

Abstract
Cardiac fibrosis is a maladaptive response to various stresses, characterized by increased interstitial collagen deposition and progressive cardiac dysfunction. The transdifferentiation of fibroblasts into myofibroblasts is an essential process in the pathogenesis of cardiac fibrosis. SIRT3, as a mitochondrial NAD+-dependent histone deacetylase, has been demonstrated beneficial in many cardiovascular diseases. However, the specific mechanism of its protective role in cardiac fibrosis needs to be elucidated further. Here, we determined the role of SIRT3 in cardiac fibrosis by subjecting Sirt3-knockout mice to chronic AngII infusion for four weeks in vivo. In this study, the Sirt3-knockout mice developed more serious cardiac fibrosis compared to wild-type controls. In vitro, primary cardiac fibroblasts from Sirt3-knockout mice transdifferentiated into myofibroblasts spontaneously and this phenotype conversion exaggerated after AngII stimulation. The SIRT3-KO myofibroblasts secret more fibrotic mediators including TGF-β to promote cardiac fibrosis. In addition, the overexpression of SIRT3 by lentivirus transfection attenuated myofibroblasts transdifferentiation. We further demonstrated that SIRT3 directly binds to and deacetylates STAT3 to inhibit its activity. Sequentially the downstream factor, known as NFATc2, showed a reduced expression. Taken together, these results revealed that SIRT3 can protect against cardiac fibrosis by inhibiting myofibroblasts transdifferentiation via the STAT3-NFATc2 signaling pathway.
Keywords: SIRT3, transdifferentiation, myofibroblast, STAT3, NFATc2
Introduction
Cardiac fibrosis is characterized by excessive deposition of extracellular matrix with collagen as the main component. It is an internal aspect of a series of heart diseases such as myocardial infarction, hypertension and metabolic cardiomyopathy [1]. The accumulation of interstitium leads to tissue stiffness and cardiac diastolic dysfunction initially. However, persistent stress eventually results in ventricular dilation, combined diastolic and systolic heart failure [2]. Therefore how to prevent, even reverse the progress of cardiac fibrosis, is of great important significance.
Myofibroblasts rarely exist in normal myocardium, which mainly originate from the transdifferentiation of resident mesenchymal fibroblasts in response to fibrogenic stimuli. The myofibroblastic phenotype is marked by expressing α-smooth muscle actin (α-SMA), secreting massive collagen and other proteins [3]. In acute myocardial injury model, the increased proliferation and transdifferentiation of cardiac fibroblasts promote adaptive fibrosis to preserve structural integrity [4]. However, they also persist in mature infarct scars after the initial reparative process probably due to resistance to apoptosis [5]. Thus, understanding the mechanisms of fibroblasts transdifferentiation may facilitate a novel and useful strategy for managing cardiac fibrosis.
SIRT3 is an NAD+-dependent mitochondrial deacetylase, implicated in the maintenance of mitochondrial homeostasis and various cellular processes [6]. SIRT3-KO mice develop spontaneous cardiac hypertrophy and interstitial fibrosis at the age of 8 week, and can be more susceptible to hypertrophic stimuli [7]. They develop age-related hearing loss and are prone to develop cancer [8,9]. Hirschey MD et al. have demonstrated that SIRT3 deficiency accelerated the development of metabolic syndrome on a high-fat diet [10]. However, the underlying cellular and molecular mechanism by which SIRT3 mediate cardiac fibrosis needs to be further explored [11].
NFAT (nuclear factor of activated T cells) transcription factor family contains four isoforms with NFATc2 being the most abundantly expressed in the heart [12]. NFATc2-null mice subjected to pressure overload displayed reduced cardiac fibrosis [13]. STAT3 (Signal transducer and activator of transcription) activation is implicated in various organ dysfunctions, such as pulmonary fibrosis [14] and cardiac hypertrophy [15]. However, whether SIRT3 could influence the activity of NFATc2 and STAT3 during cardiac fibrosis remains to be explored.
Here, in the present study, we investigated the protective role of SIRT3 and its specific mechanism in fibroblasts transdifferentiation. To this end, our results demonstrated that the SIRT3-STAT3-NFATc2 signaling pathway was in charge of alleviating fibroblasts transdifferentiation, which could serve a protective mechanism in AngII-induced cardiac fibrosis.
Materials and methods
Animal model
The animal protocols complied with the Animal Management Rules of the Chinese Ministry of Health (Document no. 55, 2001) and were approved by the Animal Care and Use Committee of Shandong University. SIRT3-KO mice were purchased from the Jackson Laboratories. Wild-type (WT, 129S1/SvImJ) mice were purchased from the Department of Laboratory Animal Science of Peking University (Beijing, China) as controls. All animals were fed with chow diet and maintained under standard lighting (12:12 hour, day-night rhythm), temperature (20°C-22°C), and humidity (50%-60%) conditions. After 1 week of acclimatization, the 8-week old male WT and SIRT3-KO mice were randomly divided into four groups as follows: WT, SIRT3-KO, WT+AngII and SIRT3-KO+AngII, 15 mice in each group. Mice were anesthetized with 2% isoflurane in oxygen, and AngII (Sigma Aldrich, USA, dissolved in sterile ddH2O) was subcutaneously infused at the rate of 2000 ng/kg/min for 28 days by implanting of Osmotic Minipumps (Alzet micro-osmotic pump model 1007D; Alzet DURECT Corporation, Cupertino, CA, USA).
Reagents
Antibodies against SIRT3 (rabbit monoclonal) (D22A3) [5490], STAT3 (mouse monoclonal) (124H6) [9139], Phospho-Stat3 (Tyr705) (rabbit monoclonal) (D3A7) [9145], NFATc2 (rabbit monoclonal) (D43B1) [5861], Acetylated-Lysine (rabbit polyclonal) [9441] were purchased from Cell Signaling Technology (CST, UK). Primary antibodies against collagen type I (rabbit polyclonal) [ab34710], collagen type III (rabbit polyclonal) [ab7778], TGF-β1 (rabbit polyclonal) [ab92486], Fibronectin (rabbit polyclonal) [ab2413] were obtained from Abcam. GAPDH (mouse monoclonal) [BM1623], α-SMA (mouse monoclonal) [BM0002] were bought from Boster.
Histology and immunohistochemistry
Hearts were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5-μm intervals. Hematoxylin and eosin was performed using standard procedures, and Masson’s Trichrome and Picrosirius red staining were carried out to evaluate extracellular matrix deposition. For immunohistochemistry, briefly, the sections were incubated with primary antibodies against collagen I, collagen III overnight at 4°C. Then after being washed with phosphate-buffered saline, the slides were incubated with secondary antibodies at 37°C for 1 h. The results were analyzed by an automated image analysis system (Image-Pro Plus, Version 7.0, Media Cybernetics, Silver Spring, MD, USA).
Cardiac fibroblasts isolation, culture, transfection and treatment
Cardiac fibroblasts were isolated from the hearts of 1-3-day-old WT and SIRT3-KO mice as previously described [16]. In brief, the ventricular tissue was excised and digested with type II collagenase (0.08%; Sigma-Aldrich Co.). The cell suspensions were plated in a cell culture dish, and most CFs adhered to the dish after 90 minutes. The cells were then maintained in DMEM (GIBCO) containing 10% FBS and penicillin-streptomycin (100 units/mL & 100 µg/mL, Invitrogen, Carlsbad, CA) in a 5% CO2 humidified incubator at 37°C. We used CFs at passage 2 or 3. For AngII stimulated fibroblast-to-myofibroblast transformation, cardiac fibroblasts were treated with AngII (10-7 mmol/L) for 48 h. The lentivirus of SIRT3 was purchased from Shanghai Genechem Co., LTD. For all lentivirus experiments, the virus was used at a multiplicity of infection (MOI) of 10. For the RNA interference part, the cardiac fibroblasts were transfected with 50 nM of small interfering RNA (siRNA) specific for STAT3 with Lipofectamine 2000 (Invitrogen) for 48 h. The sequence of siRNASTAT3 is (5’-3’) GGGUCUCGGAAAUUUAACATTUGUUAAAUUUCCGAGACCCTT.
Immunofluorescence
Cardiac Fibroblasts were fixed in 4% paraformaldehyde for 25 min and then perforated with 0.5% Triton X-100 for 20 min. After being blocked in 1% bovine serum albumin at room temperature for 30 min, the cells were then incubated with primary antibodies including anti-α-SMA, Collagen I and NFATc2 overnight at 4°C in a humidified chamber. After that, secondary antibodies (1:200, zhongshanjinqiao, China) were added. Nuclei were stained with DAPI (Beyotime, Haimen). The images were acquired and analysed by a laser scanning confocal microscope (LSM710; Carl Zeiss, Germany).
Co-immunoprecipation
Co-immunoprecipitations were made on raft complexes, using 1 μg of STAT3 or acetylated-Lysine monoclonal antibody covalently cross-linked on pan-rabbit IgG Dynabeads. Purified rabbit IgG was used for the negative controls. Antigen capture was performed by overnight incubation of IgG-loaded beads with a raft sample. Magnetic beads carrying the immune complexes were subsequently washed five times in washing buffer (10 mM Tris-HCl, pH 7.4, NaCl 150 mM, 1% Triton X-100, 60 mM octyl-D-glucopyranoside). Precipitated proteins were eluted by boiling the beads for 5 min in RIPA buffer (150 mM NaCl, 25 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.5% sodium deoxycholate, 0.5% NP 40, 0.1% sodium dodecyl sulfate [SDS]). Samples were subjected to immunoblotting using a primary antibody against STAT3 or SIRT3 and a secondary antibody that recognized only native IgG.
Western blot analysis
Proteins were harvested from cell lysates and freshly dissected mouse hearts using cell lysis buffer. Western blot analysis was performed as described previously [17]. The PVDF membranes were blocked in 5% non-fat milk for 2 hours. Then, the blocked membranes were incubated overnight with the primary antibodies against collagen I, collagen III, TGF-β1, Fibronectin, SIRT3, STAT3, P-STAT3 (Tyr-705), NFATc2, GAPDH and α-SMA. After being washed with Tris-buffered saline containing 0.1% Tween, the membranes were incubated with the rabbit or mouse secondary antibodies for 1 hour at room temperature. The protein bands were visualized with enhanced chemiluminescence (Millipore) and the protein levels were detected using an Image Quant LAS4000 chemiluminescence reader (GE, USA). Relative protein levels were quantified by using Image J software.
Results
SIRT3 deficiency aggravated AngII-induced murine interstitial fibrosis
To investigate the role of SIRT3 in cardiac fibrosis, we subjected both the wild-type (WT) and SIRT3-knockout (SIRT3-KO) mice to chronic AngII infusion for four weeks. Firstly, we measured SIRT3 protein expression in the hearts of WT and WT+AngII group. The immunoblot analysis showed that SIRT3 levels decreased by 21% after AngII infusion compared to their WT controls (Figure 1A, 1B). And it was confirmed that SIRT3-KO mice didn’t express protein SIRT3 at all (Figure 1C). Furthermore, we compared the effect of AngII infusion on WT and SIRT3-KO mice. HE staining showed more cardiomyocytes loss and expanded cross-sections area in the hearts of mice subjected to AngII, especially the SIRT3-KO group (Figure 1D). Masson’s trichrome and Picrosirius red staining indicated that the fibrosis area fraction in SIRT3-KO mice was higher compared to WT mice at baseline and increased further after chronic AngII infusion (Figure 1E-G). The AngII-treated SIRT3-KO mice exhibited most collagen accumulation showed by immunohistochemical staining of collagen I and collagen III (Figure 2A-C). And we also confirmed this phenomenon by western blot (Figure 2D-I). Altogether, these results indicated that SIRT3 might be involved in preventing collagen deposition and cardiac fibrosis.
Figure 1.

SIRT3 deficiency aggravates AngII-induced murine interstitial fibrosis. A-C. Immunoblot analysis of the short form of the SIRT3 was performed in WT and AngII-infused WT and SIRT3-KO mice hearts. GAPDH expression was used as loading control. Bar graph shows the quantification of SIRT3 measured by densitometry analysis. D. Hematoxylin and eosin staining of cardiac sections from control or AngII-treated WT and SIRT3-KO mice showed cardiomyocytes dropout. E-G. Masson’s trichrome and Picro-Sirius red staining of the myocardium was performed to detect fibrosis. The graph showed the quantification of interstitial fibrosis in sham or AngII-treated WT and SIRT3-KO mice. (n=5) Scale bar: 20 μm. The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
Figure 2.

SIRT3-KO mice exhibit more collagen deposition. A-C. Immunostaining of collagen I and collagen III. The bar graphs showed quantitative analysis of collagen I or collagen III positive area. Scale bar: 50 μm. D-I. Western blot analysis in controls and AngII-infused WT and SIRT3-KO murine hearts. GAPDH expression was used as loading control. Bar graphs showed the quantification of Collagen I, Collagen III, Fibronectin, α-SMA and SIRT3 measured by densitometry analysis. (n=5) The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
SIRT3-KO fibroblasts transdifferentiated into myofibroblasts spontaneously
We observed more α-smooth muscle actin (α-SMA), the marker of well-differentiated myofibroblasts, expressed in the SIRT3-KO mice (Figure 2D). To further investigate the role of SIRT3 in fibroblasts transdifferentiation, we isolated primary neonatal fibroblasts from WT and SIRT3-KO mice (Figure 3A). Immunofluorescence staining of vimentin (positive) and von Willebrand factor (negative) were used to identify the fibroblasts (Figure 3B). Immunoblots and immunofluorescence analysis indicated that SIRT3-KO fibroblasts exhibited more α-SMA, collagen I and fibronection expression, which suggested fibroblasts transdifferentiate into myofibroblasts in a spontaneous way (Figure 3C-F). Then we infected fibroblasts from WT neonatal mice with LV. SIRT3, and the expression of α-SMA and collagen I decreased as SIRT3 expressed highly (Figure 3E, 3F). In summary, our data demonstrated that SIRT3 plays a vital role in inhibiting fibroblast transdifferentiation.
Figure 3.
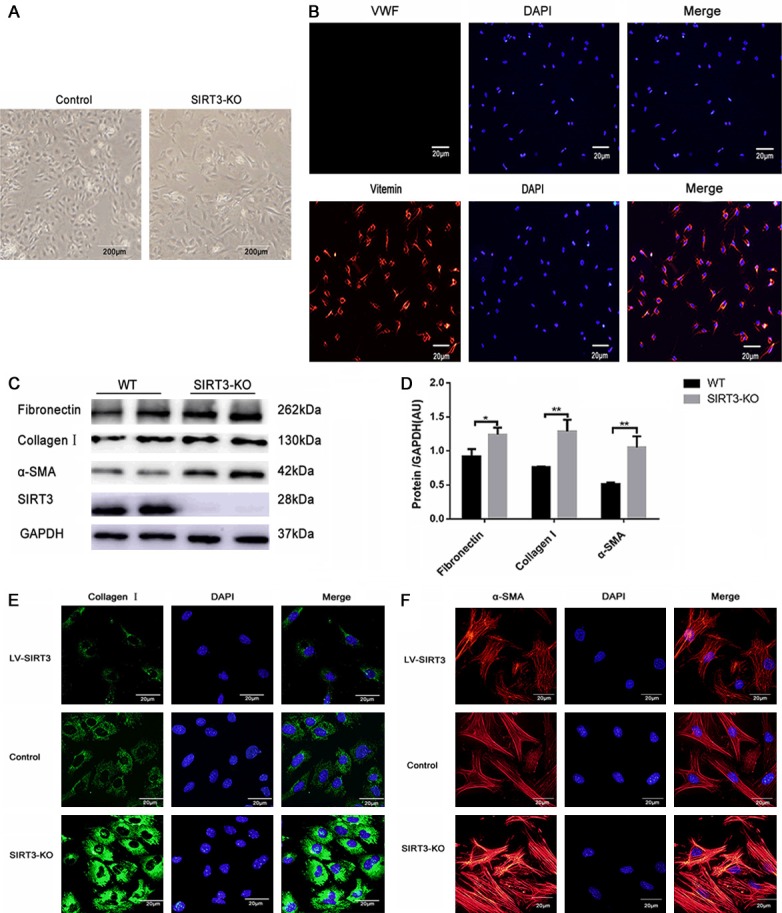
SIRT3 protected against fibroblasts transdifferentiation in vitro. A. The morphological difference of the second-passage WT and SIRT3-KO fibroblasts under an optical microscope. Scale bar: 200 μm. B. Identification of primary cultured cardiac fibroblasts (CFs) by immunofluorescence staining (green) for vimentin and negative for von Willebrand factor staining. Nuclei were stained with DAPI. Scale bar: 20 μm. C, D. Western blot analysis of fibronectin, α-SMA, and Collagen Ⅰ was performed on primary WT and SIRT3-KO cardiac myofibroblasts. GAPDH expression was used as loading control. The graphs showed the quantification of Collagen I, Fibronectin, α-SMA measured by densitometry analysis. (n=5). E, F. Representative immunofluorescence images staining for α-SMA and Collagen I. Nuclei were stained with DAPI. Scale bar: 20 μm. The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
SIRT3-KO fibroblasts acquired more evident phenotype conversion with AngII stimulation
We treated WT and SIRT3-KO fibroblasts with AngII. SIRT3-KO fibroblasts with AngII stimulation manifested maximum α-SMA expression and concomitant elevation of collagen, fibronectin (Figure 4A-E). The immunoblot analysis of SIRT3-KO murine hearts and myofibroblasts showed higher TGF-β compared to their controls (Figure 4F-I). TGF-β plays critical role in fibrogensis by promoting fibroblasts transdifferentiation. It suggested that it existed positive feedback between TGF-β stimulation and fibroblast transdifferentiation with SIRT3 deficiency.
Figure 4.

SIRT3-KO fibroblasts obtained more evident phenotype conversion after AngII stimulation. A-E. Western blot analysis of fibrotic markers was performed in controls and AngII-treated WT and SIRT3-KO cardiac fibroblasts. GAPDH expression was used as loading control. The graphs showed the quantification of Collagen I, Collagen III, Fibronectin and α-SMA measured by densitometry analysis. (n=5). F-I. Immunoblots show that SIRT3-KO murine hearts and myofibroblasts secret more fibrogenic TGF-β. The graphs showed the quantification of TGF-β. GAPDH expression was used as loading control. The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01.
SIRT3 deacetylated and inhibited the activation of STAT3
Though these results demonstrated that SIRT3 could regulate fibroblasts transdifferention, the underlying mechanism remains unclear. It noticed that there was higher ratio of p-STAT3/STAT3 in SIRT3-KO myofibroblasts, implying more STAT3 activation compared to their WT controls (Figure 5A, 5B). And SIRT3 overexpression reversed this phenomenon (Figure 5C, 5D). We proposed the hypothesis that SIRT3 could deacetylate STAT3 to inhibit its activity. We found SIRT3 deficiency was associated with elevated acetylation level of STAT3 by immunoprecipitation. Co-immunoprecipitation displayed that there exists interaction between SIRT3 and STAT3 (Figure 5E-G). It’s concluded that SIRT3 could deacetylate STAT3 and inhibit its activity.
Figure 5.

SIRT3 can deacetylate STAT3 and regulate its activity. A, B. Western blot analysis of P-STAT3 and STAT3 in WT and SIRT3-KO myofibroblasts. GAPDH expression was used as loading control. The bar graph represents protein quantification measured by densitometry analysis. (n=5). C, D. Western blot analysis of P-STAT3 and STAT3 of WT and LV.SIRT3 myofibroblasts. GAPDH expression was used as loading control. The bar graph represents protein quantification measured by densitometry analysis. (n=5). E. Immunoblot analysis of STAT3 and ac-STAT3 was compared between WT and SIRT3-KO myofibroblasts by immunoprecipitation. F. Immunoblot analysis of STAT3 and ac-STAT3 was compared between WT and LV.SIRT3 myofibroblasts by immunoprecipitation. G. Cellular extracts were immunoprecipitated with STAT3 antibody and analyzed with anti-SIRT3. The data are presented as the means ± SEM of three independent experiments.*P<0.05, **P<0.01.
NFATc2 expression and nuclear translocation were increased in SIRT3-KO myofibroblasts
Furthermore, we observed increased NFATc2 level in SIRT3-KO myofibroblasts and decreased NFATc2 level in LV. SIRT3 myofibroblasts, compared to the wild type controls (Figure 6A-D). And SIRT3 deficiency promoted nuclear translocation of transcription factor NFATc2 in fibroblasts (Figure 6E). Following the infection of si-STAT3 in SIRT3-KO fibroblasts, the NFATc2 expression was reduced markedly as well as α-SMA (Figure 6F, 6G). These results indicated STAT3-NFATc2 pathway was involved in the myofibroblasts transdifferentiation.
Figure 6.

STAT3-NFATc2 signaling pathway was involved in the SIRT3-mediated fibroblast transdifferention. A-D. Immunoblot analysis of NFATc2 in WT, SIRT3-KO and LV.SIRT3 myofibroblasts. GAPDH expression was used as loading control. The bar graph represents protein quantification measured by densitometry analysis. (n=5). E. Representative immunofluorescence images indicated the subcellular location of NFATc2 in WT and SIRT3-KO myofibroblasts. Nuclei was stained with DAPI. Scale bar: 20 μm. The data are presented as the means ± SEM of three independent experiments. *P<0.05, **P<0.01. F, G. Immunoblot analysis of NFATc2, STAT3 and α-SMA of siRNA-STAT3 groups. GAPDH expression was used as loading control. Bar graph represents protein quantification measured by densitometry analysis. (n=5).
Discussion
In our AngII-induced cardiac fibrosis model, SIRT3 protein level was downregulated, which is in line with other experimental models of cancer, obesity and diabetes [16,18,19]. The mechanism of reduced SIRT3 expression during stress is still unclear. It may be related to the change of cellular NAD/NADH ratio or reduced PGC-1α expression [10,16]. Among the seven different SIRT isoforms in mammals, SIRT3 is the only analog that has been shown to be related to human longevity extension. We also found that Sirt3-knockout mice developed aggravated cardiac interstitial fibrosis and cardiac dysfunction compared to the controls. The reduction of SIRT3 was found in aged people with sedentary life style and its suppressed activation is associated with metabolic syndrome [16,20]. Due to its important role implicated in aging-related diseases, discovery of specific activators is of great importance. Recently, a group of researchers have developed some SIRT3 activators to attenuate cardiac hypertrophy and organ fibrosis [16]. We are also looking forward to developing more specific activators and correlative human research for further verification.
In vitro, we discovered spontaneous transdifferentiation of primary SIRT3-KO fibroblasts defined by the expression of α-SMA and collagen I. The SIRT3-KO fibroblasts obtained more evident phenotype conversion after AngII stimulation. The well-transformed SIRT3-KO myofibroblasts secret more TGF-β than wild-type controls apart from apparent morphological alterations. The activated myofibroblasts produce fibronectin in response to TGF-β, thus providing scaffold for the subsequent deposition of fibrillar collagen types I and III [21,22]. Our results were in accordance with one previous discovery that SIRT3 deficiency induced TGF-β expression [23]. As a main fibrogentic mediator, TGF-β promoted fibroblasts phenotype conversion strongly in cell adhesion and integrin-dependent way [24,25]. That is, the secreted TGF-β by myofibroblasts could stimulate cardiac fibroblasts in return to form an autocrine feedback to maintain the fibrogenic microenvironment.
We tested the activity of STAT3 by measuring p-STAT3/STAT3 ratio and found higher p-STAT3/STAT3 ratio in SIRT3-KO myofibroblasts, which is in agreement with the former result in pulmonary arterial hypertension model [26]. However, it needs to be elucidated further about the specific interaction mechanism. Some study reported that STAT3 acetylation is necessary for angiotensin II-induced pro-fibrotic responses in kidney [27]. This inspired us into deeper thinking. As a mitochondrial deacetylase, SIRT3 deacetylates various kinds of proteins, thereby influencing their activities. STAT3 functions as a transcription factor, shuttling between mitochondrion, cytoplasm and nucleus. We assumed SIRT3 may deacetylate STAT3 to affect the activation. Co-immunoprecipitation analysis revealed that SIRT3 can directly bind to STAT3. Further immunoprecipitation in WT and SIRT3-KO myofibroblasts showed that the ac-STAT3 was dramatically elevated in SIRT3-KO myofibroblasts, whereas it is nearly undetectable after successful LV-SIRT3 transfection into WT fibroblasts. Based on the previous study carried out on SIRT1 and STAT3 [28], we speculated that SIRT3 may regulate the phosphorylation of STAT3 by altering its acetylation status. The limitation is that we didn’t test the precise lysine residue of STAT3 which SIRT3 interacts with, but we plan to perform mass spectrometry for further study.
Among all the four classical isoforms, the role of NFATc2 in cardiac fibroblasts transdifferentiation is ambiguous [29]. We observed increased nuclear translocation and expression of NFATc2 in the transformation of primary SIRT3-KO fibroblasts. This implies that NFATc2 is involved in the SIRT3-mediated cardiac fibroblasts transdifferentiation. Nevertheless, the mechanism by which NFATc2 was influenced remains unknown. In a previous study performed in pulmonary artery smooth muscle cells [26,30], they concluded that NFATc2 expression and activation were affected by its upstream regulator STAT3. Transfecting SIRT3-KO fibroblasts with si-STAT3 resulted in drastic downregulation of NFATc2 along with obvious decreased expression of α-SMA. Considering these findings, we put forward that STAT3-NFATc2 signaling pathway was involved in the SIRT3-mediated fibroblast transdifferentiation.
In conclusion, these results together revealed that SIRT3 can deacetylate STAT3 so as to inhibit its activity and downstream NFATc2 expression and nuclear translocation. SIRT3-STAT3-NFATc2 signaling pathway participates in alleviating myofibroblasts transdifferentiation, which plays a protective role in AngII-induced cardiac fibrosis.
Statistical analysis
All the results were obtained from at least three independent experiments. Statistical analysis was performed using GraphPad 6.0 Prism (version 5.00 for Windows, GraphPad Software). Data are presented as the mean ± standard deviation. Student’s t-test was used to assess between-group differences and the results were considered statistically significant at P<0.05.
Acknowledgements
Thanks to Dr. Xiao Wu and Na Li (The key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education and Chinese Ministry of Health, The State and Shandong Province Joint Key Laboratory of Translational Cardiovascular Medicine, Qilu Hospital of Shandong University, Jinan, Shandong, China) for technical support in digital confocal microscopy core facility. And we are also very grateful to Dr. Jie Xiao (Department of Cardiology, Qilu Hospital, Shandong University) for her kind assistance in primary cardiac fibroblasts culture. This work was supported by National Natural Science Foundation of China (grant number 81070076, 81170135, 81641013), the National 973 Basic Research Program of China (grant number 2012CB722406), and Science Foundation of Qilu hospital of Shandong (grant number 2016QLQN17).
Disclosure of conflict of interest
None.
References
- 1.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–574. doi: 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 5.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 6.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman JC, He W, Verdin E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J Biol Chem. 2012;287:42436–42443. doi: 10.1074/jbc.R112.404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV Jr, Kahn CR, Verdin E. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656. doi: 10.1038/ncomms7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 13.Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH, De Windt LJ. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 14.Pedroza M, Le TT, Lewis K, Karmouty-Quintana H, To S, George AT, Blackburn MR, Tweardy DJ, Agarwal SK. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016;30:129–140. doi: 10.1096/fj.15-273953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ, Dawn B. Deletion of Interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res. 2016;118:1918–1929. doi: 10.1161/CIRCRESAHA.116.308688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108:14608–13. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T, Li J, Liu J, Li N, Wang S, Liu H, Zeng M, Zhang Y, Bu P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-beta/Smad3 pathway. Am J Physiol Heart Circ Physiol. 2015;308:H424–434. doi: 10.1152/ajpheart.00454.2014. [DOI] [PubMed] [Google Scholar]
- 18.Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell. 2011;19:416–428. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng H, Vaka VR, He X, Booz GW, Chen JX. High-fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847–1856. doi: 10.1111/jcmm.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, Passarino G, De Benedictis G. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, Barnes JL. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horiguchi M, Ota M, Rifkin DB. Matrix control of transforming growth factor-beta function. J Biochem. 2012;152:321–329. doi: 10.1093/jb/mvs089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E, Gupta MP. SIRT3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating glycogen synthase kinase 3beta. Mol Cell Biol. 2015;36:678–692. doi: 10.1128/MCB.00586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 25.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 26.Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Cote J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation. 2011;123:1205–1215. doi: 10.1161/CIRCULATIONAHA.110.963314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni J, Shen Y, Wang Z, Shao DC, Liu J, Kong YL, Fu LJ, Zhou L, Xue H, Huang Y, Zhang W, Yu C, Lu LM. P300-dependent STAT3 acetylation is necessary for angiotensin II-induced pro-fibrotic responses in renal tubular epithelial cells. Acta Pharmacol Sin. 2014;35:1157–1166. doi: 10.1038/aps.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herum KM, Lunde IG, Skrbic B, Florholmen G, Behmen D, Sjaastad I, Carlson CR, Gomez MF, Christensen G. Syndecan-4 signaling via NFAT regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J Mol Cell Cardiol. 2013;54:73–81. doi: 10.1016/j.yjmcc.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, Michelakis ED. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014;20:827–839. doi: 10.1016/j.cmet.2014.08.011. [DOI] [PubMed] [Google Scholar]