

Abstract
Pulmonary lymphoproliferative neoplasms are rare lung tumors and account for <1% of all lung tumors. Among them, primary pulmonary lymphomas (PPL) constitute the majority, which include Non-Hodgkin’s lymphoma (NHL) that comprise of mucosa-associated lymphoid tissue lymphoma, diffuse large B-cell lymphomas and other rare types of NHL and lymphomatoid granulomatosis. HL, which arises secondary to contiguous spread from the mediastinum, is the rarest type of PPL. Other entities described within the umbrella of pulmonary lymphoproliferative neoplasms include pleural lymphomas and posttransplant lymphoproliferative disorders (PTLD) – which occurs in the poststem cell and organ transplant patients. These neoplasms although rare, have a favorable prognosis, which does not depend on disease resectability. Moreover, with its nonspecific presentation, diagnosis is challenging, which often leads to delayed diagnosis or misdiagnosis in many cases. Therefore, knowledge of this entity is important for the practicing pulmonologist. This review article aims to describe the clinical presentation, diagnosis and management of primarily the entities within PPL, as well as pleural lymphomas and PTLD.
KEY WORDS: Acquired immune-deficiency syndrome, lymphoma, primary pulmonary lymphoma, rare lung neoplasms
INTRODUCTION
The pulmonary lymphoid tissue is composed of pulmonary lymphatics and bronchus-associated lymphoid tissue (BALT).[1,2] BALT generally consists of submucosal lymphoid aggregates along the airways, which is most commonly at the bifurcation of bronchioles. BALT is not present at birth, or in the young healthy adult. However, they can appear or “be induced” secondary to chronic antigenic stimulation, such as in chronic infection, immunologic diseases such as connective tissue diseases and smoking.[1,2]
Primary lymphoid proliferative disorders of the lung arise from BALT – which include (a) nonneoplastic proliferations of lymphoid tissue which comprise nodular lymphocyte hyperplasia, reactive lymphoid hyperplasia, follicular bronchiolitis, and lymphoid interstitial pneumonias[2] and (b) neoplastic proliferations which include Non-Hodgkin’s lymphoma (NHL) types, HL, and lymphomatoid granulomatosis (LG). Among the NHL subtypes, extranodal marginal zone mucosa-associated lymphoid tissue (MALT) lymphomas are the most common entity.[2,3] Other NHLs within this spectrum include diffuse large B-cell lymphoma (DLBCL), intravascular B-cell lymphoma, and T-cell lymphomas.[1] These collectively comprise the group of primary pulmonary lymphomas (PPL).
Lymphomatous proliferation can involve lung in three ways: (1) hematogenous dissemination of NHL and HL (2) contiguous invasion from hilar or mediastinal nodes and (3) primary pulmonary (PP) involvement.[1,3] The first two entities describe the secondary involvement of the lung by lymphoma [Figures 1 and 2a, b] and are much more common than PP involvement.
Figure 1.

Bronchoscopy showing secondary involvement and encroachment into right mainstem bronchus from systemic diffuse large B cell lymphoma
Figure 2.
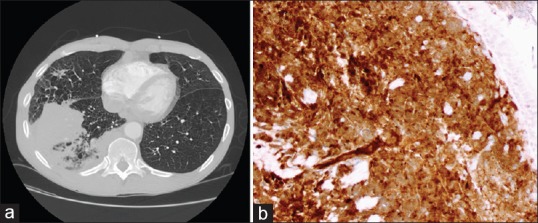
Computed tomography scan showing right lower lobe mass (a) with mediastinal adenopathy, as secondary involvement from mantle cell lymphoma. (b) Cyclin D 1 positivity on tumor cells from transbronchial biopsy, confirming Mantle cell lymphoma
PPL is defined as clonal lymphoid proliferation affecting one or both lungs (parenchyma and/or bronchi) in a patient with no extrapulmonary involvement at the time of diagnosis or the subsequent 3 months.[3,4] PPL is very rare, representing only 0.3% of all PP malignancies, <1% of all cases of NHL, and 3%–4% of extranodal NHL.[3,4] The objective of this review article is to describe the various PP lymphoproliferative neoplasms, their clinical presentations, diagnosis and management. Other pulmonary lymphoproliferative disorders such as posttransplant lymphoproliferative disorders (PTLD) and pleural lymphomas are also reviewed [Table 1].
Table 1.
Brief review of each primary pulmonary lymphoma type with clinical description, main radiographic findings, and management

MUCOSA-ASSOCIATED LYMPHOID TISSUE PRIMARY PULMONARY LYMPHOMA
Etiology
The most common type of PPL is MALT PPL that accounts for 70%–90% of diagnosed cases.[1,5,6] MALT lymphoma is a marginal zone B-cell lymphoma that represents monoclonal proliferation of lymphoid tissue in the lung and arises from a B-cell progenitor within BALT.[7] Despite the evidence of chronic antigenic stimulation leading to lymphoid hyperplasia, an association between a specific pathogen or autoimmune disease and MALT PPL has never been demonstrated.[1,6] In a large meta-analysis of patients with MALT PPL, 45% had a history of smoking, 9% had a history of exposure to toxic substances, and 19% had a known preexisting lung condition.[8,9] Approximately 10%–29% of patients diagnosed with MALT-PPL had a connective tissue disorder such as systemic lupus erythematosus, rheumatoid arthritis, Sjogren’s syndrome, or infections such as hepatitis C and human immunodeficiency virus (HIV).[1,6,8] Patients with Sjogren’s syndrome are at 6.6–44 times higher risk of developing lymphoma, of which MALT-PPL is the most common type.[10]
Clinical
The median age of MALT-PPL diagnosis is 60 years, and more recent literature suggest increased incidence in women.[9,11] About half of the patients are asymptomatic at the time of diagnosis with further investigations usually initiated after incidental abnormal pulmonary radiographs. When present, symptoms are nonspecific, such as cough, dyspnea, chest pain, and occasionally hemoptysis. The presence of fever and weight loss is particularly associated with aggressive disease forms.[3,12]
Radiography
Chest radiographic findings are not specific for MALT-PPL and vary greatly. Chest computed tomography (CT) is the imaging modality of choice. The most common findings (>70%) are multiple bilateral lung nodules with air bronchograms or areas of bronchiectasis within larger lesions.[13,14] Lesions can also be unilateral. CT attenuation can vary with lesions ranging from solid, mixed solid and ground glass, or pure ground glass.[13] Nodules or masses are in a peribronchovascular distribution with most measuring <5 cm in diameter.[3,13] Other radiological presentations described include areas of consolidation, which may be mass-like or nodular in appearance [Figure 3a]. MALT lymphoma can also infiltrate along airways resulting in small airway disease causing a mosaic attenuation pattern. Hilar and mediastinal lymphadenopathy is present in up to 30% of patients. Pleural effusions are uncommon and are seen in 10% of cases.[13,14] The CT angiogram sign, which is an enhancing vessel on a background of consolidation, may be present but is extremely nonspecific. Two retrospective analyses of histologically confirmed MALT-PPL demonstrated fluorodeoxyglucose positron emission tomography (FDG-PET) avidity in 80% of cases.[6,15] FDG-PET uptake was only significantly associated with tumor size.[15] MALT-PPL with indolent growth may have little FDG uptake; thus, FDG-PET imaging for staging remains controversial.[13]
Figure 3.

Computed tomography with mass in the right lower lobe and pleural effusion of 2 years duration. (a) The patient underwent thoracotomy and surgical excision, which demonstrated large sheets of CD 20 positive B cells (b) and confirmed as mucosa associated lymphoid tissue-primary pulmonary lymphomas. (c) H and E, under high power (×400) showing large sheets of B cells. The patient was lost to follow up
Pathology
MALT-PPL has a variable cytologic appearance, including lymphoplasmacytic, centrocyte-like, or monocytoid cells. Tumor cells spread in a lymphangitic growth pattern along bronchovascular bundles and interlobular septa [Figure 3b and c]. Immunohistochemistry (IHC) confirms a monoclonal B-cell population that stains positive for CD19, CD20, CD79a, and absence of CD5 and CD10.[3,16] If detected, CD43 co-expression with CD20, a T-cell and B-cell marker respectively, rules out a reactive process.[14,17] Flow cytometry may detect plasma cells with or without light chain restriction. Unusual features that have been reported include vascular invasion, granulomatous reaction, fibrosis, or amyloid stromal deposition.[3] Amyloid deposition can be seen in 1%–6% of cases, more commonly seen in older women.[1]
Several chromosomal translocations have been described in association with MALT lymphoma including t (11:18)/API2-MALT1, t (1:14)/BCL10-IGH, t (14:18)/IGH-MALT1, and t (3:14)/FOXP1-IGH. API2-MALT1 fusion is specific to MALT lymphoma and is the most often detected (40%) fusion abnormality in MALT-PPL.[15,18]
Diagnosis/pathology
Tissue biopsy is the gold standard for diagnosis and can be obtained through minimally invasive techniques either by bronchoscopy or CT-guided percutaneous needle biopsy, both of which have >80% diagnostic yield in recent studies.[6,12] Macroscopic views by bronchoscopy are usually normal, although abnormalities such as mucosal inflammation and bronchial stenosis may be observed.[3] In about two-thirds of MALT PPL cases, bronchoalveolar lavage (BAL) demonstrates lymphocytic alveolitis (>20% lymphocytes) which can be a specific sign when >10% B lymphocytes are seen with clonality as demonstrated by Ig gene clonal rearrangements.[3,12,13] Demonstration of fusion abnormality API2-MALT1 (t [11,18] in BAL cytology strongly supports the diagnosis. The diagnostic role of endobronchial ultrasound has not been fully evaluated. Cryobiopsy could also be a viable diagnostic tool in experienced hands.[19] Ultimately, a surgical biopsy may still be necessary to achieve a definitive diagnosis, as the tissue obtained by minimally invasive techniques may be inadequate and unable to exclude other diagnoses such as follicular bronchiolitis and lymphoid interstitial pneumonia.
Treatment/prognosis
Clinical course of MALT PPL is generally indolent, with reported 5-year overall survival (OS) rate of greater than 80%.[6,9,20] There is no standard approach to MALT PPL and therapeutic options include surgery, chemotherapy, immunotherapy, or radiotherapy. In patients who are asymptomatic and localized, a continued surveillance “watch and wait” approach may be used,[21] although radiotherapy or surgical resection may be considered depending on patient comorbidities. In locally advanced cases, management options include localized therapy such as surgical resection, radiation therapy, or systemic chemotherapy. Although localized therapy may be associated with improved progression-free survival (PFS) compared to systemic therapy – no significant difference in OS has been observed. Furthermore, PFS showed a trend in favor of patients treated with immunotherapy and immunochemotherapy versus those receiving chemotherapy alone.[9] Zucca et al. reported in MALT-PPL, the combination of rituximab plus chemotherapy may be superior to chemotherapy alone with regards to response rate and event-free survival,[9,22] which was also observed by Rummel.[9,23] Another retrospective analysis by Wei et using the combination of rituximab plus cladribine for stage IV proven unresectable MALT-PPL reported estimated 2-year PFS and 2-year OS at 80% and 100%, respectively.[11] Currently, the favored approach for advanced or metastatic MALT-PPL remains systemic chemotherapy.
Overall, the prognosis of MALT-PPL is good with 5- and 10-year survival ranging from 84% to 88%. In a study by Borie et al., age and performance status were the only two prognostic factors associated with survival.[6] The presence of an associated pleural effusion or amyloid portends a poorer prognosis.[2] As in any low-grade lymphoma, histologic transformation to diffuse large B-cell lymphoma can occur, although in a single center review by Maeshima et al., the histologic transformation was observed in 8% of MALT lymphoma, but none involved MALT-PPL.[24]
PRIMARY PULMONARY DIFFUSE LARGE B-CELL LYMPHOMA
Etiology
Diffuse large B-cell lymphoma (DLBCL) is a systemic disease in most patients. It accounts for 10%–20% of PPL and is the second-most common subtype after MALT-PPL.[2,8,25] Owing to its rapid spread to mediastinal nodes and extrathoracic sites, PP DLBCL is likely under-recognized.
About half of the PP DLBCL cases arise de novo and the other half arise from transformation of the more indolent MALT-PPL.[17] It often occurs in patients with underlying chronic infectious or inflammatory process, such as HIV infection, chronic immunosuppression, or collagen vascular disease. Among HIV-positive patients diagnosed with DLBCL, nearly all cases are Epstein–Barr virus (EBV)-positive.[1,17] PPL in this subset of patients may be related to severe immune deficiency and reactivation of latent EBV infection within the lung, as in acquired immune-deficiency syndrome (AIDS)-related primary central nervous system lymphoma.[1] There are also reported associations of PP-DLBCL diagnosis and long-term methotrexate use or solid organ transplant recipients on cyclosporine A or OKT3 immunosuppression.[3]
Clinical
The mean age of presentation is 60 years excluding HIV-positive and chronic immunosuppressed patients who can present at a younger age. Patients are usually symptomatic, with respiratory manifestations of cough, dyspnea, rarely hemoptysis, and systemic “B symptoms,” which include fever, night sweats, and weight loss.[1,3,17,25]
Radiography
PP-DLBCL can present as a single or multiple well-defined rounded solid masses on chest CT. These lesions tend to be located peripherally in the lower lobe.[8] Features of MALT-PPL and PP-DLBCL can overlap where solitary or multiple nodules or areas of consolidation can be seen. Cavitation and/or central necrosis on chest CT is seen in 50% of the cases [Figure 4a] and is a feature more common and frequent of P-DLBCL compared to MALT PPL.[8,14] Pleural effusions are more common in PP-DLBCL.[3] Hilar adenopathy may be seen. Two recent cases of PP-DLBCL described radiographic findings of consolidation of multiple pulmonary nodules with air bronchograms and halo of ground-glass shadowing at lesion margins (halo sign).[26] FDG-PET usually demonstrates metabolic activity.
Figure 4.

Computed tomography scan showing large necrotic opacity in the right upper lobe (a). Bronchoscopy with transbronchial lung biopsy revealing large population of B cells. (b) Large B cells in clusters consistent with diffuse large B-cell Lymphoma. The patient was started on cyclophosphamide, doxorubicin, vincristine and prednisone with remission achieved and is doing well at 2 years after follow-up (H and E, ×400)
Pathology
PP-DLBCL forms confluent sheets of tumor cells that destroy the normal lung parenchyma. Histologically, large sheets of dyscohesive lymphoid cells with coarse chromatin, distinct nucleoli, and abundant cytoplasm are usually described as centroblastic or immunoblastic [Figure 4b]. By IHC, tumor cells are positive for CD19, CD20, CD79a, and those of germinal center origin express CD10 and BCL-6.[1] Ki-67, proliferation index generally exceeds 40%–60%.[17,25] A particular concern in the diagnosis of DLBCL is concurrent rearrangements involving MYC and BCL2 or BCL6, termed “double hit lymphoma.” These cases show frequent extranodal involvement and are associated with poor prognosis despite aggressive chemotherapy.[17]
Diagnosis
Bronchoscopic examination is usually abnormal with budding or infiltrative stenosis of bronchi seen with possible tumoral invasion. Histologic diagnosis via minimally invasive procedures and bronchoscopy is generally easy, even with small samples due to the presence of characteristic blast-like lymphoid cells.[3] Multiple case reports demonstrating success of transbronchial lung biopsy and ultrasound-guided fine needle aspiration (FNA) in diagnosing PP-DLBCL further support this observation.[26,27,28]
Treatment/prognosis
PP-DLBCL is usually widespread by the time of diagnosis decreasing the chances of surgical resection, which can be performed in cases of localized tumors. The rate of local or distant recurrence is reported to be high and thus treatment after surgical resection is often based on combination of chemotherapy and radiation.[3,29] Chemotherapy consists of the same multi-agent regimens as those used in high-grade nodal lymphomas, including cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP).[3] A retrospective analysis of over 9,000 patients did not show a statistically significant benefit among patients with NHL who received CHOP plus rituximab (R-CHOP) versus CHOP alone, although a subset analysis demonstrated the largest impact of R-CHOP on lymphoma-related death was for DLBCL of the spleen, liver, and lung.[30]
Historically, PP-DLBCL was thought to have a worse prognosis with reported widely variable median survival times from 3 to 10 years.[3,25] However, a large case series by Neri et al. found that PP-DLBCL patients treated with conventional CHOP achieved complete response in 77 cases (94%) with 10-year PFS of 90%.[31] HIV patients and chronically immunosuppressed patients with pulmonary lymphomas have a poor prognosis usually secondary to opportunistic infections.[1]
LYMPHOMATOID GRANULOMATOSIS
Etiology
LG is a very rare extranodal neoplastic lymphoproliferative disorder that arises primarily in the lungs. First described in a case series of 80 patients by Liebow et al. in 1972, LG is a misnomer as the presence of granulomas is not typically observed and the term was assigned based on a clinical presentation similar to other granulomatoses, such as granulomatosis with polyangiitis (GPA) and eosinophilic GPA.[1] The presence of EBV involvement was demonstrated around 20 years later.[25]
LG is an angiocentric and angiodestructive disease that is driven by clonal proliferation of atypical large B-cells associated with abundant infiltrate and reactive T-cells.[14] Although part of the PPL spectrum, LG merits special consideration; the clinical features reflect systemic multiorgan disease. The current hypothesis is EBV-infected B cells release chemokines, which attract T cells and produce vascular damage. Subsequently, host impaired immunologic response permits the proliferation of EBV-infected B cells that eventually leads to the development of an independent malignant clone.[25] In addition, viral proteins that normally induce cytotoxic T-cell response, appears to be deficient in LG.[3] Lung parenchyma is the most common primary site of involvement (80%–90%) but synchronous extrapulmonary involvement is common, mainly involving skin (50%) and brain (30%).[1,12,14]
Clinical
The prevalence is unknown and much of the literature on LG derives from case series.[10] LG characteristically affects immunosuppressed patients, such as AIDS, postsolid-organ transplantation, rare hereditary T-cell deficiencies, Wiskott–Aldrich syndrome, postablation chemotherapy, autoimmune diseases, and in those with rheumatoid arthritis treated with methotrexate.[3,8] The median age at diagnosis is 30–50 years old, with a ratio of 2:1 male predominance. Rare cases of LG in immunocompetent patients have also been reported. Nearly, all patients are symptomatic (90%) for several months to years with respiratory complaints of cough and dyspnea 50%–80% of the time. Chest pain, fever, weight loss, and potentially life-threatening hemoptysis can occur.[3,12,25] Neurologic manifestations vary depending on the site of involvement.[12] A complete blood count is normal in more than 50% of cases, although lymphopenia, leukocytosis, or hypereosinophilia has been reported.[12]
Radiography
Radiography may reveal subtle nodules scattered throughout the lungs. The most common features on chest CT are multiple lung nodules ranging from 0.5 to >10 cm with preferential basal localization, occurring in approximately 80% of all cases. Anatomical-radiological studies have showed that ill-defined nodules rapidly coalesce into larger masses and undergo necrosis with resultant central cavitation, therefore mimicking granulomatosis polyangiitis or metastasis.[3] Nodules are distributed along peribronchovascular and interlobular septa.[14] FDG-PET images show avid uptake in these lesions and can also demonstrate metabolic activity in the skin, kidneys, and nodes. Central ground-glass opacities with surrounding denser consolidation (reverse halo sign) correspond to “migratory” nodules that are previous foci of active disease.[3,8,14,25] Hilar lymphadenopathy is rare.[2,25]
Pathology
Histologically, LG consists of well-circumscribed nodules of polymorphous lymphoid infiltrate involving medium-sized to small pulmonary veins and arteries with vasculitis and areas of coagulative necrosis with unremarkable surrounding lung parenchyma. The polymorphous infiltrate is composed of mainly lymphocytes that are CD3+, CD4+ T cells, occasional plasma cells and macrophages, and variable number of large atypical lymphoid cells. Morphologically, the atypical cells can resemble immunoblasts, plasmablasts, or Reed–Sternberg cells.[17,25] Well-formed granulomas are absent. The large atypical cells are B cells, positive for CD20, CD79a, CD30, and in situ hybridization for Epstein–Barr encoding region should be performed.[12]
Diagnosis
Adequate sampling of lung lesions is necessary for histologic diagnosis and proper grading of LG. Tissue samples obtained by bronchoscopy via transbronchial lung biopsy have a much lower yield due to the bronchocentric yet focal nature of LG.[1,3,12] Hypereosinophilia may be observed in the BAL but lacks specificity. Surgical lung biopsy is the preferred method of tissue sampling. However, a diagnosis from easily accessible sites such as skin may be obtained if involved.[12] Owing to the varied tumoral infiltration and EBV staining, it is recommended to biopsy all accessible sites.[12]
Treatment/prognosis
The rarity of LG precludes any therapeutic recommendations or clinical trials. Localized LG has been treated successfully with surgery or radiotherapy. Grade 1 LG [Table 2] may not require treatment and spontaneous remission has been reported.[14] Treatment for grades 1 and 2 lesions include interferon alpha-2, while grade 3 LG treatment includes high dose steroids with chemotherapy regimens such as CHOP, ICE (Ifosfamide, Carboplatin, Etoposide), and hyperCVAD (Cyclophosphamide, vincristine, doxorubicin and dexamethasone, along with methotrexate and cytarabine) or immunochemotherapy with variable success.[2,12,25]
Table 2.
Classification and subtypes of lymphomatoid granulomatosis

The natural history of LG is variable and generally associated with a poor prognosis; median survival is approximately 4 years. The clinical aggressiveness of the disease relates to the proportion of EBV + large B cells, which forms the basis of histological grading. Histologic grades 2 and 3 have poorer prognosis and higher risk of progression to malignant large B-cell lymphoma involving lymph nodes, spleen, and bone marrow.[2] Causes of death in order of frequency are respiratory insufficiency (38%–88%), hemoptysis (44%–89%), neurological complications (7%–31%), and infection (23%–38%). Favorable prognostic factors include older age, absence of symptoms, and unilateral lung involvement. Poor prognostic factors are age <25 years, neurological involvement, hepatosplenomegaly, leukopenia, persistent fever, and histologically, the number of tumoral cells and amount of necrosis.[12]
PRIMARY PULMONARY PLASMACYTOMA
Etiology
PP plasmacytoma is exceedingly rare with possibly <100 true primary lung cases reported.[25]
Clinical
PPP is seen in both sexes with equal predilection and median age of onset is 40 years. Patients are usually asymptomatic but fever, dyspnea, and hemoptysis have been reported.[3]
Radiography
The most common radiographic finding is an isolated well-circumscribed pulmonary mass or as a hilar mass with bronchial involvement.[3,25]
Diagnosis/pathology
Thoracotomy and surgical excision is necessary for diagnosis. Histologically, the tumor is composed of diffuse sheets of mature monoclonal plasma cells.[3]
Treatment/prognosis
Surgical resection is the preferred treatment for solitary tumors, with a 5-year survival of 60%.[3,25]
PULMONARY INTRAVASCULAR B-CELL LYMPHOMA
Etiology
Intravascular large B-cell lymphoma is an aggressive rare type of extranodal NHL and regarded as systemic from the outset.[1]
Clinical
Patient presentation is highly variable based on which organs are affected but primary presentation in the lungs is highly uncommon.[3]
Radiography
Pulmonary imaging usually reveals bilateral ground glass opacities, centrilobular nodules, and interstitial opacities.[3,32]
Diagnosis/pathology
Bronchoscopy with transbronchial lung biopsies can assist with diagnosis. Histology is characterized by proliferation of atypical lymphoid cells within the lumen of capillaries, arterioles, venules, and lymph ducts with little to no invasion of adjacent lung parenchyma. The intravascular invasion results in thrombotic and ischemic complications of various organs.[3]
Treatment/prognosis
Treatment is with conventional combination chemotherapy with or without rituximab, giving a complete response rate of approximately 50%.[3,32]
PRIMARY PULMONARY HODGKIN’S LYMPHOMA
Etiology
Pulmonary involvement is seen in 15%–40% of patients with HL.[33] PPHL are extremely rare, with <100 cases reported in world literature. Nodular sclerosis subtype is the most common variety of PPHL, accounting for 60%–70% of the cases. The following criteria are used to establish the diagnosis of PPHL: (1) disease must be confined to the lung (but may have minimal hilar adenopathy); (2) histologic results must be consistent with HL; and (3) exclude other pathologic conditions.[34,35]
Clinical
Clinical presentation is nonspecific, with fever, cough, and dyspnea as the most commonly reported symptoms, with a slight preponderance in young adult females (mean age 42 years).[35]
Radiography
Radiological presentations vary and may present as a single pulmonary nodule, multiple nodules, or cavitated lesions with a predilection for the upper lobes.[35,36] No radiological sign is pathognomonic for PPHL.[37]
Pathology/diagnosis
The diagnosis of PPHL can rarely be established through sputum cytology or bronchial brushings (which contain Reed–Sternberg cells).[38] Transthoracic FNA may be suggestive, however, a high index of suspicion is required, as the Reed–Sternberg cells may be misinterpreted as reactive pneumocytes.[39] Open lung biopsy with video-assisted thoracoscopy is necessary to confirm the diagnosis in the vast majority of cases.[34,40]
Treatment/prognosis
Owing to the rarity of the disease, prognostic factors are not well defined. However, multilobar involvement, age >60 years, pleural effusion, and B symptoms are associated with poor prognosis.[41] Management options include surgery in single lesions, while multi-agent chemotherapy is recommended for bilateral and diffuse disease.[41,42]
T-CELL LYMPHOMAS OF LUNG
Etiology
T-cell lymphomas, also known as non-B-cell lymphomas are extremely rare with only a handful of cases reported in world literature. Most of the reported cases are from the Far East.[25,43] Pulmonary anaplastic large cell lymphoma (P-ALCL) is the most commonly reported subtype.[25] Other T-cell lymphomas reported include PP peripheral T-cell lymphomas and NK/T cell lymphomas.[1] EBV is frequently seen in tumor cells of NK/T cell lymphomas.[44]
Clinical
While P-ALCL has a bimodal age distribution, NK/T cell lymphomas and pulmonary peripheral T-cell lymphomas have been mainly reported in elderly patients. Most patients are diagnosed at an advanced stage with systemic symptoms and are often clinically ill with fever, cough, and shortness of breath.[1,25,43]
Radiography
Radiographic findings seen include bilateral diffuse nodular lesions, mass-like consolidation, hilar adenopathy, and pleural effusion.[25,43,45]
Pathology
Histologically, P-ALCL is characterized by large, anaplastic lymphoid cells, hence the name-with variable morphology including plasmacytoid, sarcomatoid, or small cell features, which can lead to misdiagnosis. They are characteristically positive for a pan-T cell maker and CD-30, which is an important hallmark of P-ALCL.[1,25] Peripheral T-cell lymphomas and NK/T cell lymphomas in most cases have angiocentric and angiodestructive growth patterns,[45] with tumor cells positive for all T-cell markers and EBV in NK/T cell lymphomas.[1,43]
Diagnosis
Typically, the diagnostic workup starts with either transbronchial lung biopsy or transbronchial needle aspiration, which is insufficient, and open lung biopsy or lobectomy has been eventually required to make the diagnosis in most reported cases.[1,25,43]
Treatment/prognosis
The outcomes of patients with pulmonary non-B-cell lymphoma are much worse than pulmonary B-cell lymphoma. Currently, the most common treatment is surgical resection for localized cases or chemotherapy with CHOP, which has varying rates of success.[1,25,43]
PLEURAL LYMPHOMAS
All types of lymphoma can secondarily involve the pleura, with the most common ones in order of frequency being DLBCL (60%) and follicular lymphoma (20%), respectively.[46]
Primary effusion lymphoma (PEL) and pyothorax-associated lymphoma (PAL) are the only two primary high-grade lymphomas, which involve the pleura.[47] These rare types of lymphomas are likely caused by activation of B cells in the pleural cavity.[48] This chronic stimulation of B cells along with decrease in circulating T cells, may occur in the setting of acquired immunodeficiency or genetic causes and can lead to significant B cell proliferation, which, in turn, can lead to pleural lymphoma.[48]
PRIMARY EFFUSION LYMPHOMA
Etiology/clinical
PEL commonly arises in young to middle aged males with underlying immunodeficiency, such as AIDS,[49] and almost always involves human herpesvirus 8 (HHV-8).[50] In around 50% cases, the lymphoma cells are also infected with EBV.[47] HHV-8-infected cells undergo clonal expansion, eventually leading to neoplastic transformation through mechanisms of increased proliferation and impaired apoptosis. The role of EBV in PEL is unclear.[50] In extremely rare cases, it can also exist in the absence of HIV and HHV-8.[51] PEL usually arises in body cavities, such as the pleural space or the pericardium, hence also “body cavity lymphoma.”
Radiography
Chest CT scans will show a small degree of pleural thickening and effusions, without a tumor mass or mediastinal adenopathy [Figure 5a and b].[12]
Figure 5.
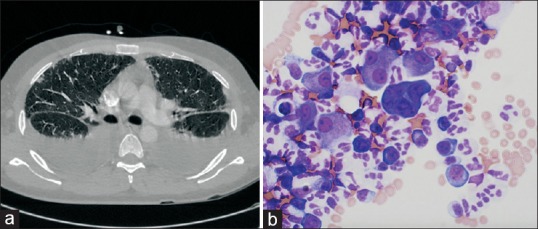
Computed tomography scan showing bilateral pleural effusions in a patient with human immunodeficiency virus/acquired immune-deficiency syndrome (a). Thoracentesis done subsequently revealed large B cells, (b) Diff-Quik stain on cytological evaluation revealing rare large lymphoid cells (×600) confirming primary effusion lymphoma
Pathology
Diagnosis is usually obtained by cytological evaluation of fluid which shows characteristic pleomorphic large cells with irregular nuclear contours, prominent nucleoli, and is positive for HHV-8. Biopsy is seldom required.[50]
Treatment/prognosis
It is generally resistant to chemotherapy drugs typically used for other NHL (CHOP) and carries a poor prognosis.[52]
PYOTHORAX-ASSOCIATED LYMPHOMA
Etiology
PAL is a newly described entity, which develops several decades after artificial pneumothorax treatment for pulmonary and pleural tuberculosis or chronic tuberculous pleural effusion. It is a high-grade NHL occurring in 2% of patients with long-standing tuberculous pleuritis. PAL is frequently EBV-associated, mainly reported in Japan and exceedingly rare in western countries.[53]
Clinical
PAL is much more common in older men and commonly described symptoms include chest pain and fever.[53]
Radiography
Chest CT scans usually reveal a mass arising within pleural cavity.[53]
Pathology
Histologically, PAL appears as a diffuse proliferation of large B cells similar to DLBCL. PAL cells also have aberrant expression of T-cell markers, such as CD2, and usually lack typical B-cell markers other than CD20.[53]
Diagnosis
As they usually present as a mass in the pleural cavity, CT-guided biopsy may obviate the need for surgical resection for evaluation with minimal complications.[54]
Treatment/prognosis
Patients appeared to respond initially to chemotherapy, but the clinical outcome is poor, with a 5-year survival of 22%.[53]
POSTTRANSPLANT LYMPHOPROLIFERATIVE DISORDER
Etiology
PTLD is a diagnosis describing the range of abnormal proliferation of B cells, ranging from benign polyclonal hyperplasia to malignant lymphomas. EBV causes B-cell proliferation resulting in PTLD in approximately 90% of cases. In the immunocompromised patient, the muted cytotoxic T-cell response allows proliferation of EBV-infected B cells to proceed unchecked. The highest risk of developing PTLD exists in EBV-naïve patients who acquire the primary infection during organ transplantation. An additional risk factor, which has been implicated in infections, is the degree of immunosuppression, especially in patients who have been exposed to antilymphocyte antibody preparations.[55]
Clinical
PTLD usually develops during the 1st year posttransplant. Fever and lymphadenopathy are the most common presenting features.[56] Incidence of PTLD following lung transplants is 2.5%–8%.[1]
Radiography
The allograft is usually involved, with a wide spectrum of radiological manifestations including solid nodules or masses, consolidation, ground-glass opacities, and interstitial disease.[57] In contrast, beyond the 1st year, intra-abdominal and disseminated forms of disease predominate.[58]
Diagnosis/pathology
Diagnosis typically requires excision biopsy, although occasionally core needle biopsies aspiration and cytology may be diagnostic. Interpretation of transbronchial lung biopsies can be difficult, as foci of PTLD may appear like the lymphocyte aggregation associated with acute cellular rejection.[59] In such cases, the diagnosis can be confirmed with in situ hybridization, or IHC staining to demonstrate the presence of EBV. An additional diagnostic tool is DNA amplification, which can demonstrate EBV viral load in the peripheral blood.[60,61]
Treatment/prognosis
The primary goal in treatment of PTLD is reducing the magnitude of immunosuppression, which allows for a partial return of the host innate cellular immunity to EBV. In up to two-thirds of patients, tumor regression occurs, but these patients need to be monitored for acute or chronic rejection, which can be precipitated by decreasing immunosuppression.[62] Immunotherapy with anti-CD20 monoclonal antibodies (rituximab) is now considered the first-line therapy for patients with rapidly progressive disease, those who cannot tolerate reduction of immunosuppression, or those who do not achieve complete remission. In solid organ transplants, rituximab is typically well-tolerated and has a complete response rate of 60%. Conversely, standard chemotherapy tends to have poorer outcomes and up to 25% of patients suffer from treatment-related morbidity.[63] While the use of antiviral agents prophylactically may reduce the subsequent risk of PTLD, there is no evidence supporting the routine use of antiviral therapy in the setting of known PTLD.[64]
CONCLUSIONS
PPL is a distinct and rare neoplasm that affects the lung. Most of these tumors arise from the BALT and of these, the most common form; MALT-PPL has an excellent prognosis. With the rise in autoimmune diseases and patients on immunosuppression for various reasons, lymphoproliferative disorders may be seen more frequently. Knowledge of this rare entity is essential to the practicing pulmonologist, as the nonspecific presentations of these entities may result in unnecessary tests and delayed diagnosis; which may affect survival. With advances in IHC and molecular biology as well as advanced bronchoscopy tools, diagnosis may be obtained with less invasive measures rather than going for more invasive procedures to clinch the diagnosis. Finally, due to the rarity of these tumors, there currently are no set guidelines on management and hence referral to specialized centers should be considered when a case of PPL is encountered in the community.[12]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.William J, Variakojis D, Yeldandi A, Raparia K. Lymphoproliferative neoplasms of the lung: A review. Arch Pathol Lab Med. 2013;137:382–91. doi: 10.5858/arpa.2012-0202-RA. [DOI] [PubMed] [Google Scholar]
- 2.Sirajuddin A, Raparia K, Lewis VA, Franks TJ, Dhand S, Galvin JR, et al. Primary pulmonary lymphoid lesions: Radiologic and pathologic findings. Radiographics. 2016;36:53–70. doi: 10.1148/rg.2016140339. [DOI] [PubMed] [Google Scholar]
- 3.Cadranel J, Wislez M, Antoine M. Primary pulmonary lymphoma. Eur Respir J. 2002;20:750–62. doi: 10.1183/09031936.02.00404102. [DOI] [PubMed] [Google Scholar]
- 4.Isaacson PG, Norton AJ. Extranodal Lymphomas. New York: Churchhill Livingstone; 1994. [Google Scholar]
- 5.Nahorecki A, Chabowski M, Straszak E, Teplicki A, Szuba A, Langfort R, et al. Primary pulmonary MALT lymphoma – Case report and literature overview. Eur Rev Med Pharmacol Sci. 2016;20:2065–9. [PubMed] [Google Scholar]
- 6.Borie R, Wislez M, Thabut G, Antoine M, Rabbat A, Couderc LJ, et al. Clinical characteristics and prognostic factors of pulmonary MALT lymphoma. Eur Respir J. 2009;34:1408–16. doi: 10.1183/09031936.00039309. [DOI] [PubMed] [Google Scholar]
- 7.Bashoura L, Eapen GA, Faiz SA. Pulmonary manifestations of lymphoma and leukemia. Clin Chest Med. 2017;38:187–200. doi: 10.1016/j.ccm.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Kligerman SJ, Franks TJ, Galvin JR. Primary extranodal lymphoma of the thorax. Radiol Clin North Am. 2016;54:673–87. doi: 10.1016/j.rcl.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Sammassimo S, Pruneri G, Andreola G, Montoro J, Steffanoni S, Nowakowski GS, et al. A retrospective international study on primary extranodal marginal zone lymphoma of the lung (BALT lymphoma) on behalf of International Extranodal Lymphoma Study Group (IELSG) Hematol Oncol. 2016;34:177–83. doi: 10.1002/hon.2243. [DOI] [PubMed] [Google Scholar]
- 10.Borie R, Wislez M, Antoine M, Fleury-Feith J, Thabut G, Crestani B, et al. Clonality and phenotyping analysis of alveolar lymphocytes is suggestive of pulmonary MALT lymphoma. Respir Med. 2011;105:1231–7. doi: 10.1016/j.rmed.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 11.Wei Z, Li J, Cheng Z, Yuan L, Liu P. A single center experience: Rituximab plus cladribine is an effective and safe first-line therapy for unresectable bronchial-associated lymphoid tissue lymphoma. J Thorac Dis. 2017;9:1081–92. doi: 10.21037/jtd.2017.03.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borie R, Wislez M, Antoine M, Cadranel J. Lymphoproliferative disorders of the lung. Respiration. 2017;94:157–75. doi: 10.1159/000477740. [DOI] [PubMed] [Google Scholar]
- 13.Cardenas-Garcia J, Talwar A, Shah R, Fein A. Update in primary pulmonary lymphomas. Curr Opin Pulm Med. 2015;21:333–7. doi: 10.1097/MCP.0000000000000180. [DOI] [PubMed] [Google Scholar]
- 14.Hare SS, Souza CA, Bain G, Seely JM, Frcpc Gomes MM, et al. The radiological spectrum of pulmonary lymphoproliferative disease. Br J Radiol. 2012;85:848–64. doi: 10.1259/bjr/16420165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albano D, Borghesi A, Bosio G, Bertoli M, Maroldi R, Giubbini R, et al. Pulmonary mucosa-associated lymphoid tissue lymphoma: 18F-FDG PET/CT and CT findings in 28 patients. Br J Radiol. 2017;90:20170311. doi: 10.1259/bjr.20170311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurtin PJ, Myers JL, Adlakha H, Strickler JG, Lohse C, Pankratz VS, et al. Pathologic and clinical features of primary pulmonary extranodal marginal zone B-cell lymphoma of MALT type. Am J Surg Pathol. 2001;25:997–1008. doi: 10.1097/00000478-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Sohani AR, Ferry JA. Lymphomas and lymphoproliferative diseases of the lung. Diagn Histopathol. 2014;20:405–14. [Google Scholar]
- 18.Fujii K, Ishibashi KI, Kato J, Kan J, Fujii K, Ito Y, et al. Cellular-level characterization of B cells infiltrating pulmonary MALT lymphoma tissues. Virchows Arch. 2016;469:575–80. doi: 10.1007/s00428-016-2012-z. [DOI] [PubMed] [Google Scholar]
- 19.Poletti V, Gurioli C, Piciucchi S, Rossi A, Ravaglia C, Dubini A, et al. Intravascular large B cell lymphoma presenting in the lung: The diagnostic value of transbronchial cryobiopsy. Sarcoidosis Vasc Diffuse Lung Dis. 2015;31:354–8. [PubMed] [Google Scholar]
- 20.Olszewski AJ, Castillo JJ. Survival of patients with marginal zone lymphoma: Analysis of the surveillance, epidemiology, and end results database. Cancer. 2013;119:629–38. doi: 10.1002/cncr.27773. [DOI] [PubMed] [Google Scholar]
- 21.Bacon CM, Du MQ, Dogan A. Mucosa-associated lymphoid tissue (MALT) lymphoma: A practical guide for pathologists. J Clin Pathol. 2007;60:361–72. doi: 10.1136/jcp.2005.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zucca E, Conconi A, Laszlo D, López-Guillermo A, Bouabdallah R, Coiffier B, et al. Addition of rituximab to chlorambucil produces superior event-free survival in the treatment of patients with extranodal marginal-zone B-cell lymphoma: 5-year analysis of the IELSG-19 Randomized Study. J Clin Oncol. 2013;31:565–72. doi: 10.1200/JCO.2011.40.6272. [DOI] [PubMed] [Google Scholar]
- 23.Rummel M, Kaiser U, Balser C, Stauch M, Brugger W, Welslau M, et al. Bendamustine plus rituximab versus fludarabine plus rituximab for patients with relapsed indolent and mantle-cell lymphomas: A multicentre, randomised, open-label, non-inferiority phase 3 trial. Lancet Oncol. 2016;17:57–66. doi: 10.1016/S1470-2045(15)00447-7. [DOI] [PubMed] [Google Scholar]
- 24.Maeshima AM, Taniguchi H, Toyoda K, Yamauchi N, Makita S, Fukuhara S, et al. Clinicopathological features of histological transformation from extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue to diffuse large B-cell lymphoma: An analysis of 467 patients. Br J Haematol. 2016;174:923–31. doi: 10.1111/bjh.14153. [DOI] [PubMed] [Google Scholar]
- 25.Piña-Oviedo S, Weissferdt A, Kalhor N, Moran CA. Primary pulmonary lymphomas. Adv Anat Pathol. 2015;22:355–75. doi: 10.1097/PAP.0000000000000090. [DOI] [PubMed] [Google Scholar]
- 26.Saitoh Y, Ohnishi-Amemiya A, Asano M, Tanaka Y, Yoshizawa S, Fujimoto H, et al. Unique radiological features of two cases of primary pulmonary diffuse large B-cell lymphoma. Thorax. 2017;72:859–60. doi: 10.1136/thoraxjnl-2017-210248. [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Xu K, Wang R, Liu X. Primary pulmonary diffuse large B-cell lymphoma on FDG PET/CT-MRI and DWI. Medicine (Baltimore) 2015;94:e1210. doi: 10.1097/MD.0000000000001210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshino N, Hirata T, Takeuchi C, Usuda J, Hosone M. A case of primary pulmonary diffuse large B-cell lymphoma diagnosed by transbronchial biopsy. Ann Thorac Cardiovasc Surg. 2015;21:396–8. doi: 10.5761/atcs.cr.14-00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JH, Lee SH, Park J, Kim HY, Lee SI, Park JO, et al. Primary pulmonary non-Hodgkin's lymphoma. Jpn J Clin Oncol. 2004;34:510–4. doi: 10.1093/jjco/hyh095. [DOI] [PubMed] [Google Scholar]
- 30.Olszewski AJ, Winer ES, Castillo JJ. Improved survival with rituximab-based chemoimmunotherapy in older patients with extranodal diffuse large B-cell lymphoma. Leuk Res. 2014;38:866–73. doi: 10.1016/j.leukres.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Neri N, Jesús Nambo M, Avilés A. Diffuse large B-cell lymphoma primary of lung. Hematology. 2011;16:110–2. doi: 10.1179/102453311X12940641877722. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Ding C, Lin Q, Yang K, Li Y, Chen S, et al. Primary intravascular large B-cell lymphoma of the lung: A review and case report. J Thorac Dis. 2014;6:E242–5. doi: 10.3978/j.issn.2072-1439.2014.08.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuentes Pradera J, Arriola Arellano E, Miguel Cisneros J, Quiroga E. Mediastinal adenopathies and pulmonary cavitated mass as onset form of Hodgkin's disease. Med Clin (Barc) 2001;117:398–9. doi: 10.1016/s0025-7753(01)72126-4. [DOI] [PubMed] [Google Scholar]
- 34.Bakan ND, Camsari G, Gur A, Ozkan G, Bayram M, Gorgulu F, et al. A21-year-old male with productive cough, hemoptysis, chest pain, and weight loss. Respiration. 2007;74:706–9. doi: 10.1159/000105539. [DOI] [PubMed] [Google Scholar]
- 35.Lluch-Garcia R, Briones-Gomez A, Castellano EM, Sanchez-Toril F, Lopez A, Brotons B, et al. Primary pulmonary Hodgkin's lymphoma. Can Respir J. 2010;17:e106–8. doi: 10.1155/2010/252746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cartier Y, Johkoh T, Honda O, Müller NL. Primary pulmonary Hodgkin's disease: CT findings in three patients. Clin Radiol. 1999;54:182–4. doi: 10.1016/s0009-9260(99)91012-7. [DOI] [PubMed] [Google Scholar]
- 37.Cooksley N, Judge DJ, Brown J. Primary pulmonary Hodgkin's lymphoma and a review of the literature since 2006. BMJ Case Rep. 2014;2014:pii: bcr2014204020. doi: 10.1136/bcr-2014-204020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kasuganti D, Cimbaluk D, Gattuso P. Reed-sternberg cells in bronchial brushings from a patient with Hodgkin's disease. Diagn Cytopathol. 2006;34:850–1. doi: 10.1002/dc.20464. [DOI] [PubMed] [Google Scholar]
- 39.Kumar R, Sidhu H, Mistry R, Shet T. Primary pulmonary Hodgkin's lymphoma: A rare pitfall in transthoracic fine needle aspiration cytology. Diagn Cytopathol. 2008;36:666–9. doi: 10.1002/dc.20872. [DOI] [PubMed] [Google Scholar]
- 40.Lachanas E, Tomos P, Fotinou M, Kalokerinou K. An unusual pulmonary cavitating lesion. Respiration. 2005;72:657–9. doi: 10.1159/000087183. [DOI] [PubMed] [Google Scholar]
- 41.Radin AI. Primary pulmonary Hodgkin's disease. Cancer. 1990;65:550–63. doi: 10.1002/1097-0142(19900201)65:3<550::aid-cncr2820650328>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 42.Yousem SA, Weiss LM, Colby TV. Primary pulmonary Hodgkin's disease. A clinicopathologic study of 15 cases. Cancer. 1986;57:1217–24. doi: 10.1002/1097-0142(19860315)57:6<1217::aid-cncr2820570626>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 43.Laohaburanakit P, Hardin KA. NK/T cell lymphoma of the lung: A case report and review of literature. Thorax. 2006;61:267–70. doi: 10.1136/thx.2004.025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oshima K, Tanino Y, Sato S, Inokoshi Y, Saito J, Ishida T, et al. Primary pulmonary extranodal natural killer/T-cell lymphoma: Nasal type with multiple nodules. Eur Respir J. 2012;40:795–8. doi: 10.1183/09031936.00123911. [DOI] [PubMed] [Google Scholar]
- 45.Chien CC, Lee HS, Lin MH, Hsieh PP. Primary extranodal natural killer/T-cell lymphoma of bronchus and lung: A case report and review of literature. Thorac Cancer. 2016;7:140–4. doi: 10.1111/1759-7714.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vega F, Padula A, Valbuena JR, Stancu M, Jones D, Medeiros LJ, et al. Lymphomas involving the pleura: A clinicopathologic study of 34 cases diagnosed by pleural biopsy. Arch Pathol Lab Med. 2006;130:1497–502. doi: 10.5858/2006-130-1497-LITPAC. [DOI] [PubMed] [Google Scholar]
- 47.Alexandrakis MG, Passam FH, Kyriakou DS, Bouros D. Pleural effusions in hematologic malignancies. Chest. 2004;125:1546–55. doi: 10.1378/chest.125.4.1546. [DOI] [PubMed] [Google Scholar]
- 48.Giardino A, O'Regan KN, Hargreaves J, Jagannathan J, Park D, Ramaiya N, et al. Primary pleural lymphoma without associated pyothorax. J Clin Oncol. 2011;29:e413–5. doi: 10.1200/JCO.2010.33.5323. [DOI] [PubMed] [Google Scholar]
- 49.Boshoff C, Weiss R. AIDS-related malignancies. Nat Rev Cancer. 2002;2:373–82. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 50.Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12:569–76. doi: 10.1634/theoncologist.12-5-569. [DOI] [PubMed] [Google Scholar]
- 51.Youngster I, Vaisben E, Cohen H, Nassar F. An unusual cause of pleural effusion. Age Ageing. 2006;35:94–6. doi: 10.1093/ageing/afj009. [DOI] [PubMed] [Google Scholar]
- 52.Castillo JJ, Shum H, Lahijani M, Winer ES, Butera JN. Prognosis in primary effusion lymphoma is associated with the number of body cavities involved. Leuk Lymphoma. 2012;53:2378–82. doi: 10.3109/10428194.2012.694075. [DOI] [PubMed] [Google Scholar]
- 53.Nakatsuka S, Yao M, Hoshida Y, Yamamoto S, Iuchi K, Aozasa K, et al. Pyothorax-associated lymphoma: A review of 106 cases. J Clin Oncol. 2002;20:4255–60. doi: 10.1200/JCO.2002.09.021. [DOI] [PubMed] [Google Scholar]
- 54.Brun V, Revel MP, Danel C, Fournier LS, Souilamas R, Frija G, et al. Case report.Pyothorax-associated lymphoma: Diagnosis at percutaneous core biopsy with CT guidance. AJR Am J Roentgenol. 2003;180:969–71. doi: 10.2214/ajr.180.4.1800969. [DOI] [PubMed] [Google Scholar]
- 55.Taylor AL, Marcus R, Bradley JA. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit Rev Oncol Hematol. 2005;56:155–67. doi: 10.1016/j.critrevonc.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 56.LaCasce AS. Post-transplant lymphoproliferative disorders. Oncologist. 2006;11:674–80. doi: 10.1634/theoncologist.11-6-674. [DOI] [PubMed] [Google Scholar]
- 57.Bligh MP, Borgaonkar JN, Burrell SC, MacDonald DA, Manos D. Spectrum of CT findings in thoracic extranodal non-Hodgkin lymphoma. Radiographics. 2017;37:439–61. doi: 10.1148/rg.2017160077. [DOI] [PubMed] [Google Scholar]
- 58.Paranjothi S, Yusen RD, Kraus MD, Lynch JP, Patterson GA, Trulock EP, et al. Lymphoproliferative disease after lung transplantation: Comparison of presentation and outcome of early and late cases. J Heart Lung Transplant. 2001;20:1054–63. doi: 10.1016/s1053-2498(01)00314-x. [DOI] [PubMed] [Google Scholar]
- 59.Blaes AH, Morrison VA. Post-transplant lymphoproliferative disorders following solid-organ transplantation. Expert Rev Hematol. 2010;3:35–44. doi: 10.1586/ehm.09.76. [DOI] [PubMed] [Google Scholar]
- 60.Stevens SJ, Verschuuren EA, Pronk I, van Der Bij W, Harmsen MC, The TH, et al. Frequent monitoring of Epstein-Barr virus DNA load in unfractionated whole blood is essential for early detection of posttransplant lymphoproliferative disease in high-risk patients. Blood. 2001;97:1165–71. doi: 10.1182/blood.v97.5.1165. [DOI] [PubMed] [Google Scholar]
- 61.Tsai DE, Nearey M, Hardy CL, Tomaszewski JE, Kotloff RM, Grossman RA, et al. Use of EBV PCR for the diagnosis and monitoring of post-transplant lymphoproliferative disorder in adult solid organ transplant patients. Am J Transplant. 2002;2:946–54. doi: 10.1034/j.1600-6143.2002.21011.x. [DOI] [PubMed] [Google Scholar]
- 62.Tsai DE, Hardy CL, Tomaszewski JE, Kotloff RM, Oltoff KM, Somer BG, et al. Reduction in immunosuppression as initial therapy for posttransplant lymphoproliferative disorder: Analysis of prognostic variables and long-term follow-up of 42 adult patients. Transplantation. 2001;71:1076–88. doi: 10.1097/00007890-200104270-00012. [DOI] [PubMed] [Google Scholar]
- 63.Elstrom RL, Andreadis C, Aqui NA, Ahya VN, Bloom RD, Brozena SC, et al. Treatment of PTLD with rituximab or chemotherapy. Am J Transplant. 2006;6:569–76. doi: 10.1111/j.1600-6143.2005.01211.x. [DOI] [PubMed] [Google Scholar]
- 64.Levine SM, Angel L, Anzueto A, Susanto I, Peters JI, Sako EY, et al. Alow incidence of posttransplant lymphoproliferative disorder in 109 lung transplant recipients. Chest. 1999;116:1273–7. doi: 10.1378/chest.116.5.1273. [DOI] [PubMed] [Google Scholar]