel GA. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab. 2012;32(7):1107–38. doi: 10.1038/jcbfm.2011.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasparov S. Are Astrocytes the Pressure-Reservoirs of Lactate in the Brain? Cell Metab. 2016;23(1):1–2. doi: 10.1016/j.cmet.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 49.De Saedeleer CJ, et al. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS One. 2012;7(10):e46571. doi: 10.1371/journal.pone.0046571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee DC, et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. doi: 10.1016/j.cell.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 51.Ruan GX, Kazlauskas A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem. 2013;288(29):21161–72. doi: 10.1074/jbc.M113.474619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vegran F, et al. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–60. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- 53.Fiaschi T, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72(19):5130–40. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 54.Zhang D, et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 2015;10(8):1335–48. doi: 10.1016/j.celrep.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Pavlides S, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 56.Rattigan YI, et al. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp Cell Res. 2012;318(4):326–35. doi: 10.1016/j.yexcr.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koukourakis MI, et al. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. 2006;66(2):632–7. doi: 10.1158/0008-5472.CAN-05-3260. [DOI] [PubMed] [Google Scholar]
- 58.Sousa CM, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–83. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang L, et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016;24(5):685–700. doi: 10.1016/j.cmet.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang CH, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162(6):1229–41. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ho PC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162(6):1217–28. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamphorst JJ, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–53. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salimian Rizi B, et al. Nitric oxide mediates metabolic coupling of omentum-derived adipose stroma to ovarian and endometrial cancer cells. Cancer Res. 2015;75(2):456–71. doi: 10.1158/0008-5472.CAN-14-1337. [DOI] [PubMed] [Google Scholar]
- 64.Grippo PJ, et al. Concurrent PEDF deficiency and Kras mutation induce invasive pancreatic cancer and adipose-rich stroma in mice. Gut. 2012;61(10):1454–64. doi: 10.1136/gutjnl-2011-300821. [DOI] [PubMed] [Google Scholar]
- 65.Zyromski NJ, et al. Obesity potentiates the growth and dissemination of pancreatic cancer. Surgery. 2009;146(2):258–63. doi: 10.1016/j.surg.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 66.Rebours V, et al. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN) Clin Cancer Res. 2015;21(15):3522–8. doi: 10.1158/1078-0432.CCR-14-2385. [DOI] [PubMed] [Google Scholar]
- 67.Lyssiotis CA, et al. Pancreatic cancers rely on a novel glutamine metabolism pathway to maintain redox balance. Cell Cycle. 2013;12(13):1987–8. doi: 10.4161/cc.25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meyer KA, et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Rep. 2016;7:144–149. doi: 10.1016/j.bbrep.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Incio J, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016;6(8):852–69. doi: 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Philip B, et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145(6):1449–58. doi: 10.1053/j.gastro.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tajan M, Vousden KH. The Quid Pro Quo of the Tumor/Stromal Interaction. Cell Metab. 2016;24(5):645–646. doi: 10.1016/j.cmet.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 73.Tardito S, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17(12):1556–68. doi: 10.1038/ncb3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang W, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–86. doi: 10.1038/ncb2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang W, et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell. 2016;165(5):1092–105. doi: 10.1016/j.cell.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8(1):74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- 78.Munn DH, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 79.Munn DH, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–72. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Munn DH, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 81.Opitz CA, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 82.Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nguyen NT, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107(46):19961–6. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Platten M, et al. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–40. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 85.Fletcher M, et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015;75(2):275–83. doi: 10.1158/0008-5472.CAN-14-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rodriguez PC, et al. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109(4):1568–73. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rodriguez PC, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171(3):1232–9. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 88.Geiger R, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell. 2016;167(3):829–842 e13. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bowles TL, et al. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int J Cancer. 2008;123(8):1950–5. doi: 10.1002/ijc.23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim RH, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69(2):700–8. doi: 10.1158/0008-5472.CAN-08-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nieman KM, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Balaban S, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1. doi: 10.1186/s40170-016-0163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chaudhri VK, et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res. 2013;11(6):579–92. doi: 10.1158/1541-7786.MCR-12-0437-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao H, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250. doi: 10.7554/eLife.10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Achreja A, et al. Exo-MFA – A 13C metabolic flux analysis to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab Eng. 2017 doi: 10.1016/j.ymben.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rogers RS, Bhattacharya J. When cells become organelle donors. Physiology (Bethesda) 2013;28(6):414–22. doi: 10.1152/physiol.00032.2013. [DOI] [PubMed] [Google Scholar]
- 97.Spees JL, et al. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J. 2008;22(4):1226–36. doi: 10.1096/fj.07-8076com. [DOI] [PubMed] [Google Scholar]
- 98.Tan AS, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21(1):81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 99.Moschoi R, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. 2016;128(2):253–64. doi: 10.1182/blood-2015-07-655860. [DOI] [PubMed] [Google Scholar]
- 100.Lorendeau D, et al. Metabolic control of signalling pathways and metabolic auto-regulation. Biol Cell. 2015;107(8):251–72. doi: 10.1111/boc.201500015. [DOI] [PubMed] [Google Scholar]
- 101.Husain Z, et al. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–95. doi: 10.4049/jimmunol.1202702. [DOI] [PubMed] [Google Scholar]
- 102.Calcinotto A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72(11):2746–56. doi: 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 103.Hutcheson J, et al. Immunologic and Metabolic Features of Pancreatic Ductal Adenocarcinoma Define Prognostic Subtypes of Disease. Clin Cancer Res. 2016;22(14):3606–17. doi: 10.1158/1078-0432.CCR-15-1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen P, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114(3):580–585. doi: 10.1073/pnas.1614035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Antonioli L, et al. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013;13(12):842–57. doi: 10.1038/nrc3613. [DOI] [PubMed] [Google Scholar]
- 106.Idzko M, et al. Nucleotide signalling during inflammation. Nature. 2014;509(7500):310–7. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58(1):58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- 108.Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36(3):293–303. doi: 10.1038/onc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hasko G, et al. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–70. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–20. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 111.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blay J, et al. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997;57(13):2602–5. [PubMed] [Google Scholar]
- 113.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sherman MH, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1620164114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guan KL, Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011;36(2):108–16. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Halestrap AP. Monocarboxylic acid transport. Compr Physiol. 2013;3(4):1611–43. doi: 10.1002/cphy.c130008. [DOI] [PubMed] [Google Scholar]
- 118.Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab. 2012;32(7):1152–66. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]