

Abstract
Ulcerative colitis (UC) patients exhibit elevated histamine, but how histamine exacerbates disease is unclear since targeting histamine 1 receptor (H1R) or H2R is clinically ineffective. We hypothesized that histamine functioned instead through the other colon-expressed histamine receptor, H4R. In humans, UC patient biopsies exhibited increased H4R RNA and protein expression over control tissue, and immunohistochemistry showed that H4R was in proximity to immunopathogenic myeloperoxidase-positive neutrophils. To characterize this association further, we employed both the oxazolone (Ox)- and dextran sulfate sodium (DSS)-induced experimental colitis mouse models and also found upregulated H4R expression. Mast cell (MC)-derived histamine and H4R drove experimental colitis, as H4R−/− mice had lower symptom scores, neutrophil-recruitment mediators (colonic IL-6, CXCL1, CXCL2), and mucosal neutrophil infiltration than wild-type (WT) mice, as did MC-deficient KitW-sh/W-sh mice reconstituted with histidine decarboxylase–deficient (HDC−/−) bone marrow–derived MCs compared to WT-reconstituted mice; adaptive responses remained intact. Furthermore, Rag2−/−×H4R−/− mice had reduced survival, exacerbated colitis, and increased bacterial translocation than Rag2−/− mice, revealing an innate protective anti-bacterial role for H4R. Taken together, colonic MC-derived histamine initiates granulocyte infiltration into the colonic mucosa through H4R, suggesting alternative therapeutic targets beyond adaptive immunity for UC.
Keywords: Mast cells, histamine 4 receptor, neutrophils, colitis, histamine
INTRODUCTION
Ulcerative colitis (UC) is a subtype of inflammatory bowel disease (IBD) with chronic symptoms of abdominal pain and bloody diarrhea secondary to colonic inflammation.1 The pathogenesis is complex, but a dysfunctional mucosal immune response is critical.2 Genome wide association studies (GWAS) and experimental animal models indicate a role for both innate and adaptive immunity, yet few therapies primarily target innate immunity.2 While these therapeutic advances in treating adaptive immune pathways have revolutionized UC patient outcomes, one in four patients still require colectomy, urging elucidation of other mechanisms that drive inflammation.3, 4
In healthy colon tissue, inflammatory cells are limited from entering the mucosa until the barrier is breached by luminal insults (e.g., bacteria).5 Neutrophils in particular enter the mucosa to kill the bacteria and initiate wound repair; the inflammation ultimately resolves.6 A hallmark of active UC is neutrophilic infiltration into the lamina propria that leads to mucosal crypt destruction and neutrophil abscess formation;7 neutrophil-derived mediators that cause tissue damage, such as myeloperoxidase (MPO), correlate with disease activity.8, 9 Although molecules critical to this type of mucosal neutrophil recruitment are well known (e.g., the pleiotropic cytokine interleukin-6 [IL-6] and chemokine CXCL1)10, the signals critical to initiating and regulating these innate processes are poorly understood.
The inflammatory cellular milieu of active UC includes a diverse group of granulocytic cells found in T helper lymphocyte type 2 diseases, including mast cells (MCs), basophils, and eosinophils.2, 11 Of these, MCs are uniquely tissue resident and thus poised as early sensors of luminal insults, capable of signaling innate and adaptive immune responses.12, 13 Activated MCs rapidly release granules containing preformed mediators such as the vasoactive amine, histamine.12, 14 Although the precise roles of MCs and histamine in UC pathogenesis remain unclear, previous studies implicate both.15 For MCs, several reports note correlations between active UC and increased mucosal MC density.16–18 Although results from experimental colitis models in MC-deficient animals vary, the overwhelming evidence suggests an important role for MCs and their mediators in driving pathogenesis, reviewed by Boeckxstaens in 2015.19 Elevated mucosal histamine as well as N-methylhistamine metabolite excretion correlate well with patient disease activity.13, 20–22 Bene et al.23 found mice lacking histidine decarboxylase (HDC)––an enzyme necessary for histidine-to-histamine conversion––exhibited reduced inflammation severity to dextran sulfate sodium (DSS)-induced colitis. Additionally, polymorphisms in diamine oxidase, an enzyme critical in histamine breakdown, may affect the outcome of immune-modulator therapeutics in IBD patients.24 Though these studies point to a role for MCs and histamine in UC, the precise mechanisms underlying the connections among MCs, histamine, and UC pathogenesis have not yet been fully determined.
Histamine functions through 4 different receptors, histamine 1 receptor (H1R)–H4R, but which receptor(s) is important in UC pathogenesis remains unknown. Small trials utilizing widely available anti-histamine blockers for H1R and H2R in IBD fail to demonstrate clear efficacy, suggesting that they may not be critical in IBD pathogenesis.25 Among the remaining histamine receptors, H3R is not expressed in the gut mucosa, and H4R is expressed in the gut mucosa by mucosal immune cells14 (including MCs, eosinophils, T cells, and dendritic cells) as well as non-immune neuroendocrine cells, enterocytes in the crypt of Lieberkuhn, neurons, and small submucosal blood vessels. Interestingly, prior work suggests that H4R contributes to IBD immunopathogenesis. Varga et al.26 found that an H4R antagonist reduced inflammatory responses in TNBS-induced rat colitis. Schirmer et al.27 found milder symptoms and histopathologic severity to DSS-induced colitis in H4R−/− BALB/c mice compared to wild-type (WT) mice or in WT mice undergoing constant pharmacological H4R blockade; the milder symptoms correlated with a broad reduction in cytokines from restimulated mesenteric lymph node (MLN) cells and serum IL-6. While these studies support a potential role for H4R and/or histamine in regulating colitis, the pathways remain unknown, and the key cellular source of histamine remains unidentified.
We demonstrate here that MCs, histamine, and H4R coordinate to promote innate immune–mediated immunopathology and damage associated with mucosal inflammation in colitis. Furthermore, our data show not only that MCs support an innate inflammatory response during colitis distinct from adaptive, T cell–associated inflammation but also that these two mechanisms are mutually cooperative in maintaining intestinal defenses.
RESULTS
H4R expression is increased in a subset of untreated patients with active UC and is found in local proximity of activated neutrophils
Histamine is increased in the mucosa of patients with active IBD, but the underlying immune mechanism to drive inflammation via histamine receptors is not well defined.28 Previous studies in humans have failed to identify a clear role for either H1R or H2R,28 despite both being found on cells in the gut. We hypothesized that H4R––the only other histamine receptor expressed in the gut––may play a critical role. We initially quantified mRNA expression levels of H1R, H2R, and H4R by real-time RT-PCR in banked clinical samples from newly diagnosed UC patients to determine whether any changes occurred in the histamine receptors compared to non-UC control participants undergoing routine surveillance colonoscopy. While UC patients and control participants had similar H1R and H2R mRNA expression in their colon, UC patients exhibited significantly higher H4R expression (Figure 1A); H4R protein levels were also higher by quantifying H4R IHC staining (Figure 1B). Since a hallmark of UC is neutrophilic infiltration and release of pre-formed MPO-containing granules, we examined MPO protein levels by IHC and found significantly higher MPO levels in UC patient colons than control colons (Figure 1C). Comparing serially sectioned IHC slides from UC patients, we noted areas in which H4R and MPO staining occurred in close proximity to each other (Figure 1D). Taken together, we observe higher H4R expression and increased MPO-positive cells in the colons of newly diagnosed, untreated UC patients compared to control participants, leading to the possibility that histamine, H4R, and granulocytic infiltration may be associated in UC.
Figure 1.
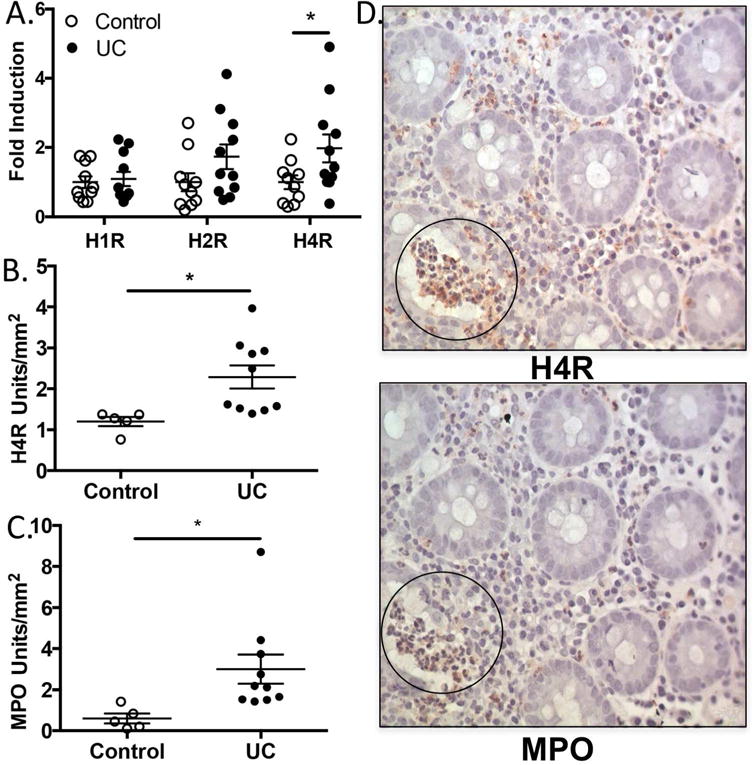
H4R is increased in active UC and regionally co-localizes with MPO+ cells. (A) Quantitative real-time PCR of RNA isolated from biopsy lysates from non-UC individuals (Control) and UC patients displayed as fold induction vs control for H1R, H2R, and H4R. (B) Relative intensity quantification of H4R IHC from non-UC individuals and UC patients displayed as relative units/mm2. (C) Relative intensity quantification of MPO IHC for non-UC individuals and UC patients in relative units/mm2. (D) Example of H4R/MPO staining on serial sections of colon tissue (circle indicates area of regional co-localization). *P < 0.05.
H4R drives the severity of experimental colitis
To explore this potential connection among histamine, H4R, granulocytic infiltration, and severity of colonic inflammation, we utilized two different, well-established experimental models of colitis. Ox-induced colitis is a T cell–driven colitis characterized by a prominent adaptive Th2 response with elevated IL-4 and IL-13 as well as a strong innate granulocytic response. DSS-induced colitis, in contrast, has a limited Th1/Th17 response and a profound innate granulocytic response. We first observed that Ox-treated WT mice exhibited significantly higher H4R expression than untreated control WT mice, mirroring our observations in the patient population (Supplemental Figure 1A). Additionally, isolated RNA from untreated murine colonic epithelial cells (>80% EPCAM-positive by flow cytometry) did not have detectable H4R expression (Supplemental Figure 1B) despite robust H1R and H2R expression, suggesting that H4R tissue expression in the colon is primarily restricted to immune cells, as has previously been described.25 To test whether H4R functionally contributed to disease pathogenesis, we examined clinical disease severity between WT and H4R-deficient knockout (H4R−/−) mice by measuring both changes in weight and degree of bloody diarrhea (disease activity index [DAI] score). In both models, H4R−/− mice had reduced weight loss over the course of 3 days compared to WT mice (Figure 2A and E) and reduced DAI scores at the peak of disease (Figure 2B and F). This protection against clinical severity in H4R−/− correlated with a decrease in tissue damage, as the colons of H4R−/− mice had significantly reduced histology scores (Figure 2C and G) compared to WT colons. Notably, among the principal components evaluated for the histology score, H4R−/− mice had significantly lower cellular infiltration and decreased overall mucosal damage in both Ox- and DSS-induced colitis (Figure 2D, H, I, and J). Taken together, H4R may play a role in driving key aspects of the inflammatory responses––in particular, tissue damage–promoting cellular infiltration––observed in both colitis models.
Figure 2.
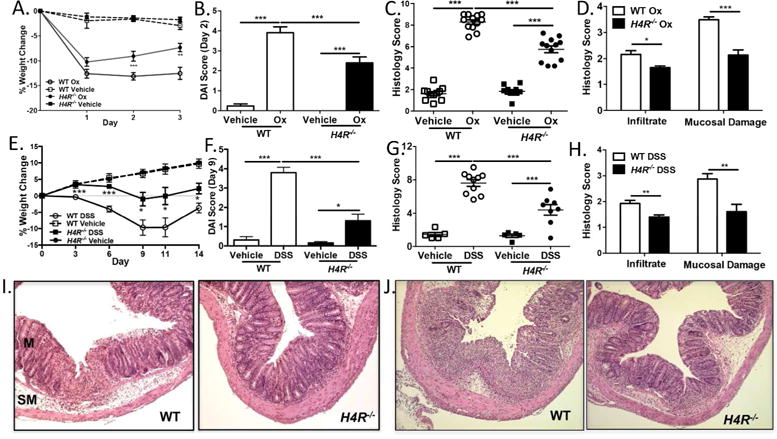
H4R drives clinical and histologic severity in Ox- and DSS-induced experimental colitis. (A–D, I) Ox-induced and (E–H, J) DSS-induced colitis experiments. (A, E) Percentage weight change in BALB/c vs H4R−/− mice (n = 16–26 mice/group). (B, F) Disease activity index (DAI) score for WT and H4R−/− mice. (C, G) Composite histology score for WT and H4R−/− mice. (D, H) Component histology score for cellular infiltrate and mucosal damage in WT and H4R−/− mice. (I, J) Representative histology in WT and H4R−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001. Data represented as mean ± SEM from 3–4 independent experiments.
H4R drives neutrophil recruitment to the mucosa
Neutrophil infiltration is a hallmark of UC; in particular, activated neutrophils attacking mucosal crypts create the characteristic neutrophil abscesses. We therefore quantified neutrophil abscess formation in the tissues of WT and H4R−/− and found that H4R−/− colons had significantly less frequent neutrophil abscesses in the mucosa (area above the submucosal space) than WT in both Ox- and DSS-induced colitis (Figure 3A and Supplemental Figure 2). These data suggest that H4R plays a role in neutrophil movement and migration into the mucosa, where neutrophils elicit the most tissue damage in colitis. Focusing on the Ox model––the model most linked to Th2-type responses observed in UC––we sought to more fully characterize this neutrophil infiltration. Consistent with neutrophil abscess frequency, H4R−/− colons contained fewer MPO-activated mucosal neutrophils compared to WT (Figure 3B and C). One possible explanation is that neutrophil migration signals are affected in the absence of H4R signaling. Indeed, H4R−/− colon lysates had significantly reduced levels of the neutrophil-attracting molecules IL-6, CXCL1, and CXCL2 than WT (Figure 3D); in DSS-induced colitis, H4R−/− colon lysates had significantly lower IL-6 and CXCL1 than WT (Supplemental Figure 3). Taken together, these data suggest that H4R plays a role upstream of recruiting neutrophils into colon tissue at least in part by regulating neutrophil-attracting chemokine/cytokine production.
Figure 3.
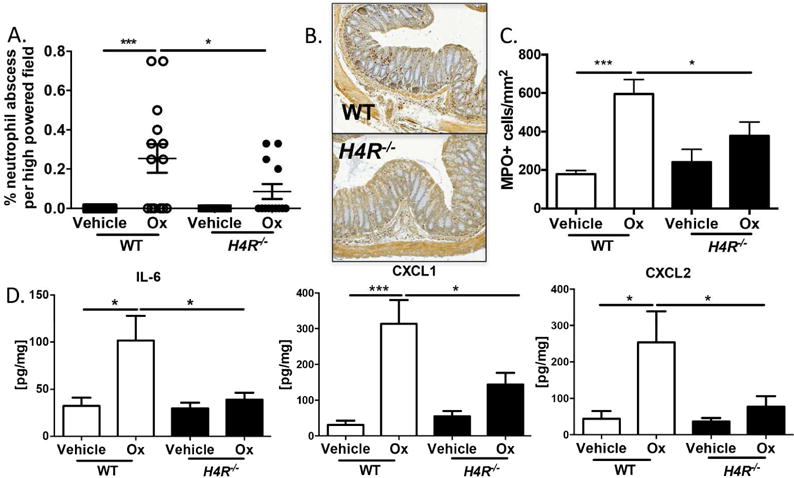
H4R signaling allows for neutrophil recruitment into the mucosa during experimental colitis. (A) Percentage neutrophil abscesses/hpf present within colon sections (3–5 sections/animal). (B) Representative MPO IHC in WT and H4R−/− Ox-treated mice. (C) Mucosal MPO+ cells/mm2 quantification in WT vs H4R−/− Ox-treated mice. (D) Colonic IL-6, CXCL1, and CXCL2 protein quantification by ELISA. *P < 0.05, ***P < 0.001. Data represented as mean ± SEM from 3 independent experiments.
MC-derived histamine drives severity of experimental colitis at least in part by recruiting neutrophils into the colonic mucosa
To next determine if histamine, the ligand for H4R, was also required for neutrophil infiltration into the colon after Ox-induced colitis, we examined Ox-induced colitis in histamine-deficient HDC−/− and WT mice and found that HDC−/− mice exhibited less severe colitis with significantly reduced weight loss, DAI scores, and histology scores (Figure 4A–C), similar to H4R−/− mice. Importantly, HDC−/− mice recapitulated the reduced neutrophil infiltration observed in the H4R−/− mice with significantly lower neutrophil abscess formation and lower IL-6, CXCL1, and CXCL2 expression (Figure 5A and B). To rule out whether this effect of histamine on exacerbating colitis worked through either of the other gut-expressed histamine receptors, we performed the Ox-induced colitis model in H1R/H2R double-knockout mice (H1R−/−×H2R−/−) and found similar weight loss, DAI scores, and histology scores between H1R−/−×H2R−/− and control WT mice (Supplemental Figure 4). Thus, the effect of histamine on pro-inflammatory clinical and histologic severity in experimental colitis is likely independent of H1R or H2R effects in the mucosa and functions instead through H4R-mediated processes.
Figure 4.
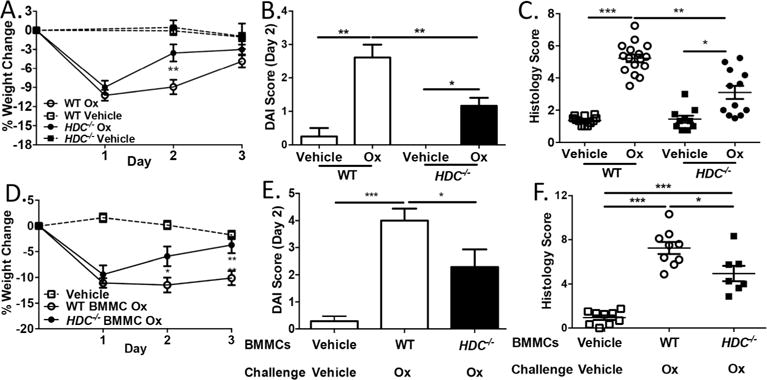
MC-derived histamine drives clinical and histologic severity of experimental colitis. (A) Percentage weight change in Ox- or vehicle-treated WT and HDC−/− mice (n = 11–24 mice/group). (B) DAI score in Ox- or vehicle-treated WT and HDC−/− mice (day 2). (C) Histology score in Ox- or vehicle-treated WT and HDC−/− mice (day 3). (D) Percentage weight change in Ox- or vehicle-treated WT or HDC−/− BMMC–reconstituted Sash mice (n = 7–10 mice/group). (E) DAI score in Ox- or vehicle-treated WT or HDC−/− BMMC–reconstituted Sash mice (day 2). (F) Histology score in Ox- or vehicle-treated WT or HDC−/− BMMC–reconstituted Sash mice (day 3). *P < 0.05, **P < 0.01, ***P < 0.001. Data represented as mean ± SEM from 3 independent experiments.
Figure 5.
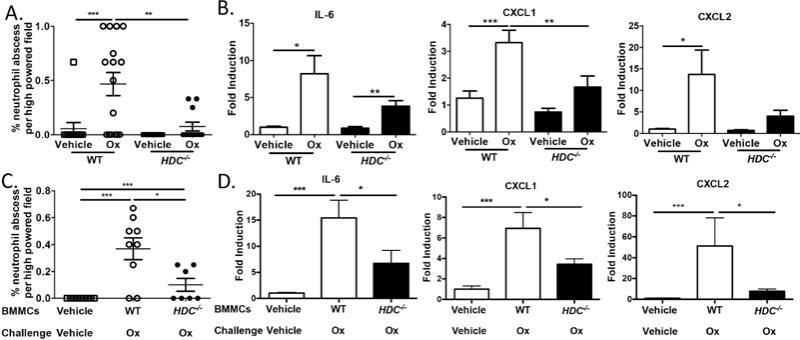
MC-derived histamine aids in neutrophil recruitment into the mucosa during experimental colitis. (A) Percentage neutrophil abscesses/hpf present within colon sections (n = 3–5 sections/animal) from Ox- or vehicle-treated WT and HDC−/− mice. (B) Quantitative real-time PCR of colon lysates for IL-6, CXCL1, and CXCL2 from Ox- or vehicle-treated WT and HDC−/− mice; fold induction shown relative to vehicle. (C) Percentage neutrophil abscesses/hpf present within colonic sections (n = 3–5 sections/animal) from Ox- or vehicle-treated WT and HDC−/− BMMC–reconstituted Sash mice. (D) Quantitative real-time PCR of colon lysates for IL-6, CXCL1, and CXCL2 from Ox- or vehicle-treated WT and HDC−/− BMMC–reconstituted Sash mice. **P < 0.05, **P < 0.01, ***P < 0.001. Data represented as mean ± SEM from 3 independent experiments.
MCs and basophils are considered the predominant histamine sources. Since MCs reside in tissues while basophils mainly circulate in blood, MC-derived histamine may play a role in coordinating neutrophil infiltration and exacerbating colitis. We tested this by reconstituting MC-deficient Sash mice with BMMCs grown from either WT or HDC−/− mice and then challenging the recipient mice with the Ox-driven model; to control for procedural stress, vehicle-reconstituted mice were first sensitized to Ox and then challenged with vehicle. Compared to WT BMMC–reconstituted Sash mice, those reconstituted with HDC−/− BMMCs exhibited decreased disease severity, neutrophil abscesses, and neutrophil-attracting molecules (Figure 4D–F, and Figure 5C and D). Collectively, MC-derived histamine plays a previously unappreciated role in regulating innate immune–mediated inflammation through H4R within the colonic mucosa.
T cell responses during experimental colitis are independent of H4R, histamine, or MC-derived histamine
The pathogenesis and severity of Ox or DSS experimental colitis is often attributed to adaptive immunity, in particular the T cell response.29–31 To test whether the histamine/H4R axis had any effect on adaptive immunity that could account for exacerbating colitis, we examined several T cell–associated responses in the colon after Ox- or DSS-induced colitis. Despite significant differences in disease severity between WT and H4R−/− mice in the Th2-associated Ox model, they exhibited similar Th2-driven IL-4 and IL-13 expression and CD4+ T cell frequency within the CD45+ pool of lamina propria mononuclear cells (LPMC) at day 3 (Figure 6A and B). Similarly, T cell–driven IL-17A was also similar between H4R−/− and WT mice in the Th1/Th17-associated DSS-induced colitis at day 9 (Figure 6C). T cell–dependent contact hypersensitivity responses (ear swelling) to Ox challenge were also similar between sensitized H4R−/− and WT mice (Supplemental Figure 5). We also observed no changes in Th2-driven IL-4 and IL-13 expression between HDC−/− and WT mice, or between WT BMMC– and HDC−/− BMMC–reconstituted Sash mice (Figure 6D and E). Taken together, the competent T cell responses observed in the H4R−/−, HDC−/−, and HDC−/− BMMC–reconstituted Sash mice suggest the histamine/H4R axis is not required for adaptive T cell responses in these colitis models.
Figure 6.
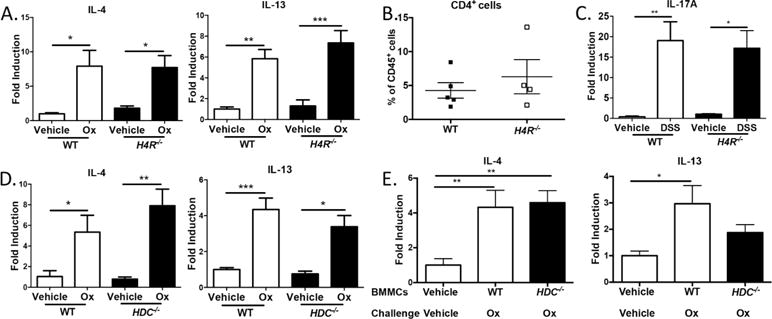
T cell responses during experimental colitis are unaffected by MC-derived histamine or H4R. (A) Quantitative real-time PCR for T cell–derived cytokines in colonic lysates from Ox-or vehicle-treated WT and H4R−/− mice (day 3). (B) Frequency of CD4+CD45+ LPMCs isolated from colon from Ox- or vehicle-treated WT and H4R−/− mice (day 3). (C) Quantitative real-time PCR for IL-17A in colonic lysates from DSS- or vehicle-treated WT and H4R−/− mice (day 9). (D) Quantitative real-time PCR from colonic lysates from Ox- or vehicle-treated WT vs HDC−/− mice (day 3). (E) Quantitative real-time PCR from colonic lysates from Ox- or vehicle-treated WT or HDC−/− BMMC–reconstituted Sash mice (day 3). *P < 0.05, **P < 0.01. Data represented as mean ± SEM from 3 independent experiments, except B (2 independent experiments).
H4R plays a key protective anti-bacterial role in innate immune signaling
Since both adaptive T cell and innate immune responses likely contribute to inflammation in colitis, we wondered what the effect of losing both adaptive immunity and this novel innate immune H4R-mediated pathway would be on mice undergoing experimental colitis. We thus crossed Rag2−/− mice (incapable of mounting adaptive immunity due to lacking T and B cells) to H4R−/− mice (Rag2−/−×H4R−/−) and performed DSS-induced colitis since Ox-induced colitis depends on T cells. We predicted that Rag2−/−×H4R−/− mice would show less disease severity than Rag2−/− mice, similar to their immunocompetent counterparts. To our surprise, Rag2−/−×H4R−/− mice exhibited more profound weight loss and lower survival rates compared to Rag2−/− mice (Figure 7A and B). Rag2−/−×H4R−/− mice had significantly more bacteria disseminated into the regional MLNs than Rag2−/− mice (Figure 7C), indicating that septicemia was the probable cause of Rag2−/−×H4R−/− death. These results demonstrate that H4R-dependent innate inflammation is required to control bacterial invasion in the absence of adaptive immunity.
Figure 7.
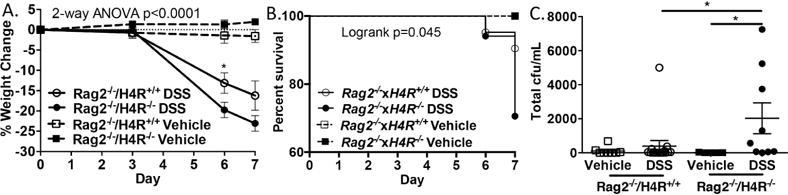
In the absence of functional adaptive immunity, H4R protects against weight loss, death, and increased bacterial translocation to the MLN after DSS-induced colitis. (A) Percentage weight change for DSS- or vehicle-treated Rag2−/− and Rag2−/−×H4R−/− mice (n = 5–17 mice/group). (B) Survival curve to day 7 in DSS- or vehicle-treated Rag2−/− and Rag2−/−×H4R−/− mice. (C) CFU counts from sterile MLN on blood agar plates on day 7 from DSS- or vehicle-treated Rag2−/− and Rag2−/−×H4R−/− mice. *P < 0.05. Data represented as mean ± SEM from 3 independent experiments.
DISCUSSION
Although histamine is elevated in the mucosa of active UC, and urinary histamine metabolites correlate with disease activity,22, 32 the mechanistic consequences of histamine release and receptor activation is lacking, limiting its consideration as a therapeutic target. Our work herein identifies a key role for MC-derived histamine release and subsequent H4R activation in driving clinical and histopathologic severity in UC-like inflammation by regulating innate neutrophil recruitment and associated mucosal injury, likely via coordinating the downstream local production of IL-6 and neutrophil-attracting chemokines.
While an early pilot study with ketotifen (an H1R blocker and MC stabilizer) showed some efficacy in a small subset of pediatric UC patients,33 the role of histamine receptors in the pathogenesis of colitis has remained unclear, with no clear role for H1R,34, 35 a proposed protective role for H2R via microbial crosstalk,35, 36 and pre-clinical evidence of a pro-inflammatory role for H4R.26, 27, 37 Our data demonstrate that mice lacking both H1R and H2R mount a competent response in experimental colitis and suggest that MC-derived histamine functions via H4R to exacerbate disease immunopathology. In patients, we noted a selective increase in H4R over the other receptors; for H1R, this is in accordance with recent work,35 but H2R upregulation previously described in a UC patient subset35 was not observed here. One explanation for this difference might be that the previous study compared inflamed to non-inflamed tissue derived from the same individual, while we compared inflamed tissue from UC patients to control tissue from non-UC patients.
Our work focuses on determining how the histamine/HR pathway drives clinical and pathologic severity through innate and/or adaptive immune mechanisms. In addition to elevated H4R transcripts in UC, we note that elevated H4R protein is found in proximity to areas of active MPO-associated cryptitis, a hallmark of UC.38 Building on previous findings,26, 27, 37 our data support the conclusion that H4R exacerbates immunopathology in experimental colitis, but the connections to innate inflammation presents a novel H4R-regulated pathway. Despite not placing mice on a histamine-free diet, our work defines a key role for endogenous MC-derived histamine in driving phenotypic severity. Although histamine levels are difficult to measure due to a lack of molecular stability in vivo, results from BMMC-reconstituted Sash mice define a signaling cascade whereby MC-derived histamine activates H4R to support neutrophil infiltration into the mucosa, likely through upstream regulation of neutrophil-attracting cytokine/chemokine production. Precisely how H4R and the release of these mediators are connected––either directly or indirectly––as well as the identity of the H4R-bearing cell (discussed further below) remains to be determined. While H4R may also function to limit the resolution of inflammation, the phenotype in H4R−/− mice occurs during the inflammatory phase of the colitis models wherein cell influx into the tissue is a critical factor. Thus, colonic histamine/H4R helps to create a neutrophil-attracting milieu that promotes neutrophil migration into the mucosa, where they are poised to damage the crypts and drive clinical severity; importantly, our data do not rule out other H4R-mediated pathways to neutrophil infiltration, such as an indirect effect on intestinal barrier function, but these mechanisms remain to be determined in future studies. Overall, these observations reflect what is known to occur in IBD: the mucosa abnormally responds to bacterial signals, directing neutrophils to invade the mucosa and cause inadvertent damage to healthy tissue, a process sustained by signals like IL-6, IL-1β, and tumor necrosis factor (TNF).10 Thus, this work is an important step forward, establishing that MCs, histamine, and H4R coordinate innate responses to drive clinical severity in IBD.
Since the loss of H4R led to partial, but not complete, restoration of WT findings compared to vehicle controls, the sufficient role of non-H4R–dependent inflammation via adaptive immune pathways remains clear. Potentially, H4R signaling could provide an alternative target for IBD treatment differing from most current IBD therapies that target adaptive immune pathways; indeed, targeting innate immunity via H4R may be a novel treatment consideration for UC patients with a limited response to T-cell or other adaptive immune therapies (i.e., glucocorticoids).
In the context of H4R promoting neutrophil-associated immunopathology, our work describes the following mechanistic scenario: 1) activated local MCs produce histamine, 2) histamine activates H4R-expressing cells, 3) H4R signals either directly or indirectly to induce chronic neutrophil-attracting chemokine release, and 4) neutrophils invade the mucosa, simultaneously causing inadvertent damage to healthy tissue and protecting against sepsis by limiting bacteria that invade the damaged tissue. Thus, neutrophils in IBD likely play dual roles as 1) damage-inducing yet 2) infection-fighting innate granulocytes. Indeed, our Rag2−/− experiments highlight an important role for H4R in protecting against bacterial invasion after mucosal barrier damage, and WT mice with experimental colitis are ultimately protected from lethal sepsis due to a compensatory balance between both innate and adaptive immunity. How can we disrupt the persistent vicious cycle of neutrophil-induced damage leading to bacterial translocation leading to more inflammation and more neutrophil recruitment? Perhaps blocking H4R may limit neutrophil invasion and “put the brakes” on perpetuating this cycle with a shutoff signal at the innate-immune level, and this may be most successful in a specific set of patients that retain a fully functioning adaptive immune system.
While not a pure Th2 disease, UC has clear evidence of Th2 activation (e.g., IL-13).39 The weight loss and pathologic inflammation still observed even after removing HDC or H4R signals suggested that histamine- and H4R-independent pathways led to inflammation persistence, likely via the well-described T cell response in IBD. Supporting this conclusion, mucosal CD4+ T cells and Th-associated cytokine (IL-4, IL-13, 1L-17) transcript levels remained intact in HDC−/− and H4R−/− mice after experimental colitis induction. Thus, in contrast to H4R’s described function to skew T cells in Th2-mediated allergic asthma,40 histamine and H4R likely do not modulate adaptive mucosal immunity in experimental colitis models.
Interestingly, Mudter et al.41 described that the pleiotropic cytokine IL-6 limited T cell apoptosis and enhanced T cell survival in Ox-induced colitis; here, however, H4R−/− mice had diminished IL-6 compared to WT during experimental colitis but exhibited equal CD4+ T cell numbers and T cell–derived cytokines in the mucosa. Thus, we propose that colonic IL-6 released downstream of H4R likely has an innate function in colitis to help recruit neutrophils via a direct or indirect mechanism. H4R can support IL-6 production in an experimental peritonitis inflammation model, as H4R blockade diminishes IL-6 production and the downstream neutrophil chemokines, CXCL1 and CXCL2.42 Moreover, IL-6 has been linked to neutrophil chemokine induction in colitis, as mice lacking gp130––an IL-6 receptor component—have reduced neutrophil chemokines after DSS colitis.43 We are actively pursuing the outstanding question of whether and which H4R-bearing cell produces IL-6 directly upon activation or through an indirect mechanism. A number of potential H4R-bearing candidate cells in the colon may participate in this process, some of which are known direct sources of IL-6 in the colon (innate cells such as MCs, neutrophils, and macrophages) and others that may play an intermediate role, like NKT cells.44 Notably, H4R blockers reduce IL-6 production in several cell types including MCs,45–47 suggesting a common pathway between MC activation and production of this important cytokine; indeed, autocrine regulation of IL-6 by histamine-producing MCs themselves might be possible, since H4R agonists can stimulate IL-6 and neutrophilic chemokines from human MCs in vitro.48
Collectively, these data suggest that MC-derived histamine and H4R help to create an environment conducive to pathogenic neutrophil infiltration into the colonic mucosa, exacerbating the symptoms of experimental colitis. Alongside prior pre-clinical work utilizing H4R blockers in UC models,26, 27, 37 our work in human UC samples and experimental mouse models lends promise towards considering H4R blockers as a therapeutic option for UC in patients that are not severely immunocompromised as well as a useful adjunct therapy to be used alongside current therapies targeting the adaptive immune system. However, more studies are needed to determine the safety of H4R blockers as induction agents or in the setting of coexisting infections based on the importance of the histamine–H4R–neutrophil innate immune axis demonstrated in our study.
METHODS
See Supplementary Methods for the following additional methods sections: Reagents, Monitoring of Disease Activity, Histology, Mast Cell Reconstitution, Real-time PCR, and Colonic Cytokine Measurement.
Patient Samples
Previously banked, IRB-approved cDNA samples and unstained formalin-fixed paraffin embedded slides were obtained from Dr. Terrence Barrett (Northwestern University, Chicago, IL). Samples were collected from healthy, consented adults (age 18–70) undergoing surveillance colonoscopy for cancer found to have benign polyps (12 non-UC control patients; mean age 44 ± 12 years; 33% male) or individuals experiencing UC symptoms undergoing diagnostic testing, not currently on UC medications (14 UC patients; mean age 44 ± 13 years; 57% male). Investigators were blinded to clinical information, including degree of disease severity for the UC patients.
Animals
Male and female (6–8 weeks old) C57BL/6J and KitW-sh/W-sh (Sash) mice (C57BL/6 background) were from the Jackson Laboratory (Bar Harbor, ME). HDC−/− mice (C57BL/6 background) were previously described.49 H4R−/− mice (BALB/c background) were previously described.50 BALB/c and Rag2−/− mice (BALB/c background) were from Taconic (Hudson, NY). Rag2−/− mice were bred with H4R−/− mice to generate Rag2−/−×H4R−/− mice; Rag2 phenotype was assessed by flow cytometry for the absence of CD3+ cells, and H4R genotype was assessed by genotyping. H1R−/−×H2R−/− double-knockout mice (C57BL/6 background) were obtained from Dr. Takeshi Watanabe (RIKEN Institute, Yokohama, Japan). All animal studies were performed under the guidelines for care and welfare by IACUC under protocols approved by the Northwestern University Animal Care and Use Committee. All procedures were performed during the light cycle. Mice were housed in micro-isolator barrier cages (Lab Products, Seaford, DE) lined with coarse-grade Aspen Sani-Chips® (Envigo, Indianapolis, IN) and provided with environmental enrichment including a Shepherd shack, Nestlet, Igloo, Safe Harbor retreat, and Nyla bones on a rotating schedule; HEPA-filtered air circulated in the cages with positive air flow. Experimental mice were fed irradiated standard mouse diet (Envigo 7912): 5.8% fat, 19.1% crude protein, and 44.3% carbohydrate. Breeder mice were fed irradiated breeder diet (Envigo 7912): 11.4% fat, 17.2% crude protein, and 45.2% carbohydrate. In an attempt to standardize the microbiome amongst all experimental mice, mice originally obtained from commercial vendors or other facilities were bred in-house for more than 3 generations to acclimate to the same environment before performing experiments.
Ox-induced colitis
Males (8–10 weeks old) were used for Ox-induced colitis in all experiments using C57BL/6, HDC−/−, BALB/c, and H4R−/− mice. For experiments with Sash mice, mice were 11–12 weeks old to allow for MC reconstitution in the gut. Mice were anesthetized, the abdomen was shaved (2 cm × 2 cm area), and 3% Ox in 100% EtOH was applied and allowed to dry. One week later, anesthetized mice received a 1% Ox in EtOH:normal saline (1:1) enema, which was given 4 cm from the anal verge with a neonatal umbilical venous catheter (Utah Medical Products, Midvale, UT). Animal was held in the head-down position for 60 seconds after enema administration.
DSS-induced colitis
DSS colitis was given to 8–10-week old female BALB/c, H4R−/−, Rag2−/−, and Rag2−/−×H4R−/− mice by administering 3.5% DSS in water ad libitum for 6 days, followed by normal water until euthanization (water bottle changed every 3 days).
H4R immunohistochemistry
Formalin-fixed, paraffin-embedded slides were stained for H4R (ab188978, Abcam, Cambridge, MA). Relative H4R protein intensity was quantified using the Tissue Gnostics System (Vienna, Austria).
Myeloperoxidase stain & quantification
Anti-MPO staining was performed on formalin-fixed colonic tissue sections (4 μm). Lamina propria and intraepithelial MPO staining was quantified for each 40× high-powered field (hpf) to determine average density of MPO-positive cells per hpf per section.
Assessment of bacterial translocation
MLNs were dissected under sterile conditions 7 days after initiating DSS colitis. Lysates were prepared in sterile PBS, and 100 μL was sterilely cultured on blood-agar plates and incubated at 37°C. Bacterial colonies were counted 96 hours later.
Statistical Analysis
Data are represented as the mean ± SEM unless otherwise noted. Statistical significance was determined using 2-tailed Student t test for 2-group comparisons or ANOVA (Dunnett’s test) for greater than 2 group differences. Two-way ANOVA (Bonferroni’s) was used to compare weight change between multiple groups over a time course. Analyses were performed using GraphPad Prism 5.0 (La Jolla, CA).
Supplementary Material
Acknowledgments
We thank Dr. Rob Thurmond and Dr. Paul Dunford at Johnson & Johnson for providing the H4R−/− mice. We also thank Dr. Takeshi Watanabe (RIKEN Institute, Yokohama, Japan) for providing the H1R−/−×H2R−/− double-knockout mice.
Grant Support/Funding: This project was financially supported in part by the National Institutes of Health (NIH) grant R01AI076456 (to P.J.B), NIH grant K08DK097721 (to J.B.W.), Northwestern University Allergy Immunology Research (NUAIR) T32 (5T32AI083216-03), and internal funding from Ann and Robert H. Lurie Children’s Hospital of Chicago.
Abbreviations
- H1R
histamine 1 receptor
- H2R
histamine 2 receptor
- H3R
histamine 3 receptor
- H4R
histamine 4 receptor
- MPO
myeloperoxidase
- Ox
oxazolone
- DSS
dextran sulfate sodium
- MC
mast cells
- WT
wild-type
- HDC
histidine decarboxylase
- ELISA
enzyme-linked immunosorbent assay
- GWAS
genome-wide association studies
- IL
interleukin
- BMMC
bone marrow–derived mast cells
- GI
gastrointestinal
- hpf
high-powered field
- LPMC
lamina propria mononuclear cells
- ANOVA
analysis of variance
- DAI
disease activity index
- MLN
mesenteric lymph node
- TNF
tumor necrosis factor
Footnotes
AUTHOR CONTRIBUTIONS
J.B.W., B.K.W., P.J.B. contributed to the overall study concept and design; J.B.W., A.S., C-L.H., R.K-B., H.S., M.Y.W., R.C., L.M.A., J.B.B., B.K.W., T.V. acquired the data; J.B.W., T.A.B., P.J.B. analyzed and interpreted the data; J.B.W., M.L.M., P.J.B. drafted the manuscript; P.J.B. critically revised the manuscript for important intellectual content; P.J.B., J.B.W., B.K.W. obtained funding; T.A.B. provided human material support; P.J.B. supervised the study
DISCLOSURES
The authors have no disclosures relevant to this work.
References
- 1.Fell JM, Muhammed R, Spray C, Crook K, Russell RK, group BIw Management of ulcerative colitis. Arch Dis Child. 2016;101(5):469–474. doi: 10.1136/archdischild-2014-307218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27. doi: 10.1038/nrgastro.2015.186. [DOI] [PubMed] [Google Scholar]
- 3.Fumery M, Duricova D, Gower-Rousseau C, Annese V, Peyrin-Biroulet L, Lakatos PL. Review article: the natural history of paediatric-onset ulcerative colitis in population-based studies. Aliment Pharmacol Ther. 2016;43(3):346–355. doi: 10.1111/apt.13478. [DOI] [PubMed] [Google Scholar]
- 4.Romano C, Syed S, Valenti S, Kugathasan S. Management of Acute Severe Colitis in Children With Ulcerative Colitis in the Biologics Era. Pediatrics. 2016;137(5) doi: 10.1542/peds.2015-1184. [DOI] [PubMed] [Google Scholar]
- 5.Brazil JC, Louis NA, Parkos CA. The role of polymorphonuclear leukocyte trafficking in the perpetuation of inflammation during inflammatory bowel disease. Inflamm Bowel Dis. 2013;19(7):1556–1565. doi: 10.1097/MIB.0b013e318281f54e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev. 2016;273(1):357–370. doi: 10.1111/imr.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5(4):354–366. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 8.Masoodi I, Kochhar R, Dutta U, Vaishnavi C, Prasad KK, Vaiphei K, et al. Fecal lactoferrin, myeloperoxidase and serum C-reactive are effective biomarkers in the assessment of disease activity and severity in patients with idiopathic ulcerative colitis. J Gastroenterol Hepatol. 2009;24(11):1768–1774. doi: 10.1111/j.1440-1746.2009.06048.x. [DOI] [PubMed] [Google Scholar]
- 9.Masoodi I, Kochhar R, Dutta U, Vaishnavi C, Prasad KK, Vaiphei K, et al. Evaluation of fecal myeloperoxidase as a biomarker of disease activity and severity in ulcerative colitis. Dig Dis Sci. 2012;57(5):1336–1340. doi: 10.1007/s10620-012-2027-5. [DOI] [PubMed] [Google Scholar]
- 10.Fonseca-Camarillo G, Yamamoto-Furusho JK. Immunoregulatory Pathways Involved in Inflammatory Bowel Disease. Inflammatory bowel diseases. 2015;21(9):2188–2193. doi: 10.1097/MIB.0000000000000477. [DOI] [PubMed] [Google Scholar]
- 11.Yadav V, Varum F, Bravo R, Furrer E, Bojic D, Basit AW. Inflammatory bowel disease: exploring gut pathophysiology for novel therapeutic targets. Transl Res. 2016 doi: 10.1016/j.trsl.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Reuter S, Stassen M, Taube C. Mast cells in allergic asthma and beyond. Yonsei Med J. 2010;51(6):797–807. doi: 10.3349/ymj.2010.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox CC, Lichtenstein LM, Roche JK. Intestinal mast cell responses in idiopathic inflammatory bowel disease. Histamine release from human intestinal mast cells in response to gut epithelial proteins. Dig Dis Sci. 1993;38(6):1105–1112. doi: 10.1007/BF01295728. [DOI] [PubMed] [Google Scholar]
- 14.Sander LE, Lorentz A, Sellge G, Coeffier M, Neipp M, Veres T, et al. Selective expression of histamine receptors H1R, H2R, and H4R, but not H3R, in the human intestinal tract. Gut. 2006;55(4):498–504. doi: 10.1136/gut.2004.061762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotlyar DS, Shum M, Hsieh J, Blonski W, Greenwald DA. Non-pulmonary allergic diseases and inflammatory bowel disease: a qualitative review. World J Gastroenterol. 2014;20(32):11023–11032. doi: 10.3748/wjg.v20.i32.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki Y, Tanaka M, Kudo H. Differentiation between ulcerative colitis and Crohn’s disease by a quantitative immunohistochemical evaluation of T lymphocytes, neutrophils, histiocytes and mast cells. Pathol Int. 2002;52(4):277–285. doi: 10.1046/j.1440-1827.2002.01354.x. [DOI] [PubMed] [Google Scholar]
- 17.Yamagata K, Tanaka M, Kudo H. A quantitative immunohistochemical evaluation of inflammatory cells at the affected and unaffected sites of inflammatory bowel disease. J Gastroenterol Hepatol. 1998;13(8):801–808. doi: 10.1111/j.1440-1746.1998.tb00736.x. [DOI] [PubMed] [Google Scholar]
- 18.King T, Biddle W, Bhatia P, Moore J, Miner PB., Jr Colonic mucosal mast cell distribution at line of demarcation of active ulcerative colitis. Dig Dis Sci. 1992;37(4):490–495. doi: 10.1007/BF01307568. [DOI] [PubMed] [Google Scholar]
- 19.Boeckxstaens G. Mast cells and inflammatory bowel disease. Current opinion in pharmacology. 2015;25:45–49. doi: 10.1016/j.coph.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Nolte H, Spjeldnaes N, Kruse A, Windelborg B. Histamine release from gut mast cells from patients with inflammatory bowel diseases. Gut. 1990;31(7):791–794. doi: 10.1136/gut.31.7.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox CC, Lazenby AJ, Moore WC, Yardley JH, Bayless TM, Lichtenstein LM. Enhancement of human intestinal mast cell mediator release in active ulcerative colitis. Gastroenterology. 1990;99(1):119–124. doi: 10.1016/0016-5085(90)91238-2. [DOI] [PubMed] [Google Scholar]
- 22.Winterkamp S, Weidenhiller M, Otte P, Stolper J, Schwab D, Hahn EG, et al. Urinary excretion of N-methylhistamine as a marker of disease activity in inflammatory bowel disease. Am J Gastroenterol. 2002;97(12):3071–3077. doi: 10.1111/j.1572-0241.2002.07028.x. [DOI] [PubMed] [Google Scholar]
- 23.Bene L, Sapi Z, Bajtai A, Buzas E, Szentmihalyi A, Arato A, et al. Partial protection against dextran sodium sulphate induced colitis in histamine-deficient, histidine decarboxylase knockout mice. J Pediatr Gastroenterol Nutr. 2004;39(2):171–176. doi: 10.1097/00005176-200408000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Martin E, Mendoza JL, Martinez C, Taxonera C, Urcelay E, Ladero JM, et al. Severity of ulcerative colitis is associated with a polymorphism at diamine oxidase gene but not at histamine N-methyltransferase gene. World J Gastroenterol. 2006;12(4):615–620. doi: 10.3748/wjg.v12.i4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neumann D, Seifert R. The therapeutic potential of histamine receptor ligands in inflammatory bowel disease. Biochem Pharmacol. 2014;91(1):12–17. doi: 10.1016/j.bcp.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Varga C, Horvath K, Berko A, Thurmond RL, Dunford PJ, Whittle BJ. Inhibitory effects of histamine H4 receptor antagonists on experimental colitis in the rat. Eur J Pharmacol. 2005;522(1–3):130–138. doi: 10.1016/j.ejphar.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 27.Schirmer B, Rezniczek T, Seifert R, Neumann D. Proinflammatory role of the histamine H4 receptor in dextrane sodium sulfate-induced acute colitis. Biochem Pharmacol. 2015;98(1):102–109. doi: 10.1016/j.bcp.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Smolinska S, Jutel M, Crameri R, O’Mahony L. Histamine and gut mucosal immune regulation. Allergy. 2014;69(3):273–281. doi: 10.1111/all.12330. [DOI] [PubMed] [Google Scholar]
- 29.Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J Surg. 1998;22(4):382–389. doi: 10.1007/s002689900401. [DOI] [PubMed] [Google Scholar]
- 30.Weigmann B, Neurath MF. Oxazolone-Induced Colitis as a Model of Th2 Immune Responses in the Intestinal Mucosa. Methods Mol Biol. 2016;1422:253–261. doi: 10.1007/978-1-4939-3603-8_23. [DOI] [PubMed] [Google Scholar]
- 31.Catana CS, Berindan Neagoe I, Cozma V, Magdas C, Tabaran F, Dumitrascu DL. Contribution of the IL-17/IL-23 axis to the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2015;21(19):5823–5830. doi: 10.3748/wjg.v21.i19.5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raithel M, Matek M, Baenkler HW, Jorde W, Hahn EG. Mucosal histamine content and histamine secretion in Crohn’s disease, ulcerative colitis and allergic enteropathy. Int Arch Allergy Immunol. 1995;108(2):127–133. doi: 10.1159/000237129. [DOI] [PubMed] [Google Scholar]
- 33.Jones NL, Roifman CM, Griffiths AM, Sherman P. Ketotifen therapy for acute ulcerative colitis in children: a pilot study. Dig Dis Sci. 1998;43(3):609–615. doi: 10.1023/a:1018827527826. [DOI] [PubMed] [Google Scholar]
- 34.Raithel M, Nagel A, Zopf Y, deRossi T, Stengel C, Hagel A, et al. Plasma histamine levels (H) during adjunctive H1-receptor antagonist treatment with loratadine in patients with active inflammatory bowel disease (IBD) Inflamm Res. 2010;59(Suppl 2):S257–258. doi: 10.1007/s00011-009-0120-9. [DOI] [PubMed] [Google Scholar]
- 35.Smolinska S, Groeger D, Perez NR, Schiavi E, Ferstl R, Frei R, et al. Histamine Receptor 2 is Required to Suppress Innate Immune Responses to Bacterial Ligands in Patients with Inflammatory Bowel Disease. Inflamm Bowel Dis. 2016;22(7):1575–1586. doi: 10.1097/MIB.0000000000000825. [DOI] [PubMed] [Google Scholar]
- 36.Gao C, Major A, Rendon D, Lugo M, Jackson V, Shi Z, et al. Histamine H2 Receptor-Mediated Suppression of Intestinal Inflammation by Probiotic Lactobacillus reuteri. MBio. 2015;6(6):e01358–01315. doi: 10.1128/mBio.01358-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fogel WA, Jochem J, Lewinski A. Influence of the H3/H4 receptor antagonist, thioperamide on regional haemodynamics in rats with trinitrobenzene sulfonic acid-induced colitis. Inflamm Res. 2007;56(Suppl 1):S21–22. doi: 10.1007/s00011-006-0510-1. [DOI] [PubMed] [Google Scholar]
- 38.Masoodi I, Tijjani BM, Wani H, Hassan NS, Khan AB, Hussain S. Biomarkers in the management of ulcerative colitis: a brief review. Ger Med Sci. 2011;9:Doc03. doi: 10.3205/000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Ueno A, Fort Gasia M, Luider J, Wang T, Hirota C, et al. Profiles of Lamina Propria T Helper Cell Subsets Discriminate Between Ulcerative Colitis and Crohn’s Disease. Inflamm Bowel Dis. 2016 doi: 10.1097/MIB.0000000000000811. [DOI] [PubMed] [Google Scholar]
- 40.Thurmond RL, Gelfand EW, Dunford PJ. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nature reviews. 2008;7(1):41–53. doi: 10.1038/nrd2465. [DOI] [PubMed] [Google Scholar]
- 41.Mudter J, Amoussina L, Schenk M, Yu J, Brustle A, Weigmann B, et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J Clin Invest. 2008;118(7):2415–2426. doi: 10.1172/JCI33227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strakhova MI, Cuff CA, Manelli AM, Carr TL, Witte DG, Baranowski JL, et al. In vitro and in vivo characterization of A-940894: a potent histamine H4 receptor antagonist with anti-inflammatory properties. Br J Pharmacol. 2009;157(1):44–54. doi: 10.1111/j.1476-5381.2009.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsumoto S, Hara T, Mitsuyama K, Yamamoto M, Tsuruta O, Sata M, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184(3):1543–1551. doi: 10.4049/jimmunol.0801217. [DOI] [PubMed] [Google Scholar]
- 44.Leite-de-Moraes MC, Diem S, Michel ML, Ohtsu H, Thurmond RL, Schneider E, et al. Cutting edge: histamine receptor H4 activation positively regulates in vivo IL-4 and IFN-gamma production by invariant NKT cells. J Immunol. 2009;182(3):1233–1236. doi: 10.4049/jimmunol.182.3.1233. [DOI] [PubMed] [Google Scholar]
- 45.Simon T, Laszlo V, Lang O, Buzas E, Falus A. Histamine regulates relevant murine dendritic cell functions via H4 receptor. Front Biosci (Elite Ed) 2011;3:1414–1424. doi: 10.2741/e343. [DOI] [PubMed] [Google Scholar]
- 46.Dunford PJ, O’Donnell N, Riley JP, Williams KN, Karlsson L, Thurmond RL. The histamine H4 receptor mediates allergic airway inflammation by regulating the activation of CD4+ T cells. J Immunol. 2006;176(11):7062–7070. doi: 10.4049/jimmunol.176.11.7062. [DOI] [PubMed] [Google Scholar]
- 47.Desai P, Thurmond RL. Histamine H(4) receptor activation enhances LPS-induced IL-6 production in mast cells via ERK and PI3K activation. Eur J Immunol. 2011;41(6):1764–1773. doi: 10.1002/eji.201040932. [DOI] [PubMed] [Google Scholar]
- 48.Jemima EA, Prema A, Thangam EB. Functional characterization of histamine H4 receptor on human mast cells. Molecular immunology. 2014;62(1):19–28. doi: 10.1016/j.molimm.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Rahman MA, Inoue T, Ishikawa T, Yatsuzuka R, Ohtsu H, Kamei C. Involvement of chemical mediators in nasal allergic responses of HDC-KO mice. European journal of pharmacology. 2007;567(3):245–251. doi: 10.1016/j.ejphar.2007.01.082. [DOI] [PubMed] [Google Scholar]
- 50.Hofstra CL, Desai PJ, Thurmond RL, Fung-Leung WP. Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. The Journal of pharmacology and experimental therapeutics. 2003;305(3):1212–1221. doi: 10.1124/jpet.102.046581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.