

Abstract
Several lines of knockout mice deficient in the genes encoding each component of the endogenous opioid system have been used for decades to clarify the specific role of the different opioid receptors and peptide precursors in many physiopathological conditions. The use of these genetically modified mice has improved our knowledge of the specific involvement of each endogenous opioid component in nociceptive transmission during acute and chronic pain conditions. The present review summarizes the recent advances obtained using these genetic tools in understanding the role of the opioid system in the pathophysiological mechanisms underlying chronic pain. Behavioural data obtained in these chronic pain models are discussed considering the peculiarities of the behavioural phenotype of each line of knockout mice. These studies have identified the crucial role of specific components of the opioid system in different manifestations of chronic pain and have also opened new possible therapeutic approaches, such as the development of opioid compounds simultaneously targeting several opioid receptors. However, several questions still remain open and require further experimental effort to be clarified. The novel genetic tools now available to manipulate specific neuronal populations and precise genome editing in mice will facilitate in a near future the elucidation of the role of each component of the endogenous opioid system in chronic pain.
Linked Articles
This article is part of a themed section on Emerging Areas of Opioid Pharmacology. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.14/issuetoc
Abbreviations
- Cas9
CRISPR‐associated protein 9
- CFA
complete Freund's adjuvant
- CNO
clozapine N‐oxide
- CRISPR
clustered regulatory interspaced short palindromic repeats
- DOP
δ‐opioid receptor
- DREADD
designed receptor exclusively activated by designed drugs
- KOP
κ‐opioid receptor
- MOP
μ‐opioid receptor
- N/OFQ
nociceptin/orfanin FQ peptide
- NOP
nociceptin/orphanin receptor
- Oprd1
gene coding for δ‐opioid receptor
- Oprk1
gene coding for κ‐opioid receptor
- Oprl1
gene coding for nociceptin/orphanin receptor
- Oprm1
gene coding for μ‐opioid receptor
- Pdyn
prodynorphin gene
- Penk
proenkephalin gene
- PKA
cAMP‐dependent protein kinase
- Pomc
proopiomelanocortin gene
- PSNL
partial sciatic nerve ligation
Introduction
The endogenous opioid system is composed of three opioid receptors, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318, and their endogenous ligands derived from three precursors, proopiomelanocortin, proenkephalin and prodynorphin. Opioid precursor genes proopiomelanocortin gene (Pomc), proenkephalin gene (Penk) and prodynorphin gene (Pdyn), respectively, were identified in the early 1980s, whereas the genes encoding for MOP (Oprm1), DOP (Oprd1) and KOP (Oprk1) were identified a decade later (Kieffer and Evans, 2009). Opioid peptides derived from Penk, mainly [http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1614] and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1613, are preferential ligands of DOP, although also show affinity for MOP. The opioid peptide derived from Pomc, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3722, shows affinity for both MOP and DOP, whereas opioid peptides derived from Pdyn, http://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=dynorphin&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&=true&submit=Search+Database and http://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=neoendorphin&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&=true&submit=Search+Database, are preferential agonists of KOP (Kosterlitz, 1985).
The endogenous opioid system plays a crucial role in the control of nociceptive responses at peripheral, spinal and supra‐spinal level (Figure 1). In physiological conditions, the neural process of encoding noxious stimuli starts with stimulation of Aδ and C nociceptors innervating peripheral tissues. These fibres synapse with projection neurons in the spinal cord dorsal horn, which send information to supra‐spinal areas. At the peripheral level, opioid peptides released by immune cells during inflammation locally inhibit pain transmission (Rittner et al., 2008). At the spinal level, stimulation of presynaptic opioid receptors from nociceptors inhibits the release of neurotransmitters. Spinal cord dorsal horn projection neurons convey inputs from peripheral and supra‐spinal projections, and post‐synaptic opioid receptors inhibit their activity. However, activation of the endogenous opioid system at spinal level can also have pro‐nociceptive effects in certain conditions, generally attributed to spinal dynorphins (Povdin et al., 2016).
Figure 1.
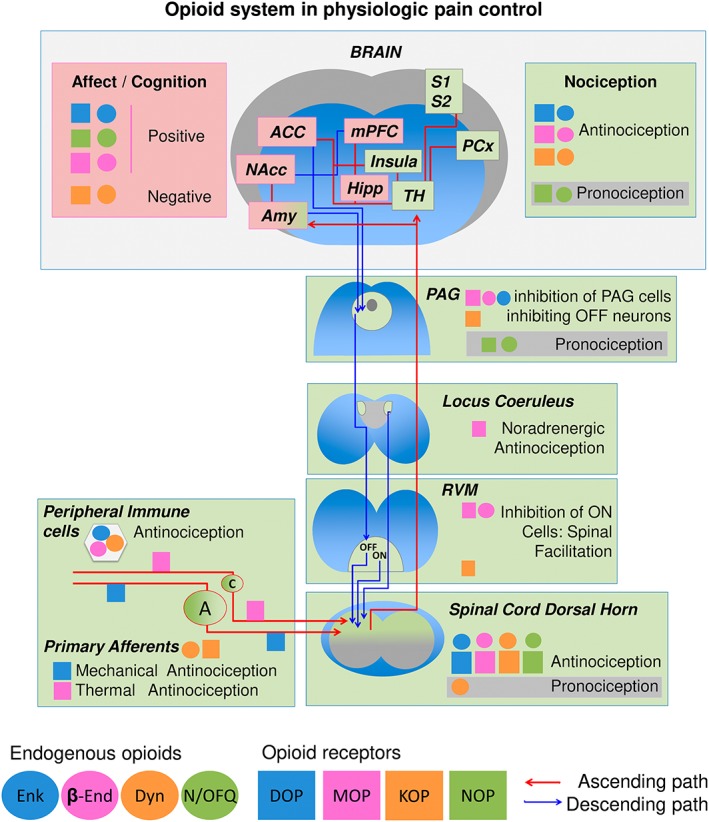
Schematic view of ascending and descending pain pathways, opioid locations and main effects in physiological conditions. A, A primary afferent fibres; ACC, anterior cingulate cortex; C, C primary afferent fibres; Enk, enkephalins; β‐end, β‐endorphin; Dyn, dynorphins; Hipp, hippocampus; mPFC, medial prefrontal cortex; NAcc, nucleus accumbens; NMDAR, N‐methyl‐D‐aspartate receptor; OFF, off cells; ON, on cells; PAG, periaqueductal gray; PCx, parietal cortex; RVM, rostral ventromedial medulla; S1 and S2, somatosensory cortex 1 and 2; TH, thalamus.
Dorsal horn neurons project their axons to the thalamus and limbic areas of the brain, and thalamic neurons connect to the somatosensory cortex S1 and S2, allowing temporal and spatial discrimination of the noxious stimuli. Activation of limbic areas results in affective and motivational consequences of pain, which, in case of sustained activation, can evolve to pain‐related anxiety, depression and cognitive impairment. Opioid receptors and peptides are expressed in these brain areas, which include the amygdala, nucleus accumbens and medial prefrontal and anterior cingulate cortices. At the same time, emotional and cognitive factors are known to modulate pain perception (Bushnell et al., 2013). While a negative emotional state can increase the unpleasantness of pain regardless of the perceived intensity, a decreased attention to the painful area can diminish the intensity of the pain sensation. This different pain modulation by cognitive and affective aspects is probably due to the different circuitry involved, being the attentional alterations associated with activity in the insula and the parietal and somatosensory cortices (Bushnell et al., 2013) and descending modulation by negative affect associated with activation of the periaqueductal gray (PAG) and the anterior cingulate and medial prefrontal cortices. Interestingly, these areas are also under control of the endogenous opioid system (Henriksen and Willoch, 2008; Lutz and Kieffer, 2013).
The PAG receives inputs from the amygdala and cortical areas and sends projections to the rostral ventromedial medulla (RVM). In the RVM, On and Off neurons have axons that reach the dorsal horn of the spinal cord to facilitate (On cells) or inhibit (Off cells) nociceptive transmission (Ossipov et al., 2010). Opioid receptor activation in the PAG inhibits the cells that keep inhibited Off cells in the RVM, whereas On cells are the only cells directly inhibited by opioids in the RVM. The endogenous opioid system also modulates the activity of noradrenergic neurons in the locus coeruleus, which also inhibit synaptic transmission in the spinal cord. Thus, at this level, opioids inhibit nociceptive transmission both by direct inhibition of On cells and disinhibition of Off cells and by increasing noradrenergic activity in the spinal cord (see Nadal et al., 2013). The opioid system also participates in the control of several other physiological functions including emotional and rewarding responses, cognition, locomotion, feeding behaviour, body temperature, endocrine, cardiovascular, respiratory and gastrointestinal functions (Terenius, 2000; Polunina and Bryun, 2013; Bodnar, 2017).
Activation of nociceptive pathways during nociceptive and acute inflammatory pain avoids further damage to the organism and has an adaptive function. However, in some circumstances, inflammatory pain persists and becomes chronic, or chronic pain can be a result of abnormal functioning of the nervous system (pathological pain). Pathological maladaptive pain is not a symptom of a disorder and can be associated with damage of the nervous system (neuropathic pain) or to conditions in which there is no neuronal damage or inflammation (dysfunctional pain) (Woolf, 2010). The modification of behavioural and physiological responses by the opioid system could influence the manifestations of acute and chronic pain.
Another receptor, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=320 (NOP), and its endogenous ligand, the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1681 peptide (N/OFQ), were also proposed to integrate the endogenous opioid system (Meunier, 1997). This receptor and endogenous peptide have been involved in nociceptive transmission, although its activation counteracts most of the classical opioid effects (Schröder et al., 2014; Kiguchi et al., 2016). Indeed, N/OFQ produces hyperalgesia in rodents when administered by intracerebroventricular route and analgesia when intrathecally administered, not suppressed by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638 (Witkin et al., 2014). The actions of the N/OFQ system are not restricted to nociceptive pathways. Indeed, NOP activation induces hypolocomotion, ataxia and loss of righting reflex, impairs spatial learning, increases feeding behaviour and regulates release of pituitary hormones (Schröder et al., 2014; Toll et al., 2016).
Different lines of genetically modified mice have been generated with mutations targeting specific components of the opioid system. These genetic models have provided invaluable tools to study the physiological role of the different opioid receptors and peptide precursors (Gavériaux‐Ruff, 2013). Constitutive deletion of a targeted gene by homologous recombination in embryonic stem cells has allowed the generation of many lines of conventional knockout mice for nearly three decades. The phenotypic characterization of these knockout mice has provided important advances in the knowledge of several physiological and pathological processes, including chronic pain. Genetic approaches targeting specific brain regions or neuronal populations are now providing additional advances in the knowledge of the specific mechanisms underlying chronic pain. Thus, the Cre‐lox P system has been used to generate conditional deletions of opioid system components in specific tissues. The generation of conditional Cre‐lox P knockouts requires the introduction of the sites for Cre recombinase (lox P sites) in direct orientation, flanking the gene of interest. These ‘floxed’ mice integrating the two loxP sites will delete the gene of interest upon delivery or expression of the Cre recombinase in the cells in which recombination is required (Morozov et al., 2003).
Knockout mice have been extensively used to evaluate the involvement of the opioid system in the control of nociceptive responses. Several reviews have summarised the advances provided by these genetic tools in pain control (Dierich and Kieffer, 2004; Nadal et al., 2013). However, these reviews are mainly focused on the role of endogenous opioid components in acute pain and the antinociceptive responses induced by different opioid compounds. While moderate to severe acute pain can be efficiently treated with opioids, opioid treatment of chronic pain has many deleterious consequences for the patients. Blunted efficacy of opioids, tolerance, hyperalgesia and addiction undermine the efficiency of these treatments (Trang et al., 2015). Understanding how the opioid system works during chronic inflammatory and neuropathic pain will be essential to optimize current treatments or to develop strategies for effective pain management. In the present review, we have summarized the recent advances obtained using knockout mice in understanding the role of the different components of the opioid system in the pathophysiological mechanisms underlying chronic pain. The models used to evaluate and mimic inflammatory and neuropathic pain conditions in the opioid knockouts are listed in Table 1. Behavioural data obtained in these models of chronic pain should be interpreted with careful consideration of the peculiarities of the behavioural phenotype of each line of knockout mice.
Table 1.
Models used to evaluate pain manifestations (A) and to mimic inflammatory and neuropathic pain conditions (B) in opioid knockout mice
| Name of the model | Principle | Response recorded | ||
|---|---|---|---|---|
| (A) Models used to evaluate pain manifestations | Mechanical sensitivity | von Frey | A battery of filaments of increasing diameter/weight is applied to the plantar surface of the hind paw | Paw withdawal threshold (weight) |
| von Frey – dynamic Plantar aesthesiometer | Automated device gradually applies a single probe against the plantar surface of the hind paw | Paw withdawal threshold (weight) | ||
| Tail pressure | Increasing pressure applied to the tail with an analgesia meter | Pressure applied when tail is withdrawn (weight) | ||
| Heat sensitivity | Plantar test | Radiant heat source applied against the plantar surface of the hind paw | Paw withdrawal latency (time) | |
| Hot tail immerson | Tail immersion in a heated water bath | Tail withdrawal latency (time) | ||
| Tail‐flick | Radiant heat source applied to the tail | Tail withdrawal latency (time) | ||
| Hot plate | Mice walking on a heated surface | Paw withdrawal latency/latency to jump/time licking, lifting, shaking the paws | ||
| Cold sensitivity | Cold plate | Mice walking on a cold surface | Difference in paw elevations ipsilateral to the injury versus contralateral | |
| Acetone drop | Application of acetone drop to plantar surface of the paw | Time licking/lifting/shaking the paw | ||
| Visceral sensitivity | Visceromotor response to colorectal distension | Colorectal distension with a balloon catheter | Electromyographic recording of abdominal contractions | |
| Memory | Object recognition memory | Exposure to novel and familiar objects in a V‐shaped maze | Score based on time exploring new object versus time exploring familiar object indicates degree of recognition memory | |
| Anxiety | Elevated plus maze | Placement on a plus‐shaped elevated maze with two open and two closed arms communicating at the intersection | Number of crossings to open/closed arms, and time spent in open/closed arms |
| Name of the model | Principle | What models? | ||
|---|---|---|---|---|
| (B) Models mimicking inflammatory or neuropathic pain conditions | Inflammatory pain | Formalin |
Formalin solution injected s.c. into dorsal or plantar paw. Two response phases: ‐early (0–5 min), direct activation nociceptors ‐late (15–60 min), tonic inflammation and central sensitization. Time licking, shaking or biting the injected paw, or number of flinches are recorded. |
Acute inflammatory pain |
| CFA | CFA injected s.c. into the plantar surface of a hindpaw. Punctate mechanical, heat and cold sensitivity develops. | Chronic inflammatory pain | ||
| Acetic acid writhing | Injection of acetic acid solution i.p. (dissolved in distilled water). Number of writhes are counted. | Visceral pain | ||
| Dextran sodium sulphate‐induced colitis | Colitis induced by adding 3% dextran sulphate sodium to the drinking water for 5 days. The visceromotor response of abdominal muscles is recorded. | Visceral pain | ||
| Zymosan A | Zymosan A s.c. injected into the plantar side of hind paw. Punctate mechanical and heat sensitivity develop. | Acute inflammatory pain | ||
| Monoiodoacetate (MIA) | Intraarticular injection of MIA in the knee joint. Punctate mechanical sensitivity, anxiety‐like behaviour and memory impairment are induced. | Osteoarthritis pain | ||
| Cartilage defects in knee joints | Osteochondral defects made with a Kirshner wire in femoral trochlear groove of a knee joint | Cartilage defects | ||
| Neuropathic pain | Sciatic nerve cuffing | Implantation of a cuff around the main branch of the sciatic nerve. Punctate mechanical sensitivity is induced. | Peripheral neuropathic pain | |
| Chronic constriction injury | 3–4 loose ligatures around sciatic nerve. Punctate mechanical and heat sensitivity develop. | Peripheral neuropathic pain | ||
| Streptozocin‐induced diabetic neuropathy | Animals treated with streptozotocin (200 mg/kg i.p.). Punctate mechanical and heat sensitivity develop. | Diabetic neuropathy | ||
| Partial sciatic nerve ligation (PSNL; Seltzer model) | Tight ligation of cranial 1/3 of sciatic nerve. Punctate mechanical, heat and cold sensitivity are induced. | Peripheral neuropathic pain | ||
| Spared nerve injury | Transection of tibial and common peroneal nerves. Punctate mechanical sensitivity is induced. | Peripheral neuropathic pain | ||
| Post‐herpetic neuralgia | s.c. inoculation of varicella zoster virus on the scarified shin. Punctate mechanical sensitivity develops. | Post‐herpetic allodynia |
Studies on knockout mice deficient in MOP, DOP and their endogenous ligands
MOP constitutive knockout mice have been obtained by disruption of exon 1 (Sora et al., 1997; Tian et al., 1997; Schuller et al., 1999), exon 2 (Matthes et al., 1996), exon 2 and 3 (Loh et al., 1998; van Rijn and Whistler, 2009) or exon 11 (Pan et al., 2009) of the Oprm1 gene in mice with C57BL/6 or mixed C57BL/6‐129S background (Figure 2). Additionally, conditional knockout with MOP exon 2 and 3 deleted in http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 (Weibel et al., 2013) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507 positive neurons (Corder et al., 2017) have also been published.
Figure 2.
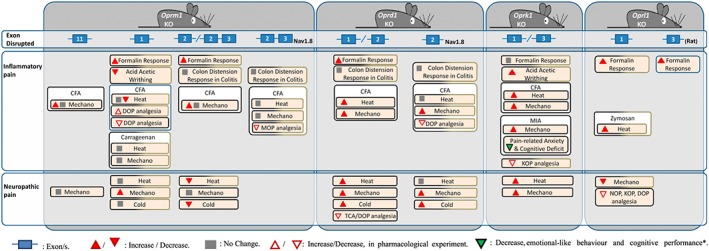
Use of opioid receptor knockout mice in chronic pain models. Cold, cold sensitivity; Heat, heat sensitivity; KO, knockout; Mechano, mechanical sensitivity; MIA, monoiodoacetate model; TCA, triciclic antidepressants. *Only results found on affective/cognitive behaviour in opioid receptor knockouts subjected to chronic pain models. Black boxes: C57BL/6 background; black‐brown boxes: C57BL/6‐129S.
Due to the extended editing of the Oprm1 gene (Pasternak and Pan, 2013), the different constructions of MOP knockouts resulted in the expression of different splice variants in the various strains generated. Exon 1 knockouts lack the canonical 7‐transmembrane (7‐TM) MOP and hence are not sensitive to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627. However, they keep the genomic sequence coding for 6 transmembrane (6‐TM) domains and were described to express 6‐TM splice variants of the MOP (Schuller et al., 1999; Lu et al., 2015). This maintained their sensitivity to other opioids, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9082, http://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=6-acetylmorphine&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&=true&submit=Search+Database, morphine‐6‐glucuronide or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1626, which still could act through the 6‐TM MOP (Schuller et al., 1999). However, exon 1 knockouts generated by other groups (Sora et al., 1997) showed decreased G‐protein activation after exposure to these opioids. Thus, it is unclear whether these other exon 1 knockouts still express the 6‐TM MOP. Interestingly, exon 11 knockouts maintain the 7‐TM forms of the MOP but cannot produce the 6‐TM variants (Pan et al., 2009). Hence, these mice were sensitive to morphine but not to morphine‐6‐glucuronide or fentanyl. Moreover, exon 11 knockouts failed to show morphine‐induced hyperalgesia, developed less tolerance and did not display hyperlocomotion after repeated morphine administration (Marrone et al., 2017), associating these negative consequences of opioid treatments with the 7‐TM MOP splice variants. On the other hand, exon 2 or exon 2–3 knockouts are expected to lack both 6 and 7‐TM variants (Pasternak and Pan, 2013) and showed loss of sensitivity to morphine and fentanyl (Matthes et al., 1996; Weibel et al., 2013). Thus, it is important to take into account the specific genetic modifications, as they may result in different behavioural outcomes depending on the phenotype being tested.
Exon 1, 2 and 2–3 knockouts showed increased sensitivity to heat, hypolocomotion and a disruption of the analgesic effects of MOP agonists (Ide et al., 2006, 2008; Pan et al., 2009; Kögel et al., 2011; Gavériaux‐Ruff, 2013). Baseline mechanical sensitivity was generally unaltered when assessing light touch sensitivity with von Frey hairs, although nociceptive responses to stronger mechanical stimuli were increased (Fuchs et al., 1999; Martin et al., 2003). Indeed, abdominal contractions in response to colorectal distension were also increased in constitutive exon 2–3 knockouts, but not in conditional http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 knockouts, suggesting a role of central MOP controlling basal colon mechanosensation (Gavériaux‐Ruff et al., 2008).
A given cognitive or emotional trait in the rodent strains could also affect the phenotypes observed in the chronic pain models. In the case of the MOP knockouts, basal anxiolytic and anti‐depressive phenotype restricted to males were reported using classical behavioural paradigms (Filliol et al., 2000), in contrast to the increased anxiety in conflict and defensive conditions, aggressiveness and deficits in social behaviour independent of the gender of these mutants (Becker et al., 2014). Other features that may indirectly affect chronic pain manifestations include cognitive impairment not associated with attentional deficits (Jamot et al., 2003; Sanders et al., 2005), increased haematopoiesis or sexual dysfunction (Tian et al., 1997).
The literature exploring chronic pain in MOP knockouts have reported increased, no difference or decreased nociceptive behaviour depending on each specific study. Although this heterogeneity suggests a lack of reproducibility at first glance, a closer inspection of the results may give some interesting conclusions. Divergent results may indicate a complex role for MOP in the pathophysiology of chronic pain that could be explained by the multiplicity of MOP knockout models or also by methodological differences.
Partly conflicting results were obtained in different models of inflammatory pain. For instance, exon 1 knockout mice showed increased response in the late phase and no change in the early phase of the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4196 test (Zhao et al., 2003), whereas exon 2 knockouts showed increased reflex responses in the early phase and no change in the late phase (Martin et al., 2003). An effect of the different genetic deletions and certain methodological differences could have favoured these divergent results. While the first study injected 2% formalin in the hindpaw and measured the number of flinches, the second one injected 5% formalin and measured the time licking, shaking or biting the paw. The significance of an increased response in each of the phases may have different interpretations (Table 1), although both studies showed antinociceptive effects of MOP.
Exon 1 MOP knockout mice showed divergent results in other models of inflammatory pain. Initial studies showed decreased number of writhings in the acetic acid model of abdominal pain (Sora et al., 1999), increased responsiveness to chemical stimulation in the formalin test (Qiu et al., 2000) and decreased heat sensitivity after complete Freund's adjuvant (CFA) (Qiu et al., 2000), suggesting different MOP modulation depending on the pain model. However, later studies in exon 1 knockouts did not detect changes in heat sensitivity after CFA (Gendron et al., 2007) or in heat and mechanical sensitivity after carrageenan‐induced inflammation (Mansikka et al., 2004). Another source of conflicting data was the antinociception mediated by DOP. Enhanced antinociceptive effects of the DOP agonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1608 ([D‐Pen2,5]‐enkephalin) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1615 were reported in exon 1 knockouts in C57BL/6–129/S background (Qiu et al., 2000), whereas reduced deltorphin‐II and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1611 antinociception was shown in a similar CFA model using C57BL/6 mice with exon 1 knockouts (Gendron et al., 2007). In this case, different strain sensitivity to the DOP agonists may have favoured this conflicting data, since previous studies already showed threefold reduced morphine sensitivity in C57BL/6 versus 129/S mice. Importantly, the reported differences in nociception between these strains could have contributed to the heterogeneity of the results in these opioid knockouts (Mogil and Wilson, 1997). Given the divergent results obtained in exon 1 knockouts, we need to know if the different exon 1 strains express similar splice variants. While some groups (Schuller et al., 1999) characterized the expression of 6‐TM MOP in these mice, this was not examined in exon 1 knockouts generated by other groups (Sora et al., 1997; Tian et al., 1997). Further studies may help to clarify these apparently conflicting results and the specific participation of 6/7‐TM splice variants in the chronic pain phenotypes.
Exon 11 knockouts (lacking 7‐TM but preserving 6‐TM variants) showed no change or marginal increases in mechanical sensitivity in the CFA model of inflammatory pain (Wieskopf et al., 2014) and unaltered mechanical sensitivity after spared nerve injury. However, these data were obtained with a dynamic plantar aesthesiometer (Table 1) and may not be directly comparable to mechanosensitivity data obtained with the other strains, generally evaluated using the von Frey filaments.
Using a similar model of inflammatory pain, exon 2 knockouts showed increased mechanical hypersensitivity after CFA (Walwyn et al., 2016). However, another study showed unchanged responses to heat and mechanical stimuli after CFA using exon 2 knockouts in mice with a similar C57BL/6 background (Gavériaux‐Ruff et al., 2008). Methodological differences may have contributed to these divergent data. The former study (Walwyn et al., 2016) estimated the 50% paw withdrawal threshold, whereas the latter (Gavériaux‐Ruff et al., 2008) used more restrictive conditions, considering the threshold as the thinnest filament eliciting 60% of positive responses. Similarly, mice lacking MOP in http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 primary afferent fibres did not show altered sensitivity to inflammatory somatic or visceral pain. However, MOP in these fibres participated in the analgesic effects of classical MOP agonists during chronic inflammation induced by CFA, but not in basal conditions (Weibel et al., 2013; Reiss et al., 2016). In the same line of results, the visceromotor response to colorectal distension after the induction of experimental colitis was similar in exon 2–3 knockouts and control mice. Hence, exon 2 or 2–3 knockouts showed no effect or antinociceptive effect of MOP in inflammatory pain conditions, depending on the specific study.
Neuropathic pain models also showed divergent data in studies conducted with exon 1 or exon 2/2–3 MOP knockouts. Exon 1 knockouts exhibited enhanced mechanical, but not heat or cold hypersensitivity after unilateral nerve injury on L5 (Mansikka et al., 2004). Again, the interpretation of these results is difficult because the expression or absence of 6‐TM MOP has not been characterized in these mice. Exon 2/2–3 knockouts showed no differences in mechanical sensitivity to von Frey hairs after cuffing the sciatic nerve (Bohren et al., 2010), whereas decreased nocifensive behaviour to thermal stimulation was observed in a model of diabetic neuropathy (Kögel et al., 2011). In the same direction, exon 2 knockouts showed attenuated sensitivity to cold after partial ligation of the sciatic nerve (PSNL) (Maldonado, 2016), suggesting pronociceptive effects of MOP. This view may be in agreement with a recent study using conditional MOP exon 2–3 knockouts in TRPV1‐positive neurons (Corder et al., 2017). The study revealed absence of morphine tolerance and morphine‐induced hyperalgesia in these mice, attributing a role to peripheral MOP in these detrimental consequences of opioid treatments (Corder et al., 2017). Hence, according to the results obtained with exon 2/2–3 knockouts, this opioid receptor may play antinociceptive roles in certain conditions of chronic inflammatory pain, whereas the results obtained with neuropathic pain models suggest a maladaptive function of MOP in abnormal pain conditions. Thus, treatments with MOP agonists might be considered during chronic inflammatory pain, and these MOP agonists would act through peripheral sites. However, the application of MOP agonists during chronic neuropathic pain may be counterproductive.
Constitutive DOP knockouts were obtained by homologous recombination of Oprd1 exon 1 (Filliol et al., 2000) or 2 (Zhu et al., 1999; van Rijn and Whistler, 2009) in mice with mixed C57BL/6–129S or complete C57BL/6 background, and new conditional models have been obtained by deletion of exon 2 in primary afferents expressing http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 channels (Gaveriaux‐Ruff et al., 2011) or in http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1067ergic neurons of the forebrain (van Rijn and Whistler, 2009; Chu Sin Chung et al., 2015). In these models, DOP appears to be critical in maintaining affective stability and cognitive performance. Indeed, mice constitutively lacking DOP showed anxious and depressive‐like phenotype (Filliol et al., 2000), and an impairment in learning and short‐term memory (Le Merrer et al., 2013). It could be expected that a basal phenotype characterized by negative affect would favour nociceptive sensitivity (Bushnell et al., 2013). However, at least in baseline conditions, nociceptive thresholds to somatic thermal or mechanical stimuli were not modified (Filliol et al., 2000; Martin et al., 2003; Nadal et al., 2006; Gavériaux‐Ruff et al., 2008). Interestingly, abdominal contractions in response to colorectal distension were increased, as in the MOP knockout animals, but this was not observed in conditional http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 knockouts, suggesting a role of central DOP on basal colon sensitivity.
The presence of DOP acquires more relevance in chronic pain conditions. Indeed, DOP knockouts developed increased thermal and mechanical sensitivity in models of chronic neuropathic (Nadal et al., 2006) and inflammatory pain (Gavériaux‐Ruff et al., 2008), suggesting a protective role of DOP for the development of these chronic pain manifestations. This antinociceptive role of DOP was also observed in the late phase of the formalin test (Martin et al., 2003) and in pharmacological experiments with constitutive DOP knockouts revealing a crucial role of this receptor for the anti‐allodynic effects of tryciclic antidepressants and DOP agonists (Benbouzid et al., 2008; Gavériaux‐Ruff et al., 2008; Gavériaux‐Ruff, 2013). On the contrary, the abdominal response to colorectal distension after experimental colitis was similar in DOP knockout and wild‐type mice (Reiss et al., 2016).
Conditional knockouts have been used to correlate the nociceptive phenotypes with DOP deletion in specific nervous system regions. Thus, DOP ablation in primary afferent fibres expressing http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=585 revealed the involvement of these receptors in chronic pain (Gaveriaux‐Ruff et al., 2011). Indeed, these conditional mutants showed unchanged heat hyperalgesia but increased mechanical allodynia after CFA and enhanced mechanical and cold hypersensitivity after PSNL (Gaveriaux‐Ruff et al., 2011). As constitutive DOP knockouts showed increased sensitivity to heat and this increase was absent in the conditional lines, this suggests that central DOP control heat hypersensitivity in chronic pain conditions. Another conditional knockout line targeting DOP in forebrain GABAergic neurons (Dlx5/6) has been generated (Chu Sin Chung et al., 2015). Surprisingly, these mice showed lower levels of anxiety, in contrast to the anxious characteristics of constitutive DOP knockouts (Filliol et al., 2000). Although the consequences of this conditional DOP deletion in the development of chronic pain have not been yet investigated, it is not yet known if the heightened chronic pain sensitivity in constitutive DOP knockouts could be influenced by their inherent depressive and anxious phenotype. However, it seems clear that DOP have an overall protective function on nociceptive sensitivity during both chronic inflammatory and neuropathic pain conditions, partly mediated through their function on primary afferent neurons. While DOP from peripheral neurons control mechanical and cold sensitivity, DOP in central structures modulate heat hypernociception.
Knockout mice deficient in the precursors of the opioid peptides acting on MOP and DOP have also been generated (Figure 3). β‐endorphin knockouts were obtained by a point mutation in exon 3 of the Pomc gene in a mixed C57BL/6‐129S background, which resulted in a premature stop codon not affecting the expression of other Pomc‐derived peptides (Rubinstein et al., 1996). Baseline thermal and mechanical nociceptive thresholds were unaltered (Gendron et al., 2007; Petraschka et al., 2007; Fell et al., 2014; Walwyn et al., 2016) or slightly increased (Mogil et al., 2000; Trigo et al., 2009). These mutants showed normal anxiety‐like behaviour (Trigo et al., 2009) and a reduced motivation for food (Hayward et al., 2002). In addition, these mice lacked stress‐induced analgesia and showed a delayed thermal hyperalgesia after forced swimming (Parikh et al., 2011).
Figure 3.
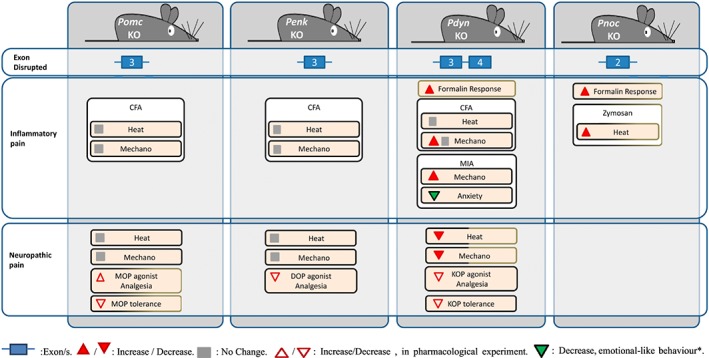
Use of endogenous opioids knockout mice in chronic pain models. Cold, cold sensitivity; Heat, heat sensitivity; KO: knockout; Mechano, mechanical sensitivity; MIA, monoiodoacetate model; TCA, triciclic antidepressants. *Only result on affective/cognitive behaviour found in the endogenous opioid knockouts subjected to chronic pain models. Black boxes: C57BL/6 background; black‐brown boxes: C57BL/6‐129S; brown box: 129S.
In chronic pain models, β‐endorphin knockout mice showed normal nociceptive sensitization to thermal and mechanical stimuli in inflammatory (Gendron et al., 2007; Walwyn et al., 2016) and peripheral neuropathy models (Petraschka et al., 2007; Niikura et al., 2008a,b; Labuz et al., 2016). However, these mice retained sensitivity to MOP agonists during neuropathic pain, whereas wild‐type mice experienced the normal tolerance to MOP stimulation. These studies suggested that an enhanced release of β‐endorphin during neuropathic pain produced stimulation of MOP and subsequent phoshorylation and desensitization, which was observed in wild‐type but not in β‐endorphin knockout mice (Petraschka et al., 2007; Niikura et al., 2008a,b; Narita et al., 2013). Hence, β‐endorphin activity would not be sufficient to modulate the nociceptive manifestations of chronic pain, although it could affect the functionality of opioid receptors.
Penk‐deficient mice were generated in C57BL/6 and DBa2 backgrounds with deletions in exon 3 of the Penk gene (König et al., 1996; Ragnauth et al., 2001). It needs to be noted that Penk deletion suppresses the production not only of enkephalins but also of different peptides coded by the same gene (peptides B, E and I) and does not fully remove the production of enkephalins, as they are also coded by the Pdyn and Pomc genes (Höllt, 1986). In spite of this, Penk deficit increased supraspinal responses to nociceptive stimulation but did not change reflex responses to heat or mechanical stimuli (Kingery et al., 2001; Gendron et al., 2007; Chen et al., 2008). Penk supression also enhanced aggressiveness and anxiety‐like behaviour (König et al., 1996; Ragnauth et al., 2001; Bilkei‐Gorzo et al., 2004), although the responses in specific tests may be strain‐dependent (Bilkei‐Gorzo et al., 2004). These mutants also showed a reduced motivation for food (Hayward et al., 2002). While enkephalin deficits are associated with increased depressive‐like behaviour and enhancing enkephalin levels with enkephalinase inhibitors showed anti‐depressant effects (Chu Sin Chung and Kieffer, 2013), depressive‐like behaviour in Penk‐deficient mice was normal under basal conditions (Bilkei‐Gorzo et al., 2007). Prominent increases of MOP and DOP in brain regions involved in emotional processing could have normalized this phenotype (Brady et al., 1999). However, acute stress provoked exacerbated anxiety and depressive‐like behaviour in these mutants, whereas stress‐induced analgesia was not modified (Ragnauth et al., 2001; Kung et al., 2010; Parikh et al., 2011). Thus, the anxious phenotype of the Penk knockout mice coexisted with increased supraspinal responses to noxious stimulation, but the magnitude of reflexive responses to nociceptive inputs was unaffected under physiological conditions.
Few studies have explored the consequences of Penk ablation in chronic pain (Labuz et al., 2016; Walwyn et al., 2016). Heat and mechanical sensitization was normal in models of inflammatory and neuropathic pain (Celik et al., 2016; Walwyn et al., 2016). Conversely, nerve‐injured mice receiving T‐lymphocytes from Penk knockout donors showed decreased anti‐allodynic response compared to mice receiving wild‐type lymphocytes (Basso et al., 2016), which could be equivalent to the reduced anti‐allodynic effect of exogenous peptides in Penk knockouts with peripheral neuropathy (Celik et al., 2016). The absence of major modifications in the nociceptive manifestations of chronic inflammatory or neuropathic pain after the genetic suppression of the main precursors of the opioid peptides acting on MOP and DOP is in contrast to the changes on these chronic pain manifestations revealed in MOP and DOP knockouts. These controversial findings have suggested the possible relevance of ligand‐independent opioid receptor constitutive activity in the control of nociceptive responses during chronic pain (Corder et al., 2013; Walwyn et al., 2016). Another plausible explanation is that endorphins and enkephalins can bind both MOP and DOP with very slight differences in affinity (Kosterlitz, 1985). Thus, each peptide type may generate similar physiological responses in the absence of the other, and this compensation may be stronger under pathological conditions that could modify their expression. According to the previous data, the aberrant affective behaviour of enkephalin knockouts did not affect nociceptive sensitization, but the emotional‐like manifestations of chronic pain have not been evaluated in these mice. Enkephalin supression may modify the affective consequences of chronic pain without altering the extent of the nociceptive sensitization, but this remains to be explored.
Studies on knockout mice deficient in KOP and their endogenous ligands
KOP knockout mice were generated by deleting exon 1 (Simonin et al., 1998) or 3 (Hough et al., 2000) of the Oprk1 gene in C57BL/6/129S or C57BL/6 background respectively (Figure 2). These mice showed normal (Simonin et al., 1998; Martin et al., 2003; Xu et al., 2004; Negrete et al., 2016) or slightly enhanced (Martin et al., 2003; Gavériaux‐Ruff et al., 2008) baseline sensitivity to mechanical and thermal stimuli. Inflammatory and nociceptive responses were unchanged in the formalin test, whereas visceral pain was increased in the writhing test (Simonin et al., 1998; Martin et al., 2003). In spite of the involvement of KOP in dysphoria, anxiety and aversive effects of stress (Land et al., 2009), constitutive deletion of the receptor had no consequences on anxiety‐ and depressive‐like behaviour (Simonin et al., 1998; Filliol et al., 2000), and spatial memory was unaffected (Jamot et al., 2003).
Removal of KOP favours a phenotype prone to nociceptive sensitization in chronic pain. These mutants showed increased heat sensitization after streptozotocin‐induced diabetic neuropathy (Rutten et al., 2014) and enhanced heat and mechanical sensitivity as well as a contralateral mirror‐image sensitization after PSNL (Xu et al., 2004). Surprisingly, the same authors showed that these knockouts displayed reduced spinal cord astrocytosis after PSNL (Xu et al., 2007). Increased and contralateral sensitization was also reported in the CFA model of inflammatory pain (Schepers et al., 2008), although another study did not show abnormal sensitivity in the same CFA model probably due to the high baseline sensitivity of the mice used (Gavériaux‐Ruff et al., 2008). The model of monoiodoacetate‐induced osteoarthritis pain also revealed enhanced mechanical sensitivity in these mutants, although anxiety‐like behaviour and cognitive impairment associated to these chronic pain manifestations were attenuated (Negrete et al., 2016). Another model of osteoarthritis showed enhanced joint damage in KOR knockouts (Wu et al., 2017), and increased formation of cancellous bone was observed in these mice (Baldock et al., 2012), suggesting that the effects of KOP deletion on chronic osteoarthritis pain could also be local. However, no histological modifications were found on the articular cartilage in KOP knockouts after monoiodoacetate (Negrete et al., 2016). Taking all the results together, KOP seem to have protective effects against nociceptive sensitization and detrimental effects promoting anxiety‐like behaviour and cognitive impairment associated with chronic pain.
Pdyn‐deficient mice were obtained by targeted disruption of exons 3 and 4 of the Pdyn gene in C57BL/6, 129S and BALB/c backgrounds (Sharifi et al., 2001; Zimmer et al., 2001; Loacker et al., 2007) (Figure 3). Pdyn gene disruption results in the ablation of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1620 several enkephalin peptides and the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1600. Baseline sensitivity to heat and mechanical stimuli were not modified (Wang et al., 2001; Zimmer et al., 2001; McLaughlin et al., 2003; Parikh et al., 2011), although certain experimental conditions have revealed increased sensitivity to mechanical (Walwyn et al., 2016) and heat stimuli (Wang et al., 2001). Furthermore, removal of the Pdyn gene decreased anxiety (Wittmann et al., 2009; Kastenberger et al., 2012), whereas depressive‐like behaviour was generally unaffected, although punctual increases were also reported (Kastenberger et al., 2012). Dynorphin deficits suppressed stress‐induced analgesia after forced swimming or social defeat in the hot tail withdrawal test (McLaughlin et al., 2003), whereas the hot plate responses were preserved after different forced swimming paradigms (Parikh et al., 2011).
Pdyn knockouts have been evaluated in different chronic pain models. Acute inflammatory pain was increased in the late phase of the formalin test (Wang et al., 2001). In contrast, mechanical hypersensitivity after chronic sciatic nerve constriction (Labuz et al., 2016) and mechanical and heat hypersensitivity after spinal nerve ligation or PSNL were decreased (Wang et al., 2001; Xu et al., 2004). In agreement, the latter study showed a reduction in KOP phosphorylation in the spinal cord accompanied by reduced astrocytosis in Pdyn knockouts after PSNL, suggesting a pronociceptive role of Pdyn gene products (Xu et al., 2007). Likewise, nerve‐injured dynorphin‐deficient mice did not develop tolerance to a KOP agonist, unlike wild‐type mice (Xu et al., 2004). Pdyn knockouts showed enhanced mechanical nociception after CFA, although this was attributed to an increased baseline sensitivity (Walwyn et al., 2016). Another study failed to show differences in pain sensitization after CFA (Gendron et al., 2007). Conversely, mice lacking dynorphin showed increased sensitization to mechanical stimuli and developed less anxiety‐like behaviour during osteoarthritis pain induced by monoiodoacetate (Negrete et al., 2016). Thus, dynorphins seem to play a complex role during chronic pain, dependent on the pain condition. Indeed, results in Pdyn knockouts suggest antinociceptive effects of Pdyn gene products in inflammatory pain conditions, whereas neuropathic pain models suggest pronociceptive effects. While Pdyn antinociception matches with the pain‐limiting effects of the KOP seen in Oprk1 knockouts, the enhanced pain phenotype in neuropathic conditions diverges from the antinociceptive role of the KOP observed in these models. Thus, dynorphins may facilitate pain sensitization through other opioid or non‐opioid receptor/s during neuropathic pain. Additionally, Pdyn gene products could participate in the affective component of chronic pain, promoting anxiety‐like behaviour.
Studies on knockout mice deficient in NOP and their endogenous ligand
Constitutive NOP knockout mice were generated by deletion of exon 2 and 3 (Clarke et al., 2001) and by deletion of exon 1 of the Oprl1 gene in a mixed C57BL/6‐129S background (Nishi et al., 1997) (Figure 2). NOP constitutive deletion did not modify acute thermal and mechanical nociception, and morphine analgesia and withdrawal signs were unaltered suggesting that NOP was not essential in these responses (Nishi et al., 1997; Mamiya et al., 2001). Interestingly, these mutants showed a consistent reduction of tolerance to morphine analgesia, revealing the involvement of NOP in the mechanisms underlying morphine tolerance (Ueda et al., 1997; Chung et al., 2006).
Knockouts deficient for the precursor of N/OFQ were generated by altering exon 2 in a C57BL/6 background (Köster et al., 1999) (Figure 3). As a consequence, these animals were lacking OFQ, OFQ2 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1681. These mutants showed an elevated basal pain threshold in the tail‐flick but developed normal stress‐induced analgesia and spatial learning performance. The mutants showed impaired adaptation to basal stress suggesting a role of N/OFQ in these responses to stress. In contrast, other authors have revealed an increased sensitivity in the tail‐flick test in N/OFQ knockouts (Chen et al., 1999; Mogil and Pasternak, 2001).
The use of both knockout lines has also provided evidence on the participation of the N/OFQ system in other physiological processes, such as locomotion (Marti et al., 2004) and emotional‐like responses (Redrobe et al., 2002; Gavioli et al., 2003). More recently, NOP knockout rats have been obtained by means of the premature stop codon using N‐ethyl‐N‐nitrosourea‐driven mutagenesis (Homberg et al., 2009). Mutant rats are more sensitive to morphine reward (Rutten et al., 2011) and showed differences in anxiety and mood‐related behaviours, locomotion and nociception, mainly in the formalin test (Rizzi et al., 2011).
The role of the N/OFQ system in chronic pain was also studied using NOP and N/OFQ knockout mice (Depner et al., 2003). These mutants showed stronger nociceptive responses in the late phase of the formalin test and increased thermal pain in the zymosan inflammatory model suggesting a protective role of the N/OFQ system during prolonged inflammatory pain. In contrast, NOP deletion prevented the appearance of postherpetic allodynia, although it did not affect the development of skin lesions and allodynia during the presence of these lesions (Sasaki et al., 2008). Therefore, the N/OFQ system may be involved in the transition from herpetic to postherpetic allodynia.
NOP‐deficient mice were also used to study the interdependency between NOP and the different opioid receptors in streptozotocin‐induced diabetic polyneuropathy (Rutten et al., 2014). Selective agonists for each opioid receptor induced smaller analgesic responses in the hot plate in MOP, DOP, KOP and NOP knockouts compared with wild‐type littermates. No evidence was shown of the interdependency of NOP and MOP suggesting the interest of its concurrent activation in chronic pain. In contrast, the antinociceptive (Lutfy et al., 2003), motor stimulatory and rewarding actions (Marquez et al., 2008) of buprenorphine were enhanced in NOP knockouts. The interaction between NOP and opioid receptors may work at circuit level as they share common signalling pathways in different anatomical regions, such as the spinal cord, PAG and RVM (Günther et al., 2018). The effect of mixed NOP/MOP agonists inhibiting tolerance and dependence compared with pure MOP agonists may be due to a chronic desensitization of NOP signalling in reward and tolerance circuits (Lufty et al., 2001), as similar results were seen after a co‐treatment of MOR receptor agonists with NOP antagonists (Chung et al., 2006).
NOP‐mediated effects on pain may be more complex than those from other opioid receptors. Its activation is followed either by pronociceptive or antinociceptive effects, depending on the administration route and pain modalities in rodents (Schröder et al., 2014). Activation of spinal NOP in models of chronic pain caused potent antihyperalgesic and antiallodynic effects that can be explained at least in part by an up‐regulation of those receptors in the spinal cord (Kiguchi et al., 2016). However, the supraspinal actions of NOP activation gave conflicting results depending on the model. While it seemed to elicit pronociceptive effects in inflammatory pain models, antinociceptive effects were observed in neuropathic pain models, whereas antagonists produced similar attenuation in both types of pain (Kiguchi et al., 2016). Additionally, preproN/OFQ and NOP receptor knockout mice showed increased inflammatory hyperalgesia in the formalin assay, but not in an acute pain assay (Depner et al., 2003). These results were similar to those obtained in NOP receptor knockout rats (Rizzi et al., 2011). In summary, these studies indicate the possibility that the NOP system behaved differently in the different pain models and that its plasticity may participate in some degree in the nociceptive responses induced by chronic pain models (Toll et al., 2016). Heterogeneous outcomes may be explained by various levels of spinal versus supraspinal participation of the NOP/NOFQ system under different pain states (Kiguchi et al., 2016).
Clinical interest of the multitarget approach
Findings obtained with these N/OFQ system knockouts and results of pharmacological studies have suggested a new therapeutic approach based on the development of opioid compounds targeting several opioid receptors, in contrast to the traditional high selectivity of agonists (Bird and Lambert, 2015). Early pharmacological studies already demonstrated beneficial effects of co‐administering several drugs acting in different opioid receptors. Thus, a reduction in the development of morphine tolerance was revealed when concomitantly administered with the DOP antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641 (Abdelhamid et al., 1991). Similarly, morphine tolerance did not appear in Penk knockouts (Nitsche et al., 2002), and several studies have revealed the blockade of morphine tolerance in DOP and NOP knockouts (Ueda et al., 1997; Zhu et al., 1999; Chung et al., 2006). In agreement, various mixed MOP/NOP ligands have demonstrated potent analgesic effects in several rodent models together with attenuation in the development of tolerance or physical dependence (Sukhtankar et al., 2013; Sobczak et al., 2014; Zielińska et al., 2015), suggesting the potential interest of using drugs simultaneously targeting NOP and MOP (Blair Journigan et al., 2014). Recently, Toll et al. (2016) have reviewed the influence of the activation or blockade of N/OFQ system on the development of opioid tolerance. Morphine tolerance is significantly reduced in NOP or ppN/N/OFQ KO mice (Ueda et al., 1997; Chung et al., 2006). Additionally, N/OFQ antibody partly reversed the tolerance associated with chronic morphine administration (Tian and Han, 2000). These results agree with the report that the co‐administration of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1691, an antagonist of the NOP, blocks tolerance development in mice (Ueda et al., 1997; Chung et al., 2006). The application of such antagonists in the ventrolateral PAG blocked the analgesic action of morphine (Scoto et al., 2010) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1647 (Parenti and Scoto, 2010). Although i.c.v. injection of N/OFQ after a daily systemic administration of morphine blocked morphine tolerance (Lufty et al., 2001), NOP antagonists also blocked the expression of tolerance to a daily injection of morphine (Zaratin et al., 2004; Chung et al., 2006). Toll et al. (2016) concluded that chronic morphine leads to up‐regulation of the brain NOP system, which attenuates morphine analgesia and can be blocked with a NOP antagonist. They suggested the need of new studies to better understand the involvement of the NOP system, given the conflicting results with agonists and antagonists (Toll et al., 2016). In this sense, a novel full MOP agonist and NOP partial agonist displaying subnanomolar affinity for both receptors, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8866, has been extensively studied at preclinical level (Bird and Lambert, 2015; Sałat et al., 2015). This drug has long‐term analgesic effects in acute and chronic pain models, and its side effects are less severe than those of standard opioid drugs. Indeed, motor coordination and respiratory function remains unaltered at high doses, and tolerance is delayed compared with morphine (Sukhtankar et al., 2013; Linz et al., 2014; Schunk et al., 2014; Raffa et al., 2017). According to http://clinicaltrials.gov (National Institutes of Health, 2017), seven Phase II clinical trials have been completed with cebranopadol. These studies analysed its analgesic effects in painful diabetic polyneuropathy, postoperative pain, severe chronic low back pain and chronic pain due to osteoarthritis of the knee. Two Phase III clinical trials were also carried out in cancer pain. Some of these clinical studies finished several years ago, but the results have not been yet published. This information would be necessary to understand the real clinical interest of targeting, with the same ligand, MOP and NOP for the treatment of acute or chronic pain in patients.
Current and future directions
Novel genetic approaches have been recently developed to manipulate specific neuronal populations in live animals as well as new techniques that facilitate the simplicity of making gene mutations in rodents. The application of these new techniques can provide important advances in the near future for understanding many neurophysiological processes. The novel genetic techniques most commonly employed to selectively manipulate specific neuronal populations are optogenetics, using channels activated by light, and chemogenetics, using engineered G‐protein coupled receptors activated by otherwise inert drugs (Whissell et al., 2016).
Optogenetic approaches have been applied to manipulate specific neuronal populations and have been recently used to study specific components of the pain circuitry (Copits et al., 2016). In these approaches, light‐sensitive proteins, called http://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?searchString=opsin&searchCategories=all&species=none&type=all&comments=includeComments&order=rank&=true&submit=Search+Database, are expressed in genetically defined neuronal populations. The majority of these opsins are light‐gated ion channels and pumps isolated from diverse microorganisms (Fenno et al., 2011). Excitatory opsins are cation‐selective and gate inward photocurrents that depolarize neurons, whereas inhibitory opsins are most frequently pumps that mediate http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2339 influx or proton efflux to silence neuronal firing (Copits et al., 2016). These optical approaches have also been developed for manipulating intracellular signalling cascades associated with G‐protein‐coupled receptors, including MOP (Siuda et al., 2015). Opsins are delivered into the nervous system through viral vectors or transgenesis (Copits et al., 2016). Cre‐dependent adeno‐associated viruses containing opsin genes are usually injected into transgenic mice where Cre‐recombinase expression is restricted to genetically defined cell types. On the other hand, crossing Cre driver transgenic mice with genetically encoded opsin mouse lines can enable specific photo‐manipulation of defined neuronal populations (Wang et al., 2016). These manipulations have already been employed to clarify the neuronal pathways involved in chronic pain control. Thus, optogenetic manipulation of the neuronal activity in the medial prefrontal cortex (Ji and Neugebauer, 2012; Zhang et al., 2015) and basolateral amygdala (Kiritoshi et al., 2016) has identified the specific contribution of these circuits in the sensory and emotional components of chronic pain. Optogenetic stimulation of cortical projections to the nucleus accumbens decreased chronic pain manifestations (Sugimura et al., 2016), whereas optogenetic stimulation of the central amygdala induced visceral pain (Crock et al., 2012). These approaches have also allowed the dissection of specific roles of serotonergic neurons of the rostral ventral medulla (Cai et al., 2014) and subpopulations of noradrenergic neurons in the locus coeruleus (Hickey et al., 2014) in pain control. An optogenetic approach has also been used to dissect the specific contribution of opioid and GABA receptors in the pre‐synaptic modulation of the nociceptive information transmitted to spinal neurons (Honsek et al., 2015). Interestingly, MOP pre‐synaptic activation mainly inhibited C‐fibres innervating lamina I spinal cord neurons, whereas activation of pre‐synaptic DOP did not modify these responses. In a similar location (spinal cord laminae I/II), another study found that a MOP agonist inhibited excitatory postsynaptic currents elicited by TRPV1 nociceptors optogenetically activated. The inhibition disappeared after removal of the MOP agonist and was followed by long‐term potentiation indicative of excitatory opioid effects. Because these effects were absent in conditional knockouts lacking MOP in TRPV1 nociceptors and correlated with suppression of opioid‐induced hyperalgesia in these mice, a fundamental role of presynaptic MOP could be defined in these processes (Corder et al., 2017). Hence, these interesting approaches are already helping to understand how opioid receptors function in specific locations of the nociceptive pathways.
The chemogenetic approach most widely used in neuroscience to selectively manipulate specific brain circuits by modifying the activity of G‐protein‐coupled receptors is the designed receptor exclusively activated by designed drugs (DREADD) technique (Whissell et al., 2016). The engineered designer receptor must first be selectively expressed in the targeted neuronal population. DREADDs are usually modified muscarinic receptors coupled to an inhibitory (Gi) or excitatory (Gq, Gs) signalling cascade with low affinity for endogenous ligands and activated by synthetic compounds, such as clozapine N‐oxide (CNO), an inert and orally available drug. The genes encoding these designer receptors are usually introduced in cells by Cre‐dependent adeno‐associated viruses restricting DREADD expression to cells that selectively express Cre. The administration of CNO will decrease or increase the activity of this specific neuronal circuit depending on the signalling cascade coupled to the designer receptor expressed (Whissell et al., 2016). These techniques have been used to clarify the specific involvement of different spinal cord neuronal populations in itch (Bourane et al., 2015), acute (Peirs et al., 2015; Saloman et al., 2016) and chronic pain (Peirs et al., 2015), and supraspinal circuits of visceral pain (Jurik et al., 2015). However, none of these studies has yet examined the function of the endogenous opioid system in chronic pain conditions.
The use of clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) technology is revolutionizing genetics in many organisms including laboratory mice. The CRISPR system is a versatile antiviral defence mechanism that provides immunity for a host bacterium against extra‐chromosomal genetic material (Mojica and Montoliu, 2016). After the acquisition of foreign DNA by the bacterium, this system involves the synthesis and maturation of CRISPR RNA followed by the formation of RNA‐Cas nuclease protein complexes that will selectively recognize and destroy the targeted foreign DNA by Cas nuclease cleavage. The CRISPR/Cas9 is a versatile system able to recognize virtually any sequence in the genome, including the mammalian genome, and introduce a controlled break in the DNA (Singh et al., 2015). This novel technology has opened a plethora of possibilities for precise genome editing in mice allowing nearly any possible change mimicking human coding variants to be introduced in the mouse genome. An important progress has been already made in targeting specificity, simplicity of making gene‐editing mice and bioinformatics design (Pulido‐Quetglas et al., 2017). Although improvements may still be needed to increase precise editing efficiency and decrease mosaicism (Singh et al., 2015), this technology provides entirely new tools extremely useful to conduct genetic mouse research faster and better. A recent study on a model of intervertebral disc degeneration (Stover et al., 2017) detected specific changes in the activity of dorsal root ganglion neurons, associated with the A kinase anchor protein AKAP150, a protein that recruits the cAMP‐dependent protein kinase (PKA) to the dendrites. These authors utilized CRISPR epigenome editing to modulate endogenous expression of AKAP150 and could erase the pathological neuronal activity while preserving normal activity. This highlights the potential use of CRISPR as a pain neuromodulatory strategy. Due to the close relationship of PKA with the processes associated to opioid receptor stimulation, it would be interesting to evaluate opioid function in these experimental conditions. However, the possibilities offered by CRISPR/Cas9 are immense.
All these novel genetic techniques will certainly provide in the near future important advances to our understanding of the neurobiological mechanisms underlying chronic pain and the specific involvement of the different components of the endogenous opioid system in these pathophysiological processes. Optogenetic or chemogenetic control of MOP expressed in genetically defined circuits could clarify their role in the nociceptive and affective manifestations of chronic pain, whereas an optimized genomic editing could avoid compensatory changes and disrupt the expression of opioid peptides with common receptor affinities to clarify their participation in chronic pain phenotypes.
Concluding remarks
Studies using knockout mice have allowed the identification the crucial role of DOP and KOP in chronic inflammatory and neuropathic pain (Figure 4). However, these approaches have not yet clarified the precise role of MOP in the development of different chronic pain conditions. Data suggest that stimulation of peripheral and central MOP could exert antinociceptive effects during certain chronic inflammatory pain conditions, but it may be counterproductive during pathological neuropathic pain or after repeated opioid treatments. The clarification of the role of the different MOP splice variants in the development of chronic pain may, as well, open new therapeutic avenues. MOP is the main opioid receptor for acute nociceptive control and the elucidation of its specific roles in chronic pain merits further studies. In contrast to these findings in opioid receptor knockouts, the suppression of the main precursors of the opioid peptides acting on MOP and DOP had no major consequences on chronic pain manifestations. This suggests either a possible role of ligand‐independent opioid receptor constitutive activity or a redundancy in the opioid system in which the presence of one opioid peptide can compensate for the absence of other with similar receptor affinity. Interestingly, β‐endorphin activity was revealed necessary for the development of morphine tolerance, whereas the effects of dynorphins could differ depending on the chronic pain condition. Another important open question is to understand the possible influence of the emotional changes revealed in these knockouts in chronic pain manifestations, as the affective aspects of chronic pain may be independent of strictly nociceptive manifestations. The N/OFQ system also seems to play an important role in specific chronic pain conditions and seems essential for the development of opioid analgesic tolerance. These findings have opened a new possible therapeutic approach consisting of mixed MOP/NOP ligands that showed beneficial effects in preclinical studies, but the results of the already completed clinical trials have not been yet published. The novel genetic tools now available to manipulate specific neuronal populations are already providing answers on the functioning of the opioid system, and quick and precise genome editing in mice will facilitate the elucidation of most of these still open questions in the pathophysiology and treatment of chronic pain.
Figure 4.
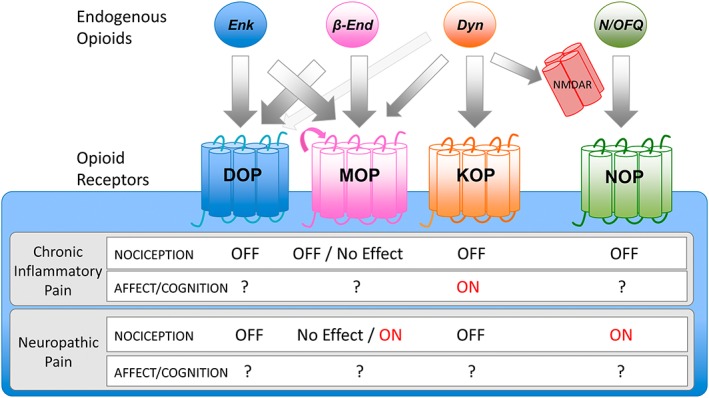
Effects of opioid receptors activating (ON) or inhibiting (OFF) chronic inflammatory or neuropathic pain manifestations, according to knockout studies. Endogenous opioids bind to opioid receptors with different affinities. Arrow thickness indicates relative affinity for the receptor. β‐end, β‐endorphin; Dyn, dynorphins; Enk, enkephalins; NMDAR, N‐methyl‐D‐aspartate receptor. Dynorphins were described to have high affinity for KOP and low for MOP and NDMA receptors (non‐opioid receptor). Pink arrow in MOP indicates possible constitutive activity independent of ligand activation. Effects of MOP are described according to exon 2/2–3 knockout studies. Additional studies are needed to characterize affective/cognitive behaviour associated with chronic pain in these knockouts.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2017), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledegments
This work was supported by the European Commission, FP7 (#HEALTH‐F2‐2013‐602891), the Spanish Ministerio de Economía y Competitividad‐MINECO (#SAF2014‐59648‐P/FEDER) and Instituto de Salud Carlos III, RETICS‐RTA (#RD12/0028/0023/FEDER), and the Generalitat de Catalunya, AGAUR (#2014‐SGR‐1547). We also acknowledge the financial support of the AGAUR (ICREA Acadèmia Award 2015) to R.M.
Maldonado R., Baños J. E., and Cabañero D. (2018) Usefulness of knockout mice to clarify the role of the opioid system in chronic pain, British Journal of Pharmacology, 175: 2791–2808, https://doi.org/10.1111/bph.14088.
Contributor Information
Rafael Maldonado, Email: rafael.maldonado@upf.edu.
David Cabañero, Email: david.cabanero@upf.edu.
References
- Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE (1991). Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J Pharmacol Exp Ther 258: 299–303. [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017. a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017. b). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldock PA, Driessler F, Lin S, Wong IPL, Shi Y, Yulyaningsih E et al (2012). The endogenous opioid dynorphin is required for normal bone homeostasis in mice. Neuropeptides 46: 383–394. [DOI] [PubMed] [Google Scholar]
- Basso L, Boué J, Mahiddine K, Blanpied C, Robiou‐du‐Pont S, Vergnolle N et al (2016). Endogenous analgesia mediated by CD4+ T lymphocytes is dependent on enkephalins in mice. J Neuroinflammation 13: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JAJ, Clesse D, Spiegelhalter C, Schwab Y, Le Merrer J, Kieffer BL (2014). Autistic‐like syndrome in mu opioid receptor null mice is relieved by facilitated mGluR4 activity. Neuropsychopharmacology 39: 2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbouzid M, Gavériaux‐Ruff C, Yalcin I, Waltisperger E, Tessier L‐H, Muller A et al (2008). Delta‐opioid receptors are critical for tricyclic antidepressant treatment of neuropathic allodynia. Biol Psychiatry 63: 633–636. [DOI] [PubMed] [Google Scholar]
- Bilkei‐Gorzo A, Michel K, Noble F, Roques BP, Zimmer A (2007). Preproenkephalin knockout mice show no depression‐related phenotype. Neuropsychopharmacology 32: 2330–2337. [DOI] [PubMed] [Google Scholar]
- Bilkei‐Gorzo A, Racz I, Michel K, Zimmer A, Klingmüller D, Zimmer A (2004). Behavioral phenotype of pre‐proenkephalin‐deficient mice on diverse congenic backgrounds. Psychopharmacology (Berl) 176: 343–352. [DOI] [PubMed] [Google Scholar]
- Bird MF, Lambert DG (2015). Simultaneous targeting of multiple opioid receptor types. Curr Opin Support Palliat Care 9: 98–102. [DOI] [PubMed] [Google Scholar]
- Blair Journigan V, Polgar WE, Khroyan TV, Zaveri NT (2014). Designing bifunctional NOP receptor–mu opioid receptor ligands from NOP‐receptor selective scaffolds. Part II. Bioorg Med Chem 22: 2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar RJ (2017). Endogenous opiates and behavior: 2015. Peptides 88: 126–188. [DOI] [PubMed] [Google Scholar]
- Bohren Y, Karavelic D, Tessier L‐H, Yalcin I, Gavériaux‐Ruff C, Kieffer BL et al (2010). Mu‐opioid receptors are not necessary for nortriptyline treatment of neuropathic allodynia. Eur J Pain 14: 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourane S, Duan B, Koch SC, Dalet A, Britz O, Garcia‐Campmany L et al (2015). Gate control of mechanical itch by a subpopulation of spinal cord interneurons. Science 350: 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady LS, Herkenham M, Rothman RB, Partilla JS, König M, Zimmer AM et al (1999). Region‐specific up‐regulation of opioid receptor binding in enkephalin knockout mice. Brain Res Mol Brain Res 68: 193–197. [DOI] [PubMed] [Google Scholar]
- Bushnell MC, Ceko M, Low LA (2013). Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci 14: 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y‐Q, Wang W, Hou Y‐Y, Pan ZZ (2014). Optogenetic activation of brainstem serotonergic neurons induces persistent pain sensitization. Mol Pain 10: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik MÖ, Labuz D, Henning K, Busch‐Dienstfertig M, Gaveriaux‐Ruff C, Kieffer BL et al (2016). Leukocyte opioid receptors mediate analgesia via Ca(2+)‐regulated release of opioid peptides. Brain Behav Immun 57: 227–242. [DOI] [PubMed] [Google Scholar]
- Chen TC, Cheng YY, Sun WZ, Shyu BC (2008). Differential regulation of morphine antinociceptive effects by endogenous enkephalinergic system in the forebrain of mice. Mol Pain 4: 1741–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Inoue M, Hopkins E, Pellegrino MJ, Novakowki R, Mogil JS et al (1999). Nociceptin/OFQ gene knock‐out reveals an antinociceptive role for the nociceptin/OFQ in the mouse. Soc Neurosci Abstr 25: 1472. [Google Scholar]
- Chu Sin Chung P, Boehrer A, Stephan A, Matifas A, Scherrer G, Darcq E et al (2015). Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80‐induced seizures. Behav Brain Res 278: 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Sin Chung P, Kieffer BL (2013). Delta opioid receptors in brain function and diseases. Pharmacol Ther 140: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Pohl S, Zeng J, Civelli O, Reinscheid RK (2006). Endogenous orphanin FQ/nociceptin is involved in the development of morphine tolerance. J Pharmacol Exp Ther 318: 262–267. [DOI] [PubMed] [Google Scholar]
- Clarke S, Chen Z, Hsu MS, Pintar J, Hill R, Kitchen I (2001). Quantitative autoradiographic mapping of the ORL 1, μ‐, δ‐ and κ‐receptors in the brains of knockout mice lacking the ORL1 receptor gene. Brain Res 906: 13–24. [DOI] [PubMed] [Google Scholar]
- Copits BA, Pullen MY, Gereau RW (2016). Spotlight on pain: optogenetic approaches for interrogating somatosensory circuits. Pain 157: 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y et al (2013). Constitutive μ‐opioid receptor activity leads to long‐term endogenous analgesia and dependence. Science 341: 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR et al (2017). Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med 23: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crock LW, Kolber BJ, Morgan CD, Sadler KE, Vogt SK, Bruchas MR et al (2012). Central amygdala metabotropic glutamate receptor 5 in the modulation of visceral pain. J Neurosci 32: 14217–14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depner UB, Reinscheid RK, Takeshima H, Brune K, Zeilhofer HU (2003). Normal sensitivity to acute pain, but increased inflammatory hyperalgesia in mice lacking the nociceptin precursor polypeptide or the nociceptin receptor. Eur J Neurosci 17: 2381–2387. [DOI] [PubMed] [Google Scholar]
- Dierich A, Kieffer BL (2004). Knockout mouse models in pain research. Methods Mol Med 99: 269–299. [DOI] [PubMed] [Google Scholar]
- Fell GL, Robinson KC, Mao J, Woolf CJ, Fisher DE (2014). Skin β‐endorphin mediates addiction to UV light. Cell 157: 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno L, Yizhar O, Deisseroth K (2011). The development and application of optogenetics. Annu Rev Neurosci 34: 389–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F et al (2000). Mice deficient for delta‐ and mu‐opioid receptors exhibit opposing alterations of emotional responses. Nat Genet 25: 195–200. [DOI] [PubMed] [Google Scholar]
- Fuchs PN, Roza C, Sora I, Uhl G, Raja SN (1999). Characterization of mechanical withdrawal responses and effects of mu‐, delta‐ and kappa‐opioid agonists in normal and muopioid receptor knockout mice. Brain Res 821: 480–486. [DOI] [PubMed] [Google Scholar]
- Gavériaux‐Ruff C (2013). Opiate‐induced analgesia: contributions from mu, delta and kappa opioid receptors mouse mutants. Curr Pharm Des 19: 7373–7381. [DOI] [PubMed] [Google Scholar]
- Gavériaux‐Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL (2008). Inflammatory pain is enhanced in delta opioid receptor‐knockout mice. Eur J Neurosci 27: 2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaveriaux‐Ruff C, Nozaki C, Nadal X, Hever XC, Weibel R, Matifas A et al (2011). Genetic ablation of delta opioid receptors in nociceptive sensory neurons increases chronic pain and abolishes opioid analgesia. Pain 152: 1238–1248. [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Marzola G, Guerrini R, Bertorelli R, Zucchini S, De Lima TCM et al (2003). Blockade of nociceptin/orphanin FQ‐NOP receptor signalling produces antidepressantlike effects: pharmacological and genetic evidences from the mouse forced swimming test. Eur J Neurosci 17: 1987–1990. [DOI] [PubMed] [Google Scholar]
- Gendron L, Pintar JE, Chavkin C (2007). Essential role of mu opioid receptor in the regulation of delta opioid receptor–mediated antihyperalgesia. Neuroscience 150: 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther T, Dasgupta P, Mann A, Miess E, Kiewer A, Fritzwanker S et al (2018). Targeting multiple opioid receptors – improved analgesics with reduced side effects? Br J Pharmacol. 175: 2857–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward MD, Pintar JE, Low MJ (2002). Selective reward deficit in mice lacking beta‐endorphin and enkephalin. J Neurosci 22: 8251–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen G, Willoch F (2008). Imaging of opioid receptors in the central nervous system. Brain 131: 1171–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey L, Li Y, Fyson SJ, Watson TC, Perrins R, Hewinson J et al (2014). Optoactivation of locus ceruleus neurons evokes bidirectional changes in thermal nociception in rats. J Neurosci 34: 4148–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höllt V (1986). Opioid peptide processing and receptor selectivity. Annu Rev Pharmacol Toxicol 26: 59–77. [DOI] [PubMed] [Google Scholar]
- Homberg JR, Mul JD, de Wit E, Cuppen E (2009). Complete knockout of the nociceptin/orphanin FQ receptor in the rat does not induce compensatory changes in mu, delta and kappa opioid receptors. Neuroscience 163: 308–315. [DOI] [PubMed] [Google Scholar]
- Honsek SD, Seal RP, Sandkühler J (2015). Presynaptic inhibition of optogenetically identified VGluT3+ sensory fibres by opioids and baclofen. Pain 156: 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J et al (2000). Improgan, a cimetidine analog, induces morphine‐like antinociception in opioid receptor‐knockout mice. Brain Res 880: 102–108. [DOI] [PubMed] [Google Scholar]
- Ide S, Minami M, Ishihara K, Uhl GR, Satoh M, Sora I et al (2008). Abolished thermal and mechanical antinociception but retained visceral chemical antinociception induced by butorphanol in mu‐opioid receptor knockout mice. Neuropharmacology 54: 1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide S, Minami M, Ishihara K, Uhl GR, Sora I, Ikeda K (2006). Mu opioid receptordependent and independent components in effects of tramadol. Neuropharmacology 51: 651–658. [DOI] [PubMed] [Google Scholar]
- Jamot L, Matthes HWD, Simonin F, Kieffer BL, Roder JC (2003). Differential involvement of the mu and kappa opioid receptors in spatial learning. Genes Brain Behav 2: 80–92. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V (2012). Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain 5: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurik A, Auffenberg E, Klein S, Deussing JM, Schmid RM, Wotjak CT et al (2015). Roles of prefrontal cortex and paraventricular thalamus in affective and mechanical components of visceral nociception. Pain 156: 2479–2491. [DOI] [PubMed] [Google Scholar]
- Kastenberger I, Lutsch C, Herzog H, Schwarzer C (2012). Influence of sex and genetic background on anxiety‐related and stress‐induced behaviour of prodynorphin‐deficient mice. PLoS One 7: e34251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ (2009). Opioid receptors: from binding sites to visible molecules in vivo . Neuropharmacology 56: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Ding H, Ko MC (2016). Central N/OFQ‐NOP receptor system in pain modulation. Adv Pharmacol 75: 217–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingery WS, Sawamura S, Agashe GS, Davies MF, Clark JD, Zimmer A (2001). Enkephalin release and opioid receptor activation does not mediate the antinociceptive or sedative/hypnotic effects of nitrous oxide. Eur J Pharmacol 427: 27–35. [DOI] [PubMed] [Google Scholar]
- Kiritoshi T, Ji G, Neugebauer V (2016). Rescue of impaired mGluR5‐driven endocannabinoid signaling restores prefrontal cortical output to inhibit pain in arthritic rats. J Neurosci 36: 837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kögel B, De Vry J, Tzschentke TM, Christoph T (2011). The antinociceptive and antihyperalgesic effect of tapentadol is partially retained in OPRM1 (μ‐opioid receptor) knockout mice. Neurosci Lett 491: 104–107. [DOI] [PubMed] [Google Scholar]
- König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ et al (1996). Pain responses, anxiety and aggression in mice deficient in pre‐proenkephalin. Nature 383: 535–538. [DOI] [PubMed] [Google Scholar]
- Köster A, Montkowski A, Schulz S, Stübe EM, Knaudt K, Jenck F et al (1999). Targeted disruption of the orphanin FQ/nociceptin gene increases stress susceptibility and impairs stress adaptation in mice. Proc Natl Acad Sci U S A 96: 10444–10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosterlitz HW (1985). The Wellcome Foundation lecture, 1982. Opioid peptides and their receptors. Proc R Soc London Ser B, Biol Sci 225: 27–40. [DOI] [PubMed] [Google Scholar]
- Kung J‐C, Chen T‐C, Shyu B‐C, Hsiao S, Huang A (2010). Anxiety‐ and depressivelike responses and c‐fos activity in preproenkephalin klockout mice: oversensitivity hypothesis of enkephalin deficit‐induced posttraumatic stress disorder. J Biomed Sci 17: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuz D, Celik MÖ, Zimmer A, Machelska H (2016). Distinct roles of exogenous opioid agonists and endogenous opioid peptides in the peripheral control of neuropathy‐triggered heat pain. Sci Rep 6: 32799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D et al (2009). Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A 106: 19168–19173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Merrer J, Rezai X, Scherrer G, Becker JAJ, Kieffer BL (2013). Impaired hippocampus‐dependent and facilitated striatum‐dependent behaviors in mice lacking the δ opioid receptor. Neuropsychopharmacology 38: 1050–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M et al (2014). Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther 349: 535–548. [DOI] [PubMed] [Google Scholar]
- Loacker S, Sayyah M, Wittmann W, Herzog H, Schwarzer C (2007). Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain 130: 1017–1028. [DOI] [PubMed] [Google Scholar]
- Loh HH, Liu HC, Cavalli A, Yang W, Chen YF, Wei LN (1998). mu opioid receptor knockout in mice: effects on ligand‐induced analgesia and morphine lethality. Brain Res Mol Brain Res 54: 321–326. [DOI] [PubMed] [Google Scholar]
- Lu Z, Xu J, Rossi GC, Majumdar S, Pasternak GW, Pan YX (2015). Mediation of opioid analgesia by a truncated 6‐transmembrane GPCR. J Clin Invest 125: 2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lufty K, Hossain SM, Khaliq I, Maidment NT (2001). Orphanin FQ/nociceptin attenuates the development of morphine tolerance in rats. Br J Pharmacol 134: 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W et al (2003). Buprenorphine‐induced antinociception is mediated by mu‐opioid receptors and compromised by concomitant activation of opioid receptor‐like receptors. J Neurosci 23: 10331–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz PE, Kieffer BL (2013). Opioid receptors: distinct roles in mood disorders. Trends Neurosci 36: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R (2016). Opioid receptors and drug‐seeking behaviour. In INRC Meeting, (Bath, United Kingdom), p 12.
- Mamiya T, Noda Y, Ren X, Nagai T, Takeshima H, Ukai M et al (2001). Morphine tolerance and dependence in the nociceptin receptor knockout mice. J Neural Transm 108: 1349–1361. [DOI] [PubMed] [Google Scholar]
- Mansikka H, Zhao C, Sheth RN, Sora I, Uhl G, Raja SN (2004). Nerve injury induces a tonic bilateral mu‐opioid receptor‐mediated inhibitory effect on mechanical allodynia in mice. Anesthesiology 100: 912–921. [DOI] [PubMed] [Google Scholar]
- Marquez P, Nguyen AT, Hamid A, Lutfy K (2008). The endogenous OFQ/N/ORL‐1 receptor system regulates the rewarding effects of acute cocaine. Neuropharmacology 54: 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone GF, Le Rouzic V, Varadi A, Xu J, Rajadhyaksha AM, Majumdar S et al (2017). Genetic dissociation of morphine analgesia from hyperalgesia in mice. Psychopharmacology (Berl) 234: 1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Mela F, Veronesi C, Guerrini R, Salvadori S, Federici M et al (2004). Blockade of nociceptin/orphanin FQ receptor signaling in rat substantia nigra pars reticulate stimulates nigrostriatal dopaminergic transmission and motor behavior. J Neurosci 24: 6659–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Matifas A, Maldonado R, Kieffer BL (2003). Acute antinociceptive responses in single and combinatorial opioid receptor knockout mice: distinct mu, delta and kappa tones. Eur J Neurosci 17: 701–708. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I et al (1996). Loss of morphine‐induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu‐opioid‐receptor gene. Nature 383: 819–823. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton‐Popovici M, Chavkin C (2003). Kappa opioid receptor antagonism and prodynorphin gene disruption block stress‐induced behavioral responses. J Neurosci 23: 5674–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier JC (1997). Nociceptin/orphanin FQ and the opioid receptor‐like ORL1 receptor. Eur J Pharmacol 340: 1–15. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Grisel JE, Hayward MD, Bales JR, Rubinstein M, Belknap JK et al (2000). Disparate spinal and supraspinal opioid antinociceptive responses in beta‐endorphin‐deficient mutant mice. Neuroscience 101: 709–717. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Pasternak GW (2001). The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev 53: 381–415. [PubMed] [Google Scholar]
- Mogil JS, Wilson SG (1997). Nociceptive and morphine antinociceptive sensitivity of 129 and C57BL/6 inbred mouse strains: implications for transgenic knock‐out studies. Eur J Pain 1: 293–297. [DOI] [PubMed] [Google Scholar]
- Mojica FJM, Montoliu L (2016). On the origin of CRISPR‐Cas technology: from prokaryotes to mammals. Trends Microbiol 24: 811–820. [DOI] [PubMed] [Google Scholar]
- Morozov A, Kellendonk C, Simpson E, Tronche F (2003). Using conditional mutagenesis to study the brain. Biol Psychiatry 54: 1125–1133. [DOI] [PubMed] [Google Scholar]
- Nadal X, Baños J‐E, Kieffer BL, Maldonado R (2006). Neuropathic pain is enhanced in delta‐opioid receptor knockout mice. Eur J Neurosci 23: 830–834. [DOI] [PubMed] [Google Scholar]
- Nadal X, La Porta C, Andreea Bura S, Maldonado R (2013). Involvement of the opioid and cannabinoid systems in pain control: new insights from knockout studies. Eur J Pharmacol 716: 142–157. [DOI] [PubMed] [Google Scholar]
- Narita M, Imai S, Nakamura A, Ozeki A, Asato M, Rahmadi M et al (2013). Possible involvement of prolonging spinal μ‐opioid receptor desensitization in the development of antihyperalgesic tolerance to μ‐opioids under a neuropathic pain‐like state. Addict Biol 18: 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institutes of Health . ClinicalTrials.gov. Accessed January 16, 2017.
- Negrete R, García Gutiérrez MS, Manzanares J, Maldonado R (2016). Involvement of the dynorphin/KOR system on the nociceptive, emotional and cognitive manifestations of joint pain in mice. Neuropharmacology 116: 315–327. [DOI] [PubMed] [Google Scholar]
- Niikura K, Narita M, Narita M, Nakamura A, Okutsu D, Ozeki A et al (2008a). Direct evidence for the involvement of endogenous beta‐endorphin in the suppression of the morphineinduced rewarding effect under a neuropathic pain‐like state. Neurosci Lett 435: 257–262. [DOI] [PubMed] [Google Scholar]
- Niikura K, Narita M, Okutsu D, Tsurukawa Y, Nanjo K, Kurahashi K et al (2008b). Implication of endogenous beta‐endorphin in the inhibition of the morphine‐induced rewarding effect by the direct activation of spinal protein kinase C in mice. Neurosci Lett 433: 54–58. [DOI] [PubMed] [Google Scholar]
- Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T et al (1997). Unrestrained nociceptive response and disregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. EMBO J 16: 1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche JF, Schuller AGP, King MA, Zengh M, Pasternak GW, Pintar JE (2002). Genetic dissociation of opiate tolerance and physical dependence in delta‐opioid receptor‐1 and preproenkephalin knock‐out mice. J Neurosci 22: 10906–10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Dussor GO, Porreca F (2010). Central modulation of pain. J Clin Invest 120: 3779–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y‐X, Xu J, Xu M, Rossi GC, Matulonis JE, Pasternak GW (2009). Involvement of exon 11‐associated variants of the mu opioid receptor MOR‐1 in heroin, but not morphine, actions. Proc Natl Acad Sci U S A 106: 4917–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti C, Scoto GM (2010). The functional antiopioid action of the ventrolateral periaqueductal gray nociceptin/orphanin FQ and nociceptin receptor system underlies DAMGO analgesic tolerance. Pharmacology 86: 138–144. [DOI] [PubMed] [Google Scholar]
- Parikh D, Hamid A, Friedman TC, Nguyen K, Tseng A, Marquez P et al (2011). Stress‐induced analgesia and endogenous opioid peptides: the importance of stress duration. Eur J Pharmacol 650: 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW, Pan YX (2013). Mu opioids and their receptors: evolution of a concept. Pharmacol Rev 65: 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, Williams S‐PG, Zhao X, Walsh CE, Gedeon JY, Cagle NE et al (2015). Dorsal horn circuits for persistent mechanical pain. Neuron 87: 797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petraschka M, Li S, Gilbert TL, Westenbroek RE, Bruchas MR, Schreiber S et al (2007). The absence of endogenous beta‐endorphin selectively blocks phosphorylation and desensitization of mu opioid receptors following partial sciatic nerve ligation. Neuroscience 146: 1795–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polunina AG, Bryun EA (2013). Neuropsychological functions of μ‐ and δ‐opioid systems. ISRN Addict 2013: 674534: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povdin S, Yaksch T, Hook V (2016). The emerging role of spinal dynorphin in chronic pain: a therapeutic perspective. Annu Rev Pharmacol Toxicol 56: 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido‐Quetglas C, Aparicio‐Prat E, Arnan C, Polidori T, Hermoso T, Palumbo E et al (2017). Scalable design of paired CRISPR guide RNAs for genomic deletion. PLoS Comput Biol 13: e1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Sora I, Ren K, Uhl G, Dubner R (2000). Enhanced delta‐opioid receptormediated antinociception in mu‐opioid receptor‐deficient mice. Eur J Pharmacol 387: 163–169. [DOI] [PubMed] [Google Scholar]
- Raffa RB, Burdge G, Gambrah J, Kinecki HE, Lin F, Lu B et al (2017). Cebranopadol: novel dual opioid/NOP receptor agonist analgesic. J Clin Pharm Ther 42: 8–17. [DOI] [PubMed] [Google Scholar]
- Ragnauth A, Schuller A, Morgan M, Chan J, Ogawa S, Pintar J et al (2001). Female preproenkephalin‐knockout mice display altered emotional responses. Proc Natl Acad Sci U S A 98: 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redrobe JP, Calo' G, Regoli D, Quirion R (2002). Nociceptin receptor antagonists display antidepressant‐like properties in the mouse forced swimming test. Naunyn Schmiedebergs Arch Pharmacol 365: 164–167. [DOI] [PubMed] [Google Scholar]
- Reiss D, Ceredig RA, Secher T, Boué J, Barreau F, Dietrich G et al (2016). Mu and delta opioid receptor knockout mice show increased colonic sensitivity. Eur J Pain 21: 623–634. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Whistler JL (2009). The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol Psychiatry 66: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittner HL, Brack A, Stein C (2008). Pain and the immune system. Br J Anaesth 101: 40–44. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Molinari S, Marti M, Marzola G, Calo’ G (2011). Nociceptin/orphanin FQ receptor knockout rats: in vitro and in vivo studies. Neuropharmacology 60: 572–579. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japón M, Chan EC, Allen RG, Low MJ (1996). Absence of opioid stress‐induced analgesia in mice lacking beta‐endorphin by site‐directed mutagenesis. Proc Natl Acad Sci U S A 93: 3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten K, De Vry J, Bruckmann W, Tzschentke TM (2011). Pharmacological blockade or genetic knockout of the NOP receptor potentiates the rewarding effect of morphine in rats. Drug Alcohol Depend 114: 253–256. [DOI] [PubMed] [Google Scholar]
- Rutten K, Tzschentke TM, Koch T, Schiene K, Christoph T (2014). Pharmacogenomic study of the role of the nociceptin/orphanin FQ receptor and opioid receptors in diabetic hyperalgesia. Eur J Pharmacol 741: 264–271. [DOI] [PubMed] [Google Scholar]
- Sałat K, Jakubowska A, Kulig K (2015). Cebranopadol: a first‐in‐class potent analgesic agent with agonistic activity at nociceptin/orphanin FQ and opioid receptors. Expert Opin Investig Drugs 24: 837–844. [DOI] [PubMed] [Google Scholar]
- Saloman JL, Scheff NN, Snyder LM, Ross SE, Davis BM, Gold MS (2016). Gi‐ DREADD expression in peripheral nerves produces ligand‐dependent analgesia, as well as ligand‐independent functional changes in sensory neurons. J Neurosci 36: 10769–10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Kieffer BL, Fanselow MS (2005). Deletion of the mu opioid receptor results in impaired acquisition of Pavlovian context fear. Neurobiol Learn Mem 84: 33–41. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Takasaki I, Andoh T, Shiraki K, Takeshima H, Takahata H et al (2008). Nociceptin‐receptor deficiency prevents postherpetic pain without effects on acute herpetic pain in mice. Neuroreport 19: 83–86. [DOI] [PubMed] [Google Scholar]
- Schepers RJ, Mahoney JL, Gehrke BJ, Shippenberg TS (2008). Endogenous kappaopioid receptor systems inhibit hyperalgesia associated with localized peripheral inflammation. Pain 138: 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder W, Lambert DG, Ko MC, Koch T (2014). Functional plasticity of the N/OFQ‐NOP receptor system determines analgesic properties of NOP receptor agonists. Br J Pharmacol 171: 3777–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ et al (1999). Retention of heroin and morphine‐6 beta‐glucuronide analgesia in a new line of mice lacking exon 1 of MOR‐1. Nat Neurosci 2: 151–156. [DOI] [PubMed] [Google Scholar]
- Schunk S, Linz K, Frormann S, Hinze C, Oberbörsch S, Sundermann B et al (2014). Discovery of Spiro[cyclohexane‐dihydropyrano[3,4‐b]indole]‐amines as potent NOP and opioid receptor agonists. ACS Med Chem Lett 5: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoto GM, Aricò G, Iemolo A, Ronsisvalle G, Parenti C (2010). Selective inhibition of the NOP receptor in the ventrolateral periaqueductal gray attenuates the development and the expression of tolerance to morphine‐induced antinociception in rats. Peptides 31: 696–700. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U (2001). Generation of dynorphin knockout mice. Brain Res Mol Brain Res 86: 70–75. [DOI] [PubMed] [Google Scholar]
- Simonin F, Valverde O, Smadja C, Slowe S, Kitchen I, Dierich A et al (1998). Disruption of the kappa‐opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa‐agonist U‐50,488H and attenuates morphine withdrawal. EMBO J 17: 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Schimenti JC, Bolcun‐Filas E (2015). A mouse geneticist's practical guide to CRISPR applications. Genetics 199: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuda ER, Copits BA, Schmidt MJ, Baird MA, Al‐Hasani R, Planer WJ et al (2015). Spatiotemporal control of opioid signaling and behavior. Neuron 86: 923–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak M, Cami‐Kobeci G, Sałaga M, Husbands SM, Fichna J (2014). Novel mixed NOP/MOP agonist BU08070 alleviates pain and inhibits gastrointestinal motility in mouse models mimicking diarrhea‐predominant irritable bowel syndrome symptoms. Eur J Pharmacol 736: 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sora I, Li XF, Funada M, Kinsey S, Uhl GR (1999). Visceral chemical nociception in mice lacking mu‐opioid receptors: effects of morphine, SNC80 and U‐50,488. Eur J Pharmacol 366: R3–R5. [DOI] [PubMed] [Google Scholar]
- Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM et al (1997). Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine‐induced analgesia. Proc Natl Acad Sci U S A 94: 1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2017). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover JD, Farhang N, Berrett KC, Gertz J, Lawrence B, Bowles RD (2017). CRISPR epigenome editing of AKAP150 in DRG neurons abolishes degenerative IVD‐induced neuronal activation. Mol Ther. pii: S1525‐0016(17)30272‐1 25: 2014–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura YK, Takahashi Y, Watabe AM, Kato F (2016). Synaptic and network consequences of monosynaptic nociceptive inputs of parabrachial nucleus origin in the central amygdala. J Neurophysiol 115: 2721–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhtankar DD, Zaveri NT, Husbands SM, Ko M‐C (2013). Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/μ‐opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J Pharmacol Exp Ther 346: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenius L (2000). From opiate pharmacology to opioid peptide physiology. Ups J Med Sci 105: 1–15. [DOI] [PubMed] [Google Scholar]
- Tian JH, Han JS (2000). Functional studies using antibodies against orphanin FQ/nociceptin. Peptides 21: 1047–1050. [DOI] [PubMed] [Google Scholar]
- Tian M, Broxmeyer HE, Fan Y, Lai Z, Zhang S, Aronica S et al (1997). Altered hematopoiesis, behavior, and sexual function in mu opioid receptor‐deficient mice. J Exp Med 185: 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Bruchas MR, Calo G, Cox BM, Zaveri NT (2016). Nociceptin/Orphanin FQ receptor structure, signalling, ligands, functions, and interactions with opioid system. Pharmacol Rev 68: 419–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Al‐Hasani R, Salvemini D, Salter MW, Gutstein H, Cahill CM (2015). Pain and poppies: the good, the bad, and the ugly of opioid analgesics. J Neurosci 35: 13879–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo JM, Zimmer A, Maldonado R (2009). Nicotine anxiogenic and rewarding effects are decreased in mice lacking beta‐endorphin. Neuropharmacology 56: 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Yamaguchi T, Tokuyama S, Inoue M, Nishi M, Takeshima H (1997). Partial loss of tolerance liability to morphine analgesia in mice lacking the nociceptin receptor gene. Neurosci Lett 237: 136–138. [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Chen W, Kim H, Minasyan A, Ennes HS, McRoberts JA et al (2016). Sustained suppression of hyperalgesia during latent sensitization by μ‐, δ‐, and κ‐opioid receptors and 2A adrenergic receptors: role of constitutive activity. J Neurosci 36: 204–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Bélanger E, Paquet M‐E, Côté DC, De Koninck Y (2016). Probing pain pathways with light. Neuroscience 338: 248–271. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U et al (2001). Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci 21: 1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, Filliol D et al (2013). Mu opioid Receptors on primary afferent Nav1.8 neurons contribute to opiate‐induced analgesia: insight from conditional knockout mice. PLoS One 8: e74706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whissell PD, Tohyama S, Martin LJ (2016). The use of DREADDs to deconstruct behavior. Front Genet 7: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieskopf JS, Pan YX, Marcovitz J, Tuttle AH, Majumdar S, Pidakala J et al (2014). Broad‐spectrum analgesic efficacy of IBNtxA is mediated by exon 11‐associated splice variants of the mu‐opioid receptor gene. Pain 55: 2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Statnick MA, Rorick‐Kehn LM, Pintar JE, Ansonoff M, Chen Y et al (2014). The biology of nociceptin/orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol Ther 141: 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann W, Schunk E, Rosskothen I, Gaburro S, Singewald N, Herzog H et al (2009). Prodynorphin‐derived peptides are critical modulators of anxiety and regulate neurochemistry and corticosterone. Neuropsychopharmacology 34: 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ (2010). What is this thing called pain? J Clin Invest 120: 3742–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Zhang S, Shkhyan R, Lee S, Gullo F, Eliasberg CD et al (2017). Kappa opioid receptor signaling protects cartilage tissue against posttraumatic degeneration. JCI Insight 2: e88553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C (2007). Sciatic nerve ligation‐induced proliferation of spinal cord astrocytes is mediated by opioid activation of p38 mitogen‐activated protein kinase. J Neurosci 27: 2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ et al (2004). Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci 24: 4576–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P et al (2004). Modification of nociception and morphine tolerance by the selective opiate receptor‐like orphan receptor antagonist (−)‐cis‐1‐methyl‐7‐[[4‐(2,6‐dichlorophenyl)piperidin‐1‐yl]methyl]‐6,7,8,9‐tetrahydro‐5H‐benzocyclohepten‐5‐ol (SB‐612111). J Pharmacol Exp Ther 308: 454–461. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gadotti VM, Chen L, Souza IA, Stemkowski PL, Zamponi GW (2015). Role of prelimbic GABAergic circuits in sensory and emotional aspects of neuropathic pain. Cell Rep 12: 752–759. [DOI] [PubMed] [Google Scholar]
- Zhao C‐S, Tao Y‐X, Tall JM, Donovan DM, Meyer RA, Raja SN (2003). Role of μ‐opioid receptors in formalin‐induced pain behavior in mice. Exp Neurol 184: 839–845. [DOI] [PubMed] [Google Scholar]
- Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP et al (1999). Retention of supraspinal delta‐like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 24: 243–252. [DOI] [PubMed] [Google Scholar]
- Zielińska M, Haddou TB, Cami‐Kobeci G, Sałaga M, Jarmuż A, Padysz M et al (2015). Anti‐inflammatory effect of dual nociceptin and opioid receptor agonist, BU08070, in experimental colitis in mice. Eur J Pharmacol 765: 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Valjent E, Konig M, Zimmer AM, Robledo P, Hahn H et al (2001). Absence of delta −9‐tetrahydrocannabinol dysphoric effects in dynorphin‐deficient mice. J Neurosci 21: 9499–9505. [DOI] [PMC free article] [PubMed] [Google Scholar]