

Abstract
Photochemical precursors that produce dA• and dG(N2-H)• are needed to investigate their reactivity. The synthesis of two 1,1-diphenylhydrazines (1, 2) and their use as photochemical sources of dA• and dG(N2-H)• is presented. Trapping studies indicate production of these radicals with good fidelity, and 1 was incorporated into an oligonucleotide via solid phase synthesis. Cyclic voltammetric studies show that reduction potentials of 1 and 2 are lower than those of widely-used “hole sinks”, e.g., 8-oxodGuo and 7-deazadGuo, to investigate DNA-hole transfer processes. These molecules could be useful: (a) as sources of dA• and dG(N2-H)• at specific sites in oligonucleotides, and, (b) as “hole sinks” for the study of DNA-hole transfer processes.
Graphical abstract

Nucleic acid oxidation is an important and complex collection of chemical processes.1,2 Valuable mechanistic information concerning how individual chemotherapeutic agents damage DNA, which could lead to more effective therapy, was gleaned by examining their reactivity with nucleic acids.3–5 A variety of tools have also been used to understand the more chemically diverse nucleic acid damage pathways induced by γ-radiolysis.6 Independent generation of reactive intermediates involved in nucleic acid oxidation is useful for understanding how these biopolymers are damaged.7 We wish to report the generation of the nitrogen centered radicals, 2′-deoxyadenosin-N6-yl radical (dA•) and 2′-deoxyguanosin-N2-yl radical (dG(N2-H)•) via photodissociation of the corresponding hydrazines (1, 2).
Photochemistry has proven to be very useful for independently generating nucleic acid radicals in nucleosides, as well as in DNA and RNA.6,7 Most of the nucleic acid radicals studied via photochemical generation are C-centered radicals and the Norrish Type I photocleavage reaction of ketones has been used most frequently to generate them, although aryl sulfides and aryl selenides have also been employed.8–10 Nucleobase π-radicals (Scheme 1) also play important roles in nucleic acid oxidation, such as in electron transfer, and the direct (dG(N1-H)•, dG(N2-H)•) and indirect (dA•) effects of ionizing radiation.6,11,12 Furthermore, dG(N1-H)• and dG(N2-H)• are irreversibly formed from deprotonation of the dG radical cation involved in DNA charge transfer.13 Progress in understanding the reactivity of these radicals, particularly within the biopolymers, has lagged behind that of C-centered radicals. A small number of approaches for generating neutral purine radicals have been described.14,15 However, some are incompatible with solid-phase oligonucleotide synthesis, and photolysis of others does not provide high mass balances.16,17 Herein we describe an alternative method to produce dA• involving photodissociation of 1, and the application of this approach to provide the first example of dG(N2-H)• generation.
Scheme 1.
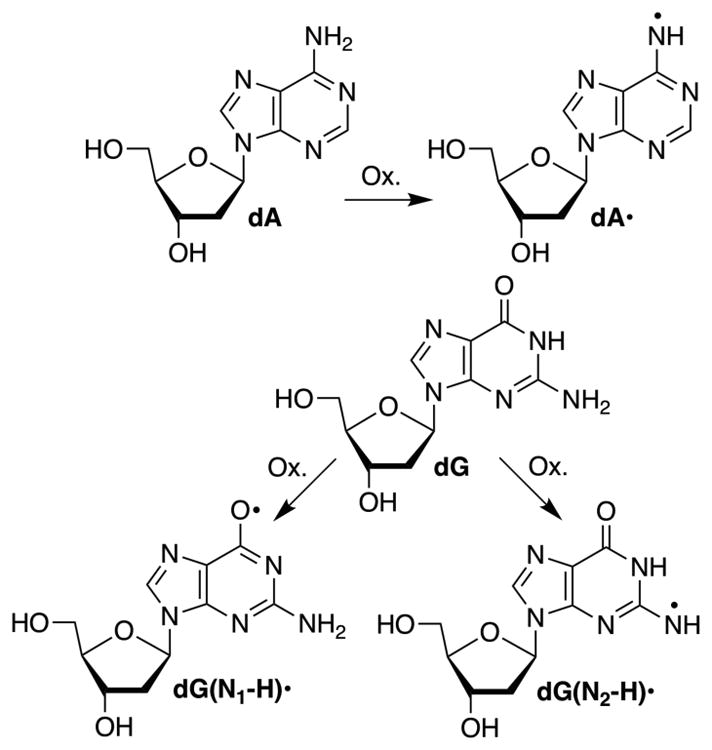
Neutral Purine Radical Formation.
We speculated that photolysis of hydrazines could result in cleavage of the relatively weak N-N bond. Furthermore, we anticipated that the use of aryl hydrazines would enable us to carry out photolysis at wavelengths of light (350 nm) that would minimize nucleic acid degradation. Condensed phase spectroscopy experiments demonstrate that photoexcited tetraphenyl hydrazine dissociates to form two diphenyl aminyl radicals.18–21 Although other tetraaryl hydrazines have been shown to undergo photodissociation, examples of differently substituted hydrazines are rare, and studies on the reactivity of diffusively free aminyl radicals derived from them are lacking.21,22 Cage recombination appears to be a major pathway in these examples. N,N-Diphenyl hydrazines were also attractive as precursors, because the concomitantly produced diphenyl aminyl radical (Ph2N•) was expected to be less reactive than many radicals, due to the low BDE of the N-H bond in Ph2NH (<85 kcal/mol).23,24

We set out to prepare 1 via nucleophilic aromatic substitution, which has been used to prepare less highly substituted hydrazine N6-purines.25,26 using protected We initially chose Lakshman’s O6-(benzotriazol-1-yl)-2′-deoxyinosine (3).27 Reaction of 3 with phenyl hydrazine (10 equiv.) provided the expected N,N′-disubstituted hydrazine (4) in 99% yield. In contrast, no reaction was observed between 3 and 1,1-diphenyl hydrazine (6, 10 equiv.), or its hydrochloride salt under a variety of conditions. These included temperatures as high as 80 °C, the presence of Cs2CO3 and/or DABCO, in either THF, MeOH, or dimethoxyethane (80 °C). Reacting 6 with 7, which readily reacts with variety of arylamines28,29, did not yield desired product. Similarly, sulfonate 8, which undergoes nucleophilic aromatic substitution when activated by DABCO30, also did not react with 6.
Following these unsuccessful efforts, we turned our attention to Buchwald-Hartwig amination chemistry. A variety of 6-substituted purines and N6-amino substituted adenosines have been prepared from 6-bromopurine 7 using Pd catalysts31,32 Use of Pd(OAc)2 and BINAP ligand gave only trace amounts of the desired product (5). However, switching to Xantphos as ligand and Pd2dba2 as catalyst produced 5 in 48% yield (Scheme 2).33 Desilylation provided the candidate photochemical hydrazine precursor 1.
Scheme 2.

Diphenylhydrazine Synthesis.
Hydrazine 1 had a λmax = 267 nm (ε = 1.81 ± 0.01 × 104 M−1cm−1) in H2O, but its absorption tailed into the >300 nm region at the concentration (100 μM) at which it was present during photolysis. Hydrazine 1 was completely consumed after photolysis (Rayonet Photoreactor equipped with λmax = 350 nm lamps) for 4 h. 2′-Deoxyadenosine (dA) was the major product detected under all irradiation conditions (Table 1). Thymidine was used as an internal standard in all experiments, and was shown to be stable to the reaction conditions (Figure S9). The only other molecule detected was formal recombination product 10 (Scheme 3).
Table 1.
Reactivity of dA• from irradiated 1.
| % yield | ||||
|---|---|---|---|---|
| [1] (μM) | red. (mM) | dA | 10 | % mass bal. |
| 100 | - | 64 ± 2 | 9 ± 1 | 73 ± 2 |
| 100 | Fe+2 (10) | 65 ± 1 | 6 ± 1 | 71± 1 |
| 100 | BME (10) | 57 ± 2 | 8 ± 1 | 65 ± 3 |
| 100 | PhSH (100) | 72 ± 1 | 10 ± 1 | 81 ± 1 |
| 100 | PhSH (10) | 67 ± 2 | 11 ± 1 | 78 ± 2 |
| 50 | PhSH (10) | 72 ± 1 | 8 ± 1 | 80 ± 2 |
| 25 | PhSH (10) | 75 ± 1 | 9 ± 1 | 84 ± 1 |
Average ± std. dev. of 3 experiments.
Scheme 3.

Product Formation From Photolysis of 1.

Product 10 was initially identified by LC/MS analysis in the photolysate of 1. It exhibited (Figure 1a) a molecular ion identical to that of 1, but in contrast to the hydrazine (Figure S1), fragmentation of the diphenylamino group was not observed. Attempted synthesis of 10 via a variety of Pd coupling conditions of the protected 8-bromo-2′-deoxyadenosine with diphenylamine was unsuccessful.34 Consequently, support for the assignment of 10 was obtained via LC/MS analysis of photolyzed 8-2H-1. The deuterated hydrazine was prepared from 1 via Et3N-catalyzed D2O exchange.35 The recombination product formed from 8-2H-1 (Figure 1b) lacked deuterium, consistent with the proposed structure (10). Yields of 10 were roughly estimated by synthesizing 11 and measuring its extinction coefficient at 260 nm.34 This value was compared to that of 1 (whose response factor was independently measured) to approximate its yield by HPLC (Table 1).
Figure 1.

Mass spectrometric identification of recombination product 10 formed from photolysis of (a) 1 (b) 8-2H-1.
The major product from photolysis of 1 was dA. Overall, the effects of reductants on the yields of dA are similar to those observed when dA• was generated from 11.15 For instance, β-mercaptoethanol (BME) was an inefficient trap compared to ferrous ion and thiophenol (PhSH), presumably due to polarity mismatching, as previously discussed.15 Furthermore, there was an inverse relationship between dA yield and concentration of 1. Studies on 12 attributed these observations to dA• reduction by the photochemical precursor. Evidence in support of hydrazine 1 serving as a viable reductant for dA• is obtained from cyclic voltammetry (Figure 2, Figure S2). The observed reduction potential illustrates that 1 is more readily oxidized than dG, the most electron rich native nucleotide, 7-deazadGuo, and even 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodGuo), a DNA lesion that is susceptible to further oxidation (Figure 2, Figures S3–5).36–39 However, the yield of dA in the absence of any exogenous reductant is >50%, indicating that a species other than 1 must reduce dA•. We tentatively suggest that in addition to providing 10, the radical pair also undergoes electron transfer (Scheme 3). Finally, the yield of 10 is more or less independent of reductant or its concentration, and substrate concentration, suggesting that it could result from a radical pair process. However, due to the expected stability of Ph2N•,23 we cannot rule out selective reaction of diffusively free aminyl radical with dA•.
Figure 2.

Reduction potentials of nucleoside radical cations versus NHE in CH3CN.
The success of 1 in generating dA• led us to apply the same strategy for generating 2′-deoxyguanosin-N2-yl radical (dG(N2-H)•). dG(N2-H)• represents one of the two one-electron oxidized tautomers of dG (Scheme 1).40 The pKa values of the N1- and N2- protons of the guanine radical cation (G•+) in dG, are believed to be ~3.9 and ~4.7 respectively.12,40 From the closeness of these pKa values, it is expected that deprotonation from N1 as well as N2 of G•+ should be competitive with each other. dG(N1-H)• is believed to be the major neutral purine radical formed upon deprotonation, following one-electron oxidation of the nucleoside dG and in ssD-NA.40,41 However, in dsDNA, owing to the base pairing, G•+ deprotonation occurs from N1 and N2 to produce dG(N1-H)• and dG(N2-H)•.42 Therefore, independent generation of dG(N1-H)• and dG(N2-H)• could help the study of the properties of these radicals and their role in DNA damage.
The requisite hydrazine (2) was prepared from the O6-benzyl protected, disilylated dG (9, Scheme 2).43 The Pd coupling reaction proceeded in slightly higher yield (63%) than the analogous reaction did for the preparation of 1. Precursor 2 was obtained following desilylation and hydrogenolysis.
Hydrazine 2 is slightly more readily oxidized than the adenine analogue (1). Cyclic voltammetry indicated that the reduction potential for 2 was 0.77 V vs. NHE (Figure 2, Figure S3). Photoconversion of 2 was qualitatively less efficient than that for 1, requiring 8 h photolysis for almost complete conversion instead of 4 h. Importantly, photolysis of 2 provided strong evidence for the formation of dG(N2-H)• (Table 2). Comparisons of the yield of dG and mass balance in photolysates of 2 under various conditions with the corresponding mass balances and dA yields from 1 (Table 1) indicate subtle differences in the two systems. For instance, the yield of dG (and mass balance) in the absence of exogenous reducing agent were closer to 50%, which would be the maximum yield if 2 is the sole reducing agent of dG(N2-H)•. This could reflect slower electron transfer within the respective radical pair formed upon photodissociation, and correlates with the smaller thermodynamic driving force for dG(N2-H)• reduction. The mass balances are modestly higher from the photolyses of 2 than from 1 in the presence of Fe+2, BME, or PhSH. This too could be a consequence of greater thermodynamic stability of dG(N2-H)• compared to dA•, which results in slower reactions with its respective precursor and Ph2N•.
Table 2.
Reactivity of dG(N2-H)• from irradiated 2.
| [2] (μM) | red. (mM) | % yield dGa | % mass bal.a |
|---|---|---|---|
| 100 | - | 54 ± 3 | 54 ± 3 |
| 100 | Fe+2 (10) | 67 ± 3 | 73 ± 1 |
| 100 | BME (10) | 69 ± 1 | 75 ± 1 |
| 100 | PhSH (10) | 68 ± 2 | 74 ± 2 |
| 50 | PhSH (10) | 74 ± 3 | 74 ± 3 |
| 25 | PhSH (10) | 78 ± 1 | 82 ± 1 |
Average ± std. dev. of 3 experiments.
Hydrazines 1 and 2 are compatible with solid phase oligonucleotide synthesis conditions. This was further demonstrated by introducing 1 into an oligonucleotide (13) via its corresponding phosphoramidite. The TM of a dodecameric duplex containing 1 (14a, 29.2 °C) was decreased significantly compared to an otherwise identical one containing dA (14b, 49.6 °C) in place of 1. This does not detract from the utility of 1 as a tool for producing dA• in DNA since the lifetime of the radical (4.0 ± 1.0 ms) is long compared to conformational changes of the helix.44
In summary, in response to a dearth of methods for generating aminyl radicals photochemically, we demonstrate that hydrazines are suitable photochemical precursors for π-aminyl purine radicals (dA•, dG(N2-H)•). The precursors are also compatible with solid phase oligonucleotide synthesis conditions, indicating that they will be useful for elucidating the reactivity of these important reactive intermediates within nucleic acids. Furthermore, the redox potentials of 1 and 2 are found to be far lower than those of 8-oxo-dGuo and 7-deazadGuo (Figure 2, Figures S4,5), which are used extensively as DNA hole transfer probes.45 Consequently, 1 and 2 may be much better hole traps than either 8-oxodGuo or 7-deazadGuo and would be excellent probes for the investigation of hole transfer in DNA.
Supplementary Material
Acknowledgments
We are grateful for generous financial support from the National Institute of General Medical Science (GM-054996) to MMG. AA and MDS acknowledge financial support from the National Cancer Institute (CA-045424) and the Oakland University Research Excellence Funds (CBR). LZ thanks Johns Hopkins University for the Glen E. Meyer ‘39 Fellowship.
Footnotes
All experimental details and spectroscopic data for all new compounds (PDF). The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Pitié M, Pratviel G. Chem Rev. 2010;110:1018–1059. doi: 10.1021/cr900247m. [DOI] [PubMed] [Google Scholar]
- 2.Gates KS. Chem Res Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy DR, Lu J, Shen B, Beerman TA. Proc Nat Acad Sci USA. 2007;104:17632–17637. doi: 10.1073/pnas.0708274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy B, Hecht SM. J Am Chem Soc. 2014;136:4382–4393. doi: 10.1021/ja500414a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Stubbe J. Nature Rev Cancer. 2005;5:102–112. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]
- 6.Dizdaroglu M, Jaruga P. Free Rad Res. 2012;46:382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg MM. Adv Phys Org Chem. 2016;50:119–202. doi: 10.1016/bs.apoc.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giese B, Beyrich-Graf X, Erdmann P, Giraud L, Imwindelried P, Müller SN, Schwitter U. J Am Chem Soc. 1995;117:6146–6147. [Google Scholar]
- 9.Paul R, Greenberg MM. J Am Chem Soc. 2015;137:596–599. doi: 10.1021/ja511401g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.San Pedro JMN, Greenberg MM. J Am Chem Soc. 2014;136:3928–3936. doi: 10.1021/ja412562p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cadet J, Douki T, Ravanat JL. Acc Chem Res. 2008;41:1075–1083. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]
- 12.Adhikary A, Becker D, Sevilla MD. In: Applications of EPR in Radiation Research. Lund A, Shiotani M, editors. Springer International Publishing; Heidelberg, New York, London: 2014. pp. 299–352. [Google Scholar]
- 13.Yoshioka Y, Kitagawa Y, Takano Y, Yamaguchi K, Nakamura T, Saito I. J Am Chem Soc. 1999;121:8712–8719. [Google Scholar]
- 14.Kuttappan-Nair V, Samson-Thibault F, Wagner JR. Chem Res Toxicol. 2010;23:48–54. doi: 10.1021/tx900268r. [DOI] [PubMed] [Google Scholar]
- 15.Zheng L, Griesser M, Pratt DA, Greenberg MM. J Org Chem. 2017;82:3571–3580. doi: 10.1021/acs.joc.7b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaloudis P, Paris C, Vrantza D, Encinas S, Perez-Ruiz R, Miranda MA, Gimisis T. Org & Biomol Chem. 2009;7:4965–4972. doi: 10.1039/b909138f. [DOI] [PubMed] [Google Scholar]
- 17.Vrantza D, Kaloudis P, Leondiadis L, Gimisis T, Vougioukalakis GC, Orfanopoulos M, Gasparutto D, Cadet J, Encinas S, Paris C, Miranda MA. Helv Chim Acta. 2006;89:2371–2386. [Google Scholar]
- 18.Hirata Y, Niga Y, Ohta M, Takizawa M, Okada T. Res Chem Intermed. 1995;21:823–836. [Google Scholar]
- 19.Lenderink E, Duppen K, Wiersma DA. Chem Phys Lett. 1992;194:403–409. [Google Scholar]
- 20.Hyde MG, Reid GD, Beddard GS. Chem Phys Lett. 1992;190:130–134. [Google Scholar]
- 21.Neugebauer FA, Bamberger S. Chem Ber. 1974;107:2362–2382. [Google Scholar]
- 22.Schlosser K, Steenken S. J Am Chem Soc. 1983;105:1504–1506. [Google Scholar]
- 23.Pratt DA, DiLabio GA, Valgimigli L, Pedulli GF, Ingold KU. J Am Chem Soc. 2002;124:11085–11092. doi: 10.1021/ja026289x. [DOI] [PubMed] [Google Scholar]
- 24.Ingold KU, Pratt DA. Chem Rev. 2014;114:9022–9046. doi: 10.1021/cr500226n. [DOI] [PubMed] [Google Scholar]
- 25.Kolarski D, Szymanski W, Feringa BL. Org Lett. 2017;19:5090–5093. doi: 10.1021/acs.orglett.7b02361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Too K, Brown DM, Bongard E, Yardley V, Vivas L, Loakes D. Bioorg Med Chem. 2007;15:5551–5562. doi: 10.1016/j.bmc.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 27.Bae S, Lakshman MK. J Am Chem Soc. 2007;129:782–789. doi: 10.1021/ja064682n. [DOI] [PubMed] [Google Scholar]
- 28.Nair V, Richardson SG. J Org Chem. 1980;45:3969–3974. [Google Scholar]
- 29.Veliz EA, Beal PA. J Org Chem. 2001;66:8592–8598. doi: 10.1021/jo016078v. [DOI] [PubMed] [Google Scholar]
- 30.Lakshman MK, Ngassa FN, Keeler JC, Dinh YQV, Hilmer JH, Russon LM. Org Lett. 2000;2:927–930. doi: 10.1021/ol005564m. [DOI] [PubMed] [Google Scholar]
- 31.Lakshman MK, Hilmer JH, Martin JQ, Keeler JC, Dinh YQV, Ngassa FN, Russon LM. J Am Chem Soc. 2001;123:7779–7787. doi: 10.1021/ja0107172. [DOI] [PubMed] [Google Scholar]
- 32.Lakshman MK, Keeler JC, Hilmer JH, Martin JQ. J Am Chem Soc. 1999;121:6090–6091. [Google Scholar]
- 33.Buchwald SL, Wagaw S, Geis O. 9943643A2. WO. 1999
- 34.Szombati Z, Baerns S, Marx A, Meier C. ChemBioChem. 2012;13:700–712. doi: 10.1002/cbic.201100573. [DOI] [PubMed] [Google Scholar]
- 35.Huang X, Yu P, LeProust E, Gao X. Nucleic Acids Res. 1997;25:4758–4763. doi: 10.1093/nar/25.23.4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steenken S, Jovanovic SV, Bietti M, Bernhard K. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 37.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 38.Thapa B, Schlegel HB. J Phys Chem A. 2015;119:5134–5144. doi: 10.1021/jp5088866. [DOI] [PubMed] [Google Scholar]
- 39.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Nucleic Acids Res. 2004;32:6565– 6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adhikary A, Kumar A, Munafo SA, Khanduri D, Sevilla MD. Phys Chem Chem Phys. 2010;12:5353–5368. doi: 10.1039/b925496j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adhikary A, Kumar A, Becker D, Sevilla MD. J Phys Chem B. 2006;110:24171–24180. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rokhlenko Y, Cadet J, Geacintov NE, Shafirovich V. J Am Chem Soc. 2014;136:5956–5962. doi: 10.1021/ja412471u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harwood EA, Hopkins PB, Sigurdsson ST. J Org Chem. 2000;65:2959–2964. doi: 10.1021/jo991501+. [DOI] [PubMed] [Google Scholar]
- 44.Banyasz A, Ketola TM, Muñoz-Losa A, Rishi S, Adhikary A, Sevilla MD, Martinez-Fernandez L, Improta R, Markovitsi D. J Phys Chem Lett. 2016;7:3949–3953. doi: 10.1021/acs.jpclett.6b01831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genereux JC, Barton JK. Chem Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.