

Abstract
RasGRP comprises a family of guanine nucleotide exchange factors, regulating the dissociation of GDP from Ras GTPases to enhance the formation of the active GTP-bound form. RasGRP1 possesses REM (Ras exchange), GEF (catalytic), EF-hand, C1, SuPT (suppressor of PT), and PT (plasma membrane-targeting) domains, among which the C1 domain drives membrane localization in response to diacylglycerol or phorbol ester and the PT domain recognizes phosphoinositides. The homologous family member RasGRP3 shows less plasma membrane localization. The objective of this study was to explore the role of the different domains of RasGRP3 in membrane translocation in response to phorbol esters. The full-length RasGRP3 shows limited translocation to the plasma membrane in response to PMA, even when the basic hydrophobic cluster in the PT domain, reported to be critical for RasGRP1 translocation to endogenous activators, is mutated to resemble that of RasGRP1. Moreover, exchange of the C-termini (SuPT-PT domain) of the two proteins had little effect on their plasma membrane translocation. On the other hand, while the C1 domain of RasGRP3 alone showed partial plasma membrane translocation, truncated RasGRP3 constructs, which contain the PT domain and are missing the REM, showed stronger translocation, indicating that the REM of RasGRP3 was a suppressor of its membrane interaction. The REM of RasGRP1 failed to show comparable suppression of RasGRP3 translocation. The marked differences between RasGRP3 and RasGRP1 in membrane interaction necessarily will contribute to their different behavior in cells and are relevant to the design of selective ligands as potential therapeutic agents.
Keywords: RasGRP3, RasGRP1, REM domain, SPR analysis, membrane translocation
Graphical abstract

Introduction
Ras signaling pathways, which play a prominent role both in cancer and in cell physiology, are regulated by guanine nucleotide exchange factors (RasGEFs) and GTPase activating proteins (RasGAPs). Ras cycles between an inactive GDP-bound and an active GTP-bound form, with its regulators shifting the balance between these two states [1, 2, 3]. RasGEFs cause release of GDP from the inactive RasGDP complex, allowing the binding of GTP to form the active RasGTP complex; RasGAPs enhance hydrolysis of bound GTP, generating the inactive RasGDP complex. The active RasGTP complex functions by associating with its downstream effectors, causing their allosteric activation. Ras family members are membrane-bound signal-transducing proteins and their attachment to specific cellular membranes allows them to confine, concentrate, and organize networks of signal detectors and transmitters [4].
There are three major families of Ras-specific nucleotide exchange factors in humans, the SOS proteins, the Ras guanine nucleotide releasing proteins (RasGRP), and the Ras guanine nucleotide releasing factors (RasGRF) [5]. Unlike SOS, the RasGRPs are not ubiquitously expressed but display restricted and overlapping patterns of tissue localization. Additionally, while sharing many structural domains, they differ somewhat in specificity for Ras family members and mechanisms of activation [5]. In vertebrates, four RasGRP family members have been described. RasGRP1, RasGRP3, and RasGRP4 can all activate Ras, while RasGRP2 functions as a GEF for the small GTPase Rap. The prominent biological role of RasGRP2 is in platelet signaling [6, 7]; RasGRP4 is largely restricted to mast cells [8]. Most attention has been focused on RasGRP1 and RasGRP3, which are critically involved in signaling in T and B lymphocytes [9, 10, 11, 12, 13]. RasGRP1 has been associated with human autoimmune disease [14, 15, 16] and cancer [17, 18], while RasGRP3 has been shown to play a role in immune function [5], in human melanoma [19, 20] and in prostate cancer [21, 22].
A feature distinguishing the RasGRP family of RasGEFs is the presence of a C1 domain. C1 domains represent the recognition domain for the lipophilic second messenger diacylglycerol (DAG) [23, 24, 25] and are likewise the target for the phorbol esters, which function as ultrapotent DAG analogs [26]. C1 domains are present in six other families of critical signaling proteins as well, including protein kinase C, the chimaerins, and myotonic dystrophy-related Cdc42-binding kinase (MRCK) [27]. The diverse biological functions of these proteins have led to great interest in the development of C1 domain directed compounds as potential therapeutic agents, with ingenol 3-mebutate approved by the FDA for actinic keratosis, bryostatin 1 in clinical trials for cancer and dementia, and prostratin of interest for combination therapy in HIV-AIDs.
DAG / phorbol esters interact with C1 domains by inserting into a hydrophilic cleft surrounded by a hydrophobic rim [28]. The ligand completes the hydrophobic surface, promoting its translocation and insertion into the membrane and, depending upon the protein, promoting conformational change in the protein by breaking the intramolecular contacts occupied by the unliganded C1 domain [see, for example, 29]. Importantly, the C1 domain – ligand – membrane interaction has highly complex pharmacology, both because the lipid headgroups provide part of the pharmacophoric interactions with the ligand and because the complex is influenced by the interactions of the C1 domain surface with the lipid membrane. Using combinatorial chemistry, we have shown that DAG-lactones with relatively minor structural differences can show great diversity of biological consequence [30]. We likewise have generated DAG-lactones with moderate selectivity for RasGRP1/3 relative to PKC [31, 32].
C1 domains are important regulatory elements of RasGRP1 and RasGRP3. They were shown to be essential for the transforming activity of RasGRP1 in rat cells [33] as well as for the biological activity of RasGRP1 and RasGRP3 in immune cells [5]. As with protein kinase Cs, they also confer membrane translocation of RasGRPs in response to phorbol ester treatment [34]. Emerging evidence suggests, however, that the elements contributing to membrane interaction of RasGRP are substantially more complicated.
All exchange factors for Ras-GTPases contain a conserved GEF domain which directly catalyzes displacement of GDP from Ras, thus enabling its replacement by GTP [35]. In the catalytic module (REM; Ras exchange motif and GEF; catalytic domain; Figure 1) [36, 37, 38, 39, 40], the REM domain was shown to provide positive feedback regulation by binding Ras-GTP and then allosterically enhancing the activity of the GEF domain in SOS proteins [41, 42]. In RasGRP1, the catalytic module is followed by an EF domain with a predicted pair of EF hands (EF1 and EF2 modules) [43, 44, 45], the C1 domain, and C-terminal SuPT (suppressor of PT) and PT (plasma membrane-targeting) domains (Figure 1). The EF hands control plasma membrane targeting by counteracting the SuPT domain which negatively regulates the PT domain [46]. The plasma membrane targeting domain is directly responsible for targeting RasGRP1 to the plasma membrane in some cell types [47]. In the PT domain, a small segment enriched in basic and hydrophobic residues, the BHC motif has been identified [48]. A similar motif has been found in other proteins that bind directly to plasma membranes enriched in anionic phospholipids, particularly phosphoinositides [49, 50, 51, 52].
Figure 1. Domain structure of RasGRPs.

RasGRPs are complex proteins with REM (Ras exchange motif), GEF (catalytic domain), EF-hand (Ca2+ binding domain), C1 (binding domain for the lipid second messenger DAG, mediates membrane translocation of RasGRP1), SuPT (suppressor of PT), and PT (plasma membrane-targeting) domains. The catalytic module of RasGRPs includes the REM and GEF domains, followed by the regulatory module containing the EF domain, C1 domain and the C-terminal segment. The REM, GEF and C1 domains are important for targeting RasGRP1 to the endomembrane and plasma membrane. The EF hand and SuPT domain only affect plasma membrane targeting of RasGRP1. The PT domain is sufficient and essential for antigen receptor-induced plasma membrane targeting. The constructs used in the study are shown.
For plasma membrane targeting of RasGRP1, the activation of PI3K is important because it generates PI(3,4)P2 or PI(3,4,5)P3. In the absence of PI3K activation, there is insufficient negative charge at the plasma membrane to support stable electrostatic binding of RasGRP1 via its BHC. The PI(3,4)P2 or PI(3,4,5)P3 provides the additional clustered negative charges that drive electrostatic binding of the BHC to the membrane, enabling insertion of aromatic side chains into the bilayer. Binding of the BHC to phosphoinositides generated by PI3K can synergize with binding of the C1 domain to DAG generated by PLCγ to maximize plasma membrane targeting of RasGRP1 (48). Since mutations leading to constitutive activation of PI3K, generating PI(3,4,5)P3, or inactivation of PTEN, a phosphatase which degrades PI(3,4,5)P3, are both commonly seen in cancer [53, 54], an expectation is that cancers expressing these mutations would lead to enhanced stimulation of RasGRP1, if present, with downstream activation of Ras.
RasGRP3 has the broadest substrate selectivity of all the RasGRP family members, being able to activate H-Ras, R-Ras, and Rap-1 [55, 56]. An important aspect of RasGRP3 regulation is its binding to the second messenger diacylglycerol. This binding is usually accompanied by subcellular redistribution of RasGRP3, which is believed to contribute to co-localization with its substrates [57]. In fact, subcellular redistribution in response to diacylglycerol and its ultrapotent analogs, the phorbol esters, is one of the hallmarks of activation. The process of translocation is orchestrated by a combination of factors, among them the nature and lipophilicity of the ligand [58] and the affinity for phospholipids.
In the present paper, we have examined the role of different domains, including the basic/hydrophobic cluster in the PT domain of RasGRP3, in phosphoinositide recognition and membrane translocation in response to phorbol esters. We show that the difference between the phosphoinositide binding (PT) domain of RasGRP3 and that of RasGRP1 cannot account for the failure of RasGRP3 to be targeted to the plasma membrane by phorbol ester. Our results clearly show that the N-terminus (REM domain) of RasGRPs is a critical element contributing to the difference between RasGRP1 and RasGRP3 in their ability to translocate to the plasma membrane. This difference in membrane translocation between RasGRP1 and RasGRP3 further emphasizes the substantial divergence in the regulation of these homologous proteins and potentially in their selectivity in living cells for lipophilic ligands.
Experimental Procedures
Materials
Phorbol 12-myristate 13-acetate (PMA) was purchased from LC Laboratories (Woburn, MA). Dimethyl sulfoxide (DMSO) was purchased from Sigma – Aldrich (St. Louis, MO). LNCaP human prostate cancer cells, HEK 293 human embryonic kidney cells, fetal bovine serum (FBS), RPMI 1640 medium, L-glutamine, and Eagle’s Minimum Essential (EMEM) medium were from American Type Culture Collection (Manassas, VA). Reagents used for culturing bacteria (LB Broth, LB agar plates with different selection of antibiotics, etc.) were from K-D Medical, Inc. (Columbia, MD). The oligonucleotide primers used for polymerase chain reaction (PCR), and site-directed mutagenesis were obtained from Invitrogen (Carlsbad, CA) and Integrated DNA technologies, Inc. (Coralville, IA). Ras activation assays were performed by using the Ras Activation Assay Kit from Cell Biolabs, Inc. (San Diego, CA). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), 1,2-dioleoyl-sn-glycero-3-[phosphoinositol-3,4,5-triphosphate] (PIP3) were purchased from Avanti Polar Lipids (Alabaster, AL). Reagents for expression and purification of glutathione S-transferase (GST) fusion proteins and maltose binding protein (MBP) were obtained from Thermo Fisher Scientific (Waltham, MA) and New England Biolabs (Ipswich, MA), respectively. For Surface Plasmon Resonance (SPR), all experiments were performed with a Biacore 3000 with optical biosensor at 25°C. CM-5, L1 sensor chips, EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide), NHS (N-hydroxysuccinimide), and P20 surfactant, buffers, NBS-EP and NBS-N, and the GST-capture kit were obtained from GE Healthcare (Piscataway, MJ).
Construction of GFP-tagged full-length RasGRP1/3 and RasGRP3 truncated constructs
The full-length RasGRP1 (NCBI Accession # NM_005739.3) and RasGRP3 (NCBI Accession # NM_001139488.1) cDNAs were amplified by PCR using specific primers (forward primer: 5′-AATaagcttGGCACCCTGGGCAAGGCGAGA-3′ and reverse primer: 3′-AATggatccCTAAGAACAGTCACCCTGCTCCA-5′; HindIII and BamHI sites; forward primer: 5′-CATctcgagGGATCAAGTGGCCTTGGGAAAG-3′ and reverse primer: 3′-AATccgcggTCAGCCATCCTCACCATCCTGT-5′; XhoI and SacII sites, respectively indicated by lower case letters, were incorporated to facilitate cloning) and subcloned into the pEGFP-C3 plasmid (BD Biosciences Clontech, Palo Alto, CA) generating pEGFP-C3-RasGRP1/3 with an N-terminal GFP tag. The full-length cDNA clone of RasGRP3 served as a template to generate the recombinant truncated constructs. The truncated versions of RasGRP3 were subcloned into the pEGP-C3 vector using the HindIII and BamHI sites. The DNA fragments of the PCR were purified with the QIAquick PCR purification kit (Qiagene, Inc., Valencia, CA) and afterwards digested. After an additional step of purification, the fragments were finally ligated into the GFP-containing pEGFP-C3 plasmid using the restriction sites. The integrity of the inserts was verified by DNA sequencing, which was performed by the Genomics Core (Center for Cancer Research, NCI).
Site-directed mutagenesis of the plasma membrane targeting (PT) domain of the full-length RasGRP3
Point mutations of the amino acid residues were introduced using the GeneTailorR and GeneArtR site-directed mutagenesis system (Invitrogen) according to the manufacturer’s instructions. To generate the mutants of RasGRP3, the above mentioned wild type full-length construct in pEGFP-C3 was used. Single (A651V) and double (D647K/K648R) mutations were introduced in one step, and triple (D647K/K648R/A651V) mutations were generated in a stepwise fashion using the single mutant as template. The presence of mutations was verified by DNA sequencing (Genomics Core, Center for Cancer Research, NCI).
Construction of GFP-tagged RasGRP1 and RasGRP3 chimeras by overlap extension PCR
To generate REM domain or C-terminal RasGRP1-RasGRP3 chimeras the full-length RasGRP1 and RasGRP3 served as templates. The necessary fragments were amplified separately by extension PCR using specific primers. The DNA fragments of the PCR were purified with the QIAquick PCR purification kit (Qiagene, Inc.) and afterwards 15 PCR cycles were run without primers in the overlap PCR reaction; then, the end primers were added to the overlap PCR reaction in the purification PCR section. Fragments with the correct size were purified again with the QIAquick PCR purification kit and, with use of BamHI and HindIII restriction enzymes, subcloned into the pEGFP-C3 plasmid (BD Biosciences Clontech) to generate pEGFP-C3-RasGRP1/3 chimeras with an N-terminal GFP tag. The integrity of the inserts was verified by DNA sequencing, which was performed by the Genomics Core (Center for Cancer Research, NCI).
Construction of the GST-tagged C1 domain and the REM domain of RasGRP1/3
To generate a recombinant RasGRP1/3 C1 domain and REM domain fused to glutathione S-transferase (GST), PCR amplification of the appropriate sequence was performed. The full-length cDNA clone of RasGRP1/3 served as a template. The DNA fragments of the PCR were purified with the QIAquick PCR purification kit (Qiagene, Inc.) and ligated into the GST-containing pGEX-2T. In the case of the RasGRP1 REM domain the BamHI and XhoI restriction enzymes were used to subclone into the pGEX-5X1 plasmid (GE Healthcare, Pittsburgh, PA).
Expression in BL21 cells and purification of the GST-tagged C1 and REM domains of RasGRP1/3
The C1 domain and REM motif of RasGRP1/3 in the pGEX-2T and pGEX-5X1 plasmids were transformed into BL21 (DE3) One Shot chemically competent E. coli (Invitrogen). Transformants were grown in LB broth medium (K-D Medical) at 37°C until the optical density of the bacterial suspension reached 0.6–0.8. Expression of the GST fusion proteins was induced with 0.3 mM isopropyl O-D-thiogalactopyranoside (Thermo Fisher Scientific) for 4 h at 37°C or 6 h at room temperature (C1 domains and REM motifs, respectively). Bacterial cells were subjected to B-PER bacterial protein extraction reagent (Thermo Fisher Scientific) or lysis buffer (150 mM NaCl, 50 mM Tris buffer, pH 7.4). The expressed GST-tagged C1 and REM proteins were purified using a GST Spin Purification Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Purification efficiency was evaluated by SDS-PAGE analysis. Purified proteins were stored in 20% glycerol at −80°C.
Construction of MBP-tagged full-length wild type RasGRP1/3 and REM chimeras of RasGRP1/3
The full-length RasGRP1/3 and REM chimeras of RasGRP1/3 cDNAs were amplified by PCR using specific primers and, with the restriction enzymes NdeI, BamHI, they were subcloned into a pMAL-c5x plasmid (New England Biolabs) generating pMAL-c5x-RasGRP1/3 and REM chimeras of RasGRP1/3 with an N-terminal MBP tag.
Expression in BL21 cells and purification of the MBP-tagged full-length RasGRP1/3 and REM chimeras of RasGRP1/3
The full-length RasGRP1/3 and the REM chimeras in the pMAL-c5x plasmid were transformed into BL21 (DE3) One Shot chemically competent E. coli (Invitrogen). Transformants were grown in LB broth medium (K-D Medical) at 37°C until the optical density of the bacterial suspension reached 0.5–0.6. Expression of the MBP fusion protein was induced with 0.3 mM isopropyl O-D-thiogalactopyranoside (Thermo Fisher Scientific) for 6 h at room temperature. The expressed MBP-tagged proteins were purified using the pMAL™ Protein Fusion and Purification System according to the manufacturer’s instructions. Purification efficiency was evaluated by SDS-PAGE analysis. Purified proteins were stored in 20% glycerol at −80°C.
Confocal analysis of GFP-labeled RasGRP1/3 proteins
LNCaP cells were plated at a density of 100,000 cells/plate on Ibidi μ-dishes (Ibidi, LLC, Verona, WI) and subcultured at 37°C in RPMI 1640 medium supplemented with 10% FBS and 2 mM L-glutamine. After 48 h in culture, cells were transfected with GFP-tagged recombinant constructs using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer’s recommendations. After 24 hours, the cells were treated with 1000 nM of PMA in confocal medium (Dulbecco’s Modified Eagle Medium without phenol red supplemented with 1% FBS), and time-lapse images were collected every 30 s using the Zeiss AIM software. Imaging was performed with a Zeiss LSM 510 confocal microscopy system (Carl Zeiss, Inc.) with an Axiovert 100 M inverted microscope operating with a 25 mW argon laser tuned to 488 nm. A 63×1.4 NA Zeiss Plan-Apochromat oil-immersion objective was used together with varying zooms (1.4 to 2×). Imaging was performed in the Center for Cancer Research Confocal Microscopy Core facility.
Quantification of Confocal Images
Two regions of 4 μm2 each were selected in each cell as follows: one in the cytoplasm, and one in the cell membrane. Mean intensities in the selected regions were calculated using the Zeiss AIM software at the different time points; the ratio of the intensities for membrane/cytoplasm was then calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation.
Pan-Ras Activation Assay
For assays of Ras activation, cells (non-transfected HEK 293 cells used as control or HEK293 cells expressing RasGRP1, RasGRP3, the different chimeras or the indicated truncated mutants) were lysed in a 1X assay buffer (Pan-Ras Activation Assay Kit, Cell Biolabs, Inc.). Lysates were centrifugated at 14,000 × g for 10 min at 4°C. The resulting supernatants were incubated for 60 min at 4°C with 40 μL Raf-1 RBD Agarose beads. After incubation, beads were collected and washed three times with 1X assay buffer. Proteins were then eluted from the beads with Laemmli sample buffer, separated by electrophoresis and analyzed by immunoblotting using anti-Pan-Ras antibody (Pan-Ras activation assay kit, Cell Biolabs, Inc.). The signal was developed by enhanced chemiluminescence (Amersham) and imaged on Amersham Hyperfilm™ ECL.
Homology modeling and amino acid sequence alignment
The homology modeling of RasGRP1 and RasGRP3 was carried out using the automated homology model generation module in ExPaSy SWISS-MODEL, a web-based protein model builder (http://swissmodel.expasy.org/workspace/ [59, 60, 61]. The crystal structure of RasGRP1 (Protein Databank code 4L9M) [45] showed 60.78% sequence identity homology with RasGRP3 and was used as the template. The homology modeling of the REM and C-terminal domains were performed separately on the selected structures. The amino acid sequence alignment was performed using the NCBI/ BLAST program.
SPR analysis
Liposome preparation
Large unilamellar vesicles with a diameter of 100 nm (liposomes) were prepared for SPR measurements. The control vesicles contained 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; 80 mole %) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS; 20 mole %) or POPS; 100 mole %. The PMA-liposomes contained 4 mole % of PMA and the PIP3-liposomes were made by the addition of 10 mole % of PIP3 to the control liposomes. Aliquots of lipids (POPC (48 μL, 10 mg/mL) and POPS (12 μL, 10 mg/mL) for control liposomes, plus PMA (3 μL of 10 mM) for PMA liposomes, plus PIP3 (6 μL, 1 mg/mL) for PIP3 liposomes were mixed and dried under a stream of nitrogen, then were resuspended in HBS-N buffer (600 μL) to give a final concentration of 1 mg/mL. The samples were vortexed for 30 s, were subjected to four freeze-thaw cycles by placing them alternately in a 42°C water bath and in dry ice, and then were extruded 46 times through two-stacked 0.1 μm pore polycarbonate filters using a LipoFast microextruder (Sigma-Aldrich) to form liposomes. The liposomes were diluted tenfold when applied to the Biacore.
Biacore experiments using the CM-5 chip
Anti-GST antibody (4700–5200 resonance units) was covalently bound to the surface of each of four flow cells of the chip, according to the manufacturer’s protocol, in HBS-EP buffer. Later, HBS-EP buffer was used as running buffer at a flow rate of 20 μL/min. The GST-tagged PKCδ C1b domain and the GST-tagged C1 domains of RasGRP1/3 (150 μL) were injected over the surface of flow cells 1, 2 and 4, respectively. The captured proteins gave signals between 60 and 150 RU. Flow cell 3, coated with anti-GST antibody, served as a reference surface and flow cell 1 containing GST-δC1b PKC domain was used as a positive control surface in each experiment. A tenfold-diluted liposome mixture (240 μL; final lipid concentration of 100 μg/mL) was injected over all four flow cells. Kinetics of liposome association to and dissociation from the GST-δC1b PKC domain and the RasGRP1/3 C1 domains were acquired for 10 min. After each round of injection, bound liposomes and captured proteins were completely removed by consecutive passage of glycine (10 mM, pH 2) and NaOH (10 mM) for 1 min each. All experiments were repeated three times.
Biacore experiments using the L1 chip
Control liposomes and PMA- and PIP3-liposomes of the same concentration were immobilized on flow cell 2, flow cell 3 and flow cell 4, respectively, of an L1 chip using HBS-N buffer as running buffer. Before the injection of liposomes, the flow cells were washed with 20 mM CHAPS for 1 min at a flow rate of 20 μL/min. The lipid surface was prepared by the injection of 100 μg/mL liposomes at a flow rate of 5 μL/min, followed by a 1 min injection of 10 mM NaOH and a 1 min injection of 40 mM n-octyl β-D-glucopyranoside (OG) at a flow rate 20 μL/min to remove the non-bound liposomes. 240 μL of purified RasGRP1/3 C1 domains, REM motifs and the full-length RasGRP1/3 and chimera proteins were injected over the surfaces at a flow rate of 20 μL/min. The dissociation was monitored for 10 min. The lipid surface was regenerated with 20 mM CHAPS and 40 mM OG at a flow rate 20 μL/min for 1 min each. The sensorgram of the control (flow cell 1) was subtracted from the sensorgram of the samples and the difference in resonance units was plotted against the time.
RESULTS
The phosphoinositide binding (plasma membrane targeting, PT) domain alone is not responsible for lack of targeting of RasGRP3 to the plasma membrane
Depending on the quality and intensity of signaling from receptors, RasGRP1 can be specifically targeted to the plasma membrane or to the endomembranes such as Golgi or the endoplasmic reticulum. The plasma membrane targeting domain makes a critical contribution to targeting RasGRP1 to the plasma membrane in some cell types. We compared the translocation pattern of RasGRP3 with that of RasGRP1 in live cells using confocal microscopy. We prepared fusion constructs between GFP and wild type RasGRP1/3. The constructs were transfected into the LNCaP human prostate cancer cell line. This cell line was chosen because it has a loss-of-function mutated PTEN, which results in markedly elevated phosphoinositide levels. The translocation of the overexpressed full-length wild type GFP-RasGRP1/3 was visualized by confocal microscopy after the addition of PMA. Images were taken every 30 s. Time points at 0, 5, 10 min after PMA addition are illustrated (Figure 2A). Data at these time points as well as at 2 min were quantitated (Figure 2B). In the presence of 1 μM PMA, RasGRP1 started to translocate to the plasma membrane after 2 min (Figure 2B), with further enhancement after 5 min (Figure 2A row 1). In our experiments RasGRP3 translocated to the endomembranes in the presence of PMA, with quite limited association with the plasma membrane (Figure 2A row 2).
Figure 2. Translocation in response to PMA of the GFP-tagged wild type RasGRP1/3 as well as RasGRP3 with a mutated PT domain in living LNCaP cells.

Cells expressing GFP-tagged wild type RasGRP1/3 and RasGRP3 in which the basic hydrophobic cluster was mutated to resemble that of RasGRP1 were treated with 1 μM PMA and the living cells were imaged by confocal microscopy as a function of time. A) Images shown are representative of three to five independent experiments. B) The ratios of the intensities for membrane/cytoplasm were calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation. Values represent the mean of the independent experiments. Bars ± SEM. *P<0.05, **P<0.005 by student’s t-test. C) Amino acid residues in the basic-hydrophobic cluster in the PT domain of RasGRP3 that were mutated to resemble those in RasGRP1 are color coded (green).
In the plasma membrane targeting (PT) domain of RasGRP1, a small segment enriched in basic hydrophobic residues, termed the BHC motif, has been identified that binds directly to the plasma membrane enriched in anionic phospholipids [48]. This unstructured basic/hydrophobic cluster can mediate membrane binding by presenting positive charges that electrostatically interact with the strongly negatively charged phosphoinositide headgroups; additionally, it has aromatic and long aliphatic side chains that insert into the lipid bilayer [49, 50, 51, 52]. There is a conserved cluster of residues (three arginines or lysines) at positions 719–721 in the PT domain of RasGRP1, missing from RasGRP3. In the corresponding sequence of RasGRP3 this conserved cluster is disrupted by an acidic residue (aspartic acid). Substituting the amino acids (Asp647, Lys648 and Ala651) of RasGRP3 with the corresponding residues (Lys720, Arg721 and Val724) of RasGRP1 in the PT domain of full-length RasGRP3 had little effect on its weak plasma membrane translocation in response to PMA (Figure 2C and 2A row 3).
In contrast, truncation of the RasGRP3 construct resulted in significant enhancement of the plasma membrane translocation in response to PMA. Removal of the REM domain allowed RasGRP3 to translocate to the plasma membrane in response to 1 μM PMA (Figure 3A row 1 and Figure 3B). Further truncations in the RasGRP3 construct, deleting the GEF and EF hands but leaving the PT domain and the lipid second messenger DAG binding C1 domain, resulted in even stronger translocation (Figure 3A row 2, 3 and 3B). Interestingly, the isolated GFP-tagged C1 domain of RasGRP3 showed only partial plasma membrane translocation in response to PMA (Figure 3A row 4 and 3B), along with translocation to the nuclear and endomembranes. After translocation, there was some tendency for the constructs to transfer back from the plasma membrane to the interior of the cells. The live cell imaging confirmed that differences in the PT (plasma membrane targeting) domain alone are not the major factor explaining the difference in localization to the plasma membrane of RasGRP1 compared to RasGRP3 in response to phorbol esters. Unexpectedly, removal of the N-terminus (REM domain) of RasGRP3 markedly enhanced its plasma membrane association in response to phorbol ester.
Figure 3. Translocation in response to PMA of the GFP-tagged truncated clones of RasGRP3 in living LNCaP cells.

Cells expressing GFP-tagged truncated RasGRP3 constructs were treated with 1 μM PMA and the living cells were imaged by confocal microscopy as a function of time. A) Images shown are representative of three to five independent experiments. B) The ratio of the intensities for membrane/cytoplasm was calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation. Values represent the mean of the independent experiments. Bars ± SEM. *P<0.05, **P<0.005, ***P=0.0002, ****P<0.0001 by student’s t-test relative to the 0 time controls. The graphic portrays the expressed domains in the various RasGRP3 truncated constructs.
Comparison of the N- and C-termini of RasGRPs by Swiss-Model homology modelling and amino acid sequence alignment showed differences in the structure of the REM and SuPT-PT domains
To develop further insights into the different translocation patterns of RasGRP1 and RasGRP3 we used amino acid sequence alignment to compare each domain in RasGRP3 with that of RasGRP1. Comprehensive sequence alignment of the RasGRP3 GEF, C1 domain and EF hands with those of RasGRP1 showed a high percentage of identities, which was corroborated by Swiss-Model homology/comparative modelling in which RasGRP1 served as the “template” and RasGRP3 as a “target”. In contrast, comparison of the REM domains of RasGRP3 and RasGRP1 showed substantial differences in their amino acid sequence and structure, with only 41–44 percent identities (Figure 4A). Comparison of the C-termini of RasGRP3 and RasGRP1 showed even greater differences (32 percent amino acid identity) and no identity using Swiss-Model comparative modelling (Figure 4B). The latter difference may be of particular structural importance, because the C-terminus of RasGRP1 forms a parallel coiled coil structure promoting dimerization [45]; this structure is unique to the RasGRP1 member of the RasGRP family in vertebrates.
Figure 4. Comparison of N- (A) and C-termini (B) of RasGRPs by Swiss-Model homology modelling and amino acid sequence alignment.
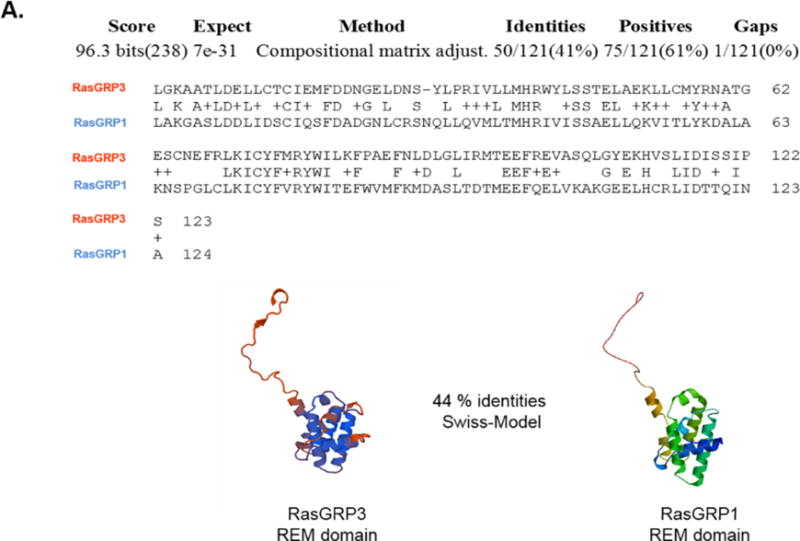

RasGRP1 served as a “template” and RasGRP3 as a “target” in homology/comparative modelling methods (Swiss-Model) and in amino acid sequence alignment studies by NCBI/BLAST. A) Comparison of the N-terminus (REM domain) of RasGRP3 and RasGRP1. B) Comparison of the C-terminus (SuPT-PT domains) of RasGRP3 and RasGRP1.
Exchange of the REM domain to that of RasGRP1 enhanced, and exchange of the SuPT-PT domain to that of RasGRP1 did not change, the translocation of RasGRP3 to the plasma membrane
To assess the contribution of the N- and C-termini of RasGRP3 to its pattern of translocation in response to PMA, we designed chimeric constructs of RasGRP3 and RasGRP1. We replaced the REM domain of RasGRP3 with the corresponding REM domain of RasGRP1. Similarly, we replaced the SuPT-PT domain of RasGRP3 with the corresponding SuPT-PT domain of RasGRP1. For comparison, we made the reciprocal chimeras with RasGRP1. The four constructs, GFP-RasGRP1(REM)-RasGRP3; GFP-RasGRP3(REM)-RasGRP1; GFP-RasGRP3-RasGRP1(SuPT-PT) and GFP-RasGRP1-RasGRP3(SuPT-PT) were transfected into the LNCaP human prostate cancer cell line and their translocation was visualized by confocal microscopy after the addition of 1 μM PMA. Substituting the REM domain of RasGRP3 with that of RasGRP1 markedly enhanced the translocation of RasGRP3 to the plasma membrane (Figure 5A row 1 and 5C). Strikingly, the strong plasma membrane translocation of RasGRP1 was not blocked by the reciprocal exchange, where its REM domain was replaced with that of RasGRP3 (Figure 5A row 2 and 5C). The SuPT-PT chimeras confirmed our previous results that the SuPT-PT domain alone is not primarily responsible for the lack of membrane targeting of RasGRP3 to the plasma membrane. The RasGRP3 chimera with the SuPT-PT domain from RasGRP1 did not translocate to the plasma membrane. The RasGRP1 chimera with the SuPT-PT domain from RasGRP3 did so (Figure 5B and C). Our results clearly show that the N-terminus (REM domain) of RasGRP3 is a critical element contributing to the difference between RasGRP3 and RasGRP1 in its ability to translocate to the plasma membrane in response to PMA.
Figure 5. Translocation in response to PMA of the GFP-tagged REM domain (A) and SUPT-PT domain (B) chimeras of RasGRP1/3 in living LNCaP cells.

Cells expressing GFP-tagged chimeras of RasGRP3 and RasGRP1 were treated with 1 μM PMA and the living cells were imaged by confocal microscopy as a function of time. Images shown are representative of three to five independent experiments. C) The ratios of the intensities for membrane/cytoplasm were calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation. Values represent the mean of the independent experiments. Bars ± SEM. *P<0.05, **P<0.005, ****P<0.0001 by student’s t-test relative to the 0 time controls. In the chimeric constructs, the domains from RasGRP3 are in grey; those from RasGRP1 are in blue.
PIP3 recognition by the REM domain might influence the binding of RasGRPs to different membranes
The patterns of membrane translocation in vivo will naturally be influenced by many factors. To explore differences in the lipid sensitivity of RasGRP1/3 proteins we performed surface plasmon resonance (SPR) experiments. We measured the binding of the C1 domain, the REM domain, and full-length RasGRP1/3 and RasGRP1/3 chimeras to phospholipids in the absence and presence of PMA and PIP3. Two approaches were used. First, the C1 domains of RasGRP1/3 and PKCδ were immobilized to generate the stationary phase, and liposomes containing either 20–80 % POPS-POPC or 20–80 % POPS-POPC together with PMA (4 mole %) or PIP3 (10 mole %) were injected over the surface (data not shown). Second, a reciprocal approach was used when liposomes were immobilized to the surface of the L1 chip to generate the stationary phase, and the C1 domains of RasGRP1/3 and PKCδ were injected over the surface (Figure 6A–B). All three C1 domains bound best to the PMA containing liposomes. For the C1 domains of RasGRP1 and RasGRP3, the PIP3 containing liposomes also supported enhanced binding, although not to the level of the PMA containing liposomes. Next, we tested the interaction of the RasGRP1/3 REM domains with lipids. The 100:0 % POPS-POPC liposomes with no ligand, with PMA and with PIP3 were immobilized as the stationary phase and the purified RasGRP1/3 REM domain proteins were injected over the surface. Appreciable lipid binding was found in the presence of PIP3 for either the RasGRP1 or RasGRP3 REM domain (Figure 6C). Comparing the binding of wild type full-length RasGRP1/3 with the REM chimeras to the phospholipids with no ligand, with PMA and with PIP3 we got a suprising result. None of the proteins bound to the PMA containing liposomes better than to control liposomes although we measured high affinity binding of radioactive PDBu in vitro (data not shown). One possible explanation for these results could be that PMA was lost from the surface bound liposomes into the mobile phase. Consistent with the in vivo translocation, the chimera with the REM domain of RasGRP1 replacing the REM domain of RasGRP3 showed enhanced binding compared to RasGRP3 (p=0.03); likewise, the chimera with the REM domain of RasGRP3 replacing the REM domain of RasGRP1 showed enhanced binding compared to RasGRP1 (p=0.02, Figure 6D). Inclusion of PIP3 enhanced the lipid binding significantly for the full-length RasGRP3 (p = 0.002) and for the RasGRP1 chimera with the RasGRP3 REM domain (p = 0.05). Binding also appeared to be increased for the RasGRP3 chimera with the RasGRP1 REM domain, but this did not reach statistical significance, similar to the limited PIP3 induced increase in binding for wild type RasGRP1.
Figure 6. SPR analysis of the binding of RasGRP1/3 C1 and REM domains and the full-length wild-type and chimeric proteins to liposomes.

(A) A SPR sensorgram showing binding of the RasGRP1 C1 domain to 20 % POPS/80 % POPC liposomes immobilized to the surface of the L1 chip in the presence of no ligand, PMA, or PIP3. The graph represents the average of three independently performed experiments. The maximum binding of the RasGRP1/3 C1 (B) and REM domains (C) and the full-length (D) proteins to liposomes containing, as indicated, PS-PC (20 % POPS/80 % POPC) or PS (100 % POPS/0 % POPC) in the presence of no ligand, PMA (4 mol %), or PIP3 (10 %) was measured. The bar graph displays the mean ± SEM from three independent experiments. *P<0.05, **P<0.005, ***P<0.0005 by student’s t-test relative to PS or PS-PC alone. #P<0.05, #P<0.005 by student’s t-test for chimera relative to wild-type under the same lipid conditions. RasGRP1 and its domains are shown in blue, RasGRP3 and its domains are shown in grey. RU: resonance units.
Exchange of REM domain impaired the Ras activation in the presence of PMA
RasGRP1/3 is a guanyl nucleotide exchange factor for Ras. A Ras-GTPase pulldown assay was performed to compare guanine nucleotide exchange activity of wild-type RasGRP1/3, their REM and SuPT-PT chimeras, and the truncated RasGRP3 constructs in HEK293 cells in the presence of 1 μM PMA. The cells were transfected with these GFP-tagged constructs. At 24 h posttransfection the cells were treated with PMA for 30 min. A representative blot is illustrated in Figure 7A; quantitation of the replicated experiments is presented in Figure 7B. In the presence of PMA, cells transfected with wild-type RasGRP1/3 yielded 15-fold higher levels of activated Ras compared to control nontransfected HEK293 cells. Truncated RasGRP3 constructs missing the Ras exchange domain or the whole catalytic module caused substantially reduced guanyl exchange compared to that of the wild-type RasGRP3 (p<0.02 and p<0.003, respectively), as expected since they should only have non-specific effects. Exchange of the REM domain in RasGRP3 with that of RasGRP1 resulted in a significant decrease in Ras activation (p<0.05). A lesser decrease occurred with the REM domain of RasGRP3 replacing that of RasGRP1, although that did not reach statistical significance (p=0.07). Exchange of the SuPT-PT domains in RasGRP1/3 did not affect the activation of Ras (Figure 7A–B). These results support the model that the REM domain plays an important role in the membrane interactions of RasGRP3, potentially by maintaining it in a closed conformation with decreased membrane affinity as well as by providing some level of membrane interactions itself.
Figure 7. Activation of Ras by RasGRP3 constructs after PMA treatment in HEK 293 cells.

HEK 293 cells were transfected with wild type RasGRP1, RasGRP3, or different mutants of RasGRP1/3. After 24 h, cells were treated with 1 μM PMA (30 min). Untransfected HEK293 cells served as a negative control. The cells were then lysed, and the levels of activation of the endogenous Ras protein were evaluated by pull-down of the activated Ras-GTP and detection by immunoblotting with anti-pan RAS antibody. A) A representative immunoblot is illustrated. (B) Band intensities were quantitated for each of 3 independent experiments. Values represent the mean ± SEM. *P< 0.05 by student’s t-test.
DISCUSSION
The mechanistic role of RasGRP family members as upstream regulators of Ras, coupled with the demonstrated functional role in the immune system and in cancer, makes them potential therapeutic targets. Efforts to understand their regulation have highlighted the prominent role of membrane lipids. Indirectly, the lipophilic second messenger DAG stimulates the phosphorylation of RasGRP1 at threonine 184 (T184) and of RasGRP3 at threonine 133 (T133) through its activation of the novel PKC isoforms, while the combination of DAG and elevated intracellular calcium drives phosphorylation of these sites by the classic PKC isoforms [5]. These phosphorylations markedly enhance Ras activation by the RasGRPs but are not absolutely required. Directly, DAG interacts with the C1 domains of RasGRP1/3, promoting their membrane localization to bring them into proximity with their substrate, Ras. Additionally, calcium binding to the EF hands contributes to enhancing the accessibility of the C1 domains, at least in RasGRP1, further promoting translocation [45]. PIP3 represents a second critical regulatory lipid. It is elevated in many cancers either through constitutively active phosphatidylinositol 3-kinase or through inactivation of its degradative pathway as a result of PTEN mutation. The report that RasGRP1 bound PIP3 through its PT domain [48], which is divergent from that of RasGRP3, suggested that RasGRP3 might show markedly different behavior depending on the membrane environment.
In the present work, one focus was on the importance of the PT domain in phosphoinositide recognition and membrane translocation of RasGRP3. RasGRP3 translocates to endomembranes and to the nuclear envelope [62]. In our confocal experiments, we likewise observed this same translocation pattern in response to PMA, which contrasted with the plasma membrane translocation of RasGRP1 in the same cells under the same conditions. In RasGRP1, the basic/hydrophobic cluster of amino acids within the PT domain is responsible for binding phosphoinositides through electrostatic interaction with polyanionic phosphoinositide headgroups as well as through insertion of a tryptophan into the lipid bilayer. This basic/hydrophobic cluster in RasGRP3 is disrupted by replacement with an acidic amino acid (aspartic acid), which suggested that this residue might be responsible for the different translocation pattern between RasGRP1 and RasGRP3. In our experiments, mutation of this residue did not have any substantial effect on the plasma membrane translocation in response to PMA. Surprisingly, removal of Ras exchange motif (REM) from RasGRP3 allowed RasGRP3 to translocate to the plasma membrane, and further truncations in the RasGRP3 construct promoted even stronger translocation to the plasma membrane in live cells. Our data show that the REM domain of RasGRP3 negatively impacts plasma membrane translocation of RasGRP3. In its absence, RasGRP3 plasma membrane translocation is enhanced. Despite the differences between RasGRP3 and RasGRP1 in the PT basic/hydrophobic cluster, the RasGRP3 PT domain still contributes to the plasma membrane translocation of RasGRP3, indicated by the enhanced localization of the C1-SuPT-PT construct of RasGRP3 as compared to the RasGRP3 C1 domain alone. A critical role was played by the C1 domain in the translocation, since in all cases the translocation was driven by the addition of phorbol ester.
To further clarify the role of the RasGRP3 REM domain in inhibiting plasma membrane translocation, we used surface plasmon resonance experiments to measure lipid binding directly. First, we confirmed that phorbol ester caused enhanced association of the isolated RasGRP3 and RasGRP1 C1 domains and that the inclusion of PIP3 in control membranes had a modest effect, reflecting the greater negative charge of the surface. Exchange of the REM domains between RasGRP3 and RasGRP1 enhanced their interaction with membrane surfaces compared to the wild type proteins, suggesting that the substituted REM domains were not able to fully replace the function of the endogenous domain in limiting lipid binding. By themselves, the REM domains of both RasGRP3 and RasGRP1 could bind to a lipid surface and this binding was enhanced in membranes with the elevated surface charge provided by inclusion of PIP3 in the membranes. Taken together, these results do not provide support for a unique role of the RasGRP1 REM domain in directly conferring plasma membrane activity. Rather, it fits better with the model that the REM domains are constraining membrane association and that the swapped REM domains cannot efficiently provide the intramolecular interactions that maintain this constraint.
The measurements of functional guanyl exchange activity further support this interpretation. Both chimeras with swapped REM domains were less effective in stimulating Ras activation, even though the RasGRP3 chimera containing the RasGRP1 REM domain translocated to the plasma membrane. Since the plasma membrane is where Ras localizes, this translocation would have been expected to enhance activity were the chimera fully functional [63]. Finally, neither of the chimeras with the swapped SuPT-PT domain showed reduced activity, indicating that at least under these conditions functional activity was maintained, consistent with the lack of change in localization that we observed with these chimeras. Of course, modest changes might have been missed depending on experimental conditions.
In our studies, it should be emphasized that we characterized membrane translocation in response to PMA. Elegant studies by Newton and coworkers [64] have shown that the binding of DAG and phorbol ester to the C1 domain of protein kinase C isoforms as well as protein kinase C translocation in response to this binding is differently dependent on structural features of the C1 domain. In particular, the presence of Tyr versus Trp at position 22 of the C1 domain reduces DAG binding affinity and response but has much less effect in the case of phorbol ester. It is also the case that different phorbol esters may cause different patterns of translocation depending on the specific derivative [65]. Such differences both highlight the potential opportunities for drug design and, conversely, provide a note of caution on over-generalization from the behavior of a specific ligand.
We conclude that the N-terminus (REM domain) of RasGRP3 is a critical element contributing to the difference between RasGRP1 and RasGRP3 in their ability to translocate to the plasma membrane. This difference in membrane translocation between RasGRP1 and RasGRP3, in turn, should imply important differences in their regulation and, potentially, in their ligand and/or substrate selectivity.
Highlights.
The N-terminus of RasGRP1/3 is a critical element in plasma membrane translocation.
The RAS exchange (REM) domain is a suppressor of membrane translocation for RasGRP3.
Differences in membrane interaction of RasGRP1/3 will contribute to different behavior in cells.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Z1A BC 005270).
Abbreviations
- SPR
Surface Plasmon Resonance
- RasGEF
Ras guanine nucleotide exchange factor
- POPS
1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- POPC
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- REM
Ras exchange motif
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None of the authors had any conflict of interest to declare.
References
- 1.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294(5545):1299–304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 2.Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773(8):1177–95. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kholodenco BN, Hancock JF, Kolch W. Signaling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11(6):414–26. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone JC. Regulation and function of the RasGRP family of Ras activators in blood cells. Genes Cancer. 2011;2(3):320–34. doi: 10.1177/1947601911408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canault M, Ghalloussi D, Grosdidier C, Guinier M, Perret C, Chelghoum N, Germain M, Raslova H, Peiretti F, Morange PE, Saut N, Pillois X, Nurden AT, Cambien F, Pierres A, van den Berg TK, Kuijpers TW, Alessi MC, Tregouet DA. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211(7):1349–1362. doi: 10.1084/jem.20130477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefanini L, Bergmeier W. CalDAG-GEF1 and platelet activation. Platelets. 2010;21(4):239–243. doi: 10.3109/09537101003639931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reuther GW, Lambert QT, Rebhun JF, Caligiuri MA, Quilliam LA, Der CJ. RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J Biol Chem. 2002;277(34):30508–14. doi: 10.1074/jbc.M111330200. [DOI] [PubMed] [Google Scholar]
- 9.Aiba Y, Oh-hora M, Kiyonaka S, Kimura Y, Hijikata A, Mori Y, Kurosaki T. Activation of RasGRP3 by phosphorylation of Thr-133 is required for B cell receptor-mediated Ras activation. Proc Natl Acad Sci U S A. 2004;101(47):16612–7. doi: 10.1073/pnas.0407468101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brodie C, Steinhart R, Kazimirsky G, Rubinfeld H, Hyman T, Ayres JN, Hur GM, Toth A, Yang D, Garfield SH, Stone JC, Blumberg PM. PKCdelta associates with and is involved in the phosphorylation of RasGRP3 in response to phorbol esters. Mol Pharmacol. 2004;66(1):76–84. doi: 10.1124/mol.66.1.76. [DOI] [PubMed] [Google Scholar]
- 11.Coughlin JJ, Stang SL, Dower NA, Stone JC. RasGRP1 and RasGRP3 regulate B cell proliferation by facilitating B cell receptor-Ras signaling. J Immunol. 2005;175(11):7179–84. doi: 10.4049/jimmunol.175.11.7179. [DOI] [PubMed] [Google Scholar]
- 12.Roose JP, Mollenauer M, Gupta VA, Stone J, Weiss A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol Cell Biol. 2005;25(11):4426–41. doi: 10.1128/MCB.25.11.4426-4441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Limnander A, Depeille P, Freedman TS, Liou J, Leitges M, Kurosaki T, Roose JP, Weiss A. STIM1, PKC-δ and RasGRP set a threshold for proapoptotic Erk signaling during B cell development. Nat Immunol. 2011;12(5):425–33. doi: 10.1038/ni.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qu HQ, Grant SF, Bradfield JP, Kim C, Frackelton E, Hakonarson H, Polychronakos C. Association of RASGRP1 with type 1 diabetes is revealed by combined follow-up of two genome-wide studies. J Med Genet. 2009;46(8):553–4. doi: 10.1136/jmg.2009.067140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plagnol V, Howson JM, Smyth DJ, Walker N, Hafler JP, Wallace C, Stevens H, Jackson L, Simmonds MJ, Bingley PJ, Gough SC, Todd JA. Type 1 Diabetes Genetics Consortium, Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PLoS Genet. 2011;7(8):e1002216. doi: 10.1371/journal.pgen.1002216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, Horita T, Kataoka H, Atsumi T, Koike T. Defective expression of Ras guanyl nucleotide-releasing protein 1 in a subset of patients with systemic lupus erythematosus. J Immunol. 2007;179(7):4890–900. doi: 10.4049/jimmunol.179.7.4890. [DOI] [PubMed] [Google Scholar]
- 17.Sharma A, Fonseca LL, Rajani C, Yanagida JK, Endo Y, Cline JM, Stone JC, Ji J, Ramos JW, Lorenzo PS. Targeted deletion of RasGRP1 impairs skin tumorigenesis. Carcinogenesis. 2014;35(5):1084–91. doi: 10.1093/carcin/bgu016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ksionda O, Melton AA, Bache J, Tenhagen M, Bakker J, Harvey R, Winter SS, Rubio I, Roose JP. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. x. Oncogene. 2016;35(28):3658–68. doi: 10.1038/onc.2015.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang D, Tao J, Li L, Kedei N, Tóth ZE, Czap A, Velasquez JF, Mihova D, Michalowski AM, Yuspa SH, Blumberg PM. RasGRP3, a Ras activator, contributes to signaling and the tumorigenic phenotype in human melanoma. Oncogene. 2011;30(45):4590–600. doi: 10.1038/onc.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Wu Q, Depeille P, Chen P, Thornton S, Kalirai H, Coupland SE, Roose JP, Bastian BC. RasGRP3 mediates MAPK pathway activation in GNAQ mutant uveal melanoma. Cancer Cell. 2017;31(5):685–696.e6. doi: 10.1016/j.ccell.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang D, Kedei N, Li L, Tao J, Velasquez JF, Michalowski AM, Tóth BI, Marincsák R, Varga A, Bíró T, Yuspa SH, Blumberg PM. RasGRP3 contributes to formation and maintenance of the prostate cancer phenotype. Cancer Res. 2010;70(20):7905–17. doi: 10.1158/0008-5472.CAN-09-4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng X, Hu Z, Wang Z, Tao J, Lu T, Yang C, Lee B, Ye Z. Upregulation of RasGRP3 expression in prostate cancer correlates with aggressive capabilities and predicts biochemical recurrence after radical prostatectomy. Prostate Cancer Trostatic Dis. 2014;17(2):119–25. doi: 10.1038/pcan.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das J, Rahman GM. C1 domains: structure and ligand-binding properties. Chem Rev. 2014;114(24):12108–31. doi: 10.1021/cr300481j. [DOI] [PubMed] [Google Scholar]
- 24.Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997;6(2):477–80. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colon-Gonzalez F, Kazanietz MG. C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta. 2006;1761(8):827–37. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Blumberg PM, Kedei N, Lewin NE, Yang D, Czifra G, Pu Y, Peach ML, Marquez VE. Wealth of opportunity – the C1 domain as a target for drug development. Curr Drug Targets. 2008;9(8):641–52. doi: 10.2174/138945008785132376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kazanietz MG. Targeting protein kinase C and “non-kinase” phorbol ester receptors: emerging concepts and therapeutic implications. Biochim Biophys Acta. 2005;1754(1–2):296–304. doi: 10.1016/j.bbapap.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 28.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81(6):917–24. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 29.Sosa MS, Lewin NE, Choi SH, Blumberg PM, Kazanietz MG. Biochemical characterization of hyperactive beta2-chimaerin mutants revealed an enhanced exposure of C1 and Rac-GAP domains. Biochemistry. 2009;48(34):8171–8. doi: 10.1021/bi9010623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duan D, Sigano DM, Kelley JA, Lai CC, Lewin NE, Kedei N, Peach ML, Lee J, Abeyweera TP, Rotenberg SA, Kim A, El Kazzouli S, Chung JU, Young HA, Young MR, Baker A, Colburn NH, Haimovitz-Friedman A, Truman JP, Parrish DA, Deschamps JR, Perry NA, Surawski RJ, Blumberg PM, Marquez VE. Conformationally constrained analogues of diacylglycerol. 29. Cells sort diacylglycerol-lactone chemical zip codes to produce diverse and selective biological activities. J Med Chem. 2008;51(17):5198–220. doi: 10.1021/jm8001907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pu Y, Perry NA, Yang D, Lewin NE, Kedei N, Braun DC, Choi SH, Blumberg PM, Garfield SH, Stone JC, Duan D, Marquez VE. A novel diacylglycerol-lactone shows marked selectivity in vitro among C1 domains of protein kinase C (PKC) isoforms alpha and delta as well as selectivity for RasGRP compared with PKC alpha. J Biol Chem. 2005;280(29):27329–38. doi: 10.1074/jbc.M414132200. [DOI] [PubMed] [Google Scholar]
- 32.El Kazzouli S, Lewin NE, Blumberg PM, Marquez VE. Conformationally constrained analogues of diacylglycerol. 30. An investigation of diacylglycerol-lactones containing heteroaryl groups reveals compounds with high selectivity for Ras guanyl nucleotide-releasing proteins. J Med Chem. 2008;51(17):5371–86. doi: 10.1021/jm800380b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebinu JO, Bottorff DA, Chan EYW, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280(5366):1082–6. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- 34.Johnson JE, Goulding RE, Ding Z, Partovi A, Anthony KV, Beaulieu N, Tazmini G, Cornell RB, Kay RJ. Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem J. 2007;406(2):223–36. doi: 10.1042/BJ20070294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–77. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119(3):393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Gureasko J, Galush WJ, Boykevisch S, Sondermann H, Bar-Sagi D, Groves JT, Kuriyan J. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat Struct Mol Biol. 2008;15(5):452–61. doi: 10.1038/nsmb.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gureasko J, Kuchment O, Makino DL, Sondermann H, Bar-Sagi D, Kuriyan J. Role of the histone domain in the autoinhibition and activation of the Ras activator Son of Sevenless. Proc Natl Acad Sci USA. 2010;107(8):3430–5. doi: 10.1073/pnas.0913915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439(7076):625–8. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- 40.Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455(7209):124–7. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 41.Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, Kuriyan J. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci U S A. 2006;103(45):16692–7. doi: 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112(5):685–95. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 43.Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405(2):199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharya S, Bunick CG, Chazin WJ. Target selectivity in EF-hand calcium binding proteins. Biochim Biophys Acta. 2004;1742(1–3):69–79. doi: 10.1016/j.bbamcr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 45.Iwig JS, Vercoulen Y, Das R, Barros T, Limnander A, Che Y, Pelton JG, Wemmer DE, Roose JP, Kuriyan J. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. Elife. 2013;2:e00813. doi: 10.7554/eLife.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tazmini G, Beaulieu N, Woo A, Zahedi B, Goulding RE, Kay RJ. Membrane localization of RasGRP1 is controlled by an EF-hand, and by the GEF domain. Biochim Biophys Acta. 2009;1793(3):447–61. doi: 10.1016/j.bbamcr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 47.Fuller DM, Zhu M, Song X, Ou-Yang CW, Sullivan SA, Stokne JC, Zhang W. Regulation of RasGRP1 function in T cell development and activation by its unique tail domain. PLoS One. 2012;7(6):e38796. doi: 10.1371/journal.pone.0038796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zahedi B, Goo HJ, Beaulieu N, Tazmini G, Kay RJ, Cornell RB. Phosphoinositide 3-kinase regulates plasma membrane targeting of the Ras-specific exchange factor RasGRP1. J Biol Chem. 2011;286(14):12712–23. doi: 10.1074/jbc.M110.189605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gambhir A, Hangyás-Mihályné G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J. 2004;86(4):2188–207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314(5804):1458–61. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi S, Pryciak PM. Identification of novel membrane-binding domains in multiple yeast Cdc42 effectors. Mol Biol Cell. 2007;18(12):4945–56. doi: 10.1091/mbc.E07-07-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W, Crocker E, McLaughlin S, Smith SO. Binding of peptides with basic and aromatic residues to bilayer membranes: phenylalanine in the myristoylated alanine-rich C kinase substrate effector domain penetrates into the hydrophobic core of the bilayer. J Biol Chem. 2003;278(24):21459–66. doi: 10.1074/jbc.M301652200. [DOI] [PubMed] [Google Scholar]
- 53.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 54.Milella M, Falcone I, Conciatori F, Cests Icani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015;5:24. doi: 10.3389/fonc.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamashita S, Mochizuki N, Ohba Y, Tobiume M, Okada Y, Sawa H, Nagashima K, Matsuda M. CalDAG-GEFIII activation of Ras, R-ras, and Rap1. J Biol Chem. 2000;275(33):25488–93. doi: 10.1074/jbc.M003414200. [DOI] [PubMed] [Google Scholar]
- 56.Rebhun JF, Castro AF, Quilliam LA. Identification of guanine nucleotide exchange factors (GEFs) for the Rap1 GTPase. Regulation of MR-GEF by M-Ras-GTP interaction. x. J Biol Chem. 2000;275(45):34901–8. doi: 10.1074/jbc.M005327200. [DOI] [PubMed] [Google Scholar]
- 57.Lorenzo PS, Kung JW, Bottorff DA, Garfield SH, Stone JC, Blumberg PM. Phorbol esters modulate the Ras exchange factor RasGRP3. Cancer Res. 2001;61(3):943–9. [PubMed] [Google Scholar]
- 58.Braun DC, Cao Y, Wang S, Garfield SH, Hur GM, Blumberg PM. Role of phorbol ester localization in determining protein kinase C or RasGRP3 translocation: real-time analysis using fluorescent ligands and proteins. Mol Cancer Ther. 2005;4(1):141–50. [PubMed] [Google Scholar]
- 59.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. x. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 60.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;(Suppl 1):S162–73. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 61.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–8. doi: 10.1093/nar/gku340. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okamura SM, Oki-Idouchi CE, Lorenzo PS. The exchange factor and diacylglycerol receptor RasGRP3 interacts with dynein light chain 1 through its C-terminal domain. J Biol Chem. 2006;281(47):36132–9. doi: 10.1074/jbc.M605093200. [DOI] [PubMed] [Google Scholar]
- 63.Magee T, Marshall C. New insights into the interaction of Ras with the plasma membrane. Cell. 1999;98(1):9–12. doi: 10.1016/S0092-8674(00)80601-7. [DOI] [PubMed] [Google Scholar]
- 64.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282(2):826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 65.Wang QJ, Bhattacharyya D, Garfield S, Nacro K, Marquez VE, Blumberg PM. Differential localization of protein kinase C δ by phorbol esters and related compounds using a fusion protein with green fluorescent protein. J Biol Chem. 1999;274(52):37233–37239. doi: 10.1074/jbc.274.52.37233. [DOI] [PubMed] [Google Scholar]