

Abstract
Alkenyl N-pivaloylhydroxamates undergo an Ir(III)-catalyzed diamination of the alkene with simple exogenous secondary amines under extraordinarily mild reaction conditions. The regioselectivity of the diamination is controlled by the solvent and the electronics of the cyclopentadienyl (Cpx) ligand on Ir. Based on a set of mechanistic experiments, we propose that the relative rates of Ir(V)-nitrenoid formation versus attack on the amido-Ir-coordinated alkene by the exogenous amine determine the outcome of the reaction.
Graphical abstract

Nitrogen-containing molecules are heavily represented among FDA-approved drugs,1 and thus synthetic methods that can quickly and efficiently incorporate nitrogen are greatly desired. Among these methods, catalytic alkene diamination has proven to be an attractive yet challenging transformation.2 Intermolecular diamination reactions have been developed by Shi and others, although these frequently suffer from limited amine scope or lack of distinction between the two amines.3 One strategy to address this shortcoming is to perform diamination reactions wherein one of the amines is tethered. Michael has published reports of Pd(II)-catalyzed alkene diamination using NFSI as the exogenous nitrogen source (eq 1),4 which was later rendered intermolecular in a report by Muñiz.3e More recently, Cu(II) complexes have proven to be competent diamination catalysts that expand the scope of the exogenous amine. Wang has described a Cu(II)-catalyzed diamination of N-alkoxy alkenyl amides with O-benzoylhydroxylamines5 (eq 2) and an azidoiodinane6. Yu7 (eq 3) and Chemler8 (eq 4) have also recently reported similar Cu(II)-catalyzed alkene diamination reactions using tethered alkenyl N-oxy amines. In these examples with Cu(II), the reactivity is limited to 5-exo and the activating N–O bond is retained in the product. Blakey has shown that hypervalent iodine can effect 6-endo cyclizations of alkenyl sulfonamides with trifluoromethanesulfonimide.9 Herein we describe two mild Ir(III)-catalyzed alkene diamination methods utilizing unactivated functionally diverse secondary amines and a traceless N-pivaloylhydroxamate oxidant (eq 5) that deliver complementary γ- and δ-lactams. We demonstrate that the two methods proceed by complementary reaction pathways largely controlled by the solvent and exogenous additive.
For our initial investigations, we tethered the alkene to an amine with a three-carbon linker that would result in a favorable five- or six-membered ring formation. We had previously used this strategy to enable a challenging Rh(III) allylic C–H activation.10 Moreover, we were inspired to use an alkene-tethered N-pivaloylhydroxamate (1) for this reaction because it has proven to be a versatile and robust oxidizing directing group for analogous Rh(III)-catalyzed alkene carboamination reactions.11 We felt that in this application they would enable installation of two distinct nitrogen units across an alkene due to their orthogonal reactivity versus simple amines. A preliminary screen of catalysts and exogenous amines revealed that Cp*Rh(III) complexes are ineffective at promoting this transformation, but analogous Cp*Ir(III) complexes, previously demonstrated by Chang12 to tolerate amines and anilines in C-H amination, are competent catalysts. Based on these initial results, we conscripted N-methylaniline (1a, Table 1) to be the model amine for our optimization studies.
Table 1.
Reaction Optimizationa

| ||||
|---|---|---|---|---|
|
| ||||
| entry | solvent | additive | yield (%)b | rr (γ:δ)c |
| 1 | TFE | 2 eq. CsOPiv | 57 | 1:12 |
| 2 | HFIP | 2 eq. CsOPiv | 30 | 2:1 |
| 3 | HFIP | none | 45 | 9:1 |
| 4d | HFIP | none | 62g | 14:1 |
| 5 | TFE | 2 eq. Cs2CO3 | 45 | <1:20 |
| 6 | TFE | 2 eq. KHCO3 | 60 | 1:7 |
| 7d | TFE/H2Oe | 2M KHCO3f | 76g | <1:20 |
Conditions: 1a (1.0 eq.), 2a (2.0 eq.), [Cp*IrCl2]2 (2.5 mol %), 18 h.
Yield of dominant isomer as determined by 1H NMR.
Determined by analysis of 1H NMR of the unpurified reaction mixture.
0.12 mmol 1a, 1.2 eq. 2a.
1:1 ratio.
Concentration in H2O.
Isolated yield of dominant isomer.
In an examination of solvents (see SI for details), we were pleasantly surprised to find that [Cp*IrCl2]2 in both trifluoroethanol (TFE) and hexafluoroisopropanol (HFIP) promotes productive catalysis, but with opposite regioselectivity (entries 1 and 2). Speculating that the increased acidity of HFIP relative to TFE is the source of regiodivergence, we determined that removal of the base and lowering of the amine loading delivers γ-lactam 3aa in good yield and improved regioselectivity (62 %, 14:1 γ:δ, entry 4). On the other hand, the use of Cs2CO3 presents a drastic improvement in regioselectivity for δ-lactam 4aa but is detrimental to the yield (entry 5), a situation that is rectified with the less basic KHCO3 (entries 6 and 7). Ultimately, we found that use of 2 M aqueous KHCO3 as a cosolvent, coupled with lowering of the amine loading, gives 4aa in superior yield and regioselectivity (76%, >20:1 δ:γ, entry 7).
With optimized reaction conditions in hand, we examined the scope of each reaction. With regard to the γ-lactam scope (Scheme 2), we found that inclusion of substituents on the arene of varying electronics (methoxy, bromo, fluoro, chloro) is tolerated with reasonable yield and good to excellent regioselectivity (3ab – 3ae). The reaction is tolerant of moderate steric bulk near the nitrogen as anilines 2e bearing a 2-chloro group and 2f bearing an N-benzyl group are converted to the corresponding products 3ae and 3af in 64 and 76 % yields respectively and >20:1 rr (γ:δ) for both. Next, we investigated the effect of substitution on the lactam ring, and found that alkyl substituents at the β-position are compatible with the reaction (3ba –3da); however, this substitution has little influence on the stereochemistry of the formation of the adjacent C–N bond, resulting in poor diastereoselectivity of the reaction. Disappointingly, we found that α-substitution on the lactam is not well-tolerated for this regioisomer (vide infra).13
Scheme 2.

γ-Lactam Scopea
aConditions: 1 (1.0 eq.), 1.2 eq. 2, 2.5 mol % [Cp*IrCl2]2, HFIP, 21 °C for 16 h. Yields are of isolated γ-lactam only. Regioisomeric ratios reported as γ:δ.
Next, we turned our attention to the scope of δ-lactams, shown in Scheme 3, and found that this reaction is tolerant of a wider scope of amines. Similar to the γ-lactam scope, we found that a variety of electronically diverse substituents on the aniline are tolerated (methoxy, fluoro, and bromo), and the corresponding lactams (4ab – 4ad) are delivered in good yield (69 to 76%) and excellent regioselectivity (all >20:1 δ:γ) (Scheme 3a). Furthermore, dialkyl amines with a variety of functional groups are also competent in the reaction (Scheme 3b). N-benzyl and N-dimethoxybenzyl protected amines 2i and 2m were found to couple with 1a productively, giving the corresponding products 4ai and 4am in moderate yield (57 % and 60 %, respectively) and regioselectivity (8.9:1 and 5.5:1, respectively). Amines 2k and 2l are also converted to the corresponding products 4ak and 4al in moderate yield (47% and 50%, respectively) and regioselectivity. Impressively, amidine-containing piperazine 4aj, derived from the antidepressant amoxapine (2j), is delivered in excellent yield (79%) and regioselectivity (>20:1).
Scheme 3.

δ-Lactam Secondary Amine Scopea
aYields are of isolated δ-lactam only. Regioisomeric ratios reported as δ:γ. Conditions: 1a (1.0 eq., 0.12 mmol, 0.3 M,), 1.2 eq. 2, 2.5 mol % [Cp*IrCl2]2, 1:1 TFE: 2 M aq. KHCO3, 21 °C for 16 h. b5.0 mol % [Cp*IrCl2]2.
We subsequently interrogated the effect of substitution on the lactam ring, but were disappointed to find that unlike in the γ-lactam scope, β-substituted alkenyl amides 1b – 1d did not engage in productive reactivity. When we tested α-methyl alkenyl amide 1e under the standard reaction conditions, we were encouraged to see some of the corresponding lactam 4ea (Scheme 4a, entry 1), but the reaction is plagued by the formation of urea 5ea. Changing the counterion of the catalyst presents no improvement (entries 2 and 3). However, we found that use of the more electron-deficient [Cp*pCF3IrCl2]2 suppresses the formation of 5ea (entry 5) and delivers 4ea in good isolated yield (74%) and excellent regioselectivity (>20:1) (Scheme 4b).14 α-substitution also confers a high degree of diastereoselectivity to the reaction (>20:1). This high diastereoselectivity is strongly suggestive of a chair-like transition state for the diastereomer-determining C–N bond formation between the alkene and 2a. α-substituted δ-lactams 4fa and 4ga are also delivered in good yield (67 and 53%, respectively) with >20:1 rr and dr.
Scheme 4.

α-Substituted Lactam Optimization and Scope
aConditions: 0.030 mmol 1e (1.0 eq.), 1.2 eq. 2a, 2.5 mol % [CpxIrCl2]2, 2.0 eq CsOPiv, TFE, 21 °C for 16 h. bDetermined by 1H NMR. c5.0 mol %. dIsolated yield using 0.12 mmol 1e. eConditions: 0.12 mmol 1 (1.0 eq.), 1.2 eq. 2a, 2.5 mol % [CppCF3IrCl2]2, 2.0 eq CsOPiv, TFE, 21 °C for 16 h. Yields are of isolated δ-lactam only. Regioisomeric ratios reported as δ:γ.
We were puzzled by the formation of significant amounts of urea 5ea from 1e when 1a leads to little to no urea formation under both HFIP and TFE reaction conditions. When we examined 1e in the standard reaction conditions for γ-lactam synthesis, urea 5ea is predominantly formed (Scheme 5a). Crossover experiments demonstrate that none of the products – urea, γ-, or δ-lactam – interconvert under the reaction conditions and are thus not intermediates to each other.15 Considering that this undesired urea formation involves a migration of the alkyl chain of 1e onto the amide nitrogen, we postulate that 5ea is formed via a Lossen rearrangement followed by trapping of the isocyanate by 2a (Scheme 5a). When the reaction is run in the absence of catalyst, no product or urea is observed, demonstrating that this Lossen rearrangement is catalyst-dependent.16 Therefore, we postulate that this rearrangement proceeds through an Ir(V) nitrenoid.17 Considering that a higher ratio of urea to lactam is observed in the γ-lactam reaction conditions than the δ-lactam conditions, and that δ-lactam formation is heavily favored over both γ-lactam and urea formation when using the more electron-poor [CpIr*pCF3Cl2]2, we posit that in the product-determining step, oxidation of an Ir(III)-amido complex to an Ir(V)-nitrenoid causes γ-lactam formation.
Scheme 5.
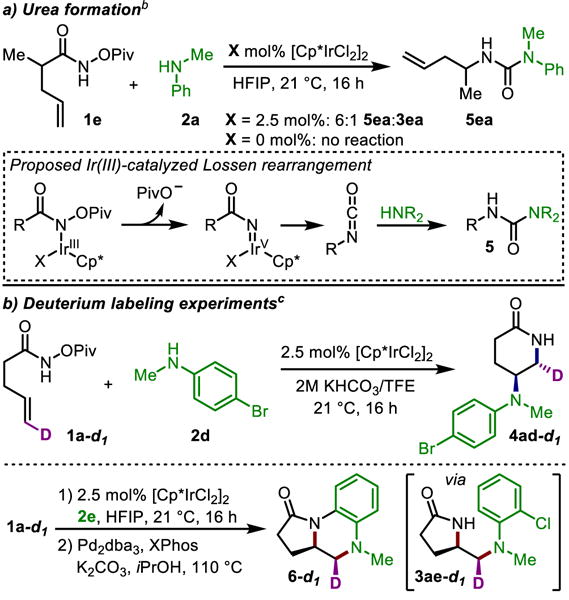
Mechanistic Experimentsa
aSee Supporting Information for reaction details and yields. bUrea 5ea is also observed in approximately 1:1.6 ratio with lactam 4ea when using the standard conditions for δ-lactam formation. See Supporting Information for more details. cRelative stereochemistry determined by 1H NMR and comparison to the non-deuterated spectra. See Supporting Information for more details.
To probe whether the exogenous amine is installed by migratory insertion or reductive elimination from the Ir versus nucleophilic attack of the carbon, we synthesized alkenyl amide trans-1a-d1 (Scheme 5b), and subjected it to the complementary reaction conditions. Under both TFE and HFIP conditions, we determined that the two nitrogen units add anti across the double bond.18
Based on these results we propose the following mechanism (Scheme 6). Active catalyst I coordinates the amide alkene of 1 to generate II. From here, hydrogen bonding of the relatively acidic solvent to amine 2 slows nucleophilic attack and enables Ir(V)-nitrenoid III to form. Migratory insertion of the Ir(V)-nitrenoid onto the alkene closes the lactam ring and generates Ir(V) bicycle IV. Nucleophilic attack of 2 then opens the four-membered iridacycle and reduces the metal to Ir(III), generating V.19 Protodemetallation generates γ-lactam 3 and regenerates the active catalyst. Conversely, the basicity of TFE/2 M KHCO3 when using it as solvent mollifies hydrogen bonding to 2 and speeds up nucleophilic attack, allowing seven-membered iridacycle VI to form. Alternatively, the electron-deficiency of [Cp*pCF3IrCl2]2 slows down oxidation of the catalyst to III. Nitrene formation followed by reductive coupling, or alternatively reductive elimination followed by oxidative insertion into the N–O bond closes the lactam ring and generates VII. Protodemetallation generates δ-lactam 4 and regenerates active catalyst I.
Scheme 6.

Proposed Mechanisma
aFor clarity, oxidation states and Lossen rearrangement are omitted, 1a is shown, and OPiv and Cl, when on Ir, are abbreviated as X. See supporting information for the postulated Lossen rearrangement mechanism.
In conclusion, we have described a novel, regiodivergent alkene diamination reaction that proceeds under mild conditions, wherein two orthogonal nitrogen units are installed across the double bond and the choice of solvent and additive determine the regioselectivity. Investigation of the mechanism revealed that this reaction can proceed via two distinct pathways, likely involving Ir nitrenoid intermediates. Efforts at extending this reactivity are currently underway.
Supplementary Material
Scheme 1.

Alkene Diamination
Acknowledgments
We thank NIGMS (GM80442) for support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary figures, experimental details, characterization data, and NMR spectra. (PDF)
ORCID
Tomislav Rovis: 0000-0001-6287-8669
Notes:
The authors declare no competing financial interests.
References
- 1.Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.For reviews, see:; (a) Cardona F, Goti A. Nat Chem. 2009;1:269. doi: 10.1038/nchem.256. [DOI] [PubMed] [Google Scholar]; (b) De Jong S, Nosal DG, Wardrop DJ. Tetrahedron. 2012;68:4067. doi: 10.1016/j.tet.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Muñiz K, Martínex C. J Org Chem. 2013;78:2168. doi: 10.1021/jo302472w. [DOI] [PubMed] [Google Scholar]; (d) Zhu Y, Cornwall RG, Du H, Zhao B, Shi Y. Acc Chem Res. 2014;47:3665. doi: 10.1021/ar500344t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Du H, Zhao B, Shi Y. J Am Chem Soc. 2007;129:762. doi: 10.1021/ja0680562. [DOI] [PubMed] [Google Scholar]; (b) Wang B, Du H, Shi Y. Angew Chem Int Ed. 2008;47:8224. doi: 10.1002/anie.200803184. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao B, Peng X, Cui S, Shi Y. J Am Chem Soc. 2010;132:11009. doi: 10.1021/ja103838d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Röben C, Souto JA, González Y, Lishchynskyi A, Muñiz K. Angew Chem Int Ed. 2011;50:9478. doi: 10.1002/anie.201103077. [DOI] [PubMed] [Google Scholar]; (e) Muñiz K, Kirsch J, Chávez P. Adv Synth Catal. 2011;353:689. [Google Scholar]; (f) Olson DE, Su JY, Roberts DA, Du Bois J. J Am Chem Soc. 2014;136:13506. doi: 10.1021/ja506532h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Danneman MW, Hong KB, Johnston JN. Org Lett. 2015;17:2558. doi: 10.1021/acs.orglett.5b01177. [DOI] [PubMed] [Google Scholar]; (h) Muñiz K, Barreiro L, Romero RM, Martínez C. J Am Chem Soc. 2017;139:4354. doi: 10.1021/jacs.7b01443. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sibbald PA, Michael FE. Org Lett. 2009;11:1147. doi: 10.1021/ol9000087. [DOI] [PubMed] [Google Scholar]; (b) Ingalls EL, Sibbald PA, Kaminsky W, Michael FE. J Am Chem Soc. 2013;135:8854. doi: 10.1021/ja4043406. [DOI] [PubMed] [Google Scholar]
- 5.Shen K, Wang Q. Chem Sci. 2015;6:4279. doi: 10.1039/c5sc00897b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen K, Wang Q. J Am Chem Soc. 2017;139:13110. doi: 10.1021/jacs.7b06852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu R-H, Wei D, Han B, Yu W. ACS Catal. 2016;6:6525. [Google Scholar]
- 8.Khoder ZM, Wong CE, Chemler SR. ACS Catal. 2017;7:4775. doi: 10.1021/acscatal.7b01362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong A, Blakey SB. Synthesis. 2012;44:1190. [Google Scholar]
- 10.Archambeau A, Rovis T. Angew Chem Int Ed. 2015;54:13337. doi: 10.1002/anie.201504150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Guimond N, Gorelsky SI, Fagnou K. J Am Chem Soc. 2011;133:6449. doi: 10.1021/ja201143v. [DOI] [PubMed] [Google Scholar]; (b) Rakshit S, Grohmann C, Besset T, Glorius F. J Am Chem Soc. 2011;133:2350. doi: 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]; (c) Hyster TK, Knörr L, Ward TR, Rovis T. Science. 2012;338:500. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hyster TK, Rovis T. Synlett. 2013;24:1842. doi: 10.1055/s-0033-1339510. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hyster TK, Dalton DM, Rovis T. Chem Sci. 2015;6:254. doi: 10.1039/c4sc02590c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Semakul N, Jackson KE, Paton RS, Rovis T. Chem Sci. 2017;8:1015. doi: 10.1039/c6sc02587k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Piou T, Romanov-Michailidis F, Romanova-Michaelides M, Jackson KE, Semakul N, Taggart TD, Newell BS, Rithner CD, Paton RS, Rovis T. J Am Chem Soc. 2017;139:1296. doi: 10.1021/jacs.6b11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Kim H, Shin K, Chang S. J Am Chem Soc. 2014;136:5904. doi: 10.1021/ja502270y. [DOI] [PubMed] [Google Scholar]; (b) Kim H, Chang S. ACS Catal. 2015;5:6665. [Google Scholar]
- 13.Dialkyl amines participate under these conditions with modest selectivities for the γ-lactam but proceed in low yields (~6:1 rr, ~20% yield).
- 14.[Cp*pCF3IrCl2]2 was found to be incompatible with alkyl amines. Additionally, we found 4aa to be the major product when this catalyst was used in HFIP. See supporting information for more details.
- 15.See supporting information for experiment details.
- 16.No product was observed when catalyst was omitted in either HFIP or TFE/2M KHCO3 conditions.
- 17.For a thorough theoretical and experimental examination into Ir(V)-nitrenoid formation using Cp*Ir(III) complexes, see:; Park Y, Heo J, Baik M-H, Chang S. J Am Chem Soc. 2016;138:14020. doi: 10.1021/jacs.6b08211. [DOI] [PubMed] [Google Scholar]
- 18.Widenhoefer has used a similar deuterium-labeling strategy to determine that a Pd(II)-catalyzed alkene hydroalkylation with a diketone proceeds through an outer sphere anti-carbometalation pathway. See:; Qian H, Widenhoefer RA. J Am Chem Soc. 2003;125:2056. doi: 10.1021/ja0293002. [DOI] [PubMed] [Google Scholar]
- 19.Reductive elimination of IV can be envisioned to deliver a bicyclic acyl aziridine, which one might expect would lead to ring-opening to deliver one or both of the observed products. However, this aziridine has never been isolated, has resisted our own attempts at isolation and cannot account for the effect of catalyst structure on regioselectivity; see SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.