

Abstract
New therapies are needed for patients with Hodgkin and Non-Hodgkin lymphomas that are resistant to standard therapies. Lack of response to standard cytotoxic chemotherapy and relapse after autologous stem cell transplant are especially poor prognostic indicators. Chimeric antigen receptor (CAR) T cells are emerging as a novel treatment for these patients. In this review, we discuss the efficacy, toxicity, limitations, and future directions of CAR T-cell therapy for lymphoma. Clinical trial data of anti-CD19 CAR T cells have demonstrated potent activity against multiple B-cell lymphoma subtypes, including diffuse large B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma. Importantly, anti-CD19 CAR T cells have impressive activity against chemotherapy-refractory lymphoma. Some patients with chemotherapy-refractory diffuse large B-cell lymphoma have obtained durable complete remissions lasting 2 years or more after anti-CD19 CAR T-cell infusions. CAR T cells are associated with significant toxicities, including cytokine-release syndrome and neurologic toxicity. New target antigens for CAR T-cell therapies of lymphoma, including CD20 and CD22 for B-cell lymphomas and CD30 for Hodgkin and T-cell lymphomas, are being studied. CAR T cells are being tested in industry-sponsored multicenter clinical trials, and CAR T cells will likely become a standard lymphoma therapy soon. Future directions include improved toxicity management, CAR design changes aimed at improving the efficacy to toxicity ratio of CAR T cells, and combination therapies of CAR T cells plus other novel agents.
Keywords: chimeric antigen receptor, Hodgkin lymphoma, Non-Hodgkin lymphoma
New therapies are needed for relapsed and refractory lymphoma
Non-Hodgkin lymphoma (NHL) is a heterogeneous malignancy that comprises approximately 4% of all cancers in the United States1. While the development of the anti-CD20 monoclonal antibody rituximab has improved treatment outcomes for multiple B-cell NHL subtypes2–4, prognoses for patients with chemotherapy-refractory or multiply-relapsed B-cell NHL remain poor5–8. The standard approach for DLBCL patients with residual or relapsed lymphoma after first-line therapy is salvage chemotherapy followed by auto-HSCT9, 10. Diffuse large B-cell lymphoma (DLBCL) is curable with front-line anthracycline-based regimens, but a substantial number of patients have relapsed or primary-refractory lymphoma6, 11. Primary-refractory DLBCL, which is defined as a response of less than partial remission to a first-line regimen, has especially poor outcomes; only 23–29% of patients with DLBCL that is refractory to first-line chemotherapy respond to second-line treatment5, 12, with a median progression-free survival (PFS) of 3 months12. Patients with DLBCL refractory to second-line therapy have a median overall survival (OS) of only 4 months5, with an estimated response to third-line chemotherapy of only 14%13. The prognosis of patients with DLBCL that has relapsed after autologous stem cell transplantation (auto-HSCT) is also grim, with median OS of 10 months7, 14. While the majority of patients with follicular lymphoma (FL) respond to first-line chemo-immunotherapy15–17, these conventional therapies are not curative. A subgroup of approximately 20% of FL patients with progressive lymphoma within 24 months of initial treatment have poor long-term outcomes, with a median OS of less than 5 years8. Mantle cell lymphoma (MCL) has traditionally been managed with aggressive chemotherapy regimens followed by auto-HSCT in younger patients, an approach with significant toxicity and without curative potential18, 19. The introduction of novel targeted therapies for MCL, such as bortezomib20, lenalidomide21, and ibrutinib22, yield responses that are usually not durable beyond 2 years23.
Standard first-line therapy for peripheral T-cell lymphoma (PTCL) consists of anthracycline-based regimens24, 25. Unfortunately, the response to therapy with T-cell lymphomas is generally not as robust as it is in B-cell NHL, with a lower overall survival in T-NHL compared with B-NHL26–28. Only a minority of cases of relapsed or refractory PTCL respond to second-line therapy with targeted agents29–31.
Hodgkin lymphoma (HL) is often cured with first-line anthracycline-based regimens with or without radiation therapy, but approximately half of patients with relapsed or primary-refractory HL do not obtain durable remissions with the standard treatment of salvage chemotherapy followed by auto-HSCT32, 33. In recent years, novel therapies have provided alternatives for these patients. Brentuximab monotherapy has shown an overall response rate (ORR) of 75%34, but the 5-year PFS has been reported at 22%35. While reported ORR are high with the checkpoint inhibitors nivolumab36, 37 and pembrolizumab38, complete remissions (CRs) are the minority of the responses, and median duration of response to nivolumab is just 13.1 months39.
While allogeneic hematopoietic stem cell transplantation (allo-HSCT) offers the potential for cure for multiple lymphoma subtypes, transplant-related mortality remains high, and long-term sequelae, including chronic graft versus host disease (cGVHD), have the potential for substantial negative impact on quality of life40–43.
Chimeric Antigen Receptors
Chimeric antigen receptors (CARs) are fusion proteins that include both antigen recognition moieties and T-cell activation domains (Figure 1)44–47. The antigen-recognition domain is usually a single-chain variable fragment (scFv) derived from a monoclonal antibody44–46, 48–51. T-cell signaling domains are of two types, costimulatory domains such as CD28 and 4–1BB and T-cell activation domains such as CD3ζ48, 52–55. The gene encoding a CAR is transferred to T cells by using a gene therapy vector, which is usually a replication-incompetent gamma-retrovirus56–61, or a lentivirus62–64, although transposon systems have also been used65. Most CAR T-cell infusions are preceded by a conditioning chemotherapy regimen, because extensive evidence from murine studies indicates that recipient lymphocyte depletion enhances the activity of adoptively-transferred T cells66–69. The first preclinical studies of CAR T cells as cancer treatment were conducted in the early 1990s70, and the first preclinical studies of anti-CD19 CARs were published in 200371, 72. In this review, we will discuss the clinical results of CAR T-cell therapy for lymphoma and discuss therapeutic challenges and ongoing improvements of CAR T-cell therapy. CAR T cells have been shown to have impressive anti-lymphoma activity, but also substantial toxicity.
Figure 1: Chimeric antigen receptor structure.
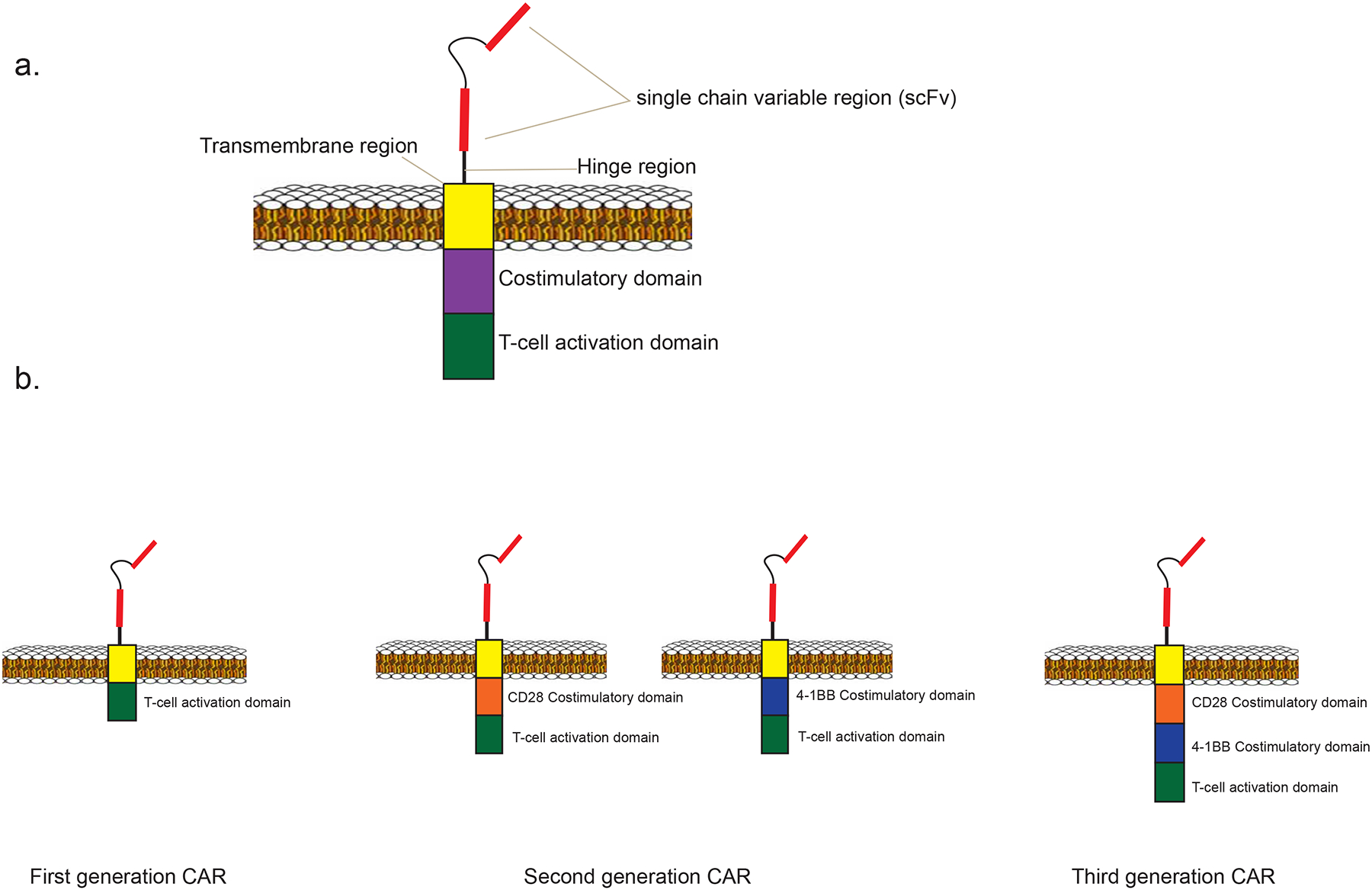
1a. A diagram is shown of a second-generation CAR, including a single chain variable region (scFv), hinge and transmembrane regions, a costimulatory domain, and a T-cell activation domain. 1b. First generation CARs incorporate a T-cell activation domain but lack a costimulatory domain. Second generation CARs include both a T-cell activation domain and a single costimulatory domain. In this example the costimulatory domain is either CD28 or 4–1BB. Third generation CARs incorporate two costimulatory domains, as well as a T-cell activation domain.
Early Clinical Results with CAR T-cell Therapy in Indolent B-cell Lymphomas
The first published report of biological activity of anti-CD19 CAR T cells in humans was published in 201073. This report described a patient with multiply-relapsed FL who was treated at the U.S. National Cancer Institute (NCI)73. This patient received a conditioning chemotherapy regimen of cyclophosphamide and fludarabine followed by an infusion of autologous T cells transduced with an anti-CD19 CAR containing a CD28 costimulatory domain; this CAR was designated FMC63–28Z48. The CAR T-cell infusion was followed by a course of high-dose interleukin-2 (IL-2)73. The patient achieved a partial remission (PR) lasting 7 months. The lymphoma then progressed, and the patient was re-treated in an identical manner with chemotherapy, anti-CD19 CAR T cells and IL-2. This patient’s lymphoma continues to be progression-free 7 years after this second treatment, and the patient is free of any toxicities74. He has received no further lymphoma therapy since his second CAR T-cell infusion in March, 2010. The patient also experienced prolonged B-cell aplasia due to the CAR T-cell-mediated eradication of all cells carrying the CD19 antigen73. This same group reported successful treatment of more patients with indolent lymphomas74. Patients were treated with cyclophosphamide and fludarabine, followed by infusion of anti-CD19 CAR T-cells and high-dose IL-274. This report confirmed that CAR T cells can eradicate lymphoma masses, as well as malignancy in the bone marrow74. This report also described a correlation between serum interferon gamma (IFNγ) and tumor necrosis factor-α and overall toxicity experienced by patients74. This syndrome of toxicity after CAR T-cell infusions that was associated with elevated serum cytokine levels was named cytokine-release syndrome (CRS)57, 58, 75.
Other early studies of anti-CD19 CAR T cells for lymphoma were also conducted. Two patients with follicular lymphoma treated on a clinical trial of T cells transduced with an anti-CD19 CAR incorporating a CD3ζ T-cell activation domain, without a costimulatory domain, did not achieve anti-lymphoma responses76. In another trial, 5 patients with relapsed or refractory B-cell NHL simultaneously received 2 different CAR T cell products. These patients received T cells transduced with a CAR containing an anti-CD19 sFv and a CD3ζ T-cell activation domain along with T cells transduced with a CAR that was identical except for addition of a CD28 costimulatory domain59. While the best malignancy response was stable disease, the CAR T cells incorporating a costimulatory domain exhibited superior in vivo expansion and persistence59.
CAR T-cell therapy for diffuse large B-cell lymphoma and indolent lymphomas
A trial that evaluated the FMC63–28Z CAR48 at the NCI was the first to demonstrate effectiveness of anti-CD19 CAR T cells against DLBCL56. Patients received lymphocyte-depleting cyclophosphamide and fludarabine chemotherapy followed by a single infusion of CAR T cells. The study included 11 patients with NHL: 4 with DLBCL not otherwise specified (NOS), 4 with PMBL, one with splenic marginal zone lymphoma, one with Richter’s transformation of chronic lymphocytic leukemia (CLL), and one with indolent NHL not otherwise specified. Infiltration of malignant lymph nodes by CAR T cells was demonstrated56. Notably, remissions were achieved in patients with chemotherapy refractory DLBCL NOS or PMBL, populations of patients with poor prognoses and few standard treatment options5, 12, 77, 78. High levels of blood CAR T-cell persistence were shown to not be necessary for sustained remissions. While B-cell aplasia was common, recovery of normal polyclonal B cells was demonstrated in absence of lymphoma recurrence56.
Thirty-two patients with NHL received infusions of T cells transduced with an anti-CD19 CAR containing a 4–1BB costimulatory domain79. The CAR T cells were produced in a defined 1:1 ratio of CD4+/CD8+ cells with the intent of administering a more homogenous and possibly more efficacious cell product. Responses were achieved in all lymphoma subtypes including DLBCL, transformed follicular lymphoma (TFL), FL, and MCL, with an ORR of 63% and a CR rate of 33%79. Twenty patients receiving fludarabine as part of their conditioning chemotherapy achieved an ORR of 72% and a CR rate of 50%, compared to an ORR of 50% and CR rate of 8% among 12 patients not receiving fludarabine79. Patients receiving fludarabine-containing conditioning chemotherapy regimens experienced greater CAR T-cell expansion and higher serum levels of IL-15 and IL-7 than patients receiving chemotherapy regimens without fludarabine prior to the cell infusion79. Higher CAR T-cell doses posed added risk of CRS and neurotoxicity79. Two deaths, due to pontine hemorrhage and gastrointenstinal hemorrhage, occurred at the 2 × 107 CAR+ T cells/kg dose, leading the investigators to administer lower cell doses in all subsequent patients79. Poor CAR T-cell proliferation in patients who did not receive fludarabine was found in some patients to be associated with development of a cytotoxic T-cell immune response directed against the CAR transgene79. Similar immune responses against the CARs have been reported from other clinical trials of NHL76 and ALL64, 80.
Recent results from the NCI demonstrated that a low-dose cyclophosphamide plus fludarabine conditioning regimen administered prior to infusion of anti-CD19 CAR T cells was sufficient to deplete lymphocytes and increase recipient serum cytokines, including IL-15 and IL-761. Twenty-two patients with various NHL subtypes received low-dose cyclophosphamide and fludarabine followed by an infusion of 1, 2 or 6 × 106 CAR+ T cells/kg. The ORR was 73% and the CR rate was 55% for the total study group61. Nineteen patients had one of the various types of DLBCL, and the ORR rate among this group was 68%, with 47% CRs61. The low-dose chemotherapy used in this study would not be expected to cause significant antimalignancy activity against chemotherapy-refractory DLBCL. Anti-CD19 CAR T cells produced durable remissions in this study, with ongoing CR durations of 24 to 7 months at the time of publication61. Of the 12 CRs obtained in this study, 11 are currently ongoing61. Twelve-month PFS for all participants was 63%. This study showed that CRs were possible in patients with c-myc translocations and in so-called “double-hit” or c-myc and blc-2 translocated DLBCL61. High peak IL-15 levels were associated with high peak blood CAR+ cell levels61. Both peak blood CAR+ cell levels and high levels of serum IL-15 were associated with patients obtaining lymphoma responses of PR or CR61. Toxicities related to CRS and to neurologic effects were manageable with supportive care and intensive care in some cases. Transfusion-dependent thrombocytopenia occurred in only 2 of 22 patients, and most patients did not experience a neutrophil count less than 500/μL61. The most prominent toxicities were neurologic with 55% of patients experiencing Grade 3 or 4 neurologic toxicities that resolved in all cases61. Recovery of normal polyclonal B cells in patients in CR with absence of malignancy recurrence was demonstrated61.
Recent data from the University of Pennsylvania confirmed that anti-CD19 CAR T cells with a 4–1BB costimulatory domain (CTL019) have significant antimalignancy efficacy in various NHL subtypes, including DLBCL, FL, and MCL, with an ORR of 68% and PFS of 62% at 11.7 months81.
Recent results have demonstrated the safety and feasibility of administration of anti-CD19 CAR T cells following auto-HSCT for patients with DLBCL, TFL and MCL82. These results included data derived from two trials of anti-CD19 CAR central memory-enriched T cells82. Published clinical trial results of CARs targeting CD19 as a treatment for lymphoma are summarized in Table 1. Anti-CD19 CAR T-cell clinical trials that are currently recruiting lymphoma patients are listed in Table 2.
Table 1:
Selected published clinical trials of autologous anti-CD19 CAR T-cell therapy for patients with NHL
| Publication | Lymphoma subtypes included | Costimulatory domain | Conditioning chemotherapy | Cell dose | ORR and CR rates in NHL patients* |
|---|---|---|---|---|---|
| Jensen et al., ASBMT 201076 | FL | None | Flu 25 mg/m2 daily for 5 days starting after the first of multiple CAR T-cell infusions | 4–5 escalating doses of 1 × 108 – 2 × 109 cells/m2 | ORR: 0/2 (0%) |
| Kochenderfer et al., Blood 201073 | FL | CD28 | 60 mg/kg Cy daily for 2 days followed by Flu 25 mg/m2 daily for 5 days | 1 × 108 CAR+ T cells one day, followed by 3 × 108 CAR+ T cells the following day | ORR: 1/1 (100%) CRR: 0/1 (0%) |
| Savoldo et al., J. Clin Invest 201159 | DLBCL TFL PCNSL, systemic relapse |
One product without a costimulatory domain and one product with a CD28 costimulatory domain | None | 1–2 infusions of two simultaneous cell products in escalating doses of 2 × 107 – 2 × 108 cells/m2 | ORR: 0/5 (0%) |
| Kochenderfer et al., Blood 201274 | FL SMZL |
CD28 | 60 mg/kg Cy daily for 2 days followed by Flu 25 mg/m2 daily for 5 days | 0.3 – 3.0 × 107 CAR+ T cells/kg | ORR: 3/3 (100%)** CRR: 0/3 (0%) |
| Kochenderfer et al., JCO 201556 | DLBCL PMBL SMZL indolent NHL NOS DLBCL transformed from CLL |
CD28 | Cy total dose either 120 or 60 mg/kg followed by Flu 25 mg/m2 daily for 5 days | 1 – 5 × 106 CAR+ T cells/kg | ORR: 8/9 (89%)*** CRR: 5/9 (56%) |
| Wang et al., Blood 201682 | DLBCL MCL |
One clinical trial (NHL1) of a CAR not incorporating a costimulatory domain and one clinical trial (NHL2) of a CAR incorporating a CD28 costimulatory domain | Auto-HSCT conditioninĝ | First generation CAR trial (NHL1): 25, 50, or 100 × 106 total T cells per infusion Second generation CAR trial (NHL2): 50 or 200 × 106 CAR+ T cells per infusion |
NHL1 ORR:^^ 7/8 (88%) NHL1 CRR: 5/8 (63%) NHL2 ORR:^^^ 8/8 (100%) NHL2 CRR: 8/8 (100%) |
| Turtle et al., Sci Transl Med 201679 | Aggressive B-cell NHL TFL PMBL MCL FL |
4–1BB | One of 4 regimens:
|
2 × 105, 2 × 106, or 2 × 107 CAR+ T cells/kg | ORR: 19/30 (63%) CRR: 10/30 (33%) |
| Locke et al., Mol Ther 201783 | DLBCL | CD28 | Cy 500 mg/m2 IV daily for 3 days and Flu 30 mg/m2 daily for 3 days | 1 – 2 × 106 CAR+ T cells/kg | ORR: 5/7 (71%) CRR: 4/7 (57%) |
| Kochenderfer et al. JCO 201761 | DLBCL TFL FL PMBL MCL |
CD28 | Cy 300 mg/m2 or Cy 500 mg/m2 IV daily for 3 days and Flu 30 mg/m2 daily for 3 days | 1, 2 or 6 × 10 6 CAR+ T cells/kg | ORR: 16/22 (73%) CRR: 12/22 (55%) |
Auto-HSCT: autologous stem cell transplant. CLL: chronic lymphocytic leukemia. Cy: cyclophosphamide. CRR: completion remission rate. DLBCL: diffuse large B-cell lymphoma. Flu: fludarabine. FL: follicular lymphoma. MCL: mantle cell lymphoma. NHL: Non-Hodgkin lymphoma. NOS: Not otherwise specified. ORR: overall response rate. PMBL: primary mediastinal B-cell lymphoma. PCNSL: primary central nervous system lymphoma. SMZL: splenic marginal zone lymphoma. TFL: transformed follicular lymphoma.
Response rates take into account only patients with NHL treated on study. Patients with other B-cell malignancies treated in each trial are not included in overall response rate.
One patient’s response was not evaluable due to death on study.
Two patients’ response not evaluable, one due to death on study and one due to noncompliance with follow up.
All patients received bis-chloroethylnitrosourea, etoposide, cytarabine, and melphalan high-dose chemotherapy as conditioning and received CAR T-cell infusions following Auto-HSCT.
Prior to Auto-HSCT, 3 patients were in CR and 2 patients were in PR.
Prior to Auto-HSCT, 6 patients were in CR and 2 patients were in PR.
Table 2:
Open Clinical Trials of anti-CD19 CAR T cells for Lymphoma
| Trial identifier | Trial title | Lymphoma subtype | Center; location | Comments |
|---|---|---|---|---|
| NCT02529813 | CD19+ CAR T Cells for Lymphoid Malignancies | CD19+ B-cell NHL | University of Texas MD Anderson Cancer Center; Houston, Texas, United States | |
| NCT02030834 | Phase IIa Study of Redirected Autologous T Cells Engineered to Contain Anti-CD19 Attached to TCRz and 4–1BB Signaling Domains in Patients With Chemotherapy Relapsed or Refractory CD19+ Lymphomas | DLBCL FL TFL MCL |
University of Pennsylvania; Philadelphia, Pennsylvania, USA | |
| NCT01865617 | Laboratory Treated T Cells in Treating Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia, Non-Hodgkin Lymphoma, or Acute Lymphoblastic Leukemia | CD19+ B-cell NHL | University of Washington Cancer Consortium; Seattle, Washington, USA | |
| NCT02842138 | Autologous CD19 CAR T Cells in Relapsed or Refractory B-cell Lymphoma | CD19+ B-cell NHL | Peking University Cancer Hospital; Beijing, China | |
| NCT02976857 | A Phase 1 Study Evaluating Safety and Efficacy of C-CAR011 Treatment in DLBCL Subjects (C-CAR011) | DLBCL | People’s Hospital of Jiangsu Province; Nanjing City, Jiangsu, China | |
| NCT02081937 | CART-19 Immunotherapy in Mantle Cell Lymphoma | MCL | Chinese PLA General Hospital; Beijing, China | |
| NCT02728882 | Study Evaluating the Efficacy and Safety With CAR-T for Recurrent or Refractory Diffuse Large B Cell Lymphoma (EECBL) | DLBCL | The First Affiliated Hospital of Anhui Medical University; Hefei, Anhui, China | |
| NCT02659943 | T Cells Expressing a Fully-human Anti-CD19 Chimeric Antigen Receptor for Treating B-cell Malignancies | CD19+ B-cell NHL | National Cancer Institute; Bethesda, Maryland, USA | |
| NCT01087294 | Administration of anti-CD19-chimeric-antigen-receptor-transduced T cells from the original transplant donor to patients with recurrent or persistent B-cell malignancies after allogeneic stem cell transplantation | All B-cell NHL |
National Cancer Institute; Bethesda, Maryland, USA | Allogeneic hematopoietic stem cell transplant donors are the T-cell source. |
| NCT01430390 | In Vitro Expanded Allogeneic Epstein-Barr Virus Specific Cytotoxic T-Lymphocytes (EBV-CTLs) Genetically Targeted to the CD19 Antigen in B-cell Malignancies | CD19+ B-cell NHL | Memorial Sloan Kettering Cancer Center; New York, New York, USA | Donor-derived allogeneic CAR T cells |
| NCT02652910 | Memory-enriched CAR-T Cells Immunotherapy for B Cell Lymphoma (MeCAR) | CD19+ B-cell NHL | Xinqiao Hospital of Chongqing; Chongqing, China | |
| NCT01853631 | Activated T-Cells Expressing 2nd or 3rd Generation CD19-Specific CAR, Advanced B-Cell NHL, ALL, and CLL (SAGAN) (SAGAN) | CD19+ B-cell NHL | Houston Methodist Hospital and Texas Children’s Hospital; Houston, Texas, USA | |
| NCT02247609 | Evaluation of 4th Generation Safety-designed CAR T Cells Targeting High-risk and Refractory B Cell Lymphomas (4SCAR19273) | CD19+ B-cell NHL | Peking University Cancer Hospital; Beijing, China | |
| NCT02624258 | Pilot Study of Non-Viral, RNA-Redirected Autologous T Cells in Patients With Refractory or Relapsed Hodgkin Lymphoma | HL | Children’s Hospital of Philadelphia | |
| NCT02431988 | Evaluation of CAR19 T-cells as an Optimal Bridge to Allogeneic Transplantation (COBALT) | CD19+ DLBCL | University College London Hospital; London, United Kingdom | |
| NCT02050347 | Activated T Lymphocytes Expressing CARs, Relapsed CD19+ Malignancies Post-Allo HSCT(CARPASCIO) | CD19+ NHL | Houston Methodist Hospital and Texas Children’s Hospital; Houston, Texas, USA | |
| NCT02650999 | Phase I/II Study of Pembrolizumab in Patients Failing to Respond to or Relapsing After Anti-CD19 Chimeric Antigen Receptor Modified T Cell Therapy for Relapsed or Refractory CD19+ Lymphomas | FL DLBCL MCL |
University of Pennsylvania; Philadelphia, Pennsylvania, USA | Eligible patients must have received CTL019/CTL119 CAR T cells at the University of Pennsylvania. |
| NCT02706405 | JCAR014 and Durvalumab in Treating Patients With Relapsed or Refractory B-cell Non-Hodgkin Lymphoma | DLBCL PMBL tDLBCL |
University of Washington Cancer Consortium; Seattle, Washington, USA | |
| NCT02348216 | A Phase 1–2 Multi-Center Study Evaluating KTE-C19 in Subjects With Refractory Aggressive Non-Hodgkin Lymphoma (ZUMA-1) (ZUMA-1) | DLBCL PMBL TFL |
Multi-center study, international; sponsored by Kite Pharma, Inc. | |
| NCT02601313 | A Phase 2 Multicenter Study Evaluating Subjects With Relapsed/Refractory Mantle Cell Lymphoma (ZUMA-2) | MCL | Multi-center study, all sites in USA; sponsored by Kite Pharma, Inc. | |
| NCT02926833 | A Study Evaluating KTE-C19 in Combination With Atezolizumab in Subjects With Refractory Diffuse Large B-Cell Lymphoma (DLBCL) (ZUMA-6) | DLBCL | Multi-center study, all sites in USA; sponsored by Kite Pharma, Inc. | |
| NCT02445248 | Study of Efficacy and Safety of CTL019 in Adult DLBCL Patients (JULIET) | DLBCL | Multi-center study, international; sponsored by Novartis Pharmaceuticals | |
| NCT02631044 | Study Evaluating the Safety and Pharmacokinetics of JCAR017 in B-cell Non-Hodgkin Lymphoma (NHL) | DLBCL PMBL tDLBCL FL grade 3B MCL |
Multi-center study, all sites in USA; sponsored by Juno Therapeutics, Inc. |
DLBCL: diffuse large B-cell lymphoma. FL: follicular lymphoma. MCL: mantle cell lymphoma. NHL: Non-Hodgkin lymphoma. tDLBCL: Diffuse large B-cell lymphoma transformed from indolent histology. TFL: transformed follicular lymphoma.
Information accessed from www.clinicaltrials.gov accessed on March 23, 2017.
Multicenter Industry Sponsored Clinical Trials Show That CAR T-cell Therapies Can be Applied to Large Numbers of Patients
FMC63–28Z-expressing CAR T cells48, 56, 61 have been designated axicabtagene ciloleucel for commercial development. A phase I multicenter trial of axicabtagene ciloleucel demonstrated feasibility of cell manufacturing and high overall responses rates (71%) in a small group of 7 patients with DLBCL either refractory to last chemotherapy or relapsed within 12 months of auto-HSCT83. CRS and neurotoxicities were substantial, but could be managed across centers83. Activity was shown in both the GCB and non-GCB subtypes of DLBCL83. A phase II multicenter study of 101 patients with refractory aggressive NHL receiving axicabtagene ciloleucel reported an ORR of 82% and CR rate of 54%, demonstrating that CAR T-cell therapy can successfully be delivered to large numbers of patients84, 85. At a median follow up of 8.7 months, the ongoing response rate was 44% with a CR rate of 39%85. Early results of a multicenter trial of an anti-CD19 CAR T-cell product with a 4–1BB costimulatory domain, JCAR017, showed an ORR of 80% in 28 NHL patients with 60% attaining a CR, with no severe CRS and manageable neurologic toxicity86. CTL019, CAR T cells expressing an anti-CD19 CAR with a 4–1BB costimulatory moiety, is now being evaluated in an international multicenter study for patients with DLBCL. Interim results including 51 patients with three or more months of follow-up demonstrate an ORR of 59% and CR rate of 43%, with median duration of response not reached87.
Anti-CD19 CAR T cell therapies for lymphoma progressing after allogeneic stem cell transplantation
Patients with progressive B-cell malignancies after allo-HSCT have very limited treatment options88, 89. Relapsed B-cell malignancies after allo-HSCT is often treated with infusions of unmanipulated allogeneic lymphocytes from the original transplant donor (DLI)90, 91. DLIs have inconsistent efficacy and often cause graft-versus-host disease (GVHD), an immunologic attack against normal recipient tissues92. Transducing allogeneic T cells to express anti-CD19 CARs might have an advantage over standard DLIs by specifically targeting malignant cells. Administration of allogeneic anti-CD19 CAR T cells from an allo-HSCT recipient’s stem cell donor has been demonstrated to be feasible and safe in two clinical trials93–95.
In one trial, eight patients with acute lymphoblastic leukemia (ALL) or CLL who had prior allo-HSCT received virus-specific cytotoxic T cells transduced with an anti-CD19 CAR incorporating a CD28 costimulatory domain94. Two of 6 evaluable patients exhibited objective anti-malignancy activity94. Toxicities were mild, and, interestingly, the allogeneic anti-CD19 CAR T cells did not induce GVHD94.
Another trial of allogeneic anti-CD19 CAR T cells evaluated allogeneic T cells expressing the previously described FMC63–28Z CAR48, 93, 95. Twenty patients with various B-cell malignancies, including ALL, CLL, DLBCL, and MCL were treated with donor-derived anti-CD19 CAR T cells95. Unlike most CAR T-cell clinical trials, conditioning chemotherapy was not administered. Responses were observed in all groups95. Importantly, no patient developed acute GVHD, and only one patient had progression of previously-existing chronic GVHD95. Because this study did not include chemotherapy, it clearly demonstrated the anti-malignancy activity of anti-CD19 CAR T cells alone93, 95. The pronounced lack of GVHD seen in this trial compared with standard DLIs is compelling and not fully explained. Persistence of CAR T cells was shown to be limited to 1 to 2 months, which is the most likely explanation for the lack of GVHD95. Very rapid induction of remissions occurred on this trial; we hypothesize that remissions were induced and CAR T cells were eliminated before GVHD could occur95. Subsequently, murine studies supported this hypothesis by showing CD28-costimulated CAR T cells undergo increased activation followed by exhaustion and deletion when stimulated with CD19; deletion of the T cells protected against GVHD96. Other investigators are using gene-editing techniques to produce CAR T cells that lack T-cell receptor genes97. The goal of these studies is to use CAR T cells from unrealated cell donors that are not HLA-matched with the recipient97. In another clinical trial of anti-CD19 CAR T cells in the transplant setting, a transposon system was used to genetically-modify T cells for use as adjuvant therapy after autologous or allogeneic hematopoietic stem cell transplantation65.
Targeting lymphoma antigens other than CD19
There are 2 main reason to target antigens other than CD19 for treatment of lymphoma. First, many types of lymphoma including Hodgkin lymphoma, T-cell lymphomas, and even some cases of B-cell lymphoma, do not express CD1998. Second, treatment of CD19+ lymphomas with T cells might fail if CD19 expression is lost from some or all of the lymphoma cells after anti-CD19 CAR T-cell therapy. For these reasons, investigators are developing CARs targeting antigens other than CD1960, 76, 99–102.
The success of the CD20-targeting monoclonal antibody rituximab makes CD20 an attractive target for CAR T-cell therapy in B-cell NHL. Loss of CD20 expression following rituximab therapy103, 104 may represent a possible challenge in anti-CD20 CAR T cell therapy. Two patients with DLBCL received T cells transduced with a first-generation anti-CD20 CAR immediately following auto-HSCT, making the response to the CAR T-cell infusion difficult to evalaute76. One patient remained in an ongoing CR for 9 years, not uncommon from transplant alone, at time of publication and the other patient achieved a CR lasting 19 months76. A clinical trial was conducted using T cells transduced through plasmid electroporation to express an anti-CD20 CAR with both CD28 and 4–1BB costimulatory domains. Patients with MCL and FL received conditioning chemotherapy with cyclophosphamide and 3 infusions of anti-CD20 CAR T cells followed by 14 days of subcutaneous IL-299. Two patients with no evaluable lymphoma prior to cell infusion remained in CR, and one patient had an objective PR. CAR T cells persisted for 9 to 12 months99.
A third study was conducted with T cells transduced with a lentiviral vector to express an anti-CD20 CAR with a 4–1BB costimulatory domain100. This trial included 7 patients with heavily pretreated DLBCL, who were given variable chemotherapy conditioning regimens prior to receiving escalating doses of CAR T cells over 3–4 days100. Five out of 6 evaluable patients had an objective response100. One patient who entered the study in partial remission achieved an ongoing CR lasting 14 months at time of publication100. This patient received no conditioning chemotherapy prior to cell infusion. This patient had detectable anti-CD20 CAR T cells in the blood more than 6 months after cell infusion. CRS toxicities were observed in 4 patients, with notably delayed occurrence 3–8 weeks after cell infusion. Other notably adverse events included delayed tumor lysis syndrome and 2 episodes of gastrointestinal (GI) hemorrhage, one resulting in death, in patients with GI tract involvement of lymphoma100. Results of a phase II study of the same anti-CD20 CAR in DLBCL and indolent NHL showed an ORR of 82% and a CR rate of 55%101. Responses occurred in one patient with FL and one patient with MCL101. Clinical results are early, but anti-CD20 CAR T cells have significant therapeutic potential.
Because the kappa (κ) light chain antigen is expressed on κ-restricted B-cell lymphomas, but is not expressed on all normal B cells, κ is an attractive target for CAR T-cell therapy as complete B-cell aplasia should be avoided. In a phase I trial, 7 patients with various NHL subtypes that expressed κ-light chain, including DLBCL, TFL, MCL and lymphoplasmacytic lymphoma (LPL), were treated with a κ light chain-targeted CAR60. Cyclophosphamide conditioning was administered prior to CAR T-cell infusions when patients were not already lymphopenic60. One patient achieved a PR and 2 patients achieved CRs. One CR was ongoing 32 months after CAR T-cell infusion. No adverse events were attributable to CRS60.
The B-cell marker CD22 presents another target for B-cell malignancies. Anti-CD22 CAR T cells have undergone preclinical development and are currently being tested in early clinical trials for acute lymphoid leukemia and NHL105, 106. Clinical trials including patients with CD22-expressing NHL are ongoing (NCT02721407, NCT02315612, NCT02794961, NCT02935153, NCT02903810).
Targeting CD30 in Hodgkin Lymphoma and Peripheral T-cell lymphoma
CD30 is a transmembrane receptor and a member of the TNF receptor superfamily107. The malignant Reed-Sternberg cells of HL strongly express CD30108. The efficacy of targeting CD30 to treat HL34, 109–112 and PTCL113–115 has been demonstrated in multiple clinical trials of the antibody-drug conjugate brentuximab vedotin.116 A minority of HL patients receiving brentuximab monotherapy are progression-free at 5-year follow-up35, suggesting that CD30-targeting treatments could be improved to increase the durability of remissions.
First-generation anti-CD30 CAR T cells were developed in the 1990s. Preclinical models demonstrated lysis of CD30-expressing cell lines in vitro117, 118. Notably, the presence of soluble CD30 did not attenuate cytolysis, suggesting that CD30 that is shed into the blood from HL cells would not inhibit the efficacy of anti-CD30 CAR T cells in vivo117. Epstein Barr virus-specific cytotoxic T cells transduced with an anti-CD30 CAR have been shown to have activity against CD30+ tumor cell lines in vitro as well as in an in vivo xenograft mouse model119, 120.
A phase I clinical trial of anti-CD30 CAR T cells with a CD28 costimulatory domain for patients with HL and ALCL demonstrated responses of 1 CR and 1 PR in a group of 9 patients, 7 with HL and 2 with ALCL121, 122. No patients received conditioning chemotherapy, so the responses were clearly attributable to the anti-CD30 CAR T cells. There was no evidence of CRS122. Another phase I trial tested an anti-CD30 CAR with a 4–1BB costimulatory domain102. Eighteen patients (17 with HL and one with cutaneous ALCL) were treated on this trial; patients received a variety of chemotherapy conditioning regimens prior to cell infusion102. Seven patients had a response of PR, with a median PFS of 6 months102. CRS toxicities similar to those seen in clinical trials of anti-CD19 CAR T cells occurred. CAR T-cell persistence was less than 8 weeks in this study, but tumor biopsies showed efficient trafficking of T cells to lymphoma sites102. Published clinical trial results of CARs targeting lymphoma antigens other than CD19 are summarized in Table 3, and currently open and recruiting clinical trials of CAR T cells targeting antigens other than CD19 are listed in Table 4.
Table 3:
Selected published clinical trials of CAR T cells targeting antigens other than CD19 for patients with lymphoma
| CAR target antigen | Lymphoma subtypes included | Costimulatory domain | Conditioning chemotherapy | Cell Dose | ORR and CR rate | |
|---|---|---|---|---|---|---|
| Jensen et al., ASBMT 201076 | CD20 | DLBCL | None | Patients underwent Auto-HSCT 28–37 days prior to CAR T-cell infusion | 3 escalating doses of 1 × 108 – 1 × 109 CAR+ T cells/m2 | ORR: 2/2 (100%)* CRR: 2/2 (100%)* |
| Till et al., Blood 201299 | CD20 | MCL FL |
CD28 and 4–1BB | Cy 1000 mg/m2 on Day −2. | 3 cell infusions 2–5 days apart at incremental doses of 1 × 108, 1 × 109, and 3.3 × 109 cells/m2 | ORR: 3/3 (100%)** CRR: 2/3 (67%)** |
| Wang et al., Clin Immunol 2014100 | CD20 | DLBCL | 4–1BB | One of 4 regimens:
|
3–5 escalating daily doses of cells, total dose 0.36 – 2.35 × 107 CAR+ T cells/kg | ORR: 5/6 (83%)*** CRR: 1/6 (17%)*** |
| Zhang et al., Signal Transduction and Targeted Therapy 2016101 | CD20 | DLBCL FL MCL PCMZL |
4–1BB | One of 6 regimens: °
|
3–5 escalating daily doses of cells, total dose 0.41 – 1.46 × 107 CAR+ T cells/kg | ORR: 9/11 (82%) CRR: 6/11 (55%) |
| Ramos et al., J Clin Invest 201660 | Kappa (κ) light chain | DLBCL TFL LPL |
CD28 |
|
1.7 × 107 – 1.9 × 108 CAR+ T cells/m2 | ORR: 3/7 (43%) CRR: 2/7 (29%) |
| Wang et al., Clin Cancer Res 2017102 | CD30 | HL C-ALCL |
4–1BB | One of 7 regimens:
|
1.1 – 2.1 × 107 CAR+ T cells/kg | ORR: 7/18 (39%) CRR: 0/18 (0%) |
Auto-HSCT: Autologous stem cell transplant. C-ALCL: cutaneous anaplastic large cell lymphoma. CLL: chronic lymphocytic leukemia. CHODE: cyclophosphamide, doxorubicin, vincristine, dexamethasone, etoposide. COD: cyclophosphamide, vincristine, dexamethasone. COED: cyclophosphamide, vincristine, etoposide, dexamethasone. Cy: cyclophosphamide. CRR: completion remission rate. DLBCL: diffuse large B-cell lymphoma. EAMC: etoposide, cytarabine, mustargen, cyclophosphamide. EBV: Epstein-Barr Virus. FC: fludarabine and cyclophosphamide. ESHAP: methylprednisolone, etoposide, carboplatin, high-dose cytosine arabinoside. Flu: fludarabine. FL: follicular lymphoma. GEMC: gemcitabine, epirubicin, mustargen, cyclophosphamide. GMC: gemcitabine, mustargen, cyclophosphamide. GMMC: gemcitabine, mitoxantrone, mustargen, cyclophosphamide. MAMC: mitoxantrone, cytarabine, mustargen, cyclophosphamide. LPL: lymphoplasmacytic lymphoma. ORR: overall response rate. NHL: Non-Hodgkin lymphoma. PC: nab-paclitaxel and cyclophosphamide. PCMZL: primary cutaneous marginal zone lymphoma. TFL: transformed follicular lymphoma.
Response represents combined effect of Auto-HSCT followed by CAR T-cell infusions. Of two treated patients, one patient remained in CR for 9 years at time of publication and the other patient achieved a CR lasting 19 months.
Two patients with no evaluable disease prior to cell infusion remained in CR; one patient had an objective partial remission.
One patient not evaluable due to death on study.
Specifics of regimens not fully defined.
Table 4:
Open Clinical Trials of CAR T cells for lymphoma targeting antigens other than CD19
| Trial identifier | Trial title | CAR Antigen | Lymphoma subtype | Center; location | Comments |
|---|---|---|---|---|---|
| NCT02965157 | Pilot Study of Anti-CD20-CAR-engineered T Cells in Patients With Chemotherapy Resistant or Refractory CD20+ Lymphoma | CD20 | CD20+ B-cell lymphoma | Chinese Academy of Medical Sciences Tumor Hospital; Beijing, China | |
| NCT02721407 | Anti-CD22 CAR-T Therapy for CD19-refractory or Resistant Lymphoma Patients (MendCART) | CD22 | CD19+/CD22+ B-cell NHL | Xinqiao Hospital; Chongqing, China | Eligible patients must have received anti-CD19:TCRz:CD28 CAR-T cells at Xinqiao Hospital |
| NCT02259556 | CD30-directed Chimeric Antigen Receptor T (CART30) Therapy in Relapsed and Refractory CD30 Positive Lymphomas (CART30) | CD30 | CD30+ HL and NHL | Chinese PLA General Hospital; Beijing, China | |
| NCT03049449 | T Cells Expressing a Fully-Human Anti-CD30 Chimeric Antigen Receptor for Treating CD30-Expressing Lymphomas | CD30 | CD30+ HL and NHL | National Cancer Institute; Bethesda, Maryland, USA | |
| NCT02274584 | CAR T Cells Targeting CD30 Positive Lymphomas (4SCAR30273) | CD30 | CD30+ lymphoma | University of Florida; Gainesville, Florida, United States, Peking University Cancer Hospital; Beijing, China |
|
| NCT02690545 | Study of CD30 CAR for Relapsed/Refractory CD30+ HL and CD30+ NHL | CD30 | CD30+ HL and NHL | Lineberger Comprehensive Cancer Center at University of North Carolina - Chapel Hill; Chapel Hill, North Carolina, United States | |
| NCT02958410 | A Clinical Research of CD30-Targeted CAR-T in Lymphocyte Malignancies | CD30 | CD30+ Lymphoid malignancy | Southwest Hospital of Third Military Medical University; Chongqing, China | |
| NCT02663297 | Administration of T Lymphocytes for Prevention of Relapse of Lymphomas | CD30 | HL, CD30+ NHL, or CD30+ LPD | Lineberger Comprehensive Cancer Center at University of North Carolina - Chapel Hill; Chapel Hill, North Carolina, United States | Anti-CD30 CAR T cells are administered following autologous stem cell transplant |
| NCT02706392 | Genetically Modified T-Cell Therapy in Treating Patients With Advanced ROR1+ Malignancies | ROR1 | ROR1+ MCL | University of Washington Cancer Consortium; Seattle, Washington, USA |
HL: Hodgkin lymphoma. LPD: lymphoproliferative disorder. MCL: Mantle cell lymphoma. NHL: Non-Hodgkin lymphoma.
Information accessed from www.clinicaltrials.gov accessed on March 23, 2017.
Summary of CAR T-cell Toxicities
Toxicity is currently the biggest barrier to more effective CAR T-cell therapies for lymphoma. Toxicities associated with CAR T-cell therapies mostly resolve within 2 weeks of CAR T-cell infusion, but these short-lived toxicities can be severe, and in rare cases fatal.56, 79, 123 Moreover, toxicities hamper efforts to increase anti-malignancy efficacy of CAR T cells because approaches such as administering higher doses of CAR T cells or enhancing the activity of CAR T cells might worsen toxicity. Early CAR studies reported a syndrome with prominent signs of fever, tachycardia, and hypotension, and other toxicities after CAR T-cell infusions; this syndrome was designated cytokine-release syndrome (CRS)57, 74, 75, 124. CRS also includes many other abnormalities including but not limited to decreased cardiac ejection fraction, fatigue, delirium, dyspnea, renal failure, elevated liver enzymes, nausea, diarrhea, and elevated creatine phosphokinase indicative of muscle damage (Table 5). CRS was associated with increased levels of many serum cytokines57, 123–126 Coagulopathy and hemorrhage have been described in studies of CAR T cells in ALL63, 80. Similarly, patients with NHL receiving CAR T cells directed against varying antigen targets have experienced hemorrhagic events, including fatal gastrointestinal hemorrhange79, 100 and fatal intracranial hemorrhage79, 83. Prolonged B-cell aplasia often occurs in patients with NLH in the months following CAR T-cell infusions56, 73, 74, 79, 83. Recovery of normal polyclonal B cells in patients with NHL can occur without relapse of lymphoma56, 61, 74, 95.
Table 5:
Grade 3–4 Toxicities occurring in selected published clinical trials of CAR T-cell therapy for lymphomas*
| Clinical Trial | Target Antigen | Costimulatory Domain | Hemodynamic changes | Neurologic Toxicity | Cardiac Toxicity | Renal and Hepatic Toxicity | Cytopenias | Infections | Hemorrhage and Coagulopathic or Prothrombotic Events | Other |
|---|---|---|---|---|---|---|---|---|---|---|
| Jensen et al., ASBMT 201076 | CD19 | None, first generation CAR | None | None | None | None | None | None | None | None |
| Jensen et al., ASBMT 201076 | CD20 | None, first generation CAR | None | None | None | None | None | None | None | None |
| Kochenderfer et al., Blood 201073 | CD19 | CD28 | None | None | None | None |
|
None | None | None |
| Savoldo et al., J. Clin Invest 201159 | CD19 | One product without a costimulatory domain and one product with a CD28 costimulatory domain | None | None | None | None | None | None | None | None |
| Kochenderfer et al., Blood 201274 | CD19 | CD28 |
|
|
|
|
None |
|
None |
|
| Till et al., Blood 201299 | CD20 | CD28 and 4–1BB |
|
None | None | None |
|
Cellulitis | None | None |
| Kochenderfer et al., Blood 201393° | CD19 | CD28 | Hypotension | None | None | None | None | None | None | Headache |
| Wang et al., Clin Immunol 2014100 | CD20 | 4–1BB |
|
None | None |
|
None | None |
|
|
| Kochenderfer et al., JCO 201556 | CD19 | CD28 |
|
|
None |
|
None |
|
None |
|
| Zhang et al., Signal Transduction and Targeted Therapy 2016101 | CD20 | 4–1BB | None | None | None | Hypokalemia | None | Herpes zoster | None | None |
| Brudno et al., 201695° | CD19 | CD28 |
|
None | None |
|
|
None | None |
|
| Wang et al., Blood 201682 | CD19 | One clinical trial (NHL1) of a CAR not incorporating a costimulatory domain and one clinical trial (NHL2) of a CAR incorporating a CD28 costimulatory domain | None | None | None | None |
|
None | None | None |
| Ramos et al., J Clin Invest 201660 | Kappa (κ) light chain | CD28 | None | None | None |
|
|
None | None |
|
| Turtle et al., Sci Transl Med 201679 | CD19 | 4–1BB | Hypotension
|
|
|
None |
|
None |
|
None |
| Wang et al., Clin Cancer Res 2017102 | CD30 | 4–1BB | None | None |
|
|
None | None | None | None |
| Locke et al., Mol Ther 201783 | CD19 | CD28 |
|
|
|
|
|
|
|
|
| Kochenderfer et al. JCO 201761 | CD19 | CD28 |
|
|
|
|
|
|
|
|
ALT: alanine aminotransferase. AST: aspartate aminotransferase. Auto-HSCT: Autologous stem cell transplant. CR: complete remission. MDS: myelodysplastic syndrome. UTI: urinary tract infection.
Does not include lymphopenia or cytopenias other than anemia, thrombocytopenia, or neutropenia.. Only toxicities occurring in patients with NHL are included.
CRS grading per Lee et al. “Current concepts in the diagnosis and management of cytokine release syndrome.” Blood 2014. 124: 188–195.
Systolic dysfunction may be due to prior doxorubicin.
Both studies reporting on the same clinical trial.
Many clinical and immunological associations with CRS have been reported. The severity of CRS toxicity has been associated with peak serum CAR T-cell levels79 and serum cytokine levels, including IFN-γ56, 74, 79, interleukin-6 (IL-6)56, 79, and TNF-α74, as well as peak serum C-reactive protein (CRP)79. In clinical trials of CAR T cells for ALL, CRS severity has been associated with burden of disease63, 80. Not surprisingly, higher doses of conditioning chemotherapy worsen the severity of neutropenia and thrombocytopenia for patients with NHL receiving CAR T cells61. Clinical results suggest that higher doses of CAR T cells have been related to increased rates of severe CRS56, 79.
Neurologic toxicities including but not limited to confusion, tremors, ataxia, aphasia, obtundation, and seizures have been prominent in anti-CD19 CAR T-cell trials for lymphoma56, 61, 74, 79, 83, with reported frequencies of grade 3–5 neurologic toxicity of 25–57%61, 79, 83. Fatal cerebral edema occurred in a patient with DLBCL participating in a multicenter clinical trial of axicabtagene ciloleucel127. The exact etiology of neurologic toxicity remains unknown. Neurologic toxicity may or may not occur at the same time as CRS83, 128, suggesting differing pathophysiology. CD19 has not been shown to be expressed in the CNS56, 129. Investigators have reported some associations with presence or absence of neurological toxicity. Notably, very low rates of neurologic toxicity have been reported in a clinical trial of anti-CD19 CAR T cells given with no prior conditioning chemotherapy93, 95. Severe neurologic toxicity has been associated with higher CAR T-cell doses79. Higher peak levels of serum IL-279, IL-679, IL-1061, IL-1579, IL-1879, interferon-γ79, ferrtin79, CRP79, TIM-379, and granzyme B61, as well as higher peak blood CAR T-cell levels61, 79.
Generally, CRS is managed with supportive care and immunosuppressive agents such as the IL-6 receptor antagonist tocilizumab and corticosteroids123, 125. Tocilizumab has been used successfully to attenuate CRS-related toxicities in lymphoma patients79, 83, though CRS refractory to tocilizumab can also occur56, 123. Neurologic toxicities are sometimes preferentially managed with corticosteroids alone, due to a hypothetical risk of increased CNS IL-6 following saturation of peripheral IL-6 receptors125.
Suicide genes are being investigated as possible avenues of inducing abrupt cessation of T-cell mediated toxicities; inducible caspase-9 suicide switches are the most advanced suicide gene approach130, 131. A disadvantage of this approach is that complete elimination of all CAR T cells is likely to attenuate malignancy response, so this approach should probably be reserved for toxicities that cannot be controlled by other approaches.
Promising General Areas of Improvement for CAR T Cells
CAR T-cell therapies are in an early stage of development, and while past results are impressive, there are several areas with great potential for improvement. Gaining a better understanding of the importance of CAR T-cell persistence is an important area for future studies. The goal of CAR T-cell therapy is to provide patients with durable remissions of their malignancies. There are two general hypotheses to explain how CAR T cells could cause a lasting remission. First, a powerful T-cell response could completely eliminate all malignant cells early after infusion. In this case, long-term persistence of CAR T cells would not be needed. Second, CAR T cells are not capable of eliminating all malignant cells, but they are capable of eliminating some malignant cells. In this case persistence of CAR T cells might be important if the long-term persisting T cells could continuously detect and kill low levels of malignant cells to prevent a clinically-evident progression of malignancy. Acceptance of the second hypothesis requires that CAR T cells cannot eliminate all malignancy early after infusion, but can eliminate malignant cells long after CAR T-cell infusion at a time when CAR T-cell levels are much lower than early after infusion.61, 79 Persistence of T cells at tumor sites might be much more important than persistence of T cells in the blood, and activation-induced cell death (AICD) might be an important factor limiting persistence of CAR T cells132–134. CAR T-cell persistence in humans is widely variable61. Persistence of anti-CD19 CAR T cells of several months to years in the peripheral blood has been reported74, 79. Persistence of anti-CD19 CAR T cells for patients with NHL ranges from a mean of 20 days82 and less than 100 days56, 61 to greater than 300 days79, with an appearance of greater persistence of CAR T cells incorporating a 4–1BB costimulatory domain versus a CD28 costimulatory domain, which is consistent with preclinical models53, 135, 136. Comparison of two early phase lymphoma clinical trials of anti-CD19 CAR T cells, one with a CAR containing 4–1BB and one with a CAR containing CD28, reveals that efficacy is likely similar between costimulatory domains, with overall response rates of 72%79 versus 73%61 and CR rates of 50%79 versus 55%61 in patients receiving conditioning chemotherapy including fludarabine. While these trials had many factors in common, it is impossible to draw any firm conclusions by comparing these or other trials because the trials differ in many variables aside from the costimulatory domain used. One critical factor that is often overlooked is the importance of the hinge and transmembrane portions of CARs, so comparing CARs that have different costimulatory molecules, but also differ in other aspects is not a valid way to compare costimulatory domains133.
Immunologic rejection of CAR T cells is another potential problem with CAR T cells. Most CARs in current clinical use contain antibody variable regions from murine antibodies. These sequences are potentially immunogenic. In addition, the junctions between different CAR domains are artificial sequences that could be immunogenic. Decreasing the immunogenicity of CARs might be particularly important if multiple doses of CAR T cells are administered because immunologic memory could be generated after a first dose of CAR T cells. T-cell responses against the murine components of the single chain variable fragment (sFv) of the CAR have been reported in multiple clinical trials of anti-CD19 CARs for both ALL and NHL64, 76, 79, 80.
Loss of target antigen expression, termed “antigen escape” is a well described mechanism of malignancy progression in clinical trials of anti-CD19 CAR T cells for ALL137, and one case of CD19 negative progression in a patient with lymphoma following two infusions of anti-CD19 CAR T cells has been reported128.
Improving CAR T-cell Therapy
There are many avenues for improving CAR T-cell therapies (Figure 2). Improved CAR designs have potential to improve many of the current limitations of CAR T-cells. We have shown that holding all variables constant except changing the hinge and transmembrane regions can have an important impact on CAR T-cell cytokine production with higher levels of inflammatory cytokines produced by T cells expressing anti-CD19 CARs with hinge and transmembrane domains from CD28 compared with T cells expressing anti-CD19 CARs with CD8α hinge and transmembrane domains133. AICD is a potential factor limiting CAR T-cell persistence, and AICD is increased in T cells expressing anti-CD19 CARs with hinge and transmembrane domains from CD28 compared to T cells expressing anti-CD19 CARs with CD8α hinge and transmembrane domains133. CARs with humanized or fully human variable regions might have decreased immunogenicity, which may decrease CAR T-cell destruction by the host immune system and promote CAR T-cell expansion and persistence. Use of less immunogenic CARs may allow for greater efficacy of multiple cell infusions, and CARs with human variable regions are under development128, 138. Clinical studies are now investigating humanized and fully human CAR T cells for patients with ALL and NHL (NCT02374333, NCT02659943, NCT03049449). Bispecific CARs simultaneously targeting 2 antigens offer one possible solution to loss of antigens targeted by CARs139, 140.
Figure 2: Improvements to CAR T-cell therapy.
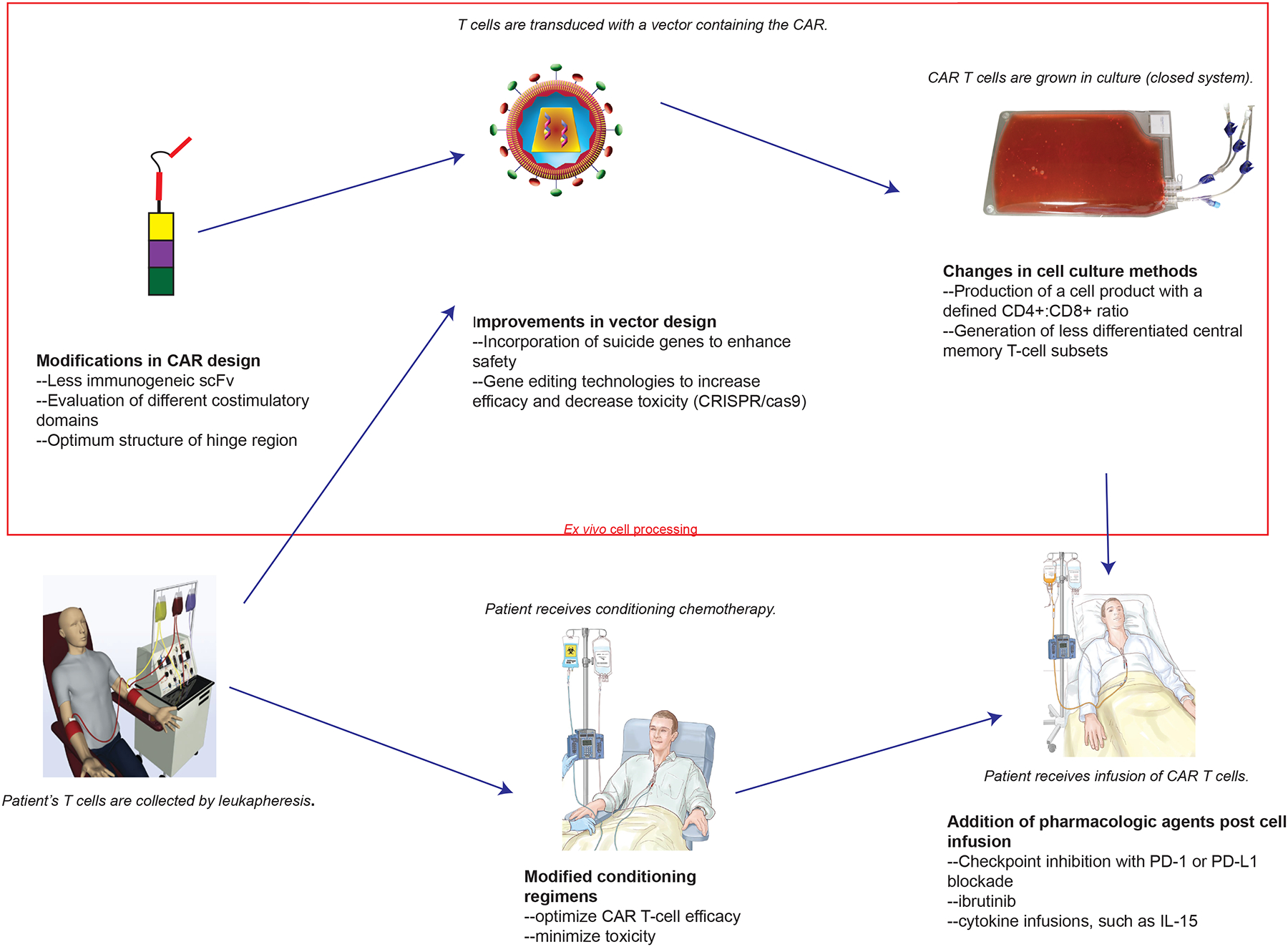
CAR T-cell therapy is being improved at multiple steps in the overall CAR T-cell therapy process. Areas of improvement include modifications in all aspects of CAR design, incorporation of different genes to enhance CAR T-cell activity or control toxicity into CAR gene therapy vectors, use of gene-editing technologies such as CRISPR/Cas9, changes to cell culture methods, optimization of conditioning regimens, and the addition of post-infusion pharmacologic agents.
Gene editing holds great promise for improving cell therapies141. For example, clustered regularly interspaced short palindromic repeats/crisper-associated system9 (CRISPR/Cas9) genome editing has allowed targeting of CAR gene integration to the T-cell receptor-α constant (TRAC) locus142. This modification decreased T-cell exhaustion; this resulted in superior anti-malignancy efficacy in a mouse leukemia model142.
Administration of CAR T-cell products composed of a defined 1:1 ratio of CD4+/CD8+ CAR T cells has been investigated in multiple clinical trials in ALL64 and NHL79. This strategy has not yet been directly compared with the use unselected cell products containing variable portions of CD4+ and CD8+ T cells in a clinical trial. Use of less differentiated T-cell subsets such as stem memory T cells is under investigation143.
Fludarabine has been used in many successful CAR clinical trials56, 64, 79, 144. There is evidence that improved lymphocyte depletion with a fludarabine-containing conditioning chemotherapy regimen may facilitate greater T-cell expansion and persistence, especially when multiple doses of T cells are administered64, 79. Patients with NHL receiving fludarabine-containing chemotherapy prior to a second cell infusion had robust anti-CD19 CAR T-cell expansion and did not develop cytotoxic T-cell responses to the sFv of the CAR. Patients receiving fludarabine-containing chemotherapy have also been reported to have improved PFS compared with patients who received cyclophosphamide alone or cyclophosphamide and etoposide79.
CAR T cell activity may be increased by the administration of pharmaceutical agents. The Bruton’s tyrosine kinase inhibitor ibrutinib has been shown to boost CAR T-cell anti-malignancy efficacy in MCL in xenograft mouse models145. Immunologic checkpoint inhibitors have clinical efficacy in HL36, 38, with some activity in NHL146. Following cell infusion, anti-CD19 CAR T cells acquire a more differentiated and exhausted phenotype, with increased expression of PD-156, 95. Interruption of the PD-1/PD-L1 pathway, through PD-1 antibody checkpoint blockade, can restore CAR T-cell effector function in in vitro models147. A response to a checkpoint inhibitor in a patient with PMBL and progressive disease following CAR T-cell infusion has been reported148. A clinical trial is underway in which patients with refractory or relapsed DLBCL, FL, or MCL following anti-CD19 CAR T-cell infusion are administered the checkpoint inhibitor pembrolizumab in an attempt to re-activate exhausted CAR T cells (NCT02650999). Administration of IL-15 after CAR T-cell infusions might be of benefit given the association of high serum IL-15 levels with remissions of lymphoma in a recent study61.
Another aspect of clinical application improvement could be to move CAR T-cell therapy earlier in the line-up of lymphoma therapies. The majority of CAR T-cell trials for lymphoma include only patients who have had multiple lines of therapy, chemotherapy-refractory lymphoma, or relapse after auto-HSCT. CAR T cells have clear antimalignancy activity in these heavily pretreated patients56, 61, 79, 83. Use of CAR T cells earlier in the course of lymphoma might increase the chance of successful treatment if for no other reason than patients are more likely to be healthy enough to undergo CAR T-cell therapy early in their courses of treatment. CAR T-cell therapy earlier in a patient’s treatment course may avoid some of the negative consequences of prolonged exposure to cytotoxic agents, such as development of myelodysplastic syndrome and other secondary malignancies, which are significant risks, especially following auto-HSCT149, 150. Another potential advantage to moving CAR T-cell therapies earlier in the course of lymphoma treatment is that patients who have never been exposed to chemotherapy might have higher blood T-cell counts and perhaps T cells that are more fit for CAR T-cell therapy. Both of these factors might facilitate CAR T-cell production.
The feasibility of administering anti-CD19 CAR T cells immediately following auto-HSCT to patients with DLBCL has been explored76, 82. The standard approach for fit patients with DLBCL and residual or relapsed lymphoma after first-line therapy is salvage chemotherapy followed by auto-HSCT9, 10. Anti-CD19 CAR T cells may eventually supplant, or be used in tandem with, auto-HSCT as best second-line therapy for DLBCL.
Conclusions
While CAR T-cell therapy for lymphoma is in its early stages, this form of adoptive immunotherapy has already been shown to have the ability to cause durable complete remissions of chemotherapy-refractory lymphoma in many patients.. Anti-CD19 CAR T cells are likely to become the new standard of care for patients with chemotherapy-refractory DLBCL given the very limited options available for these patients currently7, 12, 14. Anti-CD19 CAR T cells may also eventually supplant, or be used in tandem with, auto-HSCT as best second-line therapy for DLBCL. Anti-CD19 CAR T cells will almost certainly soon become widely-used to treat FCL and MCL. Further optimization of CAR design, improved toxicity management, and development of combination therapy will allow CAR T cells to be used safely and effectively for more patients.
Key Points.
New treatments are needed for patients with chemotherapy-refractory or multiply relapsed lymphoma.
Chimeric antigen receptor (CAR) T cells targeting CD19 have demonstrated efficacy in multiple B-cell lymphoma subtypes, with activity seen in cases of chemotherapy-refractory lymphoma. Durable remissions are possible.
Multicenter clinical trials have demonstrated that centralized cell processing is feasible, with early response rates similar to those reported in single center studies.
CARs targeting new antigens, such as CD20, CD22, CD30 and kappa light chains, are in development and will extend CAR T-cell therapy to patients with Hodgkin lymphoma, T-cell lymphoma, and CD19-negative B-cell lymphoma.
Cytokine release syndrome and neurologic toxicity are problems associated with CAR T-cell therapies for lymphoma, and reducing toxicity is a major avenue for improving CAR T-cell therapies.
CAR T-cell therapy is likely to become safer and more effective, and CAR T-cell therapies are likely to become standard treatment options for relapsed and primary chemotherapy-refractory lymphoma in the near future.
Conflict of interest:
James Kochenderfer receives research funding from cooperative research and development agreements between NCI and Kite Pharma Inc. and between the NCI and bluebird bio Inc. James Kochenderfer also has multiple patent applications related to chimeric antigen receptors. Jennifer Brudno has no relevant conflicts of interest.
Biographies
Jennifer N. Brudno, M.D. received her medical doctorate from the Emory University School of Medicine. She completed internal medicine residency training at the Johns Hopkins University Hospital. She trained in medical oncology and hematology at the National Institutes of Health. She is currently a staff clinician in the Experimental Transplantation and Immunology Branch of the National Cancer Institute. Dr. Brudno’s research focuses on clinical development of chimeric antigen receptor (CAR) T-cell therapies for hematologic malignancies.
James Kochenderfer, M.D. is a Tenure-track Investigator in the Experimental Transplantation and Immunology Branch (ETIB) of the National Cancer Institute (NCI). Dr. Kochenderfer received his M.D. from West Virginia University and then completed internal medicine training at Vanderbilt University. Next he completed a medical oncology fellowship at the M.D. Anderson Cancer Center and a hematology fellowship at Baylor College of Medicine. He then undertook a period of postdoctoral research training in T-cell immunotherapy in the laboratories of Dr. Ronald Gress and Dr. Steven Rosenberg at the National Cancer Institute. For the past 10 years, Dr. Kochenderfer has focused on a full range of chimeric antigen receptor (CAR) research from designing and constructing new CARs to preclinical testing of new CARs to conducting clinical trials. His work emphasizes developing CAR T-cell therapies for lymphoma and multiple myeloma.
Footnotes
Off-label use: The use of tocilizumab for indications not approved by the Food and Drug Administration will be discussed.
References
- 1.“About Non-Hodgkin Lymphoma.” American Cancer Society website. 2017. https://www.cancer.org/cancer/non-hodgkin-lymphoma/about/key-statistics.html. . [Google Scholar]
- 2.Coiffier B et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346, 235–42 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Marcus R et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol 26, 4579–86 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Forstpointner R et al. The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 104, 3064–71 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Elstrom RL et al. Response to second-line therapy defines the potential for cure in patients with recurrent diffuse large B-cell lymphoma: implications for the development of novel therapeutic strategies. Clin Lymphoma Myeloma Leuk 10, 192–6 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Martelli M et al. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol 87, 146–71 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Van Den Neste E et al. Outcomes of diffuse large B-cell lymphoma patients relapsing after autologous stem cell transplantation: an analysis of patients included in the CORAL study. Bone Marrow Transplant 52, 216–221 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Jurinovic V et al. Clinicogenetic risk models predict early progression of follicular lymphoma after first-line immunochemotherapy. Blood 128, 1112–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Philip T et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med 333, 1540–5 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Vose JM et al. Autologous transplantation for diffuse aggressive non-Hodgkin’s lymphoma in patients never achieving remission: a report from the Autologous Blood and Marrow Transplant Registry. J Clin Oncol 19, 406–13 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Feugier P et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol 23, 4117–26 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Telio D et al. Salvage chemotherapy and autologous stem cell transplant in primary refractory diffuse large B-cell lymphoma: outcomes and prognostic factors. Leuk Lymphoma 53, 836–41 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Seshadri T et al. Utility of subsequent conventional dose chemotherapy in relapsed/refractory transplant-eligible patients with diffuse large B-cell lymphoma failing platinum-based salvage chemotherapy. Hematology 13, 261–6 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Nagle SJ et al. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am J Hematol 88, 890–4 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Martin P et al. Patterns of delivery of chemoimmunotherapy to patients with follicular lymphoma in the United States: results of the National LymphoCare Study. Cancer 119, 4129–36 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Rummel MJ et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381, 1203–10 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Flinn IW et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 123, 2944–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisler CH et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 158, 355–62 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Geisler CH et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 112, 2687–93 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher RI et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 24, 4867–74 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Goy A et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 31, 3688–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang ML et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369, 507–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campo E & Rule S Mantle cell lymphoma: evolving management strategies. Blood 125, 48–55 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Schmitz N et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood 116, 3418–25 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Simon A et al. Upfront VIP-reinforced-ABVD (VIP-rABVD) is not superior to CHOP/21 in newly diagnosed peripheral T cell lymphoma. Results of the randomized phase III trial GOELAMS-LTP95. Br J Haematol 151, 159–66 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Armitage JO, Vose JM & Weisenburger DD Towards understanding the peripheral T-cell lymphomas. Ann Oncol 15, 1447–9 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Coiffier B et al. Peripheral T-cell lymphomas have a worse prognosis than B-cell lymphomas: a prospective study of 361 immunophenotyped patients treated with the LNH-84 regimen. The GELA (Groupe d’Etude des Lymphomes Agressives). Ann Oncol 1, 45–50 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Escalon MP et al. Prognostic factors and treatment of patients with T-cell non-Hodgkin lymphoma: the M. D. Anderson Cancer Center experience. Cancer 103, 2091–8 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Coiffier B et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30, 631–6 (2012). [DOI] [PubMed] [Google Scholar]
- 30.O’Connor OA et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33, 2492–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Connor OA et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29, 1182–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montanari F & Diefenbach C Relapsed Hodgkin lymphoma: management strategies. Curr Hematol Malig Rep 9, 284–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitz N et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 359, 2065–71 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Younes A et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 30, 2183–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen R et al. Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood 128, 1562–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ansell SM et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372, 311–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Younes A et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol 17, 1283–94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armand P et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J Clin Oncol (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Timmerman J Checkmate 205 Update with Minimum 12-Month Follow up: A Phase 2 Study of Nivolumab in Patients with Relapsed/Refractory Classical Hodgkin Lymphoma. American Society of Hematology Annual Meeting; (Abstract 1110, December 5, 2016). [Google Scholar]
- 40.Rezvani AR et al. Nonmyeloablative allogeneic hematopoietic cell transplantation in relapsed, refractory, and transformed indolent non-Hodgkin’s lymphoma. J Clin Oncol 26, 211–7 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Grigg A & Ritchie D Graft-versus-lymphoma effects: clinical review, policy proposals, and immunobiology. Biol Blood Marrow Transplant 10, 579–90 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Thomson KJ et al. Favorable long-term survival after reduced-intensity allogeneic transplantation for multiple-relapse aggressive non-Hodgkin’s lymphoma. J Clin Oncol 27, 426–32 (2009). [DOI] [PubMed] [Google Scholar]
- 43.van Kampen RJ et al. Allogeneic stem-cell transplantation as salvage therapy for patients with diffuse large B-cell non-Hodgkin’s lymphoma relapsing after an autologous stem-cell transplantation: an analysis of the European Group for Blood and Marrow Transplantation Registry. J Clin Oncol 29, 1342–8 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Eshhar Z, Waks T, Gross G & Schindler DG Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences of the United States of America 90, 720–4 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kochenderfer JN & Rosenberg SA Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol 10, 267–76 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadelain M, Brentjens R & Riviere I The basic principles of chimeric antigen receptor design. Cancer Discov 3, 388–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson LA & June CH Driving gene-engineered T cell immunotherapy of cancer. Cell Res 27, 38–58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kochenderfer JN et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. Journal of Immunotherapy 32, 689–702 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadelain M CAR therapy: the CD19 paradigm. J Clin Invest 125, 3392–400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gill S & June CH Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263, 68–89 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Jensen MC & Riddell SR Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol 33, 9–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sadelain M, Brentjens R & Riviere I The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 21, 215–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Stegen SJ, Hamieh M & Sadelain M The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 14, 499–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carpenito C et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A 106, 3360–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sadelain M, Riviere I & Riddell S Therapeutic T cell engineering. Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kochenderfer JN et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33, 540–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brentjens RJ et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science Translational Medicine 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davila ML et al. Efficacy and Toxicity Management of 19–28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med 6, 224ra25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savoldo B et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 121, 1822–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramos CA et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest 126, 2588–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kochenderfer JN et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J Clin Oncol 35, 1803–1813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turtle CJ et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kebriaei P et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest 126, 3363–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP & Rosenberg SA Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116, 3875–3886 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gattinoni L et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. Journal of Experimental Medicine 202, 907–12 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenberg SA & Restifo NP Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davila ML, Kloss CC, Gunset G & Sadelain M CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One 8, e61338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hwu P et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. Journal of Experimental Medicine 178, 361–6 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brentjens RJ et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15.[see comment]. Nature Medicine 9, 279–86 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Cooper LJ et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood 101, 1637–44 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Kochenderfer JN et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kochenderfer JN et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119, 2709–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Porter DL, BL; Kalos M et al. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. The New England Journal of Medicine 365, 725–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jensen MC et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biology of Blood and Marrow Transplantation 16, 1245–1256 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rezvani AR et al. Non-myeloablative allogeneic haematopoietic cell transplantation for relapsed diffuse large B-cell lymphoma: a multicentre experience. Br J Haematol 143, 395–403 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gisselbrecht C et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol 28, 4184–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turtle CJ et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8, 355ra116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schuster S Sustained Remissions Following Chimeric Antigen Receptor Modified T Cells Directed Against CD19 (CTL019) in Patients with Relapsed or Refractory CD19+ Lymphomas. American Society of Hematology Annual Meeting; (Abstract 183, December 6, 2015). [Google Scholar]
- 82.Wang X et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 127, 2980–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Locke FL et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther 25, 285–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Locke FL Clinical and biologic covariates of outcomes in ZUMA-1: A pivotal trial of axicabtagene ciloleucel (axi-cel; KTE-C19) in patients with refractory aggressive non-Hodgkin lymphoma (r-NHL). American Society of Clinical Oncology Annual Meeting; (Abstract 7512, June 5, 2017). . [Google Scholar]
- 85.Locke FL Primary results from ZUMA-1: a pivotal trial of axicabtagene ciloleucel (axicel; KTE-C19) in patients with refractory aggressive non-Hodgkin lymphoma (NHL). American Association for Cancer Research Annual Meeting; (Abstract CT019, April 2, 2017). . [Google Scholar]
- 86.Abramson JS CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T-cell product JCAR017 (TRANSCEND NHL 001). American Society of Clinical Oncology Annual Meeting; (Abstract 7513, June 5, 2017). . [Google Scholar]
- 87.Schuster SJ Global pivotal phase 2 trial of the CD19-targeted therapy CTL019 in adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) - an interim analysis. International Conference on Malignant Lymphoma (Abstract 7, June 14, 2017). [Google Scholar]
- 88.van den Brink MR et al. Relapse after allogeneic hematopoietic cell therapy. Biol Blood Marrow Transplant 16, S138–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spyridonidis A et al. Outcomes and prognostic factors of adults with acute lymphoblastic leukemia who relapse after allogeneic hematopoietic cell transplantation. An analysis on behalf of the Acute Leukemia Working Party of EBMT. Leukemia 26, 1211–7 (2012). [DOI] [PubMed] [Google Scholar]
- 90.Roddie C & Peggs KS Donor lymphocyte infusion following allogeneic hematopoietic stem cell transplantation. Expert Opin Biol Ther 11, 473–87 (2011). [DOI] [PubMed] [Google Scholar]
- 91.Kolb HJ Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood 112, 4371–83 (2008). [DOI] [PubMed] [Google Scholar]
- 92.Frey NV & Porter DL Graft-versus-host disease after donor leukocyte infusions: presentation and management. Best Pract Res Clin Haematol 21, 205–22 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kochenderfer JN et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122, 4129–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cruz CRY et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed After allogeneic stem cell transplant: A phase 1 study. Blood 122, 2956–2973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brudno JN et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ghosh A et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med 23, 242–249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Qasim W et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science Translational Medicine 9 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Swerdlow SH WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissue (International Agency for Research on Cancer, 2008). [Google Scholar]
- 99.Till BG et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4–1BB domains: pilot clinical trial results. Blood 119, 3940–50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol 155, 160–75 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Zhang W Treatment of CD20-directed Chimeric Antigen Receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduction and Targeted Therapy 1, 16002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang C et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin’s lymphoma: an open-label phase I trial. Clin Cancer Res (2016). [DOI] [PubMed] [Google Scholar]
- 103.Hiraga J et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood 113, 4885–93 (2009). [DOI] [PubMed] [Google Scholar]
- 104.Kennedy GA et al. Incidence and nature of CD20-negative relapses following rituximab therapy in aggressive B-cell non-Hodgkin’s lymphoma: a retrospective review. Br J Haematol 119, 412–6 (2002). [DOI] [PubMed] [Google Scholar]
- 105.Haso W et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121, 1165–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Long AH, Haso WM & Orentas RJ Lessons learned from a highly-active CD22-specific chimeric antigen receptor. Oncoimmunology 2, e23621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Durkop H et al. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell 68, 421–7 (1992). [DOI] [PubMed] [Google Scholar]
- 108.Stein H et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 66, 848–58 (1985). [PubMed] [Google Scholar]
- 109.Younes A et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363, 1812–21 (2010). [DOI] [PubMed] [Google Scholar]
- 110.Younes A et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncol 14, 1348–56 (2013). [DOI] [PubMed] [Google Scholar]
- 111.Moskowitz AJ et al. PET-adapted sequential salvage therapy with brentuximab vedotin followed by augmented ifosamide, carboplatin, and etoposide for patients with relapsed and refractory Hodgkin’s lymphoma: a non-randomised, open-label, single-centre, phase 2 study. Lancet Oncol 16, 284–92 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Moskowitz CH et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 385, 1853–62 (2015). [DOI] [PubMed] [Google Scholar]
- 113.Pro B et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 30, 2190–6 (2012). [DOI] [PubMed] [Google Scholar]
- 114.Horwitz SM et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 123, 3095–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fanale MA et al. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. J Clin Oncol 32, 3137–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Deng C, Pan B & O’Connor OA Brentuximab vedotin. Clin Cancer Res 19, 22–7 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Hombach A et al. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin’s lymphoma cells in the presence of soluble CD30. Cancer Res 58, 1116–9 (1998). [PubMed] [Google Scholar]
- 118.Hombach A et al. Characterization of a chimeric T-cell receptor with specificity for the Hodgkin’s lymphoma-associated CD30 antigen. J Immunother 22, 473–80 (1999). [DOI] [PubMed] [Google Scholar]
- 119.Savoldo B et al. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 110, 2620–30 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Di Stasi A et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113, 6392–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ramos CA, Heslop HE & Brenner MK in Annu Rev Med 165–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ramos CA Chimeric T cells for therapy of CD30+ Hodgkin and Non-Hodgkin Lymphomas. American Society of Hematology Annual Meeting; (Abstract 185, December 6, 2015). [Google Scholar]
- 123.Brudno JN & Kochenderfer JN Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science Translational Medicine 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee DW et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Brentjens R, Y.R., Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: a case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther 18, 666–668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Harris J Kite Reports Cerebral Edema Death in ZUMA-1 CAR T-Cell Trial. Onc Live. May 8, 2017. http://www.onclive.com/web-exclusives/kite-reports-cerebral-edema-death-in-zuma1-car-tcell-trial#sthash.1UENjTT7.dpufhttp://www.onclive.com/web-exclusives/kite-reports-cerebral-edema-death-in-zuma1-car-tcell-trial. (2017). [Google Scholar]
- 128.Brudno J T Cells Expressing a Novel Fully-Human Anti-CD19 Chimeric Antigen Receptor Induce Remissions of Advanced Lymphoma in a First-in-Humans Clinical Trial. American Society of Hematology Annual Meeting (Abstract 999, December 5, 2016) (2016). [Google Scholar]
- 129.Uckun FM et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood 71, 13–29 (1988). [PubMed] [Google Scholar]
- 130.Budde LE et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One 8, e82742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Diaconu I et al. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol Ther 25, 580–592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Künkele A et al. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer immunology research 3, 368–379 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Alabanza L, Pegues M, Geldres C, Shi V & Kochenderfer J The Impact of Different Hinge and Transmembrane Componenents on the Function of a Novel Fully-human Anti-CD19 Chimeric Antigen Receptor-Abstract. Molecular Therapy Volume 24 Supplement 1, S32 (2016). [Google Scholar]
- 134.Gargett T et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected from Activation-induced Cell Death by PD-1 Blockade. Molecular Therapy 24, 1135–1149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao Z et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28, 415–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Milone MC et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular Therapy: the Journal of the American Society of Gene Therapy 17, 1453–64 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kenderian SS, Porter DL & Gill S Chimeric Antigen Receptor T Cells and Hematopoietic Cell Transplantation: How Not to Put the CART Before the Horse. Biology of Blood and Marrow Transplantation 23, 235–246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sommermeyer D et al. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ruella M et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. Journal of Clinical Investigation 126, 3814–3826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schneider D et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. Journal for ImmunoTherapy of Cancer 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Osborn MJ et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Molecular Therapy 24, 570–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sabatino M et al. Generation of clinical-grade CD19-specific CAR-modified CD81 memory stem cells for the treatment of human B-cell malignancies. Blood 128, 519–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ali SA et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 128, 1688–1700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ruella M et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clin Cancer Res 22, 2684–96 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Armand P Immune checkpoint blockade in hematologic malignancies. Blood 125, 3393–400 (2015). [DOI] [PubMed] [Google Scholar]
- 147.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126, 3130–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chong EA et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129, 1039–1041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vaxman I et al. Secondary malignancies following high dose therapy and autologous hematopoietic cell transplantation-systematic review and meta-analysis. Bone Marrow Transplant 50, 706–14 (2015). [DOI] [PubMed] [Google Scholar]
- 150.Pedersen-Bjergaard J, Andersen MK & Christiansen DH Therapy-related acute myeloid leukemia and myelodysplasia after high-dose chemotherapy and autologous stem cell transplantation. Blood 95, 3273–9 (2000). [PubMed] [Google Scholar]