

Abstract
Myocardial fibrosis is closely associated with heart failure because myocardial fibrosis may cause the loss of normal cardiac function. Endoglin is a homeodimeric membrane glycoprotein, a co-receptor of transforming growth factor-β1 (TGF-β1) and β3. Endoglin is a potent mediator of profibrotic effects of angiotensin II on cardiac fibroblasts and can modulate the effect of TGF-β1 on extracellular matrix synthesis. These data indicate that endoglin plays an important role in fibrogenesis in cardiac remodeling. Endoglin induced by TGF-β1 is largely through PI-3 kinase, Akt, Smad3/4 and endoglin promoter pathways. Endoglin was upregulated in pressure- overload, volume-overload heart failure and acute myocardial infarction and was associated with myocardial fibrosis. Silencing endoglin expression could attenuate myocardial fibrosis and improve survival in animal study. Endoglin expression was increased in failing left ventricle before use of left ventricle assist device, and reduced back to control levels after use of left ventricle assist device. Targeting endoglin may provide a potentially unique and novel therapeutic approach for reducing myocardial fibrosis in patients with heart failure.
Keywords: Angiotensin II, Endoglin, MicroRNA, Myocardial fibrosis, TGF-β1
INTRODUCTION
Heart failure is one of the most common causes of hospitalization in the United States and is the predominant disease in the elderly.1,2 Heart failure is also a major cause of morbidity and mortality worldwide. Coronary heart disease and hypertension are the two major risk factors for the development of heart failure. Myocardial fibrosis, which is characteristic of all types of pathological hypertrophy, can lead to mechanical stiffness and thus contribute to contractile dysfunction.3 Myocardial fibrosis leads to the loss of normal cardiac function and is closely associated with heart failure.4 Acute myocardial infarction (AMI) caused by coronary artery occlusion is a major cause of morbidity and mortality in humans. The loss of blood flow to myocardium results in cardiac myocyte death and myocardial remodeling. Cardiac remodeling after AMI can induce myocardial fibrosis and reduce left ventricular contractility and ultimately cause heart failure.5 Modern treatment of AMI with pharmacological or mechanical reperfusion has improved survival of AMI patients. However, heart failure prevalence increases due to myocardial injury with remodeling after survival of heart attack and aging process. The failing heart is characterized by cellular and structural remodeling including cardiac myocyte hypertrophy and fibrotic deposition, which promote alterations in cardiac stiffness and function.6
Transforming growth factor-β1 (TGF-β1) plays a causal role in promoting cardiac fibrosis.7-9 TGF-β may directly participate in the progressive remodeling process in heart failure. Endoglin is a 180-kDa homeodimeric membrane glycoprotein that is a co-receptor of TGF-β1 and β3.10 It is highly expressed in vascular endothelial cells and is crucial for angiogenesis.11,12 Mutations in human endoglin on chromosome 9q34ter result in the autosomal dominant form of hereditary hemorrhagic telangiectasia type 1, a rare condition characterized by epistaxis and vascular malformation.13,14 Endoglin homozygote mutation mice die at embryonic day 10.5 as a result of impaired cardiovascular development and extraembryonic angiogenesis,15 while the endoglin heterozygote mutation mice are viable and recapitulate hereditary hemorrhagic telangiectasia type 1.16 In cardiac tissues, endoglin is expressed in endothelial cells, and fibroblast, but cardiac myocytes fail to demonstrate significant endoglin expression.17,18 Valeria et al. demonstrated that endoglin was expressed in fetal but not adult cardiac myocytes.19 The extracellular domain of endoglin can be proteolytically cleaved by matrix metalloproteinase-14 and circulates as soluble endoglin (sEng).20 Soluble endoglin could antagonize TGF-β1 signaling.18
Endoglin is a potent mediator of the profibrotic effects of angiotensin II on cardiac fibroblasts and can modulate the effect of TGF-β1 on extracellular matrix synthesis.21,22 Endoglin is upregulated in patients with renal and liver fibrosis.23 Endoglin has been shown to counteract the fibrogenic effect of TGF-β1 in renal interstitial fibrosis.24 These data indicate that endoglin plays an important role in fibrogenesis in cardiac remodeling. This review will summarize the molecular mechanism of endoglin in profibrogenic action, myocardial fibrosis and endoglin in animal model and clinical studies.
ENDOGLIN MODULATES PROFIBROGENIC ACTIONS OF Ang II AND TGF-β1
Angiotensin II (Ang II) is a pivotal mediator of myocardial fibrosis because it modulates cardiac remodeling by causing cardiac myocyte hypertrophy and myocardial fibrosis.25-27 Ang II also upregulates expression of TGF-β1 to produce myocardial fibrosis.28 Ang II and TGF-β1 increase cardiac fibroblast migration, proliferation and collagen production, the major pathophysiology of cardiac fibrosis.29 Chen et al. found that endoglin expression in cardiac fibroblasts was upregulated by Ang II.21 Ang II increased expression of endoglin mRNA and protein in a concentration and time-dependent manner.21 Ang II modulated endoglin expression via Ang II type 1 receptor because angiotensin type 1 receptor blockers abrogated Ang II-induced expression of both endoglin mRNA and protein, while the angiotensin type 2 receptor blocker had no effect. Ang II increased endoglin expression through mitogen-activated protein kinase (MAPK)p42/44 pathway. Ang II increased type 1 collagen protein expression and decreased matrix metalloproteinase-1, the collagen degrading enzyme and expression. Both effects were antagonized by a specific endoglin antibody, indicating that endoglin modulates collagen metabolism by Ang II.21
Endoglin mRNA and protein expression was significantly induced by TGF-β1 in cardiac fibroblasts.30 Collagen I protein was also significantly induced by TGF-β1. Atorvastatin has been shown to reduce endoglin expression in endothelium in apo-E deficient mice and C57BL/6J mice.31,32 Atorvastatin, a 3-hydroxy 3-methyl glutaryl-CoA reductase inhibitor, almost completely inhibited the endoglin and collagen I formation and protein expression induced by TGF-β1. Endoglin siRNA almost completely attenuated the collagen I protein expression induced by TGF-β1. TGF-β1 induced endoglin expression in cultured cardiac fibroblasts through PI-3 kinase, Akt, and Smad3 pathways. Atorvastatin attenuated the endoglin expression induced by TGF-β1.30 This finding implies that endoglin acts as a fibrogenic mediator of TGF-β1 on cardiac fibroblast. The interplay between TGF-β1 and endoglin in cardiac fibroblast is shown in Figure 1. Cardiac myocytes responded to increased mechanical load and hormonal stimulation by hypertrophic growth.33 Mechanical stretch significantly increased endoglin and collagen 1 expression in rat cardiac myoblasts.34 TGF-β1 and overexpression of microRNA-208a (miR-208a) upregulated endoglin and collagen expression in cardiac myoblasts. Mechanical stretch increased TGF-β1 secretion from cardiac myocytes.35 TGF-β1 mediates the expression of miR-208a by mechanical stretch. The interplay between mechanical stretch, TGF-β1, and miR-208a is shown in Figure 2. Endoglin induced by TGF-β1 is largely through PI-3 kinase, Akt, Smad3/4 and endoglin promoter pathways. The reduced endoglin expression resulted in less collagen I formation and less protein synthesis in cardiac fibroblasts, indicating a potential role of endoglin in cardiac remodeling.
Figure 1.
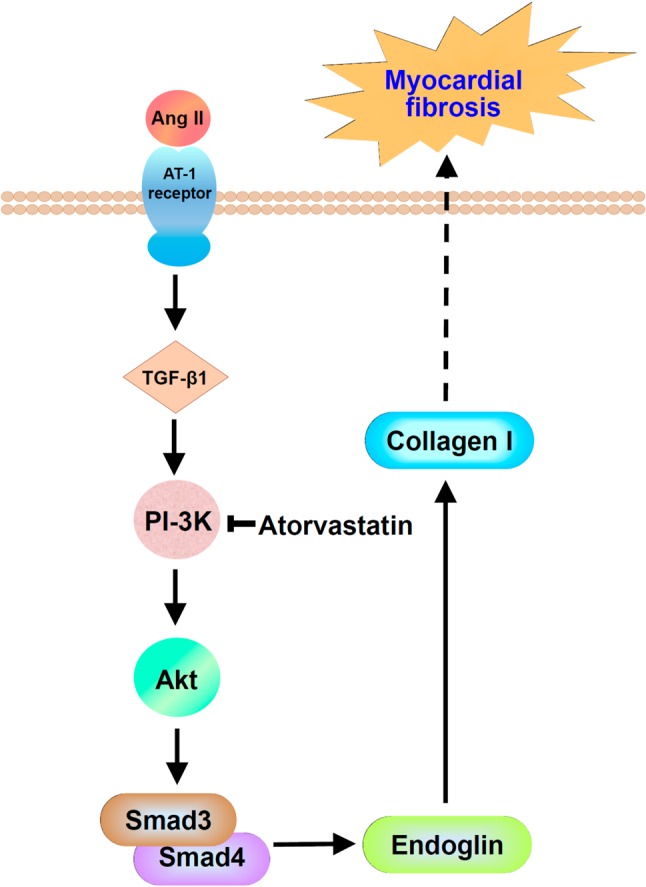
The interplay of TGF-β1 and endoglin to induce cardiac fibrosis in cardiac fibroblast. Angiotensin II (Ang II) increases TGF-β1 expression through AT1 receptor. The increased TGF-β1 activates endoglin expression through PI-3 kinase, Akt, and Smad3/4. The increased endoglin increases collagen I to induce myocardial fibrosis. Atorvastatin could inhibit PI-3 kinase phosphorylation and then reduced endoglin expression to improve myocardial fibrosis.
Figure 2.
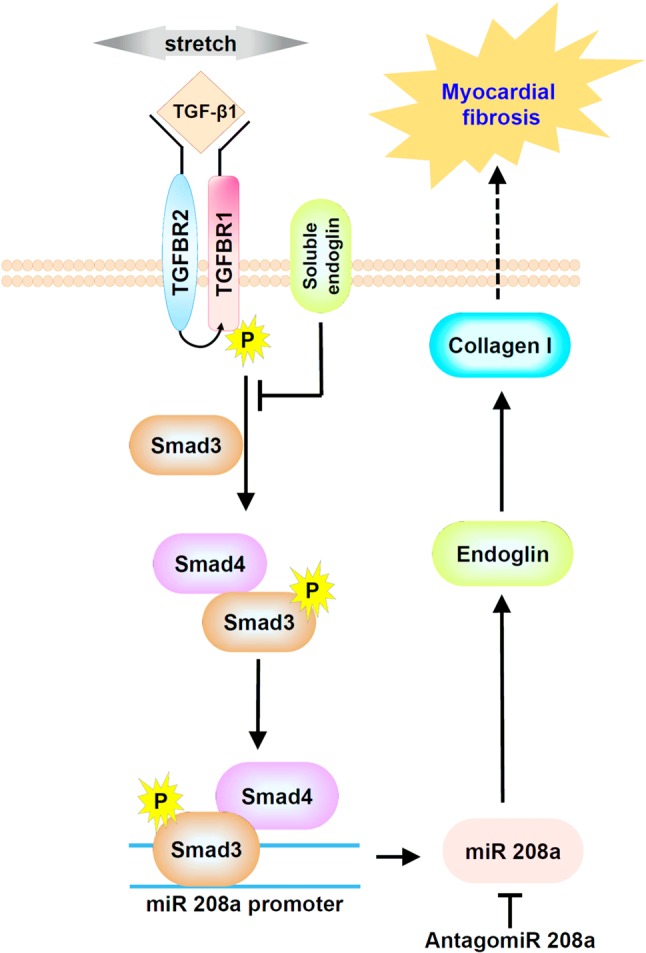
The interplay between mechanical stretch, TGF-β1, and miR-208a in cardiac myoblasts. Mechanical stretch increases TGF-β1 secretion and then TGF-β1 increases smad/3/4 phosphorylation to increase miR208a promoter activity. The increased miR208a activates endoglin and collagen I expression to induce myocardial fibrosis. The endoglin promoter sequences have specific sites that are complementary to miR-208a. Soluble endoglin could antagonize TGF-β1 signaling. Antagomir 208a could antagonize miR208a and then reduce endoglin expression to improve myocardial fibrosis.
REDUCED ENDOGLIN ACTIVITY LIMITS CARDIAC FIBROSIS IN PRESSURE OVERLOAD MODEL
The pressure overload causes concentric cardiac hypertrophy and heart failure. Transaortic constriction or aortic banding is typically used in mice or rat as pressure overload model. In the animal model of pressure overload induced by transaortic constriction, left ventricular endoglin mRNA was increased within 2 weeks and remained elevated at 4 and 10 weeks.21 Compared with wild type mice, left ventricular (LV) endoglin was lower in Eng+/- mice (endoglin-heterozygous mice). Eng+/- demonstrated preserved cardiac function and improved survival compared with wild type after transaortic constriction. Transaortic constriction induced LV hypertrophy or cardiac myocyte hypertrophy and was similar in Eng+/- and wild type mice. However, myocardial capillarity is enhanced in Eng+/- mice compared with wild type mice. Type 1 collagen mRNA and protein expression was significantly increased after transaortic constriction in wild type mice. Collagen expression was lower in heterozygous mice compared with wild type mice. Silencing endoglin expression by siRNA or endoglin antibody significantly decreased type 1 collagen mRNA and protein expression in cardiac fibroblasts. sEng significantly attenuated cardiac fibrosis and type 1 collagen expression in transaortic constriction-induced heart failure.
ENDOGLIN AND MYOCARDIAL FIBROSIS IN VOLUME OVERLOAD MODEL
Aorta-caval (AV) shunt model increased heart weight, heart weight/body weight ratio, and increased left ventricular size without increased septal and posterior wall thickness, indicating an eccentric hypertrophy which is consistent with volume overload heart failure status. AV shunt significantly increased myocardial endoglin protein expression and cardiac hypertrophic markers such as βMHC and BNP.36 Overexpression of miR-208a in the sham group without AV shunt significantly increased endoglin expression similar to the AV shunt group. Antagomir208a significantly attenuated the increase of myocardial endoglin and βMHC protein expression induced by AV shunt. Antagomir208a and pretreatment with atorvastatin in the AV shunt group significantly decreased myocardial fibrosis area induced by AV shunt. Over-expression of mutant type miR-208a in the AV shunt did not decrease the fibrosis area induced by AV shunt. The increased endoglin to induce myocardial fibrosis induced by AV shunt was mediated by miR-208a. Endoglin expression is elevated in acute volume-overload heart induced by AV shunt, indicating that endoglin plays a crucial role in pressure- and volume-overload heart failure. A miR is a small, 22-nucleotide non-protein-coding RNA that usually inhibits transcription or translation by interacting with the 3’ untranslated regions of target mRNA and promoting target mRNA degradation (gene silencing).37 However, Place et al. have demonstrated miR functioning to induce gene expression, not gene silencing.38 MiR induces gene expression by targeting specific sites in gene promoters.38 The endoglin promoter sequences have specific sites that are complementary to miR-208a.30 Figure 3 summarizes the relationship of miR-208a and endoglin in myocardial fibrosis in volume-overload heart failure model.
Figure 3.
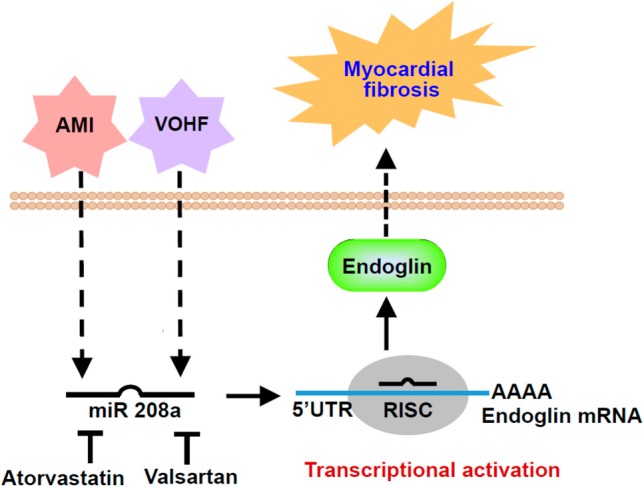
Proposed pathway of acute myocardial infarction (AMI) and volume overload heart failure (VOHF) model to induce myocardial fibrosis. AMI and VOHF could increase miR-208a expression and then activate endoglin expression to induce myocardial fibrosis. Atorvastatin and valsartan could attenuate miR-208a expression and the attenuated miR-208a reduces endoglin expression and finally improves myocardial fibrosis.
ENDOGLIN AND MYOCARDIAL FIBROSIS IN A MOUSE MODEL OF RIGHT VENTRICULAR PRESSURE OVERLOAD
Pulmonary artery constriction causes right ventricular pressure overload and subsequently induces right heart failure. Kapur et al. demonstrated that endoglin promoted TGF-β1-mediated calcineurin expression and myofibroblast conversion in right ventricular fibroblasts.39 Reduced endoglin expression in Eng+/- mice preserved indices of right ventricular diastolic function, limited right ventricular collagen accumulation, and limited myofibroblast transformation in the right ventricle in a model of angio-obliterative pulmonary hypertension induced by exposure to hypoxia and the anti-vascular endothelial growth factor compound, Sugen as compared to wild type mice. Reduced endoglin expression in Eng+/- mice also preserved right ventricle function and improved survival in surgical constriction of pulmonary artery as compared to wild type mice. Right ventricle pressure overload increased right ventricle fibrosis in wild type mice, but not in Eng+/- mice. Treatment with neutralizing endoglin antibody improved survival of mice with severe right ventricle overload as compared to treatment with IgG control. Right ventricular fibrosis was increased in wild type mice treated with IgG control, but not in mice treated with neutralizing endoglin antibody after severe right ventricular pressure overload. The animal study by Kapur et al. supports that blocking endoglin activity reverses established right ventricular fibrosis in chronic right ventricular pressure overload in mice.39
ENDOGLIN AND MYOCARDIAL FIBROSIS IN RAT MODEL OF ACUTE MYOCARDIAL INFARCTION
Rat model of acute myocardial infarction by ligating the left anterior descending artery at 7 days significantly increased myocardial endoglin and β-myosin heavy chain protein expression and over-expression of antagomir208a significantly inhibited the increase of myocardial endoglin and β-myosin heavy chain protein expression induced by myocardial infarction.40 Overexpression of antagomir208a and pretreatment of atorvastatin and valsartan in the AMI group significantly decreased the myocardial fibrosis area induced by myocardial infarction. Over-expression of miR-208a in the sham group without myocardial infarction significantly increased myocardial endoglin expression. Over-expression of antagomir208a in the myocardial infarction group significantly decreased the myocardial fibrosis area induced by myocardial infarction, indicating that miR-208a plays a crucial role in myocardial fibrosis after myocardial infarction. The increased endoglin to induce myocardial fibrosis induced by myocardial infarction was mediated by miR-208a, indicating endoglin is a downstream target of miR-208a in AMI. Hypoxia at 2.5% oxygen or overexpression of miR-208a alone without hypoxia for 2 h significantly increased endoglin promoter activity. This result indicates that endoglin expression is induced at the transcriptional level by miR-208a. When the miR-208a binding sites were mutated, the increased promoter activity induced by endoglin was abolished. The myocardial fibrosis and left ventricular dimension induced by AMI could by attenuated by endoglin siRNA and epigallocatechin-3-O-gallate.41 Figure 3 summarizes the relationship of miR-208a and endoglin in myocardial fibrosis in AMI model.
Tseliou et al. reported that cardiospheres reduced myocardial scar and collagen mass in chronic rat myocardial infarction by inhibition of TGF-β1 signaling and increased soluble endoglin.42 Soluble endoglin inhibits TGF-β1-induced cardiac fibrosis.18 Cardiospheres reduced phosphorylation of smad2/3, smad4 and TGF-β receptor 1 and TGF-β receptor 2 expression. Soluble endoglin downregulates TGF-β1/smad pathway to attenuated collagen production.
ENDOGLIN IN CLINICAL STUDIES
Endoglin expression is increased in human hearts with severe left ventricular systolic dysfunction. Endoglin was detected in the left ventricle in patients with end-stage heart failure for surgical implantation of a left ventricle assist device (LVAD). Endoglin expression was increased in the failing left ventricle at the time of LVAD, and returned back to control levels after LVAD support.21 Endoglin has been used as a noninvasive measure of left ventricular filling pressure in heart failure43 and has been regarded as a novel biomarker for acute heart failure.44 Circulating levels of sEng were elevated in patients with left ventricular dysfunction. Soluble endoglin levels also decreased in association with reduced cardiac filling pressure after diuresis. Ikemoto et al. also found that plasma endoglin could be a marker to predict cardiovascular event in patients with chronic coronary artery disease after percutaneous coronary intervention.45 The highest endoglin group patients had a significantly higher major adverse cardiovascular events (congestive heart failure, acute myocardial infarction, stroke, and sudden cardiac death) rate than the lowest endoglin group patients.
CONCLUSIONS
Control of myocardial fibrosis is essential to improving myocardial performance and preventing progressive deterioration of heart failure. Endoglin plays a crucial role in myocardial fibrosis in different models of heart failure. Therefore, targeting endoglin to prevent fibrosis and heart failure may become an important and novel therapeutic strategy to improve clinical outcome in patients with heart failure in addition to the current clinical benefits of β-adrenergic receptor antagonists, angiotensin-converting enzyme or receptor blockers and aldosterone antagonists.46
CONFLICT OF INTERESTS
None to declare.
REFERENCES
- 1.Aronow WS. Epidemiology, pathophysiology, prognosis, and treatment of systolic and diastolic heart failure. Cardiol Rev. 2006;14:108–124. doi: 10.1097/01.crd.0000175289.87583.e5. [DOI] [PubMed] [Google Scholar]
- 2.Mozaflarian D, Benzamin EJ, Go AS, et al. Heart disease and stroke statistics-2016 update. A report from the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 3.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutton MGSJ, Sharpe N. Left ventricular remodeling after myocardial infarction. Pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 5.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Phys Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 6.Benjamin IJ. Targeting endoglin, an auxiliary TGF-β coreceptor, to prevent fibrosis and heart failure. Circulation. 2012;125:2689–2691. doi: 10.1161/CIRCULATIONAHA.112.108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-β1. Mol Genetic Metabol. 2000;71:418–435. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- 8.Kuwahara F, Kai H, Tokuda K, et al. Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 9.Lim H, Zhu YZ. Role of transforming growth factor-β in the progression of heart failure. Cell Mol Life Sci. 2006;63:2584–2596. doi: 10.1007/s00018-006-6085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonsatti E, Altomonte M, Arslan P, Maio M. Endoglin (CD105): a target for anti-angiogenic cancer therapy. Curr Drug Targets. 2003;4:291–296. doi: 10.2174/1389450033491073. [DOI] [PubMed] [Google Scholar]
- 11.Van de Driesche S, Mummery CL, Wetermann CJ. Hereditary hemorrhagic telangiectasia: an update on transforming growth factor beta signaling in vasculogenesis and angiogenesis. Cardiovasc Res. 2003;58:20–31. doi: 10.1016/s0008-6363(02)00852-0. [DOI] [PubMed] [Google Scholar]
- 12.Duff SE, Li C, Garland JM, Kumar S. CD105 is important for angiogenesis: evidence and potential applications. FASEB J. 2003;17:984–992. doi: 10.1096/fj.02-0634rev. [DOI] [PubMed] [Google Scholar]
- 13.Guttmacher AE, Marchuk DA, White RI., Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- 14.McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gen for hereditary hemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 15.Li DY, Sorensen LK, Brooke BS, et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1517. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 16.Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.St-Jacques S, Cymerman U, Pece N, Letarte M. Molecular characterization and in situ localization of murine endoglin reveal that it is a transforming growth factor-beta binding protein of endothelial and stroma cells. Endocrinology. 1994;134:2645–2657. doi: 10.1210/endo.134.6.8194490. [DOI] [PubMed] [Google Scholar]
- 18.Kapur NK, Wilson S, Yunis AA, et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 2012;125:2728–2738. doi: 10.1161/CIRCULATIONAHA.111.080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valeria B, Maddalena G, Entrica V, et al. Endoglin (CD105) expression in the human heart through gestation: an immunohistochemical study. Reprod Sci. 2008;15:1018–1026. doi: 10.1177/1933719108322429. [DOI] [PubMed] [Google Scholar]
- 20.Hawinkels LJ, Kuiper P, Wiercinska E, et al. Matrix metalloproteinase-14 (MT1-MMP)-induced endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010;70:4141–4150. doi: 10.1158/0008-5472.CAN-09-4466. [DOI] [PubMed] [Google Scholar]
- 21.Chen K, Mehta JL, Li D, et al. Transforming growth factor-β receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004;95:1167–1173. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 22.Rodríguez-Barbero A, Obreo J, Alvarez-Munoz P, et al. Endoglin modulation of TGF-β1-induced collagen synthesis is dependent on ERK1/2 MAPK activation. Cell Physiol Biochem. 2006;18:135–142. doi: 10.1159/000095181. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Pozo L, Miquilena-Colina ME, Lozano-Rodriguez T, Garcia-Monzon C. Endoglin: structure, biologcial functions and role in fibrogenesis. Rev Esp Enferm Dig. 2008;100:355–360. doi: 10.4321/s1130-01082008000600008. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Pena A, Eleno N, Duwell A, et al. Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension. 2002;40:713–720. doi: 10.1161/01.hyp.0000037429.73954.27. [DOI] [PubMed] [Google Scholar]
- 25.Weber KT. Extracellular matrix remodeling in heart failure: a role for de novo angiotensin II generation. Circulation. 1997;96:4065–4082. doi: 10.1161/01.cir.96.11.4065. [DOI] [PubMed] [Google Scholar]
- 26.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium: fibrosis and the renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–1865. doi: 10.1161/01.cir.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 27.Wu L, Iwai M, Nakagani H, et al. Effect of angiotensin type 1 receptors blockade on cardiac remodeling in angiotensin II type 2 receptor null mice. Arterioscler Thromb Vasc Biol. 2002;22:49–54. doi: 10.1161/hq0102.102277. [DOI] [PubMed] [Google Scholar]
- 28.Sui Y, Zhang JQ, Zhang J, Ramires J. Angiotensin II, transforming growth factor-beta 1 and repair the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–1569. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- 29.Chung CC, Kao YH, Liou JP, Chen YI. Curcumin suppress cardiac fibroblasts activities by regulating proliferation, migration, and the extracellular matrix. Acta Cardiol Sin. 2014;30:474–482. [PMC free article] [PubMed] [Google Scholar]
- 30.Shyu KG, Wang BW, Chen WJ, et al. Mechanism of the inhibitory effect of atorvastatin on endoglin expression induced by transforming growth factor-beta 1 in cultured cardiac fibroblasts. Eur J Heart Fail. 2010;12:219–226. doi: 10.1093/eurjhf/hfq011. [DOI] [PubMed] [Google Scholar]
- 31.Pospisilova N, Semeeky V, Jamborova G, et al. Endoglin expression in hypercholesterolemia and after atorvastatin treatment in apo-E deficient mice. J Pharm Pharmaceut Sci. 2006;9:388–397. [PubMed] [Google Scholar]
- 32.Nachtigal P, Pospisilova N, Jamborova G, et al. Endothelial expression of endoglin in normocholesterolemic and hypercholesterolemic C57BL/6J mice before and after atorvastatin treatment. Can J Physiol Pharmacol. 2007;85:767–773. doi: 10.1139/y07-068. [DOI] [PubMed] [Google Scholar]
- 33.Torsoni AS, Constancio SS, Nadruz W, et al. Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circ Res. 2003;93:140–147. doi: 10.1161/01.RES.0000081595.25297.1B. [DOI] [PubMed] [Google Scholar]
- 34.Shyu KG, Wang BW, Wu GJ, et al. Mechanical stretch via transforming growth factor-β1 activates microRNA208a to regulate endoglin expression in cultured rat cardiac myoblasts. Eur J Heart Fail. 2013;15:36–45. doi: 10.1093/eurjhf/hfs143. [DOI] [PubMed] [Google Scholar]
- 35.Wang BW, Wu GJ, Cheng WP, Shyu KG. Mechanical stretch via transforming growth factor-β1 activates micro-RNA-208a to regulate hypertrophy in cultured rat cardiac myocytes. J Formos Med Assoc. 2013;112:635–643. doi: 10.1016/j.jfma.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Wang BW, Wu GJ, Cheng WP, Shyu KG. MicroRNA-208a increases myocardial fibrosis via endoglin in volume overloading heart. Plos One. 2014;9:e84188. doi: 10.1371/journal.pone.0084188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 38.Place RF, Li LC, Pookot D, et al. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105:1608–1613. doi: 10.1073/pnas.0707594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapur NK, Qiao X, Paruchuri V, et al. Reducing endoglin activity limits calcineurin and TRPC-6 expression and improves survival in a mouse model of right ventricular pressure overload. J Am Heart Assoc. 2014;3:e000965. doi: 10.1161/JAHA.114.000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shyu KG, Wang BW, Cheng WP, Lo HM. MicroRNA-208a increases myocardial endoglin expression and myocardial fibrosis in acute myocardial infarction. Can J Cardiol. 2015;31:679–690. doi: 10.1016/j.cjca.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 41.Lin CM, Chang H, Wang BW, Shyu KG. Suppressive effect of epigallocatechin-3-O-gallate on endoglin molecular regulation in myocardial fibrosis in vitro and in vivo. J Cell Mol Med. 2016;20:2045–2055. doi: 10.1111/jcmm.12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tseliou E, Reich H, de Couto G, et al. Cardiospheres reverse adverse remodeling in chronic myocardial infarction: roles of soluble endoglin and TGF-β signaling. Basic Res Cardiol. 2014;109:443–455. doi: 10.1007/s00395-014-0443-8. [DOI] [PubMed] [Google Scholar]
- 43.Kapur NK, Hefferman KS, Yunis AA, et al. Usefulness of soluble endoglin as a non-invasive measure of left ventricular filling pressure in heart failure. Am J Cardiol. 2010;106:1770–1776. doi: 10.1016/j.amjcard.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yanavitski M, Givertz MM. Novel biomarkers in acute heart failure. Curr Heart Fail Rep. 2011;8:206–211. doi: 10.1007/s11897-011-0065-5. [DOI] [PubMed] [Google Scholar]
- 45.Ikemoto T, Hojo Y, Kondo H, et al. Plasma endoglin as a marker to predict cardiovascular events in patients with chronic coronary artery diseases. Heart Vessels. 2012;27:344–351. doi: 10.1007/s00380-011-0163-z. [DOI] [PubMed] [Google Scholar]
- 46.Wang CC, Chang HY, Yin WH, et al. TSOC-HFrEF registry: a registry of hospitalized patients with decompensated systolic heart failure: description of population and management. Acta Cardiol Sin. 2016;32:400–411. doi: 10.6515/ACS20160704A. [DOI] [PMC free article] [PubMed] [Google Scholar]