

Abstract
Rett syndrome (RTT), an X-linked dominant neurodevelopmental disorder caused by mutations in MECP2, is associated with a peculiar breathing disturbance exclusively during wakefulness that is distressing, and can even prompt emergency resuscitation. Through the RTT Natural History Study, we characterized cross sectional and longitudinal characteristics of awake breathing abnormalities in RTT and identified associated clinical features. Participants were recruited from 2006 to 2015, and cumulative lifetime prevalence of breathing dysfunction was determined using the Kaplan-Meier estimator. Risk factors were assessed using logistic regression. Of 1205 participants, 1185 had sufficient data for analysis, including 922 females with classic RTT, 778 of whom were followed longitudinally for up to 9.0 years, for a total of 3944 person-years. Participants with classic or atypical severe RTT were more likely to have breathing dysfunction (nearly 100% over the lifespan) compared to those with atypical mild RTT (60-70%). Remission was common, lasting 1 year on average, with 15% ending the study in terminal remission. Factors associated with higher odds of severe breathing dysfunction included poor gross and fine motor function, frequency of stereotypical hand movements, seizure frequency, prolonged corrected QT interval on EKG, and two quality of life metrics: caregiver concern about physical health and contracting illness. Factors associated with lower prevalence of severe breathing dysfunction included higher body mass index and head circumference Z-scores, advanced age, and severe scoliosis or contractures. Awake breathing dysfunction is common in RTT, more so than seizures, and is associated with function, quality of life and risk for cardiac dysrhythmia.
Keywords: Rett syndrome, MECP2, periodic breathing, dysautonomia, Natural history study
Introduction
Rett syndrome is a debilitating neurodevelopmental disorder caused by mutations in MECP2 and characterized by a constellation of neurological and systemic symptoms that can present as early as the first year of life.[1-5] Individuals with Rett syndrome undergo developmental regression and lose expressive language and hand then cycle through periods of worsening and improvement, most of which are associated with waxing and waning comorbidities such as gastrointestinal dysfunction, seizures, and awake breathing dysfunction.[6-8] Among the varied clinical manifestations associated with Rett syndrome, awake breathing disturbances are troublesome for caregivers to tolerate and complex for clinicians to characterize and manage. Breathing abnormalities in Rett syndrome were noted in passing by Andreas Rett,[9] and Hagberg et al., found “episodic hyperpnea” in 66%.[10] After 1983, Rett syndrome was widely recognized, and Lugaresi et al., described polygraphic findings in 4 girls with alternating hyperpnea and apneic episodes associated with cyanosis and syncope, exacerbated by stress during wakefulness and resolving during sleep.[11] They postulated that this phenomenon was the opposite of that in congenital hypoventilation syndrome, and sharply contrasted from self-induced syncopal behavior resulting from the Valsalva maneuver in children with other developmental disorders.
Aside from these cross-sectional observations, the natural history of the awake breathing dysfunction in Rett syndrome remains poorly characterized. Notably, cardiorespiratory dysfunction may account for 26% to 47% of sudden, unexpected Rett syndrome-associated deaths.[12-15] As many as 14 types of breathing abnormalities have been identified in Rett syndrome that can generally be categorized into two groups: hyperventilation (shallow, fast, and/or forceful breathing), and breath-holding (apnea, Valsalva and/or apneustic breathing).[16-22] Breathing abnormalities can be further categorized through polygraphy; however, several types of dysrhythmia can present in the same individual, adding to the challenge of classifying the condition.[20] Further, although nearly all reports describe breathing abnormalities in Rett syndrome patients as disappearing during sleep, some describe these during both wakefulness and sleep.[7, 11, 16, 23-25] Although caregivers often report that breathing dysfunction worsens or improves spontaneously, the longitudinal course of breathing dysfunction and the risk factors associated with these fluctuations remain unknown.
Specific categorization of breathing dysfunction is rarely pursued in North American Rett syndrome clinics.[26] Currently no evidence-based treatments for breathing disturbances in Rett syndrome exist; however, improved breathing patterns with specific treatments suggested by several case reports may justify such detailed categorization.[27-30] Moreover, several clinical trials have used outcome measures such as the apnea index, oxygen saturation, and percentage of time with disorganized breathing to document change in respiratory function.[31, 32]
To understand cross-sectional and longitudinal characteristics of awake breathing abnormalities in Rett syndrome, we analyzed patient data on age of onset, evolution, duration, and severity of breathing disturbances in Rett syndrome obtained from the Rett Syndrome Natural History Study. Because the patterns of hyperventilation and breath-holding cease during sleep, “breathing abnormalities” hereafter refers to “awake breathing abnormalities”. We also elucidated associations between breathing disturbances and Rett syndrome-associated clinical features, MECP2 mutations, diagnosis, mortality, and quality of life to gain insights into predictors and consequences of breathing disturbances.
Materials and Methods
Participants
Participants were recruited, as described previously,[33, 34] from 2006 to 2015 through the multicenter Rett Syndrome Natural History Study at one of eight US sites and evaluated every 6 to 12 months. The Rett Syndrome Natural History Study consortium is part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, National Center for Advancing Translational Sciences. Diagnosis of Rett syndrome phenotype and epilepsy was performed by a study neurologist or geneticist (DGG, WEK, JLN, AKP, and SAS) with extensive clinical experience in Rett syndrome. Consensus criteria[35, 36] were used to categorize participants as having classic Rett syndrome, atypical Rett syndrome, or a mutation in MECP2 but not fulfilling clinical criteria for Rett syndrome (hereafter MECP2 mutation without Rett syndrome). All participants had MECP2 testing; participants with clinical Rett syndrome were included even if they lacked a mutation. Further details have been published previously, including age of symptom onset and diagnosis, and age and cause of death.[12, 33, 34, 37] History of onset and frequency of awake breathing dysfunction and associated comorbidities was recorded at the first encounter and updated at each subsequent visit. Breathing during sleep is not discussed in this manuscript. Demographic data included race and ethnicity, type of residence, and parental age. Socioeconomic data (median income and population density) were estimated using US census data based on postal address. A study physician completed a full neurological examination and an anthropometrist recorded somatic measurements.
The Motor Behavioral Assessment and Clinical Severity Score[38, 39] were administered at each visit to characterize severity of abilities, dysfunction, and comorbidities. Quality of life (QOL) was assessed for participants using the Child Health Questionnaire (CHQ-50), and for caregivers using the 36-item Short Form Health Survey (SF-36).
Parents and even physicians are often unable to distinguish categories of disordered breathing without special equipment;[7] therefore, breathing abnormalities were classified into hyperventilation (fast or deep breathing), and breath-holding (apnea, apneustic breathing, and Valsalva). Additional anomalies, such as air-swallowing and puffing were also documented. Both caregivers and physicians indicated whether abnormalities were present, and physicians estimated the frequency based on the percentage of the visit that each abnormality was present. If an abnormality began before the initial visit, age of onset was based on parental recall using a structured interview. Parents were encouraged to review home videos prior to the visit; physician documentation was used when available. If an abnormality began or ceased between two visits, the midpoint between visits was used for age of onset or cessation. Breathing disturbances were scored at a visit if present on exam or by history.
Experimental Design
Participants were analyzed in separate groups based on clinical and molecular differences. First, participants with clinical Rett syndrome were divided into classic and atypical based on diagnostic criteria. Those with MECP2 mutations without the Rett syndrome phenotype (non-Rett syndrome) were analyzed as a separate group. Those with atypical Rett syndrome were divided into mild and severe groups based on clinical severity.[2] The Clinical Severity Score in atypical Rett syndrome has a bimodal distribution, suggesting that two distinct atypical groups exist; when the breathing variable is excluded (to avoid dependent associations), the nadir between the two distributions is at 20. These two groups were divided based on this nadir as follows: an “atypical severe” category, characterized by individuals with profound developmental delay who achieve few early skills, so have fewer skills to lose during the regression period; and an “atypical mild” category in which individuals acquire language and hand use with mild or no delay, and do not experience regression in one of these domains. Ambulation in all participants was categorized in a binary (able to stand or walk, unable to stand or walk) and ordinal fashion (based on Clinical Severity Score). Other characteristics and comorbidities (e.g., seizures) were characterized based on frequency in interval since last visit or using specific parameters. Socioeconomic status was categorized into ordinal groups based on comparison to the national median income. Corrected QT interval on EKG was categorized into normal (≤450), borderline (451-≤470) and abnormal (>470). Growth parameters (height, weight, body mass index, head circumference) were categorized using both normative and Rett syndrome-specific z-scores.[34, 40, 41] The standard cutoffs of +/- 2 standard deviation (approximating 2nd and 98th percentiles) were used for normative charts and additional percentiles on Rett syndrome charts were tested, in keeping with recent recommendations.[42] Mutations of MECP2 in Rett syndrome were categorized in terms of average phenotypical severity: mild (R133C, R294X, R306C, and 3’ truncations), intermediate (T158M and other mutations), and severe (R106W, R168X, R255X, R270X, early truncations, and large deletions).[43]
Occurrence of breathing dysfunction was defined as earliest detection by a caregiver or a physician, and cessation was defined as failure of both caregiver and physician to note the abnormality. The four characteristic breathing patterns – hyperventilation, breath-holding, air-swallowing, and puffing – were each characterized in a binary fashion (those with or without the abnormality) and their frequency was defined in an ordinal fashion based on the proportion of the study visit during which the breathing dysfunction was evident; the presence of cyanosis was also recorded (Table 1). The breathing disturbance was considered active if present within the past 6-month interval. No definition for remission of a breathing abnormality has been universally accepted; therefore, we defined remission as at least 6 months without the breathing dysfunction, and subjects were categorized based on pattern of remission and relapse (Supplementary Table). We adapted the terminology “remission” commonly used in intractable epilepsy, since the pattern of breathing dysfunction is similar. Terminal remission was defined as absence of breathing dysfunction for at least 6 months at the last recorded evaluation. Most individuals had breathing dysfunction at some point, so outcome was classified based on frequency of the behavior during examination. Breathing dysfunction was classified as “severe” if hyperventilation or breath-holding occupied more than 50% of wakefulness. When breathing dysfunction is present, it dominates other behaviors and limits normal activity; breathing dysfunction this frequent would be expected to interfere with activities of daily living.
Table 1.
Breathing dysfunction based on diagnosis in Rett syndrome and related disorders.
| Diagnosis | N | Years in study (median, IQR) |
Any Hyper- ventilation by history or exam |
Hyper- ventilation 25% of time (exam) |
Hyper- ventilation 50% of time (exam) |
Hyper- ventilation 75% of time (exam) |
Hyper- ventilation 100% of time (exam) |
Any Breath- holding by history or exam |
Breath- holding 25% of time (exam) |
Breath- holding 50% of time (exam) |
Breath- holding 75% of time (exam) |
Breath- holding 100% of time (exam) |
Cyanosis Present |
Aerophagia Present |
Puffing Air or Saliva Present |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Classic Rett syndrome, female | 922 | 7.1, 4.3–8.5 | 65% | 30% | 15% | 6% | 4% | 82% | 38% | 21% | 7% | 4% | 2% | 56% | 65% |
| Atypical mild Rett syndrome, female | 73 | 5.8, 4.1–7.8 | 38% | 20% | 6% | 3% | 3% | 57% | 36% | 9% | 2% | 2% | 1% | 30% | 47% |
| Atypical severe Rett syndrome, female | 87 | 7.2, 4.5–8.4 | 62% | 28% | 14% | 5% | 4% | 78% | 47% | 10% | 4% | 4% | 3% | 47% | 65% |
| MECP2 mutation without Rett syndrome, female | 43 | 5.6, 3.3–8.2 | 11% | 5% | 1% | 1% | 0% | 11% | 3% | 0% | 0% | 0% | 0% | 6% | 9% |
| CDKL5, atypical severe Rett syndrome, female | 2 | 4.4–4.6 | 20% | 20% | 0% | 0% | 0% | 30% | 20% | 0% | 0% | 0% | 0% | 60% | 70% |
| Classic Rett syndrome, male | 1 | 4.4 | 100% | 60% | 40% | 0% | 0% | 100% | 40% | 60% | 0% | 0% | 0% | 80% | 0% |
| Atypical severe Rett syndrome, male | 1 | 0.3 | 0% | 0% | 0% | 0% | 0% | 100% | 0% | 0% | 0% | 0% | 0% | 100% | 0% |
| MECP2 mutation without Rett syndrome, male | 20 | 4.4, 2.3–6.1 | 33% | 11% | 1% | 4% | 0% | 63% | 17% | 6% | 6% | 22% | 22% | 40% | 35% |
| MECP2 duplication, male | 28 | 4.8, 3.7–7.0 | 12% | 9% | 1% | 0% | 0% | 25% | 20% | 1% | 0% | 0% | 0% | 30% | 27% |
| MECP2 duplication, female | 8 | 5.6, 3.8–8.4 | 10% | 10% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 5% | 38% |
| Total | 1184 | 6.9, 4.2–8.4 | 60% | 28% | 14% | 6% | 3% | 77% | 37% | 19% | 6% | 4% | 2% | 52% | 61% |
Statistical Analysis
Descriptive analyses were performed using mean ± SD (standard deviation) and median and interquartile range for continuous variables, frequency and percentage for categorical variables. Point prevalence (proportion with active breathing dysfunction at a particular age), period prevalence (proportion with breathing dysfunction over a certain period of time), and incidence of new onset of breathing dysfunction were calculated in those with Rett syndrome. Cumulative risk of developing breathing dysfunction was calculated using the Kaplan-Meier estimator, and the bivariate associations between cumulative risk and diagnoses, various clinical characteristics, and mutation group were tested using the log rank test. Logistic regression was used to assess the independent prognostic value for developing breathing dysfunction of Rett syndrome-relevant parameters, including MECP2 mutation group, growth, ability to walk, scoliosis, seizures, sleep disturbance, hand function, verbal and nonverbal language, frequency of stereotypies, muscle tone, reflexes, autonomic dysfunction, race, ethnicity, and socioeconomic status. Statistical analyses were performed using ArcMap Editor, Address Coder Premium, SPSS and SAS.[44-47]
Human Studies Approval
Each site maintained Institutional Review Board approval, and all data were verified by the study clinician. Parental approval for study conduct and publication of results was obtained before enrollment. The study has been registered with ClinicalTrials.gov: NCT00299312 since March 3, 2006.
Results
Overall, 1205 individuals were enrolled in the Rett Syndrome Natural History Study. Diagnosis or date of birth could not be verified for 21. Of the remaining 1185 individuals, the mean age of recruitment was 10.2 years and median age of recruitment was 6.8 years (interquartile range 3.6 to 13.9 years). The youngest subject enrolled was 0.7 years old, and the oldest was 66.5 years. The final analysis focused on 778 with classic Rett syndrome followed longitudinally for up to 9.0 years (median 5.5y, interquartile range 3.2y-7.5y) and a median of 5 visits each, for a total of 3944 person-years. Due to scarcity, only summary statistics are provided for breathing dysfunction in males with Rett syndrome or MECP2 mutation without Rett syndrome, participants with MECP2 duplication and the two individuals with atypical severe Rett syndrome due to CDKL5 mutation (Table 1, Supplementary Table). These individuals were excluded from all regression analyses. The remaining cohort of 1125 female participants included 922 with classic Rett syndrome, 73 with atypical mild Rett syndrome, 87 with atypical severe Rett syndrome, and 43 with a pathogenic mutation in MECP2 but without clinical Rett syndrome. Median age of diagnosis in classic Rett syndrome was 2.7 years (interquartile range 2.0-4.1).[37] Some subjects were only seen once (cross-sectional cases with retrospective data only), but most (87.7%, n = 985) were followed longitudinally, including 84.4% (n = 778) of those with classic Rett syndrome. None born after 1997 lived in a group home or institution. The proportion of those older than 18 years who lived in a group home was 7.3%, and in an institution 1.2%. Fifty-two of the total cohort died before the end of the study (4.6%). Most deaths were due to cardiorespiratory issues, and survival for both classic and atypical Rett syndrome was greater than 70% at 45 years.[12] Demographic and socioeconomic data were published in preceding natural history study manuscripts.[12, 48]
Breathing dysfunction in Classic Rett syndrome
At recruitment, 51.6% (466/903) of parents reported a history of hyperventilation, 67.1% (605/902) a history of breath-holding, and 47.2% (435/902) a history of air-swallowing during wakefulness. The median age of onset based on history was 3.0 y (interquartile range 2.0 – 4.0 y) for both hyperventilation and breath-holding, and 2.5 y (interquartile range 1.8 – 4.0 y) for air-swallowing. Breathing dysfunction was highly variable: some subjects developed breathing abnormalities for the first time, some who were in remission relapsed, and some with active breathing dysfunction experienced remission (Supplementary Table). The proportion with breathing dysfunction varied based on age, and peaked for both hyperventilation and breath-holding between 6 to 11 years of age (Fig. 1). However, participants were almost equally likely to experience remission of breathing dysfunction as to experience relapse at any age (Fig. 2). The one exception was the higher likelihood of remission of hyperventilation between ages 11-13 years.
Figure 1. Cross-sectional point prevalence of breathing dysregulation in classic Rett syndrome by age range.

Both hyperventilation and breath-holding reach peak cross-sectional prevalence in late childhood. Although hyperventilation frequency decreases with age, breath-holding for more than 50% of the clinic visit remains common in older individuals.
Figure 2. Remission and relapse of breathing dysfunction in classic Rett syndrome by age range.

At all ages the possibility of spontaneous emergence or remission of hyperventilation or breath-holding exists. In individual age groups, the odds of either emergence and remission of breathing dysfunction ranges from approximately 5% to 20%.
Among those who entered the study without a history of breathing dysfunction, 70% (299/428) developed hyperventilation, 82% (239/293) developed breath-holding, and 59% (273/460) developed air-swallowing. Age of onset ranged from 0.1 to 48.9 years for the 770 participants with hyperventilation, 0.1 to 66.2 years for the 851 participants with breath-holding, and 0.1 to 66.2 years for the 873 participants with either hyperventilation or breath-holding. Estimates of lifetime and age-specific proportions with breathing dysfunction were higher when calculated using Kaplan-Meier survival statistics, and surpass 90% for both (below). Many experienced remission of breathing dysfunction by the end of the study; therefore, point prevalence at annual visits ranged from 53.8% to 68.0% for hyperventilation, 76.6% to 84.3% for breath-holding, 48.7% to 63.1% for air-swallowing, and 49.2% to 68.3% for air-puffing. Severe hyperventilation or breath-holding (more than 50% of the visit) was uncommon at most visits (Fig. 1).
MECP2 Mutation, Diagnosis, and Breathing Dysfunction
Among 922 participants with classic Rett syndrome, 892 had informative data on both historical and current breathing dysfunction, and among these 864 (96.8%) had mutations in MECP2. The eight common point mutations made up 57.2% of these. Presence of breathing dysfunction was not associated with specific mutation using χ-square testing (Table 2). Moreover, severity was not associated with specific mutation in regression analysis. Mutation category was weakly associated with breath-holding onset, which was slightly delayed in the Mild Mutation group (p = .02, Fig. 3f). The only genetic factor strongly associated with breathing was the presence of a MECP2 mutation: both hyperventilation and breath-holding occurred earlier in those with a MECP2 gene mutation compared to those without a mutation (p < .01, Fig. 3c, Fig. 3d). Notably, in atypical Rett syndrome, the atypical severe group has a significantly higher proportion of individuals with MECP2 mutation (28/87) compared to the atypical mild group (10/73, p = .006). Presence of a MECP2 mutation didn’t influence breathing dysfunction in Atypical Mild Rett Syndrome, but those with Atypical Severe Rett Syndrome who lacked a mutation had a lower proportion of breathing dysfunction (p = .001).
Table 2.
MECP2 mutation types in classic Rett syndrome and proportion with hyperventilation, breath-holding, and severe breathing dysfunction.
| Type of MECP2 mutation |
R106W | R133C | T158M | R168X | R255X | R270X | R294X | R306C | C-terminal Truncation |
Large Deletion |
No mutation found |
Other mutation |
Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ever had hyperventilation | 19 (79.2%) | 32 (76.2%) | 89 (90.8%) | 86 (90.5%) | 74 (86.0%) | 39 (76.5%) | 47 (85.5%) | 50 (84.7%) | 63 (80.8%) | 62 (79.5%) | 18 (64.3%) | 165 (83.8%) | 744 (83.5%) |
| Ever had breath-holding | 23 (95.8%) | 38 (90.5%) | 94 (95.9%) | 90 (94.7%) | 80 (93.0%) | 48 (94.1%) | 49 (89.1%) | 54 (91.5%) | 71 (91.0%) | 76 (97.4%) | 25 (89.3%) | 181 (91.9%) | 829 (93.0%) |
| Severe breathing dysfunction | 10 (41.7%) | 20 (47.6%) | 50 (50.5%) | 35 (36.8%) | 38 (44.2%) | 18 (35.3%) | 29 (52.7%) | 19 (32.2%) | 35 (44.9%) | 37 (47.4%) | 6 (21.4%) | 82 (41.6%) | 379 (42.5%) |
| Total | 24 | 42 | 99 | 95 | 86 | 51 | 55 | 59 | 78 | 78 | 28 | 197 | 892 |
Figure 3. Cumulative Proportion of Hyperventilation and Breath-holding in Classic and Atypical Rett Syndrome.

The proportion of individuals who experienced hyperventilation and breath-holding overall approached 100% over the lifespan. However, diagnostic category (3a, 3b) and presence of a MECP2 mutation in classic Rett syndrome (3c, 3d) were associated with differences in cumulative risk of breathing dysfunction, whereas specific mutation type (3e, 3f) was not.
Course and predictors of breathing dysfunction in Classic and Atypical Rett syndrome
Classic Rett Syndrome
Based on Kaplan-Meier plots, in classic Rett syndrome both hyperventilation and breath-holding reached a proportion of nearly 100% over the lifespan (Fig. 3). Age of onset of breathing dysfunction increased steeply during early childhood and then began to plateau: the majority developed breath-holding or hyperventilation before age four, and 75% did so by age 8.7 years for hyperventilation and 5.6 years for breath-holding.
Weight, height, and body mass index were measured in 890 participants and head circumference in 892. Because most individuals with Rett syndrome have low anthropometric values, comparisons were made using Z-scores based on both normative and Rett syndrome-specific growth references. For Rett syndrome Z-scores, typical or low measurements for Rett syndrome (≤ 70th percentile) were compared to those with high measurements for Rett syndrome (>70th percentile). Individuals with a normal weight for Rett syndrome were more likely to have breath-holding (95.5% or 598/626) compared to those with high weight for Rett syndrome (90.2% or 238/264; p = .002), although both groups were equally likely to have hyperventilation. Height was not independently associated with hyperventilation or breath-holding. However, participants with typical or low body mass index for Rett syndrome were more likely to exhibit hyperventilation (87.6% or 536/612) and breath-holding (96.1% or 588/612) than those with high body mass index for Rett syndrome (hyperventilation: 80.2% or 223/278, p = .004; breath-holding: 89.2% or 248/278, p < .001). Considering head circumference, participants with a normal or low head circumference for Rett syndrome were more likely to have breath-holding (95.8% or 565/590) compared to those with high head circumference for Rett syndrome (90.4% or 273/302; p = .001), although both groups were equally likely to have hyperventilation.
Global Clinical Severity Scores were higher in those with breathing dysfunction. Median Clinical Severity Score (without including the breathing item) was 16.5 (interquartile range 13-24.5) in those who never had breathing dysfunction and 22.0 (interquartile range 17-28) in those with breathing dysfunction (U = 23,016, n1 = 42, n2 = 848, p = .001). Similarly, the median Motor Behavioral Assessment (without including the breathing items) was 34.5 (interquartile range 28-48.25) in those who never had breathing dysfunction and 46.0 (interquartile range 38-57) in those with breathing dysfunction (U = 24,451, n1 = 42, n2 = 850, p < .001). Because the Clinical Severity Score and Motor Behavioral Assessment are composite measures, we examined the individual severity components using logistic regression with presence of severe breathing dysfunction as the dependent variable. Severe breathing dysfunction was present in 42.3% of subjects at some point during the study (379/897). Poor walking ability, limited hand use, frequency of stereotypies, dystonia, axial hypertonia, hyperreflexia, autonomic dysfunction (dysregulation of peripheral vasoconstriction), and seizure frequency were associated with higher odds of severe breathing dysfunction, while age, severity of scoliosis, and increased contractures were independently associated with lower odds of severe breathing dysfunction (Table 3). When quality of life was examined, caregiver concern about the child’s physical health and contracting illness were associated with higher odds of severe breathing dysfunction. Numerous factors were not independently associated with severe breathing dysfunction, including race, ethnicity, MECP2 mutation category, age of diagnosis of Rett syndrome, age of onset of breathing disturbance, achievement of developmental milestones, verbal and nonverbal communication, number of types of stereotypical movements, bone fractures, frequency of hospitalization, socioeconomic status, and parental quality of life. Notably, presence of corrected QT interval prolongation was independently associated with severe breathing dysfunction in regression models.
Table 3.
Subject characteristics and Quality of Life items associated with severe breathing dysfunction. Results of logistic regression analysis.
| Variable | N | Odds ratio | 95% Confidence Interval | Sig. |
|---|---|---|---|---|
| Poor hand function | 835 | 2.0 | 1.3–2.9 | .001 |
| Impaired mobility (standing, walking) | 835 | 1.7 | 1.2–2.6 | .008 |
| Frequency of stereotypical movements | 835 | 1.6 | 1.1–2.4 | .024 |
| Dystonia severity | 835 | 1.5 | 1.2–1.8 | <.001 |
| Hyperreflexia | 835 | 1.4 | 1.1–1.8 | .002 |
| Dysautonomia (cold, discolored extremities) | 835 | 1.4 | 1.2–1.7 | <.001 |
| Axial hypertonia | 835 | 1.2 | 1.1–1.4 | .002 |
| Seizure frequency | 835 | 1.2 | 1.1–1.3 | .002 |
| Age | 835 | 1.0 | 0.9–1.0 | .005 |
| Number of joint contractures | 835 | 0.8 | 0.7–0.9 | .005 |
| Scoliosis severity | 835 | 0.7 | 0.7–0.8 | <.001 |
| Emotional worry about physical health | 818 | 1.2 | 1.0–1.4 | .022 |
| Worry about contracting frequent illness | 818 | 1.2 | 1.0–1.3 | .010 |
| EKG corrected QT interval abnormality | 513 | 2.0 | 1.3–2.9 | .001 |
| Global rigidity | 835 | 1.2 | 1.0–1.4 | .053 |
Non-significant results or results that trended to significant association are italicized.
Patterns of remission and relapse for breathing dysfunction (overall) were determined in the 778 individuals followed prospectively, and three distinct groups emerged based on (i) no history of breathing dysfunction and no emergence during the study, (ii) relapse and remission of breathing dysfunction, and (iii) intractable breathing dysfunction throughout the study (Fig. 4). Patterns of breathing dysfunction relapse and remission varied over 9 years of follow-up, and participants suffered from recurrent relapses and remissions (Supplementary Table). Although the average duration of remission was only 1 year, many experienced prolonged remission up to 8.1 years. Among the 773 individuals with active breathing dysfunction who were followed for more than 1 year after onset, 146 (18.9%) had a remission lasting at least 1 year. Likewise, 8.9% of those followed for more than 2 years (61/684) had a 2-year remission, and 2.9% of those followed for more than 5 years (12/416) had a 5-year remission. At last contact, 14.8% of those with a history of breathing dysfunction achieved terminal remission (Supplementary Table).
Figure 4. Patterns of breathing dysfunction, remission, and relapse in classic Rett syndrome.

Although very few individuals with classic Rett syndrome never experienced breathing dysfunction (green), a large proportion experienced periods of remission (blue). The largest group experienced breathing dysfunction without periods of remission.
By integrating the three patterns of breathing dysfunction over the lifespan, fluctuations in breathing dysfunction severity become clear (Fig. 5). This visual summary demonstrates that a small proportion (3% of those followed for more than 6 months) are free of breathing dysfunction over the entire lifespan. The two most common groups are an intractable group with continuous breathing dysfunction, representing 60% of the subjects followed longitudinally, and a relapsing-remitting group representing 37%. The relapsing-remitting group worsens from ages 3-12, whereas severe breathing dysfunction is rare in that group after age 12. However, the intractable group is prone to severe breathing dysfunction throughout the lifespan.
Figure 5. Longitudinal severity of breathing dysfunction in Classic Rett Syndrome.
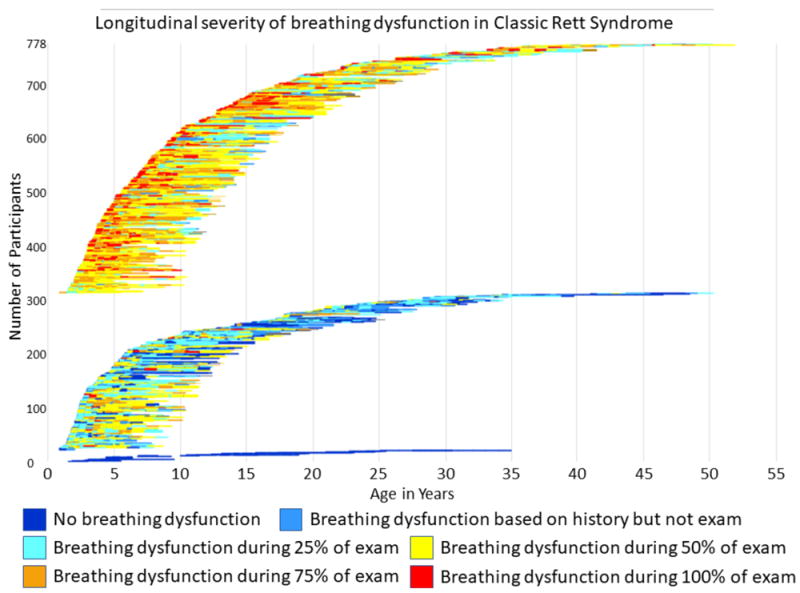
Three groups of participants were followed over time: those without breathing dysfunction (n=23), followed for as long as 7.2 years; those with remitting and relapsing episodes of breathing dysfunction, followed for as long as 8.2 years, and those with intractable breathing dysfunction throughout as long as 8.1 years of follow-up. The relapsing-remitting group exhibits a clear worsening (orange and red colors indicate presence of breathing dysfunction for 75% or more of the exam) from ages 3-12, whereas this severity of breathing dysfunction is rare in that group after age 12. However, the intractable group is prone to severe breathing dysfunction throughout the lifespan.
Atypical Rett Syndrome
The proportion of those with atypical Rett syndrome who experienced breathing dysfunction depended on the category of atypical classification. Those classified as Atypical Severe were similar to those with Classic Rett Syndrome. However, those classified as Atypical Mild showed a later onset of breathing dysfunction and a lower overall lifetime proportion: <70% ever experienced hyperventilation and <90% ever experienced breath-holding (p < .001, Fig. 3a, Fig. 3b).
Divergence of Caregiver and Physician Reporting
When we compared parental history of breathing dysfunction with physician diagnosis in Classic Rett Syndrome, physician and parent report was discordant 26.8% of the time for hyperventilation (1320/4918 observations), and 17.3% of the time for breath-holding (851/4918 observations). Because these abnormalities are paroxysmal and may not be present during physician exam, consensus was calculated for agreement about whether a breathing dysfunction was clearly present or absent. Physicians noted hyperventilation in 23.1% of cases where parents reported this was not present (1137/4918) and breath-holding in 10.1% of cases where parents reported this was not present (497/4918). When the physician found breathing dysfunction in individuals for whom parents reported no dysfunction, physicians graded this as “mild” in 14.0% for hyperventilation (688/4918) and 6.9% for breath-holding (337/4918). The percentage of discord decreased with duration of study for breath-holding, from 12.4% for those in the study for less than 2 years to 8.8% for those in the study for 2 years or longer (p = .001), but did not decrease for hyperventilation.
Discussion
Overall, the vast majority of individuals with Rett syndrome suffer from awake breathing abnormalities that follow an age-dependent pattern. Hyperventilation and breath-holding emerge by age five in two-thirds of individuals with Rett syndrome. By the 5th decade, >95% have exhibited breathing disturbances at some point, yet, only a minority experience severe breathing dysfunction. Breathing disturbances often remit over the lifespan, with 19% experiencing remission for at least 1 year, and some entering remission for over 8 years. The Rett syndrome- and MECP2 mutation-specificity of breathing abnormalities is underscored by the markedly lower frequency in males and females with MECP2 mutation but not Rett syndrome and the higher likelihood in mutation positive Rett syndrome individuals. These data add further insight into the impact and pathophysiology of breathing dysfunction in Rett syndrome, and highlight the need for additional investigations to develop relevant outcome measures, biomarkers and therapeutic interventions for these patients.
In nearly one quarter of participants, physicians were able to detect breathing dysfunction that was not recognized by parents, and in 4-7% the clinicians were unable to observe the breathing dysfunction described by parents. Over a 6-12 month span, 10-20% of individuals with breathing dysfunction will undergo spontaneous remission, and 10-20% of those without breathing dysfunction will begin to exhibit these abnormalities. Because several clinical trials have used breathing dysfunction as an outcome measure, this presents a logistical problem.[49] To be useful in a clinical trial, an outcome measure must be reliable, valid (represent the underlying construct), and associated with quality of life. Neither physician examination nor caregiver history alone provides a reliable metric of the presence of breathing dysfunction in an individual. Caregivers may under-report because they are unaware of or grow accustomed to breathing dysfunction; clinician reporting may be biased because those affected often increase the breathing dysfunction in a clinical setting, presumably due to anxiety. Moreover, current strategies for measuring severity (e.g., in-hospital polygraphy) are not clearly related to quality of life.
Because awake breathing dysfunction is one of the most prevalent comorbidities in Rett syndrome, occurring more frequently than seizures, a reliable measurement of the phenomenon is needed.[6] We demonstrate that severe breathing dysfunction is associated with poor quality of life in terms of caregiver concern over the individual’s physical health and potential frailty. Moreover, those with severe breathing dysfunction are more likely to suffer frequent stereotypic hand movements, severe motor limitations and to be non-ambulatory. Most notably, a strong association between prolonged corrected QT interval and severe breathing dysfunction in Rett syndrome may be associated with increased risk for sudden death.[50] Respiratory physiologists have suggested that one quarter of all deaths in Rett syndrome could be due to breathing dysregulation.[51] Indeed, over half of the deaths reported in the natural history study were sudden and due to unknown or presumed cardiorespiratory arrest.[12] Breathing dysfunction is therefore a meaningful target for therapeutic intervention in Rett syndrome, and research should be dedicated to bridging the gap between objective and subjective metrics of this complex clinical phenomenon. An ambulatory device could measure chest wall excursion, transcutaneous oxygenation and carbon dioxide levels, and differentiate wakefulness from sleep; such a biomarker could provide a sensitive and specific metric of change in a clinical trial when validated against physician observation and caregiver impression.
Remarkably, the cause of breathing dysfunction in Rett syndrome remains unclear. A number of neuronal mechanisms for breathing disturbances in Rett syndrome have been proposed based on different mouse models and other experimental systems.[52-54] These mechanisms include disturbances in synaptic transmission and neuromodulatory systems, notably GABAergic and glutamatergic signaling, serotoninergic and other monoaminergic signaling, and neuronal modulation by the Brain-Derived Neurotrophic Factor.[55] Moreover, oxidative stress associated with the breath-holds themselves likely contributes to produce the breathing phenotype in individuals with Rett syndrome.[18] Because of the interconnected nature of these mechanisms, aligning particular disturbances to particular mechanisms has been difficult. The complex compensatory response of neuronal networks to these disturbances results in secondary consequences, which are difficult to distinguish from the primary MeCP2 deficit. The complex pathophysiology of breathing dysfunction in Rett syndrome may help to explain inter-individual variability and intra-individual changes in the course of breathing abnormalities over the lifespan. These compensatory responses could result in improvement in some children and worsening in others.[18] Although our study offers little in terms of mechanistic explanation, participants with higher BMI Z-scores were less likely to exhibit hyperventilation and breath-holding, suggesting the mechanisms for impaired growth, believed to be in part due to dysautonomia, and breathing dysfunction share similar neurophysiological roots.
Despite the complexity of the underlying mechanisms revealed by these studies, some improvement in the breathing disturbances has been reported in animal models.[56-61] Two clinical trials, NCT02562820 and NCT02790034 have been registered to study investigational drugs to treat breathing dysfunction. The urgency to study the efficacy of therapeutics highlights the need to understand the underlying patterns of breathing dysfunction in Rett syndrome.
Nearly 30 years ago, ambitious physicians proposed that breathing dysfunction in Rett syndrome was treatable; yet, despite numerous clinical trials no clearly efficacious treatment exists Despite numerous clinical trials to treat Rett syndrome breathing dysfunction, no efficacious treatment exists. Small studies have documented improvement in respiratory dysfunction. Some practical approaches were successful; for example, gastrostomy has been used to relieve the air accumulation due to air swallowing with breath-holding, and increasing the inhaled content of CO2 has relieved hyperventilation in some patients with overbreathing.[28, 62] A study of ketogenic diet revealed dramatic improvement in hyperventilation on the diet without explanation or follow-up.[63] In a large placebo-controlled study, participants taking naltrexone experienced less breathing dysregulation than those on placebo, a finding speculated to be due to altered chemoreceptor sensitivity.[31] Case reports have reported improvement with buspirone, a serotonin 1a agonist, fluoxetine, and topiramate; however, no clinical trials have been conducted with these drugs.[29, 30, 64] None of these studies has substantially altered standard of care, and Rett syndrome experts generally agree that no effective treatment for the breathing dysfunction in Rett syndrome exists.
While breathing disturbances is one of the 11 supportive criteria used to differentiate atypical Rett syndrome from non-Rett syndrome, they are not a core component of Rett syndrome. The results of the present study support this role, since those with a MECP2 mutation who did not exhibit the clinical syndrome, were unlikely to have breathing disturbances. Notably, the opposite was true for presence of a MECP2 mutation in classic Rett syndrome. Individuals with MECP2 mutation and classic Rett syndrome or atypical severe Rett syndrome are much more likely to have breathing dysfunction compared to the other categories studied. Only 60-85% of those with atypical mild Rett syndrome express breathing dysfunction over the lifespan, and only 25-35% of those with MECP2 mutation who do not meet the clinical diagnostic criteria for Rett syndrome express breathing dysfunction. Further, paroxysmal hyperventilation and breath-holding phenotype is not unique to Rett syndrome; most notably, those with Pitt-Hopkins syndrome[65] and Joubert syndrome[66] can express periodic hyperventilation and breath-holding. In terms of risk, the presence of a MECP2 mutation in an individual with clinical Rett syndrome confers the highest risk, whereas specific mutations within MECP2 confer approximately equal risk, with the exception of slightly delayed onset of breath-holding in those with R133C, R294X, R306C and 3’ truncation mutations. These findings underscore the importance of following the consensus clinical criteria when diagnosing patients and categorizing for clinical trials, rather than clustering all individuals with a MECP2 mutation; breathing disturbances appear to be syndrome-specific rather than gene-specific.
The Rett natural history study cohort represents 10-20% of the entire US Rett syndrome population. Therefore, this should be the most accurate estimate of breathing dysfunction prevalence in Rett syndrome. Study limitations include lack of documentation of physiological breathing dysfunction and its effects on heart rate and carbon dioxide levels. Although reports of individual subjects and retrospective studies document efficacy of specific medications in treating breathing dysfunction in Rett syndrome, no systematic study has demonstrated efficacy of a specific medication to treat breathing dysfunction in Rett syndrome. No clear recommendations exist for systematic diagnosis or treatment of daytime breathing dysfunction in Rett syndrome; however, night-time breathing dysfunction should be investigated using polysomnography to exclude obstructive sleep apnea.[67]
In summary, we found that almost all individuals with classic Rett syndrome and a large proportion of those with atypical Rett syndrome eventually exhibit breathing disturbances, and although severe breathing dysfunction is relatively uncommon, these have a negative impact on quality of life. Breathing abnormalities are strongly associated with the diagnosis of Rett syndrome, and not merely with the presence of a MECP2 mutation. However, in those with Rett syndrome, the presence of a MECP2 mutation increases the likelihood of breathing dysfunction. Although both breath-holding and hyperventilation emerge in the majority by age 5 years, many do not exhibit these abnormalities until their third or fourth decade. Cross-sectionally, the prevalence of breathing disturbances in this study (60-80% across the lifespan after age two) is similar to the 66% reported by Hagberg in his original cohort.[10] However, on a longitudinal basis, severe breathing disturbance – particularly hyperventilation occupying the majority of waking hours or breath-holding that is associated with cyanosis – peaks at ages 6-11 years, and wanes in teenage years. Caregiver reports and clinician exams fail to capture the burden of breathing dysfunction over time. A critical need exists for clinical trials targeting breathing disturbances; and outcome measures will need to account for variability. Lastly, because breathing disturbances are so common in Rett syndrome, a practitioner encountering an undiagnosed individual with developmental delay and prominent hyperventilation or breath-holding during wakefulness should maintain a high index of suspicion for the diagnosis of Rett syndrome.
Supplementary Material
Acknowledgments
Support is provided by grants from the International Rett Syndrome Foundation and from the NIH (RR019478), including the Angelman, Rett, Prader-Willi syndrome consortium (U54HD61222) a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS) and the Eunice Kennedy Shriver Child Health and Human Development Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank Eric Pedrotty for statistical analysis of socioeconomic data.
Abbreviations
- CDKL5
human cyclin-dependent kinase-like 5 gene
- MeCP2
human methyl-CpG-binding protein 2
- MECP2
human methyl-CpG-binding protein 2 gene
- Mecp2
murine methyl-CpG-binding protein 2 gene
Footnotes
Financial Disclosure
Dr. Neul, Ms. Lane, Dr. Glaze, and Dr. Percy are funded by Neuren Pharmaceuticals and Newron Pharmaceuticals. Dr. Kaufmann has been a consultant on drug development trial design and/or has conducted clinical trials for Novartis, Edison, Neuren, Newron, EryDel, Marinus, Anavex, GW Pharmaceuticals, and Biohaven. He has received research support from Ipsen and Eloxx. The remaining authors have no financial relationships relevant to this article to disclose.
Conflict of Interest
No authors have conflicts of interest to disclose.
Clinical Trial Registry Number
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marschik PB, Kaufmann WE, Sigafoos J, Wolin T, Zhang D, Bartl-Pokorny KD, et al. Changing the perspective on early development of Rett syndrome. Res Dev Disabil. 2013;34:1236–9. doi: 10.1016/j.ridd.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neul JL, Lane JB, Lee HS, Geerts S, Barrish JO, Annese F, et al. Developmental delay in Rett syndrome: data from the natural history study. J Neurodev Disord. 2014;6:20. doi: 10.1186/1866-1955-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nomura Y, Segawa M, Higurashi M. Rett syndrome--an early catecholamine and indolamine deficient disorder? Brain Dev. 1985;7:334–41. doi: 10.1016/s0387-7604(85)80040-1. [DOI] [PubMed] [Google Scholar]
- 4.Nomura Y, Segawa M. Natural history of Rett syndrome. J Child Neurol. 2005;20:764–8. doi: 10.1177/08830738050200091201. [DOI] [PubMed] [Google Scholar]
- 5.Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol. 2017;13:37–51. doi: 10.1038/nrneurol.2016.186. [DOI] [PubMed] [Google Scholar]
- 6.Tarquinio DC, Hou W, Berg A, Kaufmann WE, Lane JB, Skinner SA, et al. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain. 2017;140:306–18. doi: 10.1093/brain/aww302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Julu PO, Kerr AM, Apartopoulos F, Al-Rawas S, Engerstrom IW, Engerstrom L, et al. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child. 2001;85:29–37. doi: 10.1136/adc.85.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Motil KJ, Caeg E, Barrish JO, Geerts S, Lane JB, Percy AK, et al. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr. 2012;55:292–8. doi: 10.1097/MPG.0b013e31824b6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rett A. Cerebral atrophy associated with hyperammonaemia. New York: North-Holland Publishing Co; 1977. [Google Scholar]
- 10.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–9. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 11.Lugaresi E, Cirignotta F, Montagna P. Abnormal breathing in the Rett syndrome. Brain Dev. 1985;7:329–33. doi: 10.1016/s0387-7604(85)80039-5. [DOI] [PubMed] [Google Scholar]
- 12.Tarquinio DC, Hou W, Neul JL, Kaufmann WE, Glaze DG, Motil KJ, et al. The Changing Face of Survival in Rett Syndrome and MECP2-Related Disorders. Pediatr Neurol. 2015;53:402–11. doi: 10.1016/j.pediatrneurol.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bissonnette JM, Knopp SJ. Separate respiratory phenotypes in methyl-CpG-binding protein 2 (Mecp2) deficient mice. Pediatr Res. 2006;59:513–8. doi: 10.1203/01.pdr.0000203157.31924.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerr AM, Armstrong DD, Prescott RJ, Doyle D, Kearney DL. Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):71–4. [PubMed] [Google Scholar]
- 15.Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Silvestri JM, et al. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res. 2006;60:443–9. doi: 10.1203/01.pdr.0000238302.84552.d0. [DOI] [PubMed] [Google Scholar]
- 16.Kerr AM, Julu PO. Recent insights into hyperventilation from the study of Rett syndrome. Arch Dis Child. 1999;80:384–7. doi: 10.1136/adc.80.4.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerr AM. A review of the respiratory disorder in the Rett syndrome. Brain Dev. 1992;14(Suppl):S43–5. [PubMed] [Google Scholar]
- 18.Ramirez JM, Ward CS, Neul JL. Breathing challenges in Rett Syndrome: lessons learned from humans and animal models. Respir Physiol Neurobiol. 2013;189:280–7. doi: 10.1016/j.resp.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Felice C, Signorini C, Durand T, Ciccoli L, Leoncini S, D’Esposito M, et al. Partial rescue of Rett syndrome by omega-3 polyunsaturated fatty acids (PUFAs) oil. Genes Nutr. 2012;7:447–58. doi: 10.1007/s12263-012-0285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Julu PO, Kerr AM, Hansen S, Apartopoulos F, Jamal GA. Functional evidence of brain stem immaturity in Rett syndrome. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):47–54. [PubMed] [Google Scholar]
- 21.Glaze DG, Frost JD, Jr, Zoghbi HY, Percy AK. Rett’s syndrome: characterization of respiratory patterns and sleep. Ann Neurol. 1987;21:377–82. doi: 10.1002/ana.410210410. [DOI] [PubMed] [Google Scholar]
- 22.Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Ramirez JM. Autonomic dysregulation in young girls with Rett Syndrome during nighttime in-home recordings. Pediatr Pulmonol. 2008;43:1045–60. doi: 10.1002/ppul.20866. [DOI] [PubMed] [Google Scholar]
- 23.d’Orsi G, Demaio V, Scarpelli F, Calvario T, Minervini MG. Central sleep apnoea in Rett syndrome. Neurol Sci. 2009;30:389–91. doi: 10.1007/s10072-009-0108-9. [DOI] [PubMed] [Google Scholar]
- 24.Marcus CL, Carroll JL, McColley SA, Loughlin GM, Curtis S, Pyzik P, et al. Polysomnographic characteristics of patients with Rett syndrome. J Pediatr. 1994;125:218–24. doi: 10.1016/s0022-3476(94)70196-2. [DOI] [PubMed] [Google Scholar]
- 25.Rohdin M, Fernell E, Eriksson M, Albage M, Lagercrantz H, Katz-Salamon M. Disturbances in cardiorespiratory function during day and night in Rett syndrome. Pediatr Neurol. 2007;37:338–44. doi: 10.1016/j.pediatrneurol.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Tarquinio DC. Breathless: quantifying and qualifying breathing dysfunction in Rett syndrome. In: Nues P, editor. Rett Clinics Conference Call. Web-based: International Rett Syndrome Foundation; 2014. [Google Scholar]
- 27.Julu PO, Witt Engerstrom I, Hansen S, Apartopoulos F, Engerstrom B. Treating hypoxia in a feeble breather with Rett syndrome. Brain Dev. 2013;35:270–3. doi: 10.1016/j.braindev.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Smeets EE, Julu PO, van Waardenburg D, Engerstrom IW, Hansen S, Apartopoulos F, et al. Management of a severe forceful breather with Rett syndrome using carbogen. Brain Dev. 2006;28:625–32. doi: 10.1016/j.braindev.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Andaku DK, Mercadante MT, Schwartzman JS. Buspirone in Rett syndrome respiratory dysfunction. Brain Dev. 2005;27:437–8. doi: 10.1016/j.braindev.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Gokben S, Ardic UA, Serdaroglu G. Use of buspirone and fluoxetine for breathing problems in Rett syndrome. Pediatr Neurol. 2012;46:192–4. doi: 10.1016/j.pediatrneurol.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Percy AK, Glaze DG, Schultz RJ, Zoghbi HY, Williamson D, Frost JD, Jr, et al. Rett syndrome: controlled study of an oral opiate antagonist, naltrexone. Ann Neurol. 1994;35:464–70. doi: 10.1002/ana.410350415. [DOI] [PubMed] [Google Scholar]
- 32.Khwaja OS, Ho E, Barnes KV, O’Leary HM, Pereira LM, Finkelstein Y, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci U S A. 2014;111:4596–601. doi: 10.1073/pnas.1311141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glaze DG, Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO, et al. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74:909–12. doi: 10.1212/WNL.0b013e3181d6b852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarquinio DC, Motil KJ, Hou W, Lee HS, Glaze DG, Skinner SA, et al. Growth failure and outcome in Rett syndrome: specific growth references. Neurology. 2012;79:1653–61. doi: 10.1212/WNL.0b013e31826e9a70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6:293–7. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- 36.Percy AK, Neul JL, Glaze DG, Motil KJ, Skinner SA, Khwaja O, et al. Rett syndrome diagnostic criteria: lessons from the Natural History Study. Ann Neurol. 2010;68:951–5. doi: 10.1002/ana.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarquinio DC, Hou W, Neul JL, Lane JB, Barnes KV, O’Leary HM, et al. Age of diagnosis in Rett syndrome: patterns of recognition among diagnosticians and risk factors for late diagnosis. Pediatr Neurol. 2015;52:585–91 e2. doi: 10.1016/j.pediatrneurol.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.FitzGerald PM, Jankovic J, Percy AK. Rett syndrome and associated movement disorders. Mov Disord. 1990;5:195–202. doi: 10.1002/mds.870050303. [DOI] [PubMed] [Google Scholar]
- 39.Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70:1313–21. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuczmarski MF, Kuczmarski RJ, Najjar M. Descriptive anthropometric reference data for older Americans. J Am Diet Assoc. 2000;100:59–66. doi: 10.1016/S0002-8223(00)00021-3. [DOI] [PubMed] [Google Scholar]
- 41.Cole TJ, Freeman JV, Preece MA. British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat Med. 1998;17:407–29. [PubMed] [Google Scholar]
- 42.Leonard H, Ravikumara M, Baikie G, Naseem N, Ellaway C, Percy A, et al. Assessment and Management of Nutrition and Growth in Rett Syndrome. J Pediatr Gastroenterol Nutr. 2013;57:451–60. doi: 10.1097/MPG.0b013e31829e0b65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ, Skinner SA, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51:152–8. doi: 10.1136/jmedgenet-2013-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.SAS Institute Inc. SAS Institute Inc. version 9.4 ed. Cary, NC: SAS Institute Inc.; 2016. [Google Scholar]
- 45.IBM Corp. version 22.0 ed. Armonk, NY: IBM Corp; 2013. IBM SPSS Statistics for Windows. [Google Scholar]
- 46.Esri Inc. version 10.1 ed. Redlands, CA: Esri, Inc; 2012. ArcMap Editor. [Google Scholar]
- 47.Esri Inc. version 10.1 ed. Redlands, CA: Esri, Inc; 2014. Address Coder. [Google Scholar]
- 48.Killian JT, Lane JB, Cutter GR, Skinner SA, Kaufmann WE, Tarquinio DC, et al. Pubertal development in rett syndrome deviates from typical females. Pediatr Neurol. 2014;51:769–75. doi: 10.1016/j.pediatrneurol.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katz DM, Bird A, Coenraads M, Gray SJ, Menon DU, Philpot BD, et al. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. 2016;39:100–13. doi: 10.1016/j.tins.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guideri F, Acampa M, DiPerri T, Zappella M, Hayek Y. Progressive cardiac dysautonomia observed in patients affected by classic Rett syndrome and not in the preserved speech variant. J Child Neurol. 2001;16:370–3. doi: 10.1177/088307380101600512. [DOI] [PubMed] [Google Scholar]
- 51.Julu PO, Kerr AM, Hansen S, Apartopoulos F, Jamal GA. Immaturity of medullary cardiorespiratory neurones leading to inappropriate autonomic reactions as a likely cause of sudden death in Rett’s syndrome. Arch Dis Child. 1997;77:464–5. doi: 10.1136/adc.77.5.463c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rettlike phenotype in mice. Nat Genet. 2001;27:327–31. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 53.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–6. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 54.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–7. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katz DM, Dutschmann M, Ramirez JM, Hilaire G. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. 2009;168:101–8. doi: 10.1016/j.resp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, et al. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26:10911–5. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27:10912–7. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, et al. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci. 2012;32:1803–10. doi: 10.1523/JNEUROSCI.0865-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. 2008;160:116–21. doi: 10.1016/j.resp.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 60.Abdala AP, Dutschmann M, Bissonnette JM, Paton JF. Correction of respiratory disorders in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18208–13. doi: 10.1073/pnas.1012104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 2012;32:13860–72. doi: 10.1523/JNEUROSCI.2159-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anzai Y, Ohya T. A case of effective gastrostomy for severe abdominal distention due to breathing dysfunction of Rett’s syndrome: a treatment of autonomic disorder. Brain Dev. 2001;23(Suppl 1):S240–1. doi: 10.1016/s0387-7604(01)00341-2. [DOI] [PubMed] [Google Scholar]
- 63.Haas RH, Rice MA, Trauner DA, Merritt TA, Opitz JM, Reynolds JF. Therapeutic effects of a ketogenic diet in rett syndrome. Am J Med Genet. 1986;25:225–46. doi: 10.1002/ajmg.1320250525. [DOI] [PubMed] [Google Scholar]
- 64.Goyal M, O’Riordan MA, Wiznitzer M. Effect of topiramate on seizures and respiratory dysrhythmia in Rett syndrome. J Child Neurol. 2004;19:588–91. doi: 10.1177/088307380401900804. [DOI] [PubMed] [Google Scholar]
- 65.Van Balkom ID, Quartel S, Hennekam RC. Mental retardation, “coarse” face, and hyperbreathing: confirmation of the Pitt-Hopkins syndrome. Am J Med Genet. 1998;75:273–6. doi: 10.1002/(sici)1096-8628(19980123)75:3<273::aid-ajmg9>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 66.Parisi M, Glass I. Joubert Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, et al., editors. GeneReviews(R) Seattle (WA): 1993. [PubMed] [Google Scholar]
- 67.Hagebeuk EE, Bijlmer RP, Koelman JH, Poll-The BT. Respiratory disturbances in rett syndrome: don’t forget to evaluate upper airway obstruction. J Child Neurol. 2012;27:888–92. doi: 10.1177/0883073811429859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.