

Abstract
A series of novel thio- and seleno-barbituric acid derivatives were synthesized by varying the substituents at N1 and N3 (ethyl, methyl, allyl, and phenyl), and C5 tethered with dienyl and trienyl moieties attached to substituents such as phenyl, 2-furanyl, 2-thiophenyl, 1-naphthyl, and 3-pyridyl. The cytotoxic potential of these derivatives was evaluated by using MTT assay against melanoma cell lines expressing either wild-type (CHL-1) or mutant (UACC 903) BRAF gene. Among all, 2b and 8b were identified as the most potent compounds. Both 2b and 8b inhibited viability of various melanoma cells and induced cell death as evidenced by Live and Dead assay. Western blot analysis showed that they induce PARP cleavage and inhibit anti-apoptotic Bcl-2, Bcl-xL and Survivin in a dose-dependent manner within 24 h of the treatment. Novel thiobarbituric acid analogs also inhibited viability of various other solid tumor cell lines, such as pancreatic, breast, and colon. Overall, 2b, 2d, and 8b emerged as the most effective compounds and make good leads for the development of future therapeutic agents.
Keywords: Thiobarbiturates, selenobarbiturates, anti-cancer, melanoma, apoptosis
Graphical abstract

1. Introduction
Cancer is one of the leading causes of death worldwide and accounted for 8.2 million deaths (nearly 14.6% of all deaths) in 2012.[1] Deaths from cancer worldwide are projected to continue to rise to over 13 million annually within next two decades. In the United States alone, an estimated 1,685,210 new cancer cases are diagnosed with estimated deaths of 595,690 in 2016.[2] Among all cancers, melanoma is one of the most deadly form of skin cancer with 2 million new cases annually in the United States.[2] If diagnosed and removed while it is still thin and limited to the outermost skin layer, it is almost 100% curable. However, no effective therapy is available for treatment once the cancer advances and metastasizes to other parts of the body. Commonly used drugs, dacarbazine and temozolomide, in melanoma patients provide unsatisfactory outcomes. The BRAFV600E inhibitor PLX4032 (vemurafenib)[3] (Figure 1) has proved to be a remarkable drug but develops resistance in about 7 months, and also works only on tumors with BRAFV600E mutations. Most patients require multiple lines of therapy as cancer cells acquire resistance against the chemotherapeutic agents by different mechanisms. The combination therapies with BRAF and MEK inhibitors such as Cobimetinib or Trametinib (Figure 1) also did not show much improvement.[4] Management of patients with resistance continues to present an extensive confront to researchers and oncologists. Many clinically effective anti-cancer drugs, including those for melanoma used today, suffer from serious drawbacks as majority of patients with metastatic solid tumors die after chemotherapeutic regimens failed to induce objective response or when resistance developed.[5] Therefore, to combat this resistance, still there is an impetus to identify and develop more potent therapeutic agents with fewer side effects in cancer therapy.
Figure 1.

(A) Clinically used drugs for metastatic melanoma (B) representative examples of effective anti-cancer thio-/seleno-barbituric acid compounds
In recent years, thiobarbituric acids have been reported to exhibit a broad spectrum of biological properties such as anti-convulsant,[6] anti-inflammatory,[7, 8] anti-bacterial,[9] anti-HIV,[10] anti-hypnotic,[11] anti-parkinsonian,[12] and anti-cancer activities,[13–15] including anti-melanoma properties.[16, 17] Among them some thiobarbituric acid derivatives, such as merbarone,[18] merocyanine 540[19] and its selenium analog selenomerocyanine 56[20] (Figure 1) have been identified as possible modulators of apoptosis in several cancer cells, including glioblastoma multiforme (GBM), melanoma, leukemia, and cancers of breast, lung, prostate, and cervix; albeit not particularly for cancers that are resistant to current therapy. An effective lead thiobaribituic acid analog 2a was recently identified in our laboratory from a library of 50,000 chemicals, originally screened to identify compounds that could target temozolomide (TMZ) resistant brain tumor cells. The lead compound 2a was effective on both TMZ resistant and sensitive glioma cells.[21] Interestingly, our preliminary studies showed 2a to also possess activity towards human melanoma cells. Therefore, we hypothesized that the structure of 2a can be modulated by varying substitutions at N1, N3, and C5 positions to create a potent compound for melanoma treatment. Therefore, in the present study, we conducted a structure-activity relationship (SAR) study to further optimize 2a structure to develop an efficacious novel thio-/seleno-barbituric acid compound for melanoma and possibly other solid tumors. In addition, 2a was considered as a valuable lead compound owing to its selective cytotoxicity to various cancer cells including melanoma, while sparing the normal cells. We synthesized 24 novel thio- and selenobarbituric derivatives by varying the substituents at N1, N3, and C5 tethered with dienyl and trienyl moieties attached with different substituents like phenyl, 2-furanyl, 2-thiophenyl, 1-naphthyl and 3-pyridyl. The efficacy of novel analogs was evaluated in a diverse panel of cancer cell lines and the two best identified compounds 2b and 8b, were further characterized in both wild-type and BRAFV600E mutant melanoma cells to get an insight into their mechanism of action.
2. Results and discussion
2.1. Design
The lead thiobarbituric acid analog 2a was found to be effective on both BRAF-mutant and wild-type melanoma cells. We hypothesized that careful manipulation of 2a could lead to more effective analogs for a broader range of melanomas as well as other cancers. In this regard, we modified various functionalities by manipulating substitutions at N1-, N3- and C5-positions of 2a as well as by replacing S by Se (Figure 1). While choosing substitutions, we also made sure that the resulting modified compounds satisfy structural parameters of Lipinski’s Rule-of-five[22, 23] for drug-likeness.
Our first focus was to modify 3-furan-2-yl allylidene functionality at C5 position of 2a. The modifications included replacing terminal furan ring with phenyl, thiophenyl, and naphthyl substitutions and extending the linker carbon chain length from dienyl to trienyl unit. Compounds 2b–f were thus synthesized to determine the effect of chain length and terminal aromatic ring at C5 position of 2a. Several studies have pointed out the anti-tumor and chemopreventive effects of multiple Se species in various cancers.[24–29] Recent reports by our laboratories and other groups have shown that replacement of S by Se in naturally occurring and synthetic isothiocyanates and several other compounds, in general though not always, substantially enhanced the efficacy of resulting compounds against melanoma and several other cancers.[28–32] Therefore, our second modification strategy involved replacing S in thiobarbituric acid ring by Se. We synthesized selenobarbituric acid analogs of 2a and two other effective thio-analogs 2b and 2c, to evaluate if the replacing S by Se in such scaffold would help enhance the cytotoxicity.
Finally, to determine the effect of N-substitutions on thiobarbituric acid compounds 2, we generated a series of N1-monosubstituted, N1,N3-disubstituted analogs 8a–o. The substitutions at N1 and N3 included methyl, ethyl, allyl, and phenyl groups. Overall, all the N1-substituted, N1, N3-disubstituted, and N1- and N3-unsubstituted derivatives were synthesized to study the effect of partially substituted, fully substituted, and free NH compounds with varying functionalities at C5 position.
2.2. Chemistry
Novel thio- (2 and 8) and seleno- (5) barbituric acid derivatives were prepared according to the sequence of reactions depicted in Schemes 1–3. The thiobarbituric acid analogs 2a–f were synthesized by Knoevenagel condensation of readily available thiobarbituric acid 1 with different α,β-unsaturated aldehydes in the presence of catalytic amount of pyridine in EtOH under reflux condition (Scheme 1).[33] To evaluate the difference in activity between the S compounds 2 and corresponding Se analogs, we synthesized isosteric selenium analogs of lead compound 2a and other key thiobarbituric acid analogs 2b, c. The key intermediate selenobarbituric acid 4 was synthesized in good yield by treating readily available selenourea 3 with diethylmalonate in the presence of freshly prepared sodium ethoxide in ethanol under reflux conditions.[34] Subsequently the selenobarbituric acid 4 was reacted under Knoevenagel condensation with different aldehydes in the presence of catalytic pyridine in refluxing EtOH to furnish the corresponding novel selenobarbituric acid derivatives 5a–c in good to excellent yields (Scheme 2).[33]
Scheme 1.
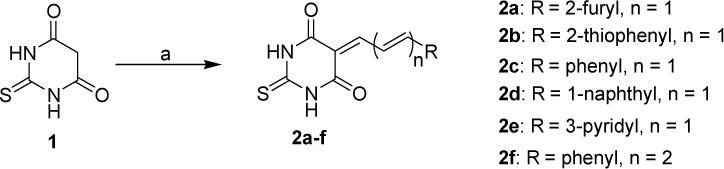
Synthesis of thiobarbituric acid analogs 2a–f. Reagents and conditions: (a) R-CH=CH-CHO, pyridine, EtOH, reflux, 12 h.
Scheme 3.

Synthesis of substituted thiobarbituric acid analogs 8a–o. Reagents and conditions: (a) Diethyl malonate, NaOEt, EtOH, reflux, 72 h; (b) R2-CH=CH-CHO, Pyridine, EtOH, reflux, 12h.
Scheme 2.

Synthesis selenobarbituric acid analogs 5a–c. Reagents and conditions: (a) Diethyl malonate, NaOEt, EtOH, reflux, 72 h; (b) R-CH=CH-CHO, pyridine, EtOH, reflux, 12 h.
To further diversify the structure activity relationship study on thiobarbitric acid derivatives, we further synthesized N1-monosubstituted and N1,N3-disubstituted thiobarbituric acid derivatives 8a–o. The precursor substituted thiobarbituric acid intermediates 7a–g were first synthesized by condensing appropriately substituted thioureas 6a–e with diethyl malonate in the presence of freshly prepared sodium ethoxide in refluxing ethanol.[34] The substituted thiobarbituric acids 7a–g were then reacted with different α,β-unsaturated aldehydes under Knoevenagel condensation to furnish the corresponding novel N1-monosubstituted and N1,N3-disubstituted thiobarbituric acid derivatives 8a–o in good to excellent yields (Scheme 2).[33] The structures of all the novel thio- and seleno-barbituric acid derivatives were confirmed by 1H NMR, 13C NMR and HRMS analysis. The compounds purity (≥98%) was analyzed by analytical high-performance liquid chromatography (HPLC) before proceeding for in vitro biological assays.
2.3. Biology
2.3.1. Evaluation of anti-proliferative activities of novel compounds against CHL-1 (BRAF wild-type) and UACC903 (BRAF-mutant) melanoma cell lines
The anti-proliferative activities of thio- and seleno-barbituric acid derivatives (2, 5, and 8) were evaluated against both BRAF-wild-type (CHL-1) and BRAF-mutant (UACC 903) melanoma cell lines. The cells were treated with various compounds for 72 h and the cell survival was determined by utilizing MTT assay. Among N1- and N3-unsubstituted compounds 2a–f, compounds 2b and 2d where furanyl ring of 2a was replaced by thiophenyl and naphthyl groups, respectively, were more effective in inhibiting viability of both CHL-1 and UACC903 cells as compared to parent compound 2a (Table 1). The phenyl or pyridyl group substitutions (compounds 2c and 2e, respectively) led to decrease in potency, 2e being the least effective. Interestingly, changing the chain from dienyl to trienyl with terminal phenyl ring (compound 2f) again restored to potency of the molecule. Notably, similar to as observed with lead compound 2a, all the new compounds 2b–f showed similar cell viability inhibition against both BRAF wild-type and mutant cells.
Table 1.
In vitro anti-proliferative activities of novel unsubstituted thio- and seleno-barbituric acid derivatives on melanoma CHL-1 & UACC 903 cancer cells (72 h treatment).a
| Compounds | Melanoma cells IC50 (μM)
|
|
|---|---|---|
| CHL-1 | UACC 903 | |
| 2a | 8.5 ± 2.7 | 8.0 ± 1.3 |
| 2b | 3.9 ± 1.3 | 4.0 ± 0.5 |
| 2c | 10.6 ± 3.6 | 9.1 ± 2.9 |
| 2d | 4.3 ± 1.6 | 4.2 ± 0.3 |
| 2e | 18.1 ± 2.3 | 18.4 ± 2.9 |
| 2f | 6.5 ± 2.1 | 6.5 ± 0.8 |
| 5a | 9.7 ± 1.9 | 2.3 ± 0.2 |
| 5b | 6.3 ± 1.2 | 7.8 ± 2.1 |
| 5c | 9.4 ± 2.3 | 13.6 ± 3.7 |
Values are the mean of triplicates of at least two independent experiments. Non-linear graphs using Prism software were used to determine Mean ± SD values.
Previous studies, both from our group[28–32] and others,[35] have shown isosteric replacement of sulfur by selenium to enhance the cytotoxicity of small molecules. Examples include increase in potency from isothiocyantes to isoselenocyantes,[30] isothioureas to isoselenoureas[28] among others, although it is not always the case.[36] Interestingly, in the present case, replacement of S in thiobarbituric acid ring of 2a by Se, led to lowering of potency of the resulting selenobarbitutic acid analog 5a (Table 1). Similarly, selenothiobarbituric acids 5b and 5c were less effective than their sulfur isosteres 2b and 2c; except that 5c showed a similar IC50 value as 2c. This indicates that presence of selenium does not necessarily increase the toxicity of a molecule but the actual effect would depend on overall molecular structure and position of the Se atom.
The N1-monosubstituted and N1,N3-disubstituted thiobarbituric acid analogs (8a–o) demonstrated variable efficacy depending on the combination of type of N-substitution(s) and C-5 moiety. Although no clear structure-activity relationship could be derived, it was observed that 8d derived by incorporating an ethyl group at N1-position of 2a resulted in loss of activity while N1,N3-diethyl analog 8j retained activity against BRAF wild-type CHL-1 cells while lost cytotoxicity against BRAF-mutated UACC903 cells. Extension of chain length at C5 from dienyl to tienyl linker (compound 8h) again restored the activity (IC50 ~7.5 μM). Similar to N1- and N3-unsubstituted compound 2b (Table 1), the compounds 8a, 8b, 8c, 8e, and 8m (Table 2) with thiophenyl terminal ring at C5 position were potent irrespective N1-monosubstitution as allyl, phenyl, methyl, ethyl or N1-allyl-N3-ethyl disubstitution. This suggested that 2-thiophenyl ring at terminal position of C5 substitution is essentially responsible for the activity which is retained irrespective of substitutions at N1 and N3. The combination of naphthyl group at C5 and N-ethyl (compound 8g) led to a substantial loss of activity (IC50 ~23 μM). N1-allyl-N3-ethyl disubstitution in combination with furanyl (5k) or phenyl (5l) also led to decrease in potency. Interestingly, compound 5j (N1-ethyl and phenyl at C5) and 5n and 5o (N1-allyl and furanyl and phenyl at C5, respectively) resulted in selective cytotoxicity towards CHL-1 cells.
Table 2.
In vitro anti-proliferative activities of N1-monosubstituted and N1,N3-disubstituted thiobarbituric acid derivatives on melanoma CHL-1 and UACC 903 cancer cells (72 h treatment).a
| Compounds | Melanoma cells IC50 (μM)
|
|
|---|---|---|
| CHL-1 | UACC 903 | |
| 8a | 7.5 ± 1.7 | 6.7 ± 1.2 |
| 8b | 3.8 ± 1.3 | 3.8 ± 0.4 |
| 8c | 4.7 ± 0.5 | 5.5 ± 1.9 |
| 8d | 13.9 ± 2.3 | 16.2 ± 3.0 |
| 8e | 4.7 ± 1.1 | 4.6 ± 0.9 |
| 8f | 6.2 ± 2.2 | 8.0 ± 1.8 |
| 8g | 23.4 ± 1.7 | 23.4 ± 3.6 |
| 8h | 7.5 ± 2.8 | 7.6 ± 3.9 |
| 8i | 8.6 ± 1.9 | 18.8 ± 2.7 |
| 8j | 8.8 ± 3.1 | 15.2 ± 2.3 |
| 8k | 18.9 ± 2.8 | 14.5 ± 1.7 |
| 8l | 20.4 ± 3.7 | 21.0 ± 3.6 |
| 8m | 6.1 ± 2.1 | 6.1 ± 1.4 |
| 8n | 8.0 ± 2.9 | 17.6 ± 2.9 |
| 8o | 8.1 ± 0.7 | 18.1 ± 3.2 |
| PLX-4032 | 13.7 ± 1.7 | 3.6 ± 1.1 |
Values are the mean of triplicates of at least two independent experiments. Non-linear graphs using Prism software were used to determine Mean ± SD values.
Overall, 2b, 2d, 2f, 5b (Table 1), 8a, 8b, 8c, 8e, 8f, 8h, and 8m (Table 2) emerged as most potent anti-proliferative compounds with IC50 lower than lead compound 2a and non-specifically targeted both UACC903 and CHL-1 cells. Among these compounds 2b and 8b were the most effective and were chosen for further studies. Importantly, unlike PLX4032, compound 2b and 8b were found equally potent in killing melanoma cells irrespective of mutation status (Table 2). As shown in Tables 1 and 2, compounds 2b and 8b inhibited growth of both CHL-1 and UACC 903 cell lines-. The fact that these compounds were equally effective in both BRAF-mutant and wild-type melanoma cells, suggest that the inhibitory activities of compounds 2b and 8b are independent of BRAF expression. Therefore, we extended our investigation to elucidate their mechanism of cell death and cell growth inhibition.
2.3.2. Compound 2b and 8b inhibited viability of a variety of melanoma cells
Based on the results of cell viability inhibition after 72 h treatment by the novel compounds against UACC903 and CHL-1 cell lines (Tables 1 and 2), the two best identified compounds 2b and 8b were selected for further screening against five human melanoma cell lines at 48 h time point. Both compounds 2b and 8b were essentially equally effective in inhibiting viability of various melanoma cells. The IC50 values for CHL-1 and UACC 903 cells at 48 h treatment were in the range of 6.5–8.1 μM (Table 3) as compared to 3.8–4.0 μM (Tables 1 and 2) for 72 h treatment. Both the compounds showed a similar potency against WM2664 cells. The compounds were slightly less effective against 1205Lu and A375M cell lines (IC50 between 10.6 and 14.8 μM, Table 3), compound 2b being slightly more potent. Figure 3 shows the cell viability curves for compounds 2b and 8b in all five melanoma cell lines at 48 h time point.
Table 3.
The IC50 of 2b and 8b in melanoma cells (48 h treatment).
| Cancer cells | 2b | 8b |
|---|---|---|
| CHL-1 | 7.2 ± 1.4 μM | 6.5 ± 2.8 μM |
| 1205Lu | 12.4 ± 2.3 μM | 13.5 ± 3.3 μM |
| A375M | 10.6 ± 3.5 μM | 14.8 ± 2.1 μM |
| UACC903 | 8.1 ± 1.9 μM | 7.6 ± 1.7 μM |
| WM2664 | 7.9 ± 2.0 μM | 9.2 ± 1.5 μM |
IC50 calculation was performed from Non-Linear regression plot using Prism software.
Figure 3.
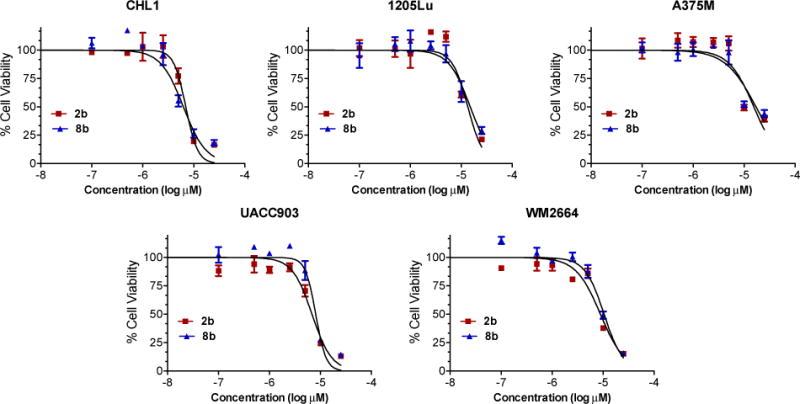
Effects of compounds 2b and 8b on various melanoma cell survival. The melanoma cancer cells (3,000 cells/well) were treated with increasing concentrations of 2b or 8b (0.5–25 μM) for 48 h. Cell viability was then analyzed by the MTT assay. Non-linear graphs are derived from triplicate values of two independent experiments and presented as Mean ± SD.
2.3.3. Compound 2b and 8b cause cell death by inducing apoptosis
Decrease of cell viability seen in MTT assay can be attributed to cell growth inhibition and/or cell death. We further confirmed our MTT assay observation by using another technique popularly known as Live/Dead assay. The Live/Dead assay determines intracellular esterase activity and plasma membrane integrity. UACC903 and CHL-1 cells were treated with 2b and 8b for 24 h. As shown in Figure 4, treatment of melanoma cells with 2b and 8b increase the number of dead cells, which reflects the cytotoxic potential of these analogs in melanoma cells. Compound 2b was more effective, in both CHL-1 and UACC903 cells, as compared to 8b. However, results from this assay suggest that 8b may potentially inhibit cell growth and induce cell death later, since it showed less death at 25 μM (25% in UACC903; 5% in CHL-1), while in MTT assay it was even more potent at reducing the cell viability for both the cell lines. Our data indicates 2b to be relatively more effective at inducing cell death than 8b.
Figure 4.

Effect of compounds 2b and 8b on melanoma cells cytotoxicity. Melanoma cells (UACC903) were treated with 25 and 50 μM concentrations of 2b and 8b for 24 h. Cells were subjected to Live/Dead analysis using microcopy as described under Experimental Section. Calcein-AM (Green) stained cells represent live cells, while ethidium bromide (Red) stained cells represent dead cells. (B) Percentage of dead cells quantified from A. Data represents mean value ±SD.
We further examined the effects of 2b and 8b on cell death using several other techniques such as Annexin V assay and measuring the activation of caspase 3/7. UACC903 cells were treated with given concentrations of 2b and 8b for 24 h. As shown in Figure 5A, vehicle-treated cells were mostly present in the bottom left quadrant. However, as the dose of 2b and 8b was escalated, cells shifted from live to early apoptotic and towards late apoptotic dead cells. Total apoptotic death obtained by Annexin V and Caspase 3/7 activity assays was similar to our Live/Dead assay. More than 70% cells were positive for both Annexin V, Caspase 3/7 activity and 7-AAD, indicating apoptotic dependent cell death. Further, necrotic death, cells only positive for 7-AAD and negative for Annexin V and Caspase 3/7 activity (cells present in the top left quadrant), was not detected. Overall, our results confirm that 2b and 8b inhibits the growth of melanoma cells by inducing apoptosis, indicated by presence of phosphatidylserine (PS) on outer membrane and positive for caspase 3/7 activity.
Figure 5.

Compounds 2b and 8b prompted apoptotic death in melanoma cells. (A), UACC 903 cells (50,000 cells/well) were treated with 2b and 8b for 24 h then cells were subjected to Annexin-V activity assay using a Muse cell analyzer. Four different populations of cells were collected: healthy cells (lower left quadrant, Annexin-V negative), early apoptotic cells (lower right corner, positive for Annexin-V and 7-ADD negative), late apoptotic/dead cells (upper right quadrant, both Annexin 7-ADD positive), and necrotic cells (only 7-ADD positive, upper left quadrant). (B) Similarly, activity of caspase 3/7 was measured using muse cell analyzer as described under method and material section.
2.3.4. Compound 2b and 8b inhibited the expression of survival proteins in melanoma
Escaping from cell death is one of the adaptations that enable cancer cells to stave off anticancer therapies. The key players in avoiding apoptosis are collectively known as survival proteins. Survival proteins mainly comprise the Bcl-2 and inhibitor of apoptosis (IAP) families.[37] The aberrant expression of these proteins is associated with a range of biological activities that promote cancer cell survival, proliferation, and resistance to therapy.[37] We determined the effect of compounds 2b and 8b on the expression of survival proteins. As shown in the Figure 6, compound 2b and 8b showed differential response on survival proteins. Treatment of these analogs induces phosphorylation of Bcl-2, which is required for its degradation, inhibits expression of anti-apoptotic proteins like Bcl-xL, Survivin and finally activates PARP cleavage. Thus, our results further support that these analogs have potential to be used as therapeutic agents.
Figure 6.

Effects of thiobarbituric acid analogs (2b, 8b) on expression of proteins associated with cell survival, and apotosis. Melanoma cells CHL-1 and UACC903 (2 × 106) were treated with the indicated concentrations of analogs for 24 h, after which Western blotting was done as described under method and material section.
2.3.5. Novel thiobarbituric acid analogs were also effective against cancers other than melanoma and were more selective towards cancer cells
To evaluate the effect of novel thiobarbituric acid analogs on cancers other than melanoma, a select effective compounds (2b, 2d, 8b, 8c, and 8e) identified from melanoma cells screen were tested against a panel of solid cancer cell lines, e.g., Panc-1 (Pancreatic cancer), MDA-MB-231 (ER/PR/HER2 negative breast cancer), MCF-7 (ER/PR/HER2 positive breast cancer), and HCT116 (colon cancer). Interestingly, compound 2d was most effective against all the cell lines tested with IC50s in the range of 3.6–7.9 μM (Table 4 and Figures 7A and B). All the compounds tested were effective against MDA-MB-231 cells with single digit IC50 values as compared to the lead compound 2a (IC50 ~16.5 μM). Compound 2b selectively inhibited viability of MDA-MB-231 and HCT-116 cells while compound 8b was selective towards Panc-1, MDA-MB-231, and HCT116 cells, being most effective against HCT116 cells (IC50 ~7.7 μM). Figure 3 shows the cell viability curves for compounds 2b and 8b in all five melanoma cell lines at 48 h time point. The cell viability curves for compounds 2b and 8b in HCT116, Panc-1, MDA-MB-231, and MCF-7 cell lines at 48 h time point are shown in Figures 7A and B. These data show that compounds 2b and 8b potently inhibited viability of most, although not all, of the tested cancer cell lines, suggesting that these compounds could be widely used against cancer cells.
Table 4.
In vitro anti-proliferative activities of selected potent thio-barbituric acid derivatives (from Table 1) on pancreatic, breast, and colon cancer cells (48 h treatment).a
| Compounds | Cancer cells (IC50, μM) | |||
|---|---|---|---|---|
|
| ||||
| Pancreatic cancer | Breast Cancer | Colon cancer | ||
|
|
||||
| Panc-1 | MDA-MB-231 | MCF-7 | HCT-116 | |
|
|
||||
| 2a | 24.4 ± 3.7 | 16.5 ± 1.8 | 27.9 ± 4.2 | 20.1 ± 3.5 |
| 2b | 19.1 ± 2.9 | 7.6 ± 2.1 | 16.2 ± 3.2 | 7.7 ± 0.6 |
| 2d | 7.9 ± 1.7 | 4.5 ± 0.9 | 4.4 ± 1.1 | 3.6 ± 1.4 |
| 8b | 8.3 ± 3.1 | 8.3 ± 2.7 | 16.5 ± 1.7 | 2.4 ± 0.9 |
| 8c | 21.0 ± 3.9 | 8.6 ± 1.3 | 23.5 ± 2.3 | 18.2 ± 1.8 |
| 8e | 20.5 ± 2.5 | 8.5 ± 2.9 | 24.0 ± 0.5 | 8.3 ± 3.2 |
Values are the mean of triplicates of at least two independent experiments. Non-linear graphs using Prism software were used to determine Mean ± SD values.
Figure 7.

Effects of compounds 2b and 8b on various solid cancer cells survival and normal cell lines. (A, B) The cancer cells (3,000 cells/wells) were treated with increasing concentrations of 2b and 8b (0.5–25 μM) for 48 h. Cell viability was then analysed by the MTT assay. Both the compounds reduced cell viability of different cancer cells including colon cancer (HCT116), pancreatic cancer (Panc-1), and breast cancer (MCF-7 and MDA-MB231) cells. Non-linear graphs are derived from triplicate values of two independent experiments and presented as Mean ±SD. (C, D) Cancer cell lines (UACC903, MCF-7 and MDA-MB-231) and normal breast cell line (MCF10A) were treated with 25 μM dose of 2b and 8b for 48 h. Cell viability was measure by MTT assay. Data presented as Mean ±SD.
To get an insight into the selectivity of these compounds towards cancer cells, we performed MTT cell viability assay at the highest concentration (25 μM) of both the compounds (2b and 8b) in normal cell line MCF10A, the human breast epithelial cell line. After 48 h treatment, the normal cell line showed ~60% cell viability while the cancer cell lines exhibited ~20%–30% cell viability (Figures 7C and D). Both the compounds were about two-fold more potent at killing breast cancer cells (MCF-7 and MDA-MB-231) and three-fold more potent towards melanoma cells (UACC903) as compared to the normal MCF10A cells. These data indicate that both the agents have some selectivity towards cancer cells than normal cells. However, in vitro studies are often not a true measure of toxicity/efficacy balance and therefore the future in vivo efficacy and toxicity studies would reveal the real therapeutic window of these compounds. Furthermore, the mechanism behind selective targeting of certain cancer cell types remains to be elucidated.
3. Conclusion
We synthesized a series of thio- and seleno-barbituric acid analogs with a variety of substitutions at N1, N3, and C5 positions. Many of these compounds inhibited viability of both BRAF wild-type (CHL-1) and mutant (UACC903) human melanoma cells. Based on the SAR study compounds 2b, 2d, and 8b were found to be the most effective analogs. Live/Dead assay showed compound 2b to be more effective than 8b in inducing cell death; causing nearly 65–70% cell death at 25 μM concentration in human melanoma cells. Both compounds 2b and 8b effectively induced apoptosis in both CHL-1 and UACC903 cell lines. The western blot analysis showed that 2b and 8b induced PARP cleavage and reduced the levels of anti-apoptotic proteins Bcl-2, Bcl-xL and Survivin in a dose-dependent manner. Further extensive studies are, however, required to reveal the mechanism of these novel thiobarbituric acid derivatives. Many of the novel thiobarbituric acid analogs were also effective in inhibiting viability of various other solid cancer cell lines, e.g., pancreatic, breast, and colon demonstrating that these compounds possess anti-proliferative ability against variety of cancer cells. Notably, compounds 2b and 8b inhibited growth of melanoma cells irrespective of mutational status of BRAF. Owing to the fact that the new compounds act independently of BRAF mechanism, they might potentially overcome the issue of resistance to currently clinically used BRAF-inhibitor PLX4032 that selectively targets BRAF-mutant tumors. Further pre-clinical studies including in vivo efficacy, toxicity, and mechanism of action, are, however, necessary to determine the future clinical potential of these compounds.
4. Experimental
4.1. Chemistry
4.1.1. General
Melting points were recorded on a Fischer-Johns melting point apparatus and are uncorrected. NMR spectra were recorded using a Bruker Avance 500 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) downfield from the internal standard. The signals are quoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublet) and dt (doublet of triplet). HRMS were determined at the Chemistry Instrumentation Center, State University of New York at Buffalo, NY. Thin-layer chromatography (TLC) was developed on aluminum-supported pre-coated silica gel plates (EM industries, Gibbstown, NJ). Column chromatography was conducted on silica gel (60–200 mesh). The substituted thioureas, e.g., N,N-diallyl- (6a), N,N-diethyl- (6b), N-allyl-N′-ethyl- (6c) and N-ethyl- (6d) thioureas were obtained from commercial sources.
4.1.2. General procedure for the synthesis of thiobarbiturates (2a–f)
To a stirred solution of 1,3-thio-barbituric acid (1) (1.0 mmol), and corresponding trans-α,β-unsaturated aldehydes (1.0 mmol) in absolute ethyl alcohol (5 mL) under nitrogen atmosphere, was added dry pyridine (0.2 mmol) and the resulting mixture was allowed to stir at reflux for 12 h. After completion of the reaction as indicated by TLC, the solvent was removed under reduced pressure. The residue thus obtained was washed with hot H2O (2 × 10 mL), cold EtOH (2 × 5 mL) and Et2O (2 × 10 mL) to afford crude thiobarbiturates (2a–f), which further recrystallized from 1,4-dioxane-water to afford pure compounds.
4.1.3. 5-(3-Furan-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (2a)
Yield 87%; reddish solid; mp 245 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 8.20 (dd, J = 12.5, 10.5 Hz, 1H), 8.03–8.00 (m, 2H), 7.62 (d, J = 12.5 Hz, 1H), 7.08 (d, J = 3.0 Hz, 1H), 6.75–6.73 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 178.84, 161.86, 161.42, 154.80, 152.35, 148.58, 139.40, 122.78, 119.65, 115.43, 114.44; HRMS calcd for C11H8N2O3S: 248.0250. Found 248.0256.
4.1.4. 5-(3-Thiophen-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (2b)
Yield 82%; brown solid; mp 270 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.29, 12.25 (2s, 2 × NH), 8.24–8.14 (m, 1H), 8.06–7.76 (m, 3H), 7.55 (s, 1H), 7.25 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 178.90, 161.84, 161.43, 154.92, 146.82, 141.48, 134.61, 133.35, 129.94, 124.21, 115.27; HRMS calcd for C11H8 N2O2S2: 264.0022. Found 264.0028.
4.1.5. 5-(3-Phenyl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (2c)
Yield 89%; yellow solid; mp 265 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 8.46 (dd, J = 13.0, 10.0 Hz, 1H), 8.06 (d, J = 9.5 Hz, 1H), 7.80 (d, J = 13.0 Hz, 1H), 7.68–7.72 (m, 2H), 7.48–7.53 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.04, 161.81, 161.31, 155.11, 154.18, 135.78, 131.96, 129.81, 129.27, 125.07, 116.44; HRMS calcd for C13H10N2O2S: 258.0457. Found 258.0461.
4.1.6. 5-(3-Naphthalen-1-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (2d)
Yield 78%; reddish brown solid; mp 247 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.36, 12.32 (2s, 2 × NH), 8.77 (d, J = 15.0, 1H), 8.61 (dd, J = 15.5, 12.0 Hz, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.33 (d, J = 12.0 Hz, 1H), 8.00–8.10 (m, 3H), 7.70–7.60 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.03, 161.91, 161.38, 155.50, 150.47, 134.00, 132.47, 132.41, 131.67, 129.32, 127.91, 127.09, 127.03, 126.41, 126.24, 123.80, 116.36; HRMS calcd for C17H12N2O2S: 308.0614. Found 308.0618.
4.1.7. 5-(3-Pyridin-3-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (2e)
Yield 72%; yellow solid; mp 274 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.37, 12.34 (2s, 2 × NH), 8.82 (d, J = 2.0 Hz, 1H), 8.62 (dd, J = 4.5, 1.5 Hz, 1H), 8.48 (dd, J = 16.0, 12.0 Hz, 1H), 8.14 (dt, J = 8.0, 2.0 Hz, 1H), 8.04 (d, J = 11.5 Hz, 1H), 7.80 (d, J = 15.5 Hz, 1H), 7.52 (dd, J = 8.0, 5.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.10, 161.70, 161.17, 154.04, 151.86, 150.52, 149.87, 135.25, 131.64, 126.63, 124.87, 117.40; HRMS calcd for C12H9N3O2S: 259.0410. Found 259.0407.
4.1.8. 5-(5-Phenyl-penta-2,4-dienylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (2f)
Yield 75%; reddish brown solid; mp 241 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.29, 12.23 (2s, 2 × NH), 8.02–7.86 (m, 2H), 7.68 (d, J = 7.0 Hz, 2H), 7.58 (dt, J = 11.0, 2.0 Hz, 1H), 7.42–7.38 (m, 4H), 7.68 (d, J = 15.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 178.85, 161.92, 161.30, 155.63, 154.99, 144.03, 136.39, 130.34, 129.48, 129.45, 129.39, 128.44, 115.28; HRMS calcd for C15H12N2O2S: 284.0614. Found 284.0613.
4.1.9. Synthesis of selenobarbituric acid analogs (5a–c)
The key intermediate 2-selenobarbarbituric acid (4) was prepared by refluxing selenourea (3) with diethylmalonate in ethanol in the presence of sodium methoxide by modification of a previously reported method.[38] Briefly, to a stirred solution of sodium ethylate [freshly prepared by dissolving sodium (0.373 g, 16.24 mmol) in absolute ethanol (10 mL)] under nitrogen atmosphere was added diethyl malonate (2.59 g, 16.24 mmol) followed by selenourea (3) (0.50 g, 4.06 mmol) and the resulting mixture was allowed to stir at reflux for 45 min. After completion of the reaction as indicated by TLC, the reaction mixture was chilled and the resulting white solid was filtered. The second crop of solid was obtained by adding Et2O (10 mL) to the filtrate. The combined solids were dissolved in ice-cold water (10 mL) and the solution was acidified with dilute HCl. The resulting solid was filtered, washed with cold water, Et2O and dried to afford 2-selenoxo-dihydro-pyrimidine-4,6-dione (4) (mp dec. >200 °C), which was pure enough to carry to the next step. The seleno-barbiturates (5a–c) were synthesized by treating 4 (1.0 mmol) with trans-α,β-unsaturated aldehydes (1.0 mmol) following the same reaction, workup, and purification sequence as described above for compound 2a–f.
4.1.10. 5-(3-Furan-2-yl-allylidene)-2-selenoxo-dihydro-pyrimidine-4,6-dione (5a)
Yield 77%; brown solid; mp 277 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.70, 12.66 (2s, 2 × NH), 8.18 (dd, J = 14.5, 12.0 Hz, 1H), 8.10–8.05 (m, 2H), 7.70 (d, J = 15.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 6.76 (dd, J = 4.0, 3.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.80, 161.33, 160.79, 155.28, 152.47, 148.79, 139.69, 123.03, 119.99, 115.71, 114.59; HRMS calcd for C11H8N2O3Se: 295.9695. Found 295.9701.
4.1.11. 2-Selenoxo-5-(3-thiophen-2-yl-allylidene)-dihydro-pyrimidine-4,6-dione (5b)
Yield 81%; brown solid; mp 275–277 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.71, 12.66 (2s, 2 × NH), 8.18–8.12 (m, 1H), 8.10–8.04 (m, 2H), 7.96 (d, J = 11.0 Hz, 1H), 7.62 (d, J = 3.5 Hz, 1H), 7.27–7.23 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.85, 161.30, 160.79, 155.42, 147.18, 141.59, 134.82, 133.63, 130.05, 124.46, 115.56; HRMS calcd for C11H8N2O2SSe: 311.9466. Found 311.9478.
4.1.12. 5-(3-Phenyl-allylidene)-2-selenoxo-dihydro-pyrimidine-4,6-dione (5c)
Yield 85%; reddish brown solid; mp 270–272 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.71, 12.74 (2s, 2 × NH), 8.42 (dd, J = 15.5, 12.0 Hz, 1H), 8.23 (d, J = 12.0 Hz, 1H), 7.86 (d, J = 15.5 Hz, 1H), 7.68–7.72 (m, 2H), 7.48–7.52 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 180.14, 161.27, 160.67, 155.58, 154.51, 135.85, 132.06, 129.86, 129.32, 125.33, 116.77; HRMS calcd for C13H10N2O2Se: 305.9902. Found 305.9899.
4.1.13. General procedure for the synthesis of thiobarbiturates (8a–o)
To a stirred solution of sodium ethylate [freshly prepared by dissolving sodium (4.0 mmol) in absolute ethyl alcohol (10 mL)] under nitrogen atmosphere was added diethyl malonate (4.0 mmol) followed by substituted thioureas 6a–d (1.0 mmol) and the resulting mixture was allowed to stir at reflux for 48 h. After completion of the reaction as indicated by TLC, the reaction mixture was diluted with H2O (10 mL). Then most of the ethyl alcohol was removed under reduced pressure. The residue was poured into cold water (10 mL), chilled and filtered. The aqueous layer was washed with Et2O (2 × 10 mL) to remove any unreacted diethyl malonate. The aqueous layer was acidified with dilute HCl and resulting solid was filtered off, washed with cold water and Et2O, and dried to afford thiobarbituric acids (8a–o). Compounds 8a–h were purified by crystallization from ethyl alcohol. The N1,N3-disubstituted compounds 8i–o were purified by flash column chromatography using silica gel (100–200 mesh) eluting with n-hexane/EtOAc (98:2) to afford the corresponding pure thiobarbiturates.
4.1.14. 1-Allyl-5-(3-thiophen-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8a)
Yield 82% (E & Z isomers); orange solid; mp 221 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.50, 12.45 (2s, 2 × NH), 8.24–8.14 (m, 1H), 8.10–8.00 (m, 2H), 7.94 (t, J = 4.0 Hz, 1H), 7.60 (t, J = 4.0 Hz, 1H), 7.27–7.23 (m, 1H), 5.99–5.79 (m, 1H), 5.20–5.10 (m, 2H), 4.91–4.87 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.36, 179.26, 161.44, 160.77, 160.28, 159.95, 156.51, 155.94, 147.72, 147.31, 141.47, 134.96, 134.88, 133.75, 133.63, 132.52, 132.43, 130.02, 124.39, 124.10, 117.33, 115.28, 115.11, 48.23, 47.90; HRMS calcd for C14H12N2O2S: 304.0335. Found 304.0345.
4.1.15. 1-Phenyl-5-(3-thiophen-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8b)
Yield 90% (E & Z isomers); orange solid; mp 287 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.62, 12.59 (2s, 2 × NH), 8.26 (dd, J = 12.5, 10.5 Hz, 1H), 8.13–8.08 (m, 2H), 8.06–8.00 (m, 3H), 7.92 (dd, J = 14.5, 4.0 Hz, 2H), 7.60–7.56 (m, 2H), 7.47–7.42 (m, 4H), 7.40–7.36 (m, 2H), 7.28–7.20 (m, 6H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 180.13, 180.03, 162.09, 161.41, 160.78, 160.45, 155.89, 155.47, 147.34, 147.11, 141.49, 139.68, 139.47, 134.81, 134.79, 133.62, 133.52, 129.98, 129.69, 129.59, 129.22, 128.61, 124.26, 124.11, 115.72, 115.55; HRMS calcd for C17H12N2O2S2: 340.0335. Found 340.03351.
4.1.16. 1-Methyl-5-(3-thiophen-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8c)
Yield 89% (E & Z isomers); orange solid; mp 245°C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.45, 12.40 (2s, 2 × NH), 8.24–8.18 (m, 1H), 8.08–8.01 (m, 2H), 7.93 (t, J = 4.0 Hz, 1H), 7.59 (t, J = 4.0 Hz, 1H), 7.26–7.23 (m, 1H), 3.50 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.84, 179.75, 162.00, 161.33, 160.33, 160.01, 156.34, 155.63, 147.44, 147.15, 141.50, 141.46, 134.84, 134.79, 133.60, 133.54, 130.00, 129.97, 124.39, 124.13, 115.36, 115.25, 33.91, 33.50; HRMS calcd for C12H10N2O2S2: 278.0178. Found 278.0184.
4.1.17. 1-Ethyl-5-(3-furan-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8d)
Yield 85% (E & Z isomers); reddish brown solid; mp 245 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.38, 12.35 (2s, 2 × NH), 8.25 (dd, J = 15.0, 12.5 Hz, 1H), 8.10–8.02 (m, 2H), 7.66 (dd, J = 15.0, 9.5 Hz, 1H), 7.12 (dd, J = 8.5, 3.5 Hz, 1H), 6.80–6.70 (m, 1H), 4.34–4.28 (m, 2H), 1.18 (q, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.13, 179.04, 161.43, 160.74, 160.29, 159.93, 156.16, 155.50, 152.41, 152.35, 148.76, 148.71, 139.97, 139.65, 122.98, 122.79, 119.92, 119.89, 115.63, 115.50, 114.53, 114.50, 41.63, 41.27, 12.75, 12.72; HRMS calcd for C13H12N2O3S: 276.0563. Found 276.0564.
4.1.18. 1-Ethyl-5-(3-thiophen-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8e)
Yield 87% (E & Z isomers); reddish solid; mp 272 °C (dec.); 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.41, 12.37 (2s, 2 × NH), 8.24–8.18 (m, 1H), 8.10–8.00 (m, 2H), 7.95–7.90 (m, 1H), 7.62–7.58 (m, 1H), 7.30–7.20 (m, 1H), 4.34–4.28 (m, 2H), 1.18 (q, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.15, 179.09, 161.41, 160.79, 160.26, 159.92, 156.31, 155.70, 147.52, 147.13, 141.50, 141.47, 134.86, 134.79, 133.64, 133.53, 129.99, 129.96, 124.40, 124.16, 115.45, 115.30, 41.64, 41.28, 12.77, 12.73; HRMS calcd for C13H12N2O2S2: 292.0335. Found 292.0331.
4.1.19. 1-Ethyl-5-(3-phenyl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8f)
Yield 92% (E & Z isomers); yellow solid; mp 256 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.45, 12.41 (2s, 2 × NH), 8.50–8.45 (m, 1H), 8.10 (dd, J = 11.5, 4.0 Hz, 1H), 7.82 (dd, J = 15.5, 10.5 Hz, 1H), 7.72–7.68 (m, 2H), 7.52–7.48 (m, 3H), 4.32–4.28 (m, 2H), 1.18 (q, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.32, 179.24, 160.71, 160.26, 159.84, 156.45, 155.86, 154.89, 154.46, 135.77, 132.10, 132.06, 129.83, 129.40, 129.34, 125.27, 125.05, 116.71, 116.54, 41.69, 41.34, 12.74, 12.72; HRMS calcd for C15H14N2O2S: 286.0771. Found 286.0774.
4.1.20. 1-Ethyl-5-(3-naphthalen-1-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8g)
Yield 81% (E & Z isomers); orange solid; mp 235 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 8.82 (t, J = 15.0 Hz, 1H), 8.64 (dd, J = 15.0, 10.0 Hz, 1H), 8.52 (dd, J = 8.0, 2.5 Hz, 1H), 8.38 (d, J = 12.0 Hz, 1H), 8.14–8.02 (m, 3H), 7.70–7.60 (m, 3H), 4.40–4.30 (m, 2H), 1.20 (q, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.29, 179.21, 161.49, 160.76, 160.35, 159.91, 156.83, 156.26, 151.09, 150.69, 134.00, 132.59, 132.52, 132.42, 131.71, 129.34, 127.93, 127.26, 127.04, 126.43, 126.37, 126.31, 123.79, 116.61, 116.41, 41.70, 41.35, 12.75; HRMS calcd for C19H16N2O2S: 336.0927. Found 336.0932.
4.1.21. 1-Ethyl-5-(5-furan-2-yl-penta-2,4-dienylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8h)
Yield 82% (E and Z isomers); reddish solid; mp 221 °C; 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 12.38, 12.33 (2s, 2 × NH), 8.02–7.96 (m, 2H), 7.90–7.88 (m, 1H), 7.62–7.55 (m, 1H), 7.12–7.00 (m, 2H), 6.96–6.92 (m, 1H), 6.70–6.68 (m, 1H), 4.35–4.25 (m, 2H), 1.20–1.10 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ [ppm]: 179.00, 160.59, 160.37, 159.79, 156.15, 156.02, 155.60, 152.57, 146.63, 146.58, 130.75, 130.67, 129.44, 129.22, 126.88, 126.85, 115.71, 115.55, 114.80, 113.78, 41.62, 41.23, 12.76, 12.74; HRMS calcd for C15H14N2O3S: 302.0720. Found 302.0725.
4.1.22. 1,3-Diethyl-5-(3-furan-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8i)
Yield 78%; brown solid; mp 156°C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.45 (dd, J = 15.5, 12.5 Hz, 1H), 8.16 (d, J = 12.5 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 7.22 (d, J = 15.0 Hz, 1H), 6.90 (d, J = 3.5 Hz, 1H), 6.60 (dd, J = 3.5, 2.0 Hz, 1H), 4.60–4.55 (m, 4H), 1.35–1.25 (m, 6H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 178.93, 160.61, 159.66, 157.57, 152.37, 147.08, 139.16, 123.96, 118.32, 114.79, 113.58, 43.72, 43.21, 12.45, 12.41; HRMS calcd for C15H16N2O3S: 304.0876. Found 304.0874.
4.1.23. 1,3-Diethyl-5-(3-phenyl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8j)
Yield 89%; reddish brown solid; mp 164 °C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.62 (dd, J = 15.5, 12.0 Hz, 1H), 8.22 (d, J = 12.0 Hz, 1H), 7.72–7.66 (m, 2H), 7.46–7.40 (m, 4H), 4.60–4.55 (m, 4H), 1.35–1.25 (m, 6H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 178.97, 160.52, 159.66, 158.48, 154.92, 135.37, 131.74, 129.30, 129.16, 125.68, 115.37, 43.77, 43.25, 12.45, 12.40; HRMS calcd for C17H18N2O2S: 314.1084. Found 314.1085.
4.1.24. 1-Allyl-3-ethyl-5-(3-furan-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8k)
Yield 92% (E and Z isomers); yellow solid; mp 143°C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.52–8.44 (m, 1H), 8.20 (dd, J = 13.0, 3.5 Hz, 1H), 7.68 (t, J = 2.5 Hz, 1H), 7.24 (dd, J = 15.0, 3.5 Hz, 1H), 6.92 (d, J = 3.5 Hz, 1H), 6.10–6.00 (m, 1H), 6.02–5.94 (m, 1H), 5.36–5.30 (m, 1H), 5.28–5.25 (m, 1H), 5.18–5.14 (m, 2H), 4.62–4.56 (m, 2H), 1.36–1.32 (m, 3H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 179.05, 179.00, 160.69, 160.57, 159.67, 159.61, 157.92, 157.89, 152.37, 147.16, 139.37, 139.36, 131.62, 131.49, 123.99, 123.93, 118.48, 118.25, 118.13, 114.59, 114.57, 113.61, 50.07, 49.56, 43.88, 43.37, 12.42, 12.37; HRMS calcd for C16H16N2O3S: 316.0876. Found 316.0875.
4.1.25. 1-Allyl-3-ethyl-5-(3-phenyl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8l)
Yield 82% (E and Z isomers); reddish solid; mp 146°C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.70–8.62 (m, 1H), 8.30–8.26 (m, 1H), 7.74–7.72 (m, 2H), 7.54–7.44 (m, 4H), 6.12–5.96 (m, 1H), 5.36–5.26 (m, 2H), 5.20–5.16 (m, 2H), 4.62–4.56 (m, 2H), 1.38–1.32 (m, 3H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 179.07, 179.02, 160.60, 160.48, 159.67, 159.61, 158.88, 158.84, 155.24, 155.22, 135.32, 131.82, 131.53, 131.41, 129.37, 129.35, 129.18, 129.17, 125.70, 125.64, 118.35, 118.16, 115.16, 115.13, 50.11, 49.61, 43.94, 43.42, 12.42, 12.37; HRMS calcd for C18H18N2O2S: 326.1084. Found 326.1086.
4.1.26. 1-Allyl-3-ethyl-5-(3-thiophen-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8m)
Yield 90% (E and Z isomers); yellow solid; mp 146°C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.46–8.38 (m, 1H), 8.20 (dd, J = 12.5, 4.0 Hz, 1H), 7.68–7.58 (m, 2H), 7.54–7.38 (m, 1H), 7.18–7.12 (m, 1H), 6.02–5.92 (m, 1H), 5.34–5.24 (m, 2H), 5.18–5.14 (m, 2H), 4.60–4.54 (m, 2H), 1.36–1.30 (m, 3H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 179.03, 178.99, 160.66, 160.54, 159.73, 159.66, 158.36, 158.31, 146.98, 144.26, 141.35, 141.33, 132.36, 132.34, 131.61, 131.48, 128.96, 128.51, 125.08, 125.02, 118.25, 118.00, 50.08, 49.53, 43.90, 43.34, 12.44, 12.37; HRMS calcd for C16H16N2O2S2: 332.0648. Found 332.0652.
4.1.27. 1,3-Diallyl-5-(3-furan-2-yl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8n)
Yield 89%; reddish brown solid; mp 135°C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.45 (dd, J = 15.0, 12.5 Hz, 1H), 8.18 (d, J = 12.5 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 7.22 (d, J = 15.0 Hz, 1H), 6.92 (d, J = 3.5 Hz, 1H), 6.60 (dd, J = 3.5, 1.5 Hz, 1H), 6.00–5.90 (m, 2H), 5.34–5.24 (m, 4H), 5.18–5.14 (m, 4H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 179.10, 160.66, 159.63, 158.28, 152.35, 147.28, 139.63, 131.50, 131.38, 123.95, 118.70, 118.29, 118.17, 114.33, 113.67, 50.22, 49.70; HRMS calcd for C15H16N2O3S: 328.0876. Found 328.0878.
4.1.28. 1,3-Diallyl-5-(3-phenyl-allylidene)-2-thioxo-dihydro-pyrimidine-4,6-dione (8o)
Yield 87%; yellow solid; mp 154 °C; 1H NMR (500 MHz, CDCl3) δ [ppm]: 8.55 (dd, J = 15.5, 12.0 Hz, 1H), 8.28 (d, J = 12.0 Hz, 1H), 7.72 (dd, J = 7.0, 1.0 Hz, 2H), 7.52–7.40 (m, 4H), 6.00–5.90 (m, 2H), 5.35–5.25 (m, 4H), 5.20–5.15 (m, 4H); 13C NMR (125 MHz, CDCl3) δ [ppm]: 179.13, 160.57, 159.62, 159.23, 155.52, 135.29, 131.90, 131.42, 131.30, 129.42, 129.18, 125.67, 118.38, 118.21, 114.94, 50.25, 49.74; HRMS calcd for C19H18N2O2S: 338.1084. Found 338.1088.
4.2. Biological evaluations
4.2.1. Reagents and antibodies
Antibodies for Western blot purposes were purchased from cell signaling [anti-PARP (9542s), p-Bcl-2 (2827s), survivin (2802s) and Bcl-xL (2764s)] and Sigma-Aldrich [β-actin (A5316)]. MTT (Thiazolyl Blue Tetrazolium Bromide) was obtained from Sigma-Aldrich (M5655−500MG).
4.2.2. Cell culture conditions
WM2664, CHl-1, A375M, MDA-MB-231, PANC-1, MCF-7, 1205Lu cell lines were maintained in DMEM medium, while HCT116 cells were maintained in McCoy’s 5A medium. Both the mediums were supplemented with 10% fetal bovine serum (FBS) and 100 units/mL of penicillin and streptomycin at 37 °C. MCF10A cells were maintained in MEGM™ Mammary Epithelial Cell Growth Medium (Lonza, CC-3151 & CC-4136) at 37 °C.
4.2.3. Cell viability assay
The effects of thio- and seleno-barbituric acid derivatives (2, 5, and 8) on the cell viability were determined by the MTT uptake method as described before.[29] Briefly, 3,000 cells were incubated with various concentrations of thiobarbiturate derivatives in triplicate in a 96-well plate for 48 h at 37 °C. MTT solution (5 mg/mL in PBS) was added to each well and incubated for 3 h at 37 °C. After 3 h, DMSO was added to dissolve the resultant formazan crystal and the optical density was measured at 570 nm using a 96-well multiscanner (Dynex Technologies, MRX Revelation; Chantilly, VA, USA). Backgrounds were subtracted at 630 nm. IC50 value for each compound was determined by at least three independent experiments and represented with a standard error (Table 1).
4.2.4. Live/dead assay
To measure apoptosis, we used the Live/Dead assay as described before.[29] Live/Dead assay determines intracellular esterase activity and plasma membrane integrity. Calcein-AM, a nonfluorescent polyanionic dye, is retained by live cells, in which it produces intense green fluorescence through enzymatic (esterase) conversion. In addition, the ethidium homodimer enters cells with damaged membranes and binds to nucleic acids, thereby producing a bright red fluorescence in dead cells. Briefly, 2 × 105 cells were incubated with mentioned amounts of 2b and 8b for 24 h at 37 °C. Cells were stained with the Live/Dead reagent (5 μM ethidium homodimer and 5 μM calcein-AM) and incubated at 37 °C for 30 min following manufacture’s protocol as described earlier. Cells were analyzed under a fluorescence microscope (Labophot-2; Nikon) as described before.6
4.2.5. Western blot analysis
To determine the effect of 2b and 8b on apoptotic proteins, whole cell lysates were prepared as previously described.[29] The whole cell lysates were resolved by SDS-PAGE. After electrophoresis, the proteins were electro-transferred to PVDF membranes, blotted with the relevant antibodies, and detected by an enhanced chemiluminescence reagent.
4.2.6. Annexin V assay
To determine the ability of 2b and 8b on apoptosis, UACC903 and CHL-1 (5 × 105) cells were treated with 25 and 50 μM with 2b and 8b for 24h. Muse Annexin V & Dead Cell kit (Millipore, Catlog No, MCH100105) was used to detect live, early apoptotic, late apoptotic and necrotic cells in both floating and adherent cells as previously described,[29] using Muse cell analyzer (EMD Millipore, Billerica, MA, USA). Muse Annexin V kit contains 7-AAD, which enters any cells that have lost their plasma membrane integrity, and hence marks dead cells. The kit also contains Annexin V dye, which stains phosphatidylserine (PS) on the outer surface of apoptotic cells. Data acquired from the equipment was analyzed using Muse 1.4 software.
4.2.7. Caspase 3/7 activity assay
To establish whether 2b and 8b induce caspase 3/7 activity, UACC903 and CHL-1 (5 × 105) cells were treated with 25 and 50 μM with 2b and 8b for 24 h. Caspase 3/7 activity was monitored using Muse Caspase-3/7 Assay kit with Muse cell analyzer (EMD Millipore, Billercia, MA, USA) as previously described.[29] Muse Caspase 3/7 kit contains a cell permeable reagent, NucView, that can detect active caspase 3/7. This reagent has a DNA binding dye that is connected to a DEVD peptide substrate, which inhibits the DNA dye from binding to the DNA. However, in presence of active caspase 3/7, DEVD peptide gets cleaved and the DNA binding dye is released and fluorescence when bound with DNA. The kit also contains 7-AAD, a dead cell marker. Muse 1.4 software was used to analyze the data.
Supplementary Material
Figure 2.

Sites of structural modification of the novel thio-/seleno-barbituric acid derivatives.
Highlights.
A series of novel thio- and seleno-barbituric acid derivatives were synthesized
The cytotoxicity was evaluated against wild-type and BRAF-mutated melanoma cells
2b and 8b were identified as the most potent compounds
2b and 8b inhibited viability of melanoma cells and induced apoptotic cell death
2b and 8b were effective towards melanoma cells with both wild-type and mutant BRAF
2b, 2d, and 8b also effectively inhibited viability of other tumor cell lines
Acknowledgments
The authors thank the Department of Pharmacology, Penn State College of Medicine, and Penn State Cancer Institute for financial support. This study was also supported partially by NIH’s NCI Grant R21 CA167406-02 & Elsa U. Pardee Foundation Grant (SYL). The authors thank Dr. Jyh-Ming Lin, Solution Phase NMR Facility at Core Research Facilities of the Penn State College of Medicine for recording NMR spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary material
Copies of the 1H NMR and 13C NMR spectra for new compounds 2a–f, 5a–c, and 8a–o.
References
- 1.Bernard WS, Christopher P. World cancer report 2014. World Health Organization; 2014. [Google Scholar]
- 2.A.C. Society. American Cancer Society. Atlanta, GA, USA: 2016. Cancer Facts & Figures 2016. [Google Scholar]
- 3.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eroglu Z, Ribas A. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Therapeutic advances in medical oncology. 2016;8:48–56. doi: 10.1177/1758834015616934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nature reviews Drug discovery. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 6.Archana, Srivastava VK, Kumar A. Synthesis of some newer derivatives of substituted quinazolinonyl-2-oxo/thiobarbituric acid as potent anticonvulsant agents. Bioorg Med Chem. 2004;12:1257–1264. doi: 10.1016/j.bmc.2003.08.035. [DOI] [PubMed] [Google Scholar]
- 7.Badawey E, El-Ashmawey I. Anti-inflammatory, analgesic and antipyretic activity of some new 1-(pyrimidin-2-yl)-3-pyrazoline-5-ones and 2-(pyrimidin-2-yl)-1, 2, 4, 5, 6, 7-hexahydro-3H-indazol-3-ones. European journal of medicinal chemistry. 1998;33:349–361. [Google Scholar]
- 8.Radwan MA, Ragab EA, Sabry NM, El-Shenawy SM. Synthesis and biological evaluation of new 3-substituted indole derivatives as potential anti-inflammatory and analgesic agents. Bioorganic & medicinal chemistry. 2007;15:3832–3841. doi: 10.1016/j.bmc.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 9.Borse BN, Shukla SR, Sonawane YA, Shankerling GS. Synthesis of some novel pyrimidinedione and pyrimidinetrione derivatives by a greener method: Study of their antimicrobial activity and photophysical properties. Synthetic Communications. 2013;43:865–876. [Google Scholar]
- 10.Al-Harbi AS, Abdel-Rahman RM, Asiri AM. Synthesis of some new fluorine substituted thiobarbituric acid derivatives as anti HIV1 and cyclin-dependent kinase 2 (CDK2) for cell tumor division: Part I. European Journal of Chemistry. 2015;6:63–70. [Google Scholar]
- 11.Srivastava VK, Kumar A. Synthesis and Anticonvulsant Activity of 1-Acetyl-5-arylidenyl-3-(2′-oxo/thio-barbiturinyl)-2-pyrazolines. Arzneimittelforschung. 2002;52:787–791. [PubMed] [Google Scholar]
- 12.Kumar P, Nath C, Agarwal J, Bhargava K, Shanker K. Substituted thiobarbituric acids as anti-parkinsonian agents. Chemischer Informationsdienst. 1984;15 [Google Scholar]
- 13.Guerin DJ, Mazeas D, Musale MS, Naguib FN, Al Safarjalani ON, el Kouni MH, Panzica RP. Uridine phosphorylase inhibitors: chemical modification of benzyloxybenzylbarbituric acid and its effects on urdpase inhibition. Bioorganic & medicinal chemistry letters. 1999;9:1477–1480. doi: 10.1016/s0960-894x(99)00238-3. [DOI] [PubMed] [Google Scholar]
- 14.Dhorajiya BD, Ibrahim AS, Badria FA, Dholakiya BZ. Design and synthesis of novel nucleobase-based barbiturate derivatives as potential anticancer agents. Medicinal Chemistry Research. 2014;23:839–847. [Google Scholar]
- 15.Bihani M, Bora PP, Verma AK, Baruah R, Boruah HP, Bez G. PPL catalyzed four-component PASE synthesis of 5-monosubstituted barbiturates: Structure and pharmacological properties. Bioorganic & medicinal chemistry letters. 2015;25:5732–5736. doi: 10.1016/j.bmcl.2015.10.088. [DOI] [PubMed] [Google Scholar]
- 16.Kong X, Qin J, Li Z, Vultur A, Tong L, Feng E, Rajan G, Liu S, Lu J, Liang Z. Development of a novel class of B-Raf v600e-selective inhibitors through virtual screening and hierarchical hit optimization. Organic & biomolecular chemistry. 2012;10:7402–7417. doi: 10.1039/c2ob26081f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penthala NR, Ponugoti PR, Kasam V, Crooks PA. 5-((1-Aroyl-1H-indol-3-yl) methylene)-2-thioxodihydropyrimidine-4, 6 (1H, 5H)-diones as potential anticancer agents with anti-inflammatory properties. Bioorganic & medicinal chemistry letters. 2013;23:1442–1446. doi: 10.1016/j.bmcl.2012.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake FH, Hofmann GA, Mong SM, Bartu JOL, Hertzberg RP, Johnson RK, Mattern MR, Mirabelli CK. In vitro and intracellular inhibition of topoisomerase II by the antitumor agent merbarone. Cancer research. 1989;49:2578–2583. [PubMed] [Google Scholar]
- 19.Tsujino I, Miyagi K, Sampson RW, Sieber F. Potentiation of the Antitumor Effect of Merocyanine 540‐mediated Photodynamic Therapy by Amifostine and Amphotericin B. Photochemistry and photobiology. 2006;82:458–465. doi: 10.1562/2005-09-02-RA-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daziano JP, Günther WH, Krieg M, Tsujino I, Miyagi K, Anderson GS, Sampson RW, Ostrowski MD, Muir SA, Bula RJ. Photochemically generated elemental selenium forms conjugates with serum proteins that are preferentially cytotoxic to leukemia and selected solid tumor cells. Photochemistry and photobiology. 2012;88:448–460. doi: 10.1111/j.1751-1097.2012.01078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SY, Slagle-Webb B, Rizk E, Patel A, Miller PA, Sung SS, Connor JR. Characterization of a novel anti-cancer compound for astrocytomas. PloS one. 2014;9:e108166. doi: 10.1371/journal.pone.0108166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug discovery today Technologies. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 24.Desai D, Kaushal N, Gandhi UH, Arner RJ, D’Souza C, Chen G, Vunta H, El-Bayoumy K, Amin S, Prabhu KS. Synthesis and evaluation of the anti-inflammatory properties of selenium-derivatives of celecoxib. Chemico-biological interactions. 2010;188:446–456. doi: 10.1016/j.cbi.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hossain SU, Sengupta S, Bhattacharya S. Synthesis and evaluation of antioxidative properties of a series of organoselenium compounds. Bioorganic & medicinal chemistry. 2005;13:5750–5758. doi: 10.1016/j.bmc.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Hwang JT, Kim YM, Surh YJ, Baik HW, Lee SK, Ha J, Park OJ. Selenium regulates cyclooxygenase-2 and extracellular signal-regulated kinase signaling pathways by activating AMP-activated protein kinase in colon cancer cells. Cancer research. 2006;66:10057–10063. doi: 10.1158/0008-5472.CAN-06-1814. [DOI] [PubMed] [Google Scholar]
- 27.Sanmartin C, Plano D, Palop JA. Selenium compounds and apoptotic modulation: a new perspective in cancer therapy. Mini reviews in medicinal chemistry. 2008;8:1020–1031. doi: 10.2174/138955708785740625. [DOI] [PubMed] [Google Scholar]
- 28.Madhunapantula SV, Desai D, Sharma A, Huh SJ, Amin S, Robertson GP. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Mol Cancer Ther. 2008;7:1297–1308. doi: 10.1158/1535-7163.MCT-07-2267. [DOI] [PubMed] [Google Scholar]
- 29.Plano D, Karelia DN, Pandey MK, Spallholz JE, Amin S, Sharma AK. Design, Synthesis, and Biological Evaluation of Novel Selenium (Se-NSAID) Molecules as Anticancer Agents. Journal of medicinal chemistry. 2016;59:1946–1959. doi: 10.1021/acs.jmedchem.5b01503. [DOI] [PubMed] [Google Scholar]
- 30.Crampsie MA, Pandey MK, Desai D, Spallholz J, Amin S, Sharma AK. Phenylalkyl isoselenocyanates vs phenylalkyl isothiocyanates: thiol reactivity and its implications. Chemico-biological interactions. 2012;200:28–37. doi: 10.1016/j.cbi.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen N, Sharma A, Nguyen N, Sharma AK, Desai D, Huh SJ, Amin S, Meyers C, Robertson GP. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer prevention research. 2011;4:248–258. doi: 10.1158/1940-6207.CAPR-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma AK, Sharma A, Desai D, Madhunapantula SV, Huh SJ, Robertson GP, Amin S. Synthesis and anticancer activity comparison of phenylalkyl isoselenocyanates with corresponding naturally occurring and synthetic isothiocyanates. Journal of medicinal chemistry. 2008;51:7820–7826. doi: 10.1021/jm800993r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanchard-Desce M, Alain V, Bedworth P, Marder S, Fort A, Runser C, Barzoukas M, Lebus S, Wortmann R. Large quadratic hyperpolarizabilities with donor–acceptor polyenes exhibiting optimum bond length alternation: correlation between structure and hyperpolarizability. Chemistry–A European Journal. 1997;3:1091–1104. [Google Scholar]
- 34.Brooker L, Keyes G, Sprague R, VanDyke R, VanLare E, VanZandt G, White FL. J Am Chem, SOC. 1951;73 [Google Scholar]
- 35.Ibanez E, Plano D, Font M, Calvo A, Prior C, Palop JA, Sanmartin C. Synthesis and antiproliferative activity of novel symmetrical alkylthio- and alkylseleno-imidocarbamates. Eur J Med Chem. 2011;46:265–274. doi: 10.1016/j.ejmech.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Sharma AK, Sk UH, He P, Peters JM, Amin S. Synthesis of isosteric selenium analog of the PPARbeta/delta agonist GW501516 and comparison of biological activity. Bioorg Med Chem Lett. 2010;20:4050–4052. doi: 10.1016/j.bmcl.2010.05.094. [DOI] [PubMed] [Google Scholar]
- 37.Pandey MK, Prasad S, Tyagi AK, Deb L, Huang J, Karelia DN, Amin SG, Aggarwal BB. Targeting Cell Survival Proteins for Cancer Cell Death. Pharmaceuticals. 2016;9 doi: 10.3390/ph9010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mautner HG, Clayton EM. 2-Selenobarbiturates. Studies of some analogous oxygen, sulfur and selenium compounds. J Am Chem Soc. 1959;81:6270–6273. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.